

Abstract
The mechanism underlying spike timing-dependent neuromodulation (STDN) was investigated in the opisthobranch mollusc Tritonia diomedea. The serotonergic dorsal swim interneurons (DSIs) dynamically modulated the synaptic output of ventral swim interneuron B (VSI); immediately after DSI stimulation, there was a potentiation of VSI synaptic strength followed by a longer-lasting synaptic depression. The potentiation phase of STDN was unaffected by spike broadening produced by the potassium channel blocker 4-aminopyridine (4-AP). In contrast, the depression phase was eliminated by 4-AP. Bath-applied serotonin (5-HT) decreased VSI spike duration and increased the magnitude of the A-current (IA), a voltage-dependent, transient, outward current. 4-AP preferentially blocked IA and prevented the spike narrowing caused by 5-HT, uncovering the full extent of 5-HT-induced synaptic potentiation. A consistent correlation was observed between IA and spike duration, but the correlation between synaptic strength and spike duration differed between preparations. Conductance-based simulations showed that the magnitude of A-current conductance could affect spike duration and gave an estimation of the change needed to produce spike narrowing. An artificial IA introduced into the VSI in the presence of 4-AP by means of the dynamic-clamp technique restored spike duration and gave a further approximation of the magnitude of modulation needed for spike narrowing. Together, these results suggest a mechanism for STDN: the DSIs release 5-HT, which causes a spike duration-independent enhancement of synaptic strength and a longer-lasting enhancement of IA that narrows the VSI spike and hence decreases VSI synaptic strength. Thus, STDN arises from the dynamics of independent intracellular signaling events.
Keywords: spike-narrowing, serotonin, opisthobranch mollusc, A-current, Tritonia diomedea, dynamic clamp
Introduction
Although tonic neuromodulatory actions have been studied extensively in relation to learning and memory (Byrne and Kandel, 1996), sensory processing (Pasztor and Bush, 1989; Nagai et al., 1996; Mercer, 1999), and motor pattern generation (Calabrese, 1998), less attention has been paid to the short-term temporal dynamics of neuromodulation (Marder and Calabrese, 1996; Kiehn and Katz, 1999; Marder and Bucher, 2001). Recently, we described an example of spike timing-dependent neuromodulation (STDN) in which activation of a serotonergic neuron evokes either heterosynaptic potentiation or depression of synaptic strength (Sakurai and Katz, 2003b). This STDN leads to a temporally biphasic and bidirectional change in synaptic strength, with a transient potentiation phase and a longer-lasting depression phase. In this study, we investigated the mechanisms underlying STDN and found evidence that the potentiation and depression phases are produced by independent downstream effects of serotonin (5-HT).
The system that we studied is the central pattern generator (CPG) for the escape swim behavior of the opisthobranch mollusc Tritonia diomedea. Within the CPG, the serotonergic dorsal swim interneurons (DSIs) serve an intrinsic neuromodulatory function by changing the excitability and synaptic strength of the other CPG neurons: cerebral cell 2 and the ventral swim interneuron B (VSI-B) (Katz, 1998; Sakurai and Katz, 2003b). There are two VSIs, VSA-A and VSI-B. DSI activity results in STDN of VSI-B synaptic strength (Sakurai and Katz, 2003b). VSI-A does not play an important role in the swim CPG (Getting, 1983b; Getting and Dekin, 1985) and was not examined in this study. VSI-B forms excitatory synaptic contacts with ventral flexion neurons (VFNs) and other follower neurons in the pedal ganglion (Sakurai and Katz, 2003b). For simplicity, we will refer to VSI-B as VSI and all VSI follower cells as VFNs.
5-HT released from the DSIs is responsible for both the potentiation and the depression phases of STDN; however, bath application of 5-HT produces variable effects (Sakurai and Katz, 2003b). One possible explanation for this variability is that bath-applied 5-HT has slower and longer-lasting kinetics than synaptically released 5-HT that may obscure differences in the onset and/or decay rates of the potentiation and depression. Bath application therefore may simultaneously produce two opposite actions that cancel each other. Here we provide evidence that supports this hypothesis by showing that the two effects of 5-HT can be separated pharmacologically.
VSI exhibits a prominent transient outward current, described as an A-current (IA) (Connor and Stevens, 1971), which was previously suggested to play an important role in the generation of the motor pattern by delaying the excitation of VSI (Getting, 1983b). In this study, we found that this current may also play a role in spike timing-dependent neuromodulation. By using 4-aminopyridine (4-AP), together with voltage-clamp and dynamic-clamp techniques, we obtained results that indicate that the DSI-evoked synaptic depression could be caused by a serotonergic enhancement of A-current that leads to spike-narrowing in VSI.
Parts of this paper have been published previously in abstract form (Sakurai and Katz, 2003a; Darghouth et al., 2004).
Materials and Methods
Preparation.
Specimens of the nudibranch T. diomedea were obtained from Living Elements (North Vancouver, British Columbia, Canada). All experiments were performed on the isolated brain preparation. Briefly, the brain, consisting of the fused cerebropleural and pedal ganglia, was removed from the animal and immediately pinned to the bottom of a Sylgard-lined chamber (1 ml) superfused with saline at 4°C. The physiological saline contained the following (in mm): 420 NaCl, 10 KCl, 10 CaCl2, 50 MgCl2, 10 d-glucose, and 10 HEPES, pH 7.4. The cell bodies of the neurons were exposed by removing the connective tissue sheath from the surface of the ganglia (Willows et al., 1973). Suction electrodes, made from polyethylene tubing, were placed on pedal nerves, including pedal nerves 3, 5, and 6. The preparation was then superfused overnight with saline at 8–10°C.
Neurons were identified by soma location, coloration, synaptic connectivity, and activity pattern at rest and during the swim motor program as described previously (Getting et al., 1980; Getting, 1981, 1983b). To identify neurons, the swim motor program was evoked by stimulating pedal nerve 3 with a train of voltage pulses (5–15 V; 1.5 ms) at 5 Hz for 1.5 s via a suction electrode. After the interneurons were identified, the bathing medium was switched to saline containing a 2.5× concentration of divalent cations, which raises the threshold for spiking and reduces spontaneous neural firing. The high-divalent cation saline also increases the size of EPSCs. The composition of the high-divalent cation saline was as follows (in mm): 285 NaCl, 10 KCl, 25 CaCl2, 125 MgCl2, 10 d-glucose, and 10 HEPES, pH 7.4 (Sakurai and Katz, 2003b). All experiments were conducted in the high-divalent cation saline superfused at 2 ml/min at 10°C.
5-HT (Sigma, St. Louis, MO) was dissolved in high-divalent cation saline at its final concentration (0.1–100 μm) just before use. 4-AP (Sigma) was dissolved in high-divalent cation saline at 100 or 200 mm, with its pH adjusted to 7.4 by adding HCl, and then aliquoted and stored at −20°C. 4-AP solution was diluted in high-divalent cation saline to final concentrations of 0.1, 1, or 2 mm just before use. Drugs were bath applied by switching the inflow lines.
Electrophysiological recordings and stimulation.
Neurons were impaled with glass microelectrodes filled with 3 m KCl (resistance, 8–15 MΩ). Axoclamp 2B amplifiers (Molecular Devices, Union City, CA) were used for all electrophysiological experiments. To examine changes in the strength of the VSI-to-VFN synapse, orthodromic action potentials were evoked by injecting current pulses (4–15 nA; 4–20 ms) into the soma through a second electrode or through the recording electrode with a balanced bridge circuit. To examine changes in VSI action potential waveforms, an antidromic action potential was elicited by applying a brief voltage pulse to a suction electrode on pedal nerve 6, stimulating the peripheral axon of VSI (see Fig. 1Ai). Spike half-width was measured as the time between the spike peak and the 50% amplitude from the resting potential in the repolarization phase. A VFN was voltage clamped at −50 mV under two-electrode voltage-clamp mode to measure VSI-evoked EPSCs. In some experiments, VSI-evoked EPSPs were recorded in a postsynaptic neuron that was held hyperpolarized at −50 to −70 mV by a constant current injected through a second electrode to prevent action potential firing.
Figure 1.
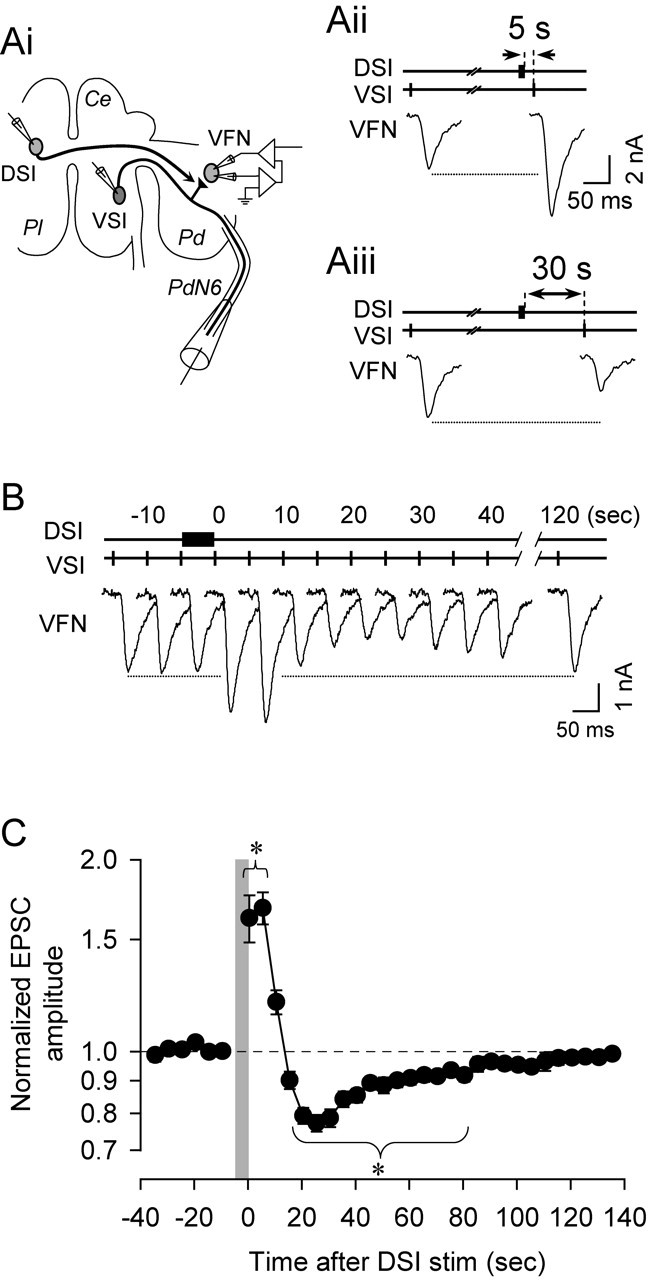
Spike timing-dependent neuromodulation of synaptic strength in the Tritonia swim network. A, Schematic diagram showing the experimental configuration (Ai) and an example of bidirectional neuromodulation of the VSI-to-VFN synapse by a DSI spike train using the protocol from Sakurai and Katz (2003b). Ai, Intracellular microelectrodes were placed in the somata of DSI, VSI, and VFN, which are located in the cerebral (Ce), pleural (Pl), and pedal (Pd) ganglia, respectively. In most experiments, a suction electrode was placed on pedal nerve 6 (PdN6) to monitor VSI impulses and for retrograde stimulation. The membrane potential of VFN was voltage clamped at −50 mV to record EPSCs evoked by VSI spikes. Aii, A DSI spike train (10 Hz; 5 s) increased the amplitude of the VSI-evoked EPSC recorded in VFN when a VSI spike was evoked 5 s after DSI. Aiii, In contrast, when the DSI spike train preceded VSI by 30 s, the VSI-evoked EPSC decreased in size. B, Revised protocol for testing the DSI neuromodulatory effects on the VSI-to-VFN synapse. VSI was stimulated at 5 s intervals by repeated brief current pulses while EPSCs were recorded from VFN under voltage clamp. A DSI spike train (10 Hz; 4.8 s) was imposed between the sixth and seventh VSI spikes. After the DSI spike train, VSI-evoked EPSCs showed a biphasic, bidirectional change in size, returning to control amplitude by 120 s. C, A plot of the normalized amplitudes of VSI-evoked EPSCs, showing a biphasic, bidirectional change after the DSI spike train indicated by the gray vertical bar. Each point represents a mean ± SEM of data obtained from 13 preparations. The EPSC amplitude was normalized to the average of the six EPSCs elicited before each DSI spike train. Asterisks indicate significant changes in EPSC amplitude from the control value at −10 s (p < 0.05; Kruskal–Wallis one-way ANOVA on ranks with a post hoc Dunn's multiple comparison procedure). stim, Stimulation.
Action potentials in DSI were elicited either intracellularly, by injecting 20 ms current pulses (7–15 nA) through a recording electrode via a balanced bridge circuit, or extracellularly, by applying voltage pulses (5–10 V; 1 ms) via a suction electrode to pedal nerve 5, which contains the axons of at least two of the three DSIs (Lynn-Bullock et al., 2002). Each intracellular current pulse evoked a single spike, allowing the spike frequency to be controlled precisely. Stimulation of pedal nerve 5 evokes the same effect as intracellular stimulation of the DSI soma (Sakurai and Katz, 2003b).
The membrane current in VSI was measured with a two-electrode voltage clamp. To reduce noise, the shaft of the voltage-recording microelectrode was coated with silver paint. The silver paint was then connected to the driven shield on the amplifier headstage. After impalement, a grounded aluminum plate was placed between the electrodes to reduce the coupling capacitance. The protocol for activating voltage-gated outward currents consisted of a series of voltage steps from the resting potential (−50 mV) or from a hyperpolarized holding potential (−90 mV) at which the conductance is deinactivated (see Fig. 4E). All voltage-clamp experiments were performed in high-divalent cation saline, which caused a shift in the activation and inactivation curves of approximately +25 mV relative to normal physiological saline because of membrane charge screening (Frankenhaeuser and Hodgkin, 1957). All current traces were processed off-line by a standard leak-subtraction protocol. A reversal potential of −70 mV was used to calculate the conductances of the potassium current.
Figure 4.
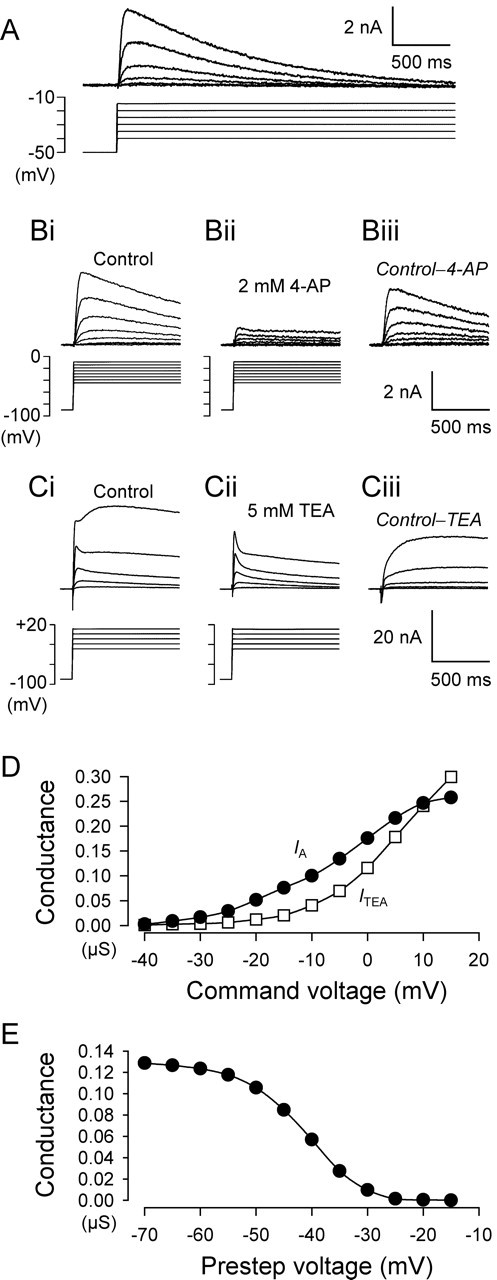
Characterization of voltage-dependent outward currents in VSI. A, VSI exhibits a large voltage-gated transient outward current with a threshold activation at approximately −35 mV that completely inactivated within 4 s. Overlaid traces show voltage-dependent transient outward currents evoked in VSI by voltage steps at 5 mV increments between −40 and −15 mV from a holding potential of −50 mV. B, 4-AP reduced the voltage-dependent transient outward currents that were evoked by voltage steps at 5 mV increments between −45 and −10 mV from a holding potential of −90 mV. Overlaid current and voltage traces were obtained in control high-divalent cation saline (Bi) and in the presence of 2 mm 4-AP (Bii). Subtraction of the traces in 4-AP from those in control saline revealed the 4-AP-sensitive component (Biii). C, Larger depolarizing steps to potentials more positive than −10 mV produced an additional delayed outward current (Ci) that was blocked by TEA, leaving a transient outward current (Cii). Subtraction of the traces in TEA from control yielded the TEA-sensitive component (Ciii). D, Plot of the maximum amplitudes of the 4-AP-sensitive conductance (IA; filled circles) and the TEA-sensitive conductance (ITEA; open squares). The activation threshold (−35 mV) for IA was more negative than that of the TEA-sensitive current (−10 mV). E, Steady-state inactivation of the 4-AP-sensitive conductance measured with a two-pulse paradigm. The conductance amplitude for a depolarization to −10 mV was plotted against the amplitude of the voltage prepulse (from −70 to −15 mV; 4 s duration).
Recordings were digitized at 3–20 kHz with a Cambridge Electronic Design (Cambridge, UK) 1401plus analog-to-digital converter. Data acquisition and analysis were performed with Spike2 software (Cambridge Electronic Design) and SigmaPlot (Jandel Scientific, San Rafael, CA). Statistical comparisons were made with a Student's t test, a paired t test, Kruskal–Wallis one-way ANOVA on ranks with a post hoc Dunn's multiple comparison procedure, or one-way or two-way ANOVA with a post hoc all pair-wise multiple comparison procedure [Fisher's least significant difference (LSD) method]. In all cases, p < 0.05 was considered significant. Results are expressed as the mean ± SEM.
Experimental protocol.
In the previous study (Sakurai and Katz, 2003b), we evoked only a single VSI spike after each DSI spike train (10 Hz; 5 s) to examine the modulatory effect of DSI on the VSI-to-VFN synapse. With this protocol (shown in Fig. 1A), trials for each DSI spike train must be repeated several times to reveal the entire biphasic–bidirectional time course of the DSI neuromodulatory action. We revised the protocol in this study so that individual VSI action potentials were evoked repeatedly at 5 s intervals for 120–180 s (see Fig. 1B). A DSI spike train (10 Hz for 4.8 s) was evoked between the seventh and eighth VSI spikes. In this way, one can monitor the biphasic–bidirectional change in the strength of the VSI-to-VFN synapse in a single trial and calculate the average responses of many trials (see Fig. 1C). The mean amplitude of the first six EPSCs or EPSPs evoked by VSI was used as a baseline for each trial; the amplitudes of all EPSCs or EPSPs were normalized to this baseline value for analysis. The effect of DSI stimulation on VSI-to-VFN synaptic strength was assayed by comparing the normalized amplitudes of the EPSCs or EPSPs evoked before and after the DSI spike train. The interval between DSI stimuli was always >2 min to prevent an accumulation of neuromodulatory actions.
Modeling VSI.
We modeled VSI with a minimal number of conductances: IA; the delayed rectifier current; the fast sodium current; and the leak current. This model was implemented in the NEURON simulation environment (Hines, 1998). The model for IA was based on voltage-clamp experiments: leak-subtracted traces from voltage-clamp experiments conducted in bath-applied 4-AP were subtracted from control traces, resulting in 4-AP-sensitive current traces (see Fig. 4A). In this study, we did not develop an empirically based model of currents other than IA because of the inefficacy of pharmacological blockers on the delayed rectifier and fast sodium currents. Instead, we relied on models and data from other molluscs (Hodgkin and Huxley, 1952; Thompson, 1977; Getting, 1983b, 1989) that were adjusted (primarily through changes in maximal conductances and time constants) to produce an optimum match to the firing responses to injected current (see Fig. 8B).
Figure 8.
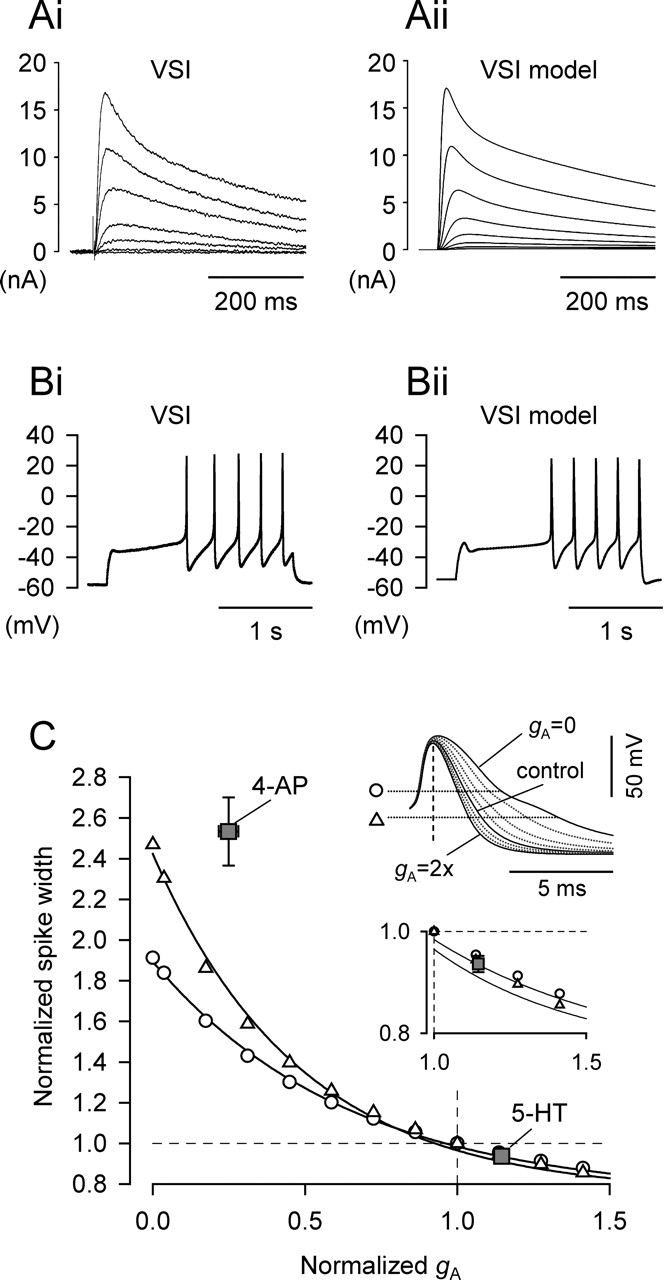
Computer simulation of VSI. A, 4-AP-sensitive current recorded from voltage-clamped VSI (Ai) and modeled IA traces produced by a minimal Hodgkin–Huxley style simulation of a VSI (Aii). The membrane potential was held at −90 mV and then stepped to various holding potentials (−50 to −15 by 5 mV). B, The simulation reproduced VSI firing in response to a 2 nA current injection. C, Effect of changes in gA on spike duration in the VSI model. The half-widths of the action potentials were measured at two spike heights (25%, triangles; 50%, circles; shown in the top inset) and plotted as a function of gA. Spike half-width and gA recorded from real VSIs in the presence of 2 mm 4-AP or 10 μm 5-HT were also plotted with gray squares with error bars (mean ± SEM) (compare Figs. 3D, 5C). The values were normalized to the control. Inset (bottom), The same graph having enlarged y-axis with limited range of x-axis.
Although the diameter of the soma of VSI ranges from 40 to 60 μm (Getting, 1983a), the cylindrical diameter of the single-compartment neuron model was set to 300 μm to account for (1) membrane invagination (Gorman and Mirolli, 1972; Mirolli and Talbott, 1972; Baxter et al., 1999) and (2) the initial segment of the axon that also was clamped. The value for resting conductance in VSI was 81.6 ± 4.8 nS (n = 21), and the capacitance was set to the standard 1 μF/cm2.
From the extracted 4-AP-sensitive traces, we qualitatively observed two components of inactivation: a fast inactivation and a slower inactivation. Two components of inactivation have been described previously and modeled in the A-currents of other species (Buchholtz et al., 1992; Huguenard and McCormick, 1992). The best fit for the A-current incorporated a single activation and two independent inactivation variables, described as follows:
where ḡA is the maximal conductance; K1 and K2 are the scaling factors for the inactivation variables (such that K1 + K2 = 1); m is the activation variable; h1 and h2 are the fast and slow inactivation state variables, respectively; V is the membrane potential; and EK is the reversal potential of potassium. We optimized fixed values for ḡA, K1, and K2 to fit the voltage-clamp traces to Equation 1 with the curve-fitting toolbox from the mathematical computing software MATLAB (MathWorks, Natick, MA). After the traces were fitted, parameters for different values of Vhold could be extracted. These parameters were then fit to curves to be used in the model of the 4-AP-sensitive current. The parameters m∞, τm, and τh2 were fit to a sigmoidal form similar to Equation 1, and τh1 was fit to a bell-shaped curve. We used a standard inactivation protocol to extract the inactivation parameters. The parameters describing the dynamics of the modeled A-current are presented in full in Table 1. The leak reversal potential was set near resting potential. The leak conductance was set to produce an input conductance of 71.4 nS.
Table 1.
Parameters for the A-current model
| τmin (ms) | τmax (ms) | a1 (ms−1) | Vhalf (mV) | Shalf(mV) | a2(ms−) | Vhalf2(mV) | Shalf2(mV) | |
|---|---|---|---|---|---|---|---|---|
| m ∞ | −33 | −14.1 | ||||||
| h ∞ | −41.4 | 5.8 | ||||||
| τm | 4 | 40 | −19.5 | 5.3 | ||||
| τ h1 | 30 | 110 | −14 | 9.0 | ||||
| τ h2 | 1.447 × 10−3 | 8.3 | −2.8 | 1.580 × 10−3 | −11.7 | 12.1 |
The h∞ parameters are for both h1 and h2.
When the activation and inactivation curves were compared with a previous model for the A-current in Tritonia (Getting, 1983b), we noted a shift of ∼25 mV. This shift was caused by our experimental conditions: measurements were made in high-divalent cation saline, which leads to charge shielding and a shift in activation and inactivation curves (Frankenhaeuser and Hodgkin, 1957; Gilly and Armstrong, 1982a,b; Hahin and Campbell, 1983). The model reflects the high-divalent cation environment.
Dynamic-clamp experiments.
A real-time Linux-based software system, called MRCI (model reference current injection) (Raikov et al., 2004), running on a Pentium III computer was used to simulate IA under dynamic clamp (Sharp et al., 1993). The sampling rate for simulating a single conductance for dynamic clamp was 20 kHz. Parameters for the Hodgkin–Huxley-type equations describing the time dependence and voltage dependence of the IA conductance were entered into the dynamic-clamp program. The membrane potential recorded from the neuron was fed directly on-line to the dynamic-clamp program via a 6052E multifunction DAQ board and a BNC2090 interface (National Instruments, Austin, TX).
The activation, inactivation, and time constant curves (m∞, h∞) are of the following form:
 |
 |
 |
 |
The equation for τh1 has the same form as the equation for τm.
Results
As shown previously (Sakurai and Katz, 2003b), a DSI spike train caused either an increase or a decrease in the size of the VSI-evoked synaptic current depending on the latency between the DSI spike train and the VSI action potential (Fig. 1Aii,Aiii). The depression was independent of the potentiation and could be evoked without previous potentiation (Fig. 1Aiii). When VSI was stimulated repeatedly at 5 s intervals, a single DSI spike train (10 Hz for 4.8 s) caused a biphasic and bidirectional change in the amplitude of the VSI-evoked EPSCs (Fig. 1B,C). Thus, both the potentiation phase and the depression phase could be observed after a single DSI spike train.
The depression phase could be pharmacologically separated from the potentiation
The depression phase of STDN, but not the potentiation phase, was eliminated by the potassium channel blocker 4-AP, transforming the biphasic modulatory action into a monophasic short-term potentiation that lasted ∼20 s (Fig. 2A). In the presence of 4-AP, there was no significant change in the amplitude of the VSI-to-VFN EPSC 25–35 s after DSI stimulation (−0.2 ± 4.5%; p = 0.49), whereas under control conditions, DSI stimulation caused a significant depression of EPSC amplitude during this time interval (−19.5 ± 3.9; p < 0.001) (Fig. 2B). There was a statistically significant difference in the extent of depression observed under control conditions and in the presence of 4-AP (p < 0.001 by two-way ANOVA with Fisher's LSD method). In contrast, the potentiation phase was unaffected by 4-AP; within 10 s after a DSI spike train, there was a significant enhancement of VSI-to-VFN EPSC amplitude both under control conditions (87.0 ± 31.6%; p < 0.05) and in the presence of 4-AP (91.1 ± 34.7%; p < 0.05) (Fig. 2B). There was no statistically significant difference in the extent of this potentiation (p = 0.90). These results suggest that the depression phase, but not the potentiation phase, of STDN is strongly influenced by an ion conductance that is blocked by 4-AP.
Figure 2.

The depression phase of STDN was selectively blocked by 4-AP. A, Averaged data showing the effects of DSI stimulation on normalized VSI-evoked EPSCs in control saline (filled circles) and in 1–2 mm 4-AP (opened triangles). Each plot shows a mean value (mean ± SEM) of pooled data obtained from six preparations. The gray vertical bar represents a 5 s, 10 Hz DSI spike train. B, Bar graph showing mean ± SEM of the normalized EPSC amplitudes before DSI stimulation and 0–10 and 25–35 s after DSI stimulation in control and 4-AP (1–2 mm) saline across preparations (n = 6). Statistical comparisons were made with two-way ANOVA with Fisher's LSD method. Asterisks indicate significant changes (p < 0.05). C, Overlaid traces of simultaneously recorded VSI action potentials and the resulting EPSCs in VFN. 4-AP (1 mm) (dashed lines) greatly broadened the VSI action potential and increased the size of the EPSC compared with control conditions (solid lines). Here, VSI action potentials were evoked by injecting current pulses (7 nA; 50 ms) into the soma through a second electrode. D, The potentiation phase of STDN was independent of spike broadening. Recordings were obtained before the DSI spike train (solid trace), 5 s after the DSI spike train (short dash), and 35 s after the DSI spike train (long dash) in control saline (Di) and in saline containing 1 mm 4-AP (Dii). The VFN traces in Di and Dii are normalized so that the EPSC amplitudes before DSI stimulation are comparable. In Dii, the three control traces are also shown at the same gain (gray traces). stim, Stimulation.
The potentiation phase is independent of spike duration
Although the extent of potentiation evoked by DSI stimulation was not affected by 4-AP, the potassium channel blocker did cause significant spike broadening and a corresponding increase in VSI-to-VFN EPSC amplitude (Fig. 2C,D). In the presence of 4-AP (1.0 mm), the duration of the VSI action potential increased, and there was a correspondingly large increase in the amplitude of the basal VSI-to-VFN EPSC (Fig. 2C) (see also Fig. 7D). Yet despite the up to 10-fold increase in VSI-to-VFN synaptic strength in the presence of 4-AP, DSI stimulation 5 s before VSI continued to potentiate the strength of that synapse (Fig. 2Dii). In contrast, the depression that would have occurred 35 s after DSI stimulation was not seen in the presence of 4-AP (Fig. 2Dii). Thus, the potentiation phase of STDN appeared to be independent of spike duration, whereas the depression phase was blocked when the spike was broadened by 4-AP.
Figure 7.
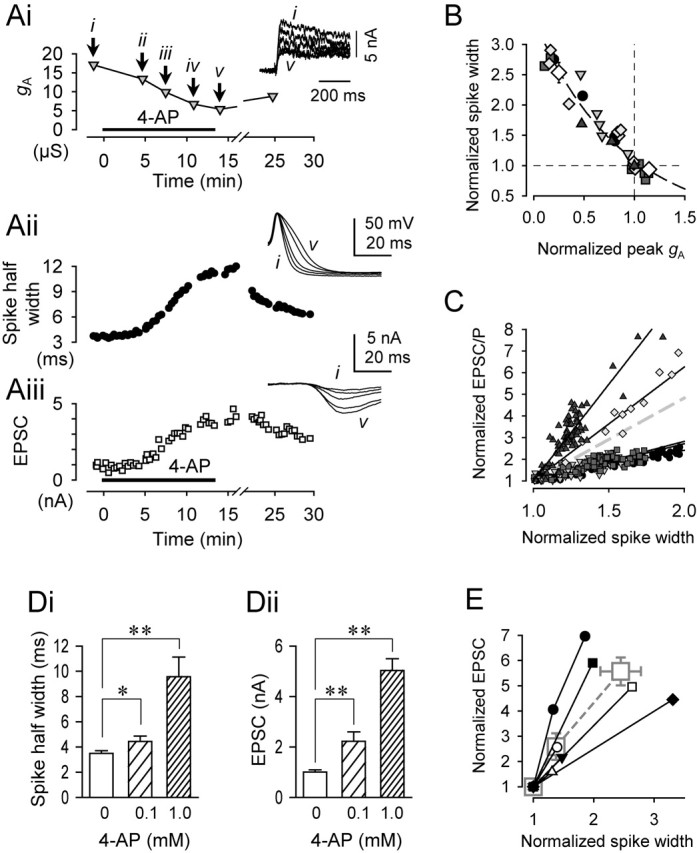
The relationship among IA, VSI spike duration, and VSI-to-VFN EPSC amplitude. A, 4-AP wash-in gradually reduced gA (Ai), increased the VSI spike half-width (Aii), and increased the amplitude of VSI-evoked EPSC (Aiii). Periodically, VSI was voltage clamped, and Vm was stepped up from −90 to −10 mV to measure IA as indicated by the arrows (i–v, top inset). While not voltage clamped, a VSI action potential (middle inset) was triggered every 20 s by the injection of a brief current pulse (10 nA; 10 ms) through a second electrode. The action potential evoked an EPSC in VFN (bottom inset). Spikes and EPSCs evoked before and after each voltage-clamp recording were averaged and overlaid in the middle and bottom insets. 4-AP (1 mm) was washed in as indicated by the horizontal bar. B, Plot of spike width as a function of normalized gA showing an inverse relationship. All data were normalized to the control values obtained before 4-AP wash-in. Data from four different preparations are fit by a single exponential curve that was constrained to pass through the point (1,1). C, Plot of the amplitudes of VSI-evoked EPSCs or EPSPs versus spike width recorded simultaneously during 4-AP wash-in. Data were normalized to an averaged control value before 4-AP wash-in. Plots are the results from five preparations, and each symbol represents data from a single preparation. Linear regression lines indicate that within each preparation, synaptic strength was linearly correlated to the spike width. The gray dashed line shows an averaged slope. D, Bar graphs of changes in the spike half-width (Di) and EPSC amplitude (Dii) in control saline (0 mm) and in the presence of 4-AP (0.1 and 1.0 mm). Asterisks indicate significant changes (*p < 0.05; **p < 0.001; one-way ANOVA with Fisher's LSD method). E, Plots of normalized changes in the spike half-width and EPSC amplitude. The values for each individual preparation are plotted separately without regard for 4-AP concentration (small symbols). Averages of the normalized EPSC amplitude and the normalized spike width measured in the control saline and in the presence of 4-AP (0.1 and 1.0 mm) are also plotted (large gray squares with dashed line).
5-HT caused spike narrowing in VSI
Although VSI spike broadening was correlated with an increase in the size of the VSI-to-VFN EPSC, we were unable to observe any effect of DSI stimulation on the duration of VSI action potentials recorded in the soma in any of the preparations examined (Fig. 2Di). The spike half-width was 3.53 ± 0.09 ms before DSI, 3.66 ± 0.09 ms 5 s after DSI, and 3.57 ± 0.12 ms at 30 s after DSI (p = 0.8; one-way ANOVA; n = 6). Similarly, in the presence of 4-AP, there was also no change in VSI spike width after DSI stimulation as observed in soma recordings (Fig. 2Dii).
We showed previously that 5-HT released from DSI mediates both the potentiation and depression phases of STDN (Sakurai and Katz, 2003b). Although DSI stimulation did not affect the shape of spikes recorded in the soma, we tested the effect of bath-applied 5-HT, which might act on extrasynaptic receptors closer to the VSI soma. Bath-applied 5-HT (10 μm) caused a reversible decrease in the half-width of the VSI spike (Fig. 3A). The effect of 5-HT was somewhat dose dependent (Fig. 3B), causing decreases of −5.1 ± 3.5% at 1 μm (n = 4), −6.6 ± 1.4% at 10 μm (n = 10), and −6.5 ± 2.9% at 100 μm (n = 6). The spike-narrowing effect of 5-HT was significant at 10 μm. This suggests that the depression phase of STDN may be caused by 5-HT released from the DSIs producing spike narrowing near the VSI terminals.
Figure 3.
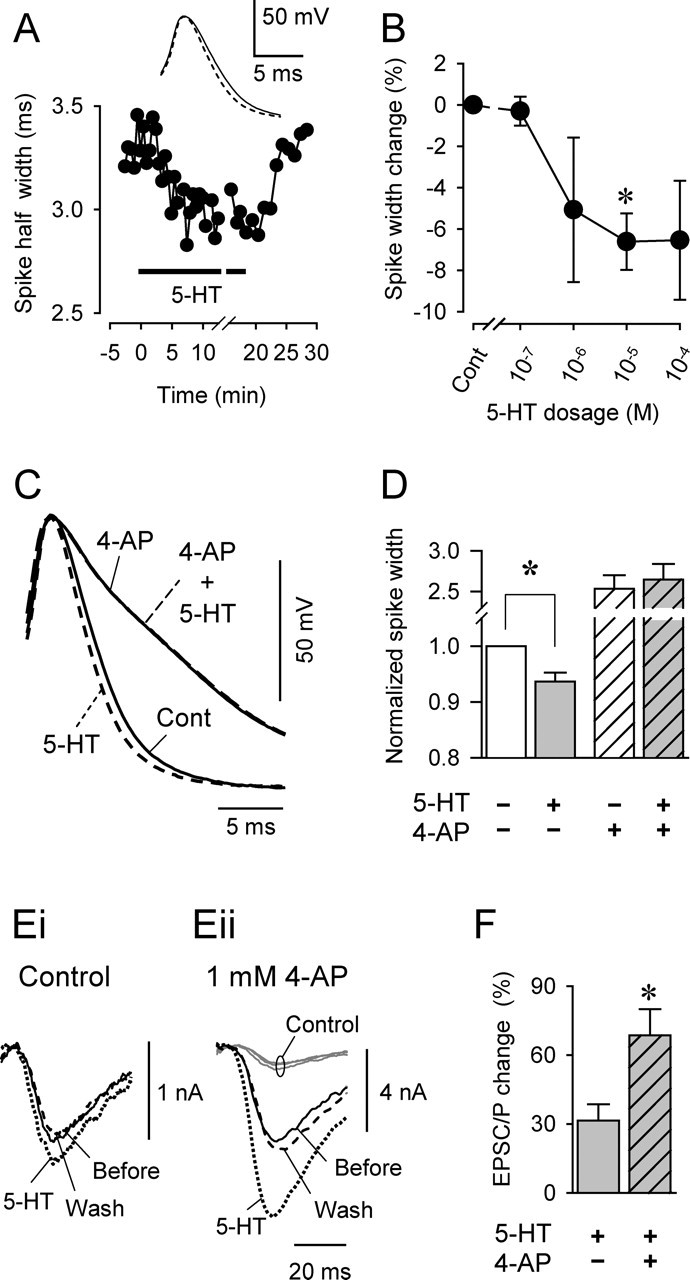
5-HT caused a 4-AP-sensitive decrease in VSI spike width and synaptic potentiation that was enhanced by 4-AP. A, Plot of the spike half-width caused by bath-applied 10 μm 5-HT (horizontal bar) shows a decrease from ∼3.3 to 3.0 ms. Action potentials were evoked repeatedly by stimulating the VSI axon extracellularly in pedal nerve 6. The retrogradely propagating potentials were recorded intracellularly in the soma. The inset shows averaged waveforms of three spikes before (solid trace) and in the presence of (dashed trace) 10 μm 5-HT. B, Dose–response relationship of 5-HT-induced spike narrowing. Asterisk indicates significant difference from control (p < 0.05; Kruskal–Wallis one-way ANOVA on ranks with a post hoc Dunn's multiple comparison procedure). C, Overlaid VSI action potentials show that 4-AP blocked 5-HT-induced spike narrowing. 5-HT (10 μm) caused spike narrowing (dashed line) compared with control (solid line). Addition of 1 mm 4-AP broadened the spike (solid line) and abolished spike-narrowing evoked by 5-HT (dashed line). D, Bar graph showing the effects of 5-HT and 4-AP on VSI spike half-width. 5-HT (10 μm) significantly reduced the spike half-width (*p < 0.05; paired t test). In 4-AP, addition of 5-HT had no further effect on the spike width. E, The effect of 5-HT is enhanced in the presence of 4-AP. Overlaid traces for VSI-evoked EPSCs in VFN were obtained before (solid) and in the presence of (dotted line) 100 μm 5-HT, and 30 min after washout of 5-HT (dashed line) in control saline (i) and in saline containing 1 mm 4-AP (ii). The control traces from i (gray lines) are shown at the same scale as the traces obtained in 4-AP to show the extent of potentiation by 4-AP. The scale bars in i and ii are different so that the EPSCs before 5-HT application appear to be the same size. F, Bar graph showing the average effects of 5-HT on VSI-evoked EPSCs or EPSPs alone or in the presence of 4-AP. 4-AP caused a significant increase in the extent of 5-HT-evoked synaptic potentiation. Asterisk indicates a significant change (p < 0.05; Student's t test).
4-AP blocked 5-HT-induced spike narrowing
The ability of 5-HT to cause spike narrowing was blocked by 4-AP. In this set of experiments, 10 μm 5-HT by itself caused the VSI spike to narrow by 6.4% (p < 0.001; paired t test; n = 11), and 4-AP broadened the VSI action potential by 158 ± 20.2% (p < 0.001; paired t test; n = 7) (Fig. 3C,D); however, in the presence of 4-AP, 5-HT no longer had a significant effect on VSI spike duration (3.1 ± 1.4%; p = 0.06; paired t test; n = 7). These results indicate that 5-HT caused spike narrowing by acting on a 4-AP-sensitive conductance in VSI. This is consistent with the hypothesis that the depression phase of STDN is caused by a serotonergic enhancement of a 4-AP-sensitive membrane conductance that leads to spike narrowing.
4-AP enhanced 5-HT-induced potentiation of the VSI-to-VFN synapse
We found previously that bath-applied 5-HT (100 μm) had inconsistent effects on the amplitude of the VSI-to-VFN synaptic current, causing synaptic potentiation in 75% of the preparations and depression or a biphasic change in the remaining preparations (Sakurai and Katz, 2003b). It was hypothesized that the variability of these effects might be caused by the summation of two opposite actions of 5-HT on the VSI-to-VFN synapse: potentiation and depression. If bath-applied 5-HT evoked opposing actions, then blocking one of these actions ought to uncover the full extent of the other. Therefore, we tested whether blocking spike narrowing with 4-AP enhanced the potentiation of VSI-to-VFN synaptic strength by 5-HT.
We found that in the presence of 4-AP, the effect of 5-HT not only became consistent (it always potentiated the VSI-to-VFN synapse), but the extent of potentiation increased (Fig. 3E,F). On average, 5-HT by itself caused a 31.5 ± 7.1% (n = 6) increase in the amplitude of VSI-evoked EPSCs or EPSPs (Fig. 3Ei,F). In the presence of 1 mm 4-AP, the extent of potentiation by 5-HT was 68.5 ± 11.4%, a significant increase (p < 0.05; Student's t test; n = 6) (Fig. 3Eii,F). This increase in potentiation occurred despite the fact that 4-AP by itself also potentiated VSI synaptic output through spike broadening and caused an increase in the basal amplitude of the VSI-to-VFN EPSC (Figs. 2C,D, 3E,F). These results support the hypothesis that 5-HT evokes two opposing actions on the VSI-to-VFN synapse: (1) potentiation, which is spike duration independent, and (2) depression, which might be caused by spike narrowing that is mediated by an enhancement of IA and therefore blocked by 4-AP.
VSI exhibits a transient voltage-dependent outward current
To determine whether the effect of 5-HT on spike width was mediated by a 4-AP-sensitive membrane conductance, we examined voltage-dependent outward currents in VSI under voltage clamp. VSI exhibited a prominent voltage-dependent transient outward current that fully inactivated after a long depolarizing step (Fig. 4A). This voltage-dependent transient current was similar in kinetics to that of the A-current, which has been described previously in this animal (Thompson, 1977; Getting, 1983b; Serrano and Getting, 1989). As expected for the A-current, the magnitude of the transient current was greatly reduced by 4-AP (Fig. 4B). Subtraction of the currents recorded before and during 4-AP reveals the 4-AP-sensitive component (Fig. 4Biii).
The other major voltage-dependent outward current expressed by VSI was a delayed rectifier type with a more positive activation threshold (Fig. 4Ci,D) (Thompson, 1977). Tetraethylammonium (TEA; 5 mm) preferentially blocked this current, revealing a transient current (Fig. 4Cii) that was blocked further by the addition of 4-AP (data not shown). Subtraction of the currents recorded before and during TEA revealed a noninactivating voltage-dependent outward current (Fig. 4Ciii).
The amplitude of the 4-AP-sensitive component could be measured separately from the TEA-sensitive component because of differences in their rates of activation and their voltage activation profiles. The more rapidly activating 4-AP-sensitive current could be detected at more negative potentials than the TEA-sensitive current, which was predominant at voltage steps more positive than −10 mV (Fig. 4D). Blockade of both outward currents by TEA and 4-AP revealed an inward current component that was activated by depolarizing steps of more than −10 mV (data not shown). Thus, outward current elicited by step commands to voltages more negative than −10 mV was caused almost exclusively by the 4-AP-sensitive transient current.
The 4-AP-sensitive current exhibited voltage-dependent inactivation typical of IA (Fig. 4E); however, the activation and inactivation curves are shifted to the right relative to other reported A-currents (Thompson, 1977; Getting, 1983b; Serrano and Getting, 1989) because of the use in this study of high-divalent cation saline, which causes charge screening.
5-HT enhanced A-current in VSI
Bath-applied 5-HT caused a small but consistent enhancement of IA (Fig. 5). The peak amplitude of IA reversibly increased by 12% in the presence of 10 μm 5-HT (Fig. 5A). 5-HT (10 and 100 μm) caused no significant changes in the input resistance (p = 0.98, n = 21 and p = 0.93, n = 16, respectively; Student's t test). 5-HT caused an increase in peak A-current conductance (gA) throughout the voltage range from the threshold potential (−35 mV) up to the maximum voltage level examined (0 mV) (Fig. 5B). This overall increase might be caused by either a decrease in the threshold voltage for activation or an increase in the maximal conductance. We were unable to resolve this because at more positive holding potentials, inward current shunted the rising phase of IA, and action potentials were elicited in distant neurites, preventing accurate measurements of IA. When IA was evoked by stepping Vm from −90 to −20 mV, 10 μm 5-HT caused an average increase in gA of 14.6 ± 1.8% (p < 0.001; paired t test; n = 14) (Fig. 5C).
Figure 5.

5-HT enhanced the A-current in VSI. A, Bath-applied 5-HT (10 μm) caused a 12% increase in the amplitude of A-current evoked by a depolarizing step to −20 mV from a holding potential of −90 mV. Inset, Averaged A-current waveforms obtained in control saline (1), in 5-HT (2), and after washout of 5-HT (3). B, Conductance–voltage relationships for A-current in VSI recorded in control saline (filled circles) and in 10 μm 5-HT (open circles). 5-HT caused an increase in A-current conductance at all voltages within the activation range. Inset, A-current responses evoked by voltage steps from −90 to −40, −30, and −20 mV. Calibration: A, B, insets, 5 nA; 200 ms. C, 4-AP blocked the 5-HT-induced enhancement of the outward current conductance. The peak amplitudes of the outward conductance were measured in the presence or absence of 10 μm 5-HT and 2 mm 4-AP. Asterisk indicates significant difference from control (p < 0.001). See Results for details.
The 5-HT-induced enhancement of IA was blocked by 4-AP (Fig. 5C). 4-AP (2 mm) reduced gA by −80.0 ± 4.1% (n = 5). In the presence of 4-AP, the addition of 10 μm 5-HT did not cause a significant change in gA (−36.3 ± 16.9%; p = 0.17; paired t test; n = 4). Thus, the 5-HT-induced enhancement of gA was blocked in 2 mm 4-AP, indicating that the increase in outward current produced by 5-HT was not likely caused by the recruitment of an additional outward conductance.
TEA had little effect on DSI modulatory action
To determine whether the depression phase was sensitive to other potassium channel blockers, we tested the effect of TEA, which also broadens presynaptic action potentials in molluscan neurons (Rosen et al., 1989; Satterlie et al., 2000) but blocks the delayed outward current (Fig. 4C). In contrast to 4-AP, TEA preferentially reduced the magnitude of the after-spike hyperpolarization rather than increasing spike duration and had inconsistent effects on the VSI-to-VFN synapse (Fig. 6A). TEA (5 mm) caused a slight increase in VSI-evoked EPSCs in three preparations, no change in one preparation, and a 29% decrease in a fifth preparation. Overall, TEA caused no significant change in EPSC amplitude (21.5 ± 19.9%; p = 0.34; n = 5).
Figure 6.

TEA had no effect on VSI synaptic strength or DSI neuromodulatory effects. A, Effect of TEA on VSI spike shape and synaptic strength. Simultaneously recorded averaged waveforms of five action potentials (top trace) and five corresponding EPSCs in VFN (bottom trace) in control saline and in 5 mm TEA are shown. TEA reduced the magnitude of the VSI spike afterhyperpolarization and caused a slight broadening of the action potential, but it had no effect on the size of VSI-evoked EPSCs in VFN. B, Overlaid traces for VSI-evoked EPSCs in VFN recorded 5 s before the DSI spike train (solid line), 5 s after the DSI spike train (dotted line), and 35 s after the DSI spike train (dashed line) in control saline (left) and in saline containing 5 mm TEA (right). All traces are shown at the same scale. C, TEA had no effect on the DSI-evoked biphasic, bidirectional change in VSI synaptic strength. Averaged data show the sequential effects of DSI stimulation on normalized VSI-evoked EPSCs in control saline (filled circles) and in 5 mm TEA (open squares). Each plot shows a mean value (mean ± SEM) of pooled data obtained from four preparations. The gray vertical bar represents a 5 s DSI spike train. stim, Stimulation.
In all cases, 5–10 mm TEA had no effect on DSI modulation of the VSI-to-VFN synapse (Fig. 6B,C). The enhancement of VSI-evoked EPSCs within 10 s of a DSI spike train was unaffected by TEA (69.1 ± 9.4% in control; 51.5 ± 9.7% in TEA; p = 0.10 by two-way ANOVA with Fisher's LSD method; n = 4). Similarly, there was no change in the depression between 25 and 35 s after DSI stimulation (−20.3 ± 3.4% in control; −17.7 ± 5.5% in TEA; p = 0.49 by two-way ANOVA with Fisher's LSD method; n = 4). Thus, the depression phase of the DSI modulatory action is not sensitive to blockade of delayed rectifier potassium channels but is sensitive to blockade of IA. This is consistent with a mechanism for the depression that involves a specific enhancement of IA.
Relationships among spike duration, A-current, and synaptic strength
To explore the relationship among spike duration, A-current, and synaptic strength, we measured these values during the wash-in of 4-AP (1 mm) to the bath. Changes in gA in VSI were monitored by periodically switching to voltage-clamp mode (Fig. 7Ai). Switching off the voltage clamp allowed VSI action potentials and the EPSCs that they evoked in VFN to be recorded (Fig. 7Aii,Aiii). The amplitude of gA decreased, whereas spike width increased, during wash-in of 4-AP. The spike duration showed an inverse relationship to gA amplitude, with the data obtained from each preparation falling along the same curve (Fig. 7B). Thus, the duration of the VSI action potential appears to be strongly influenced by gA.
As the spike broadened during 4-AP wash-in, the EPSC increased in amplitude (Fig. 7Aii,Aiii). There was a linear relationship between synaptic strength and spike duration within a given preparation (Fig. 7C); however, there were large differences in the slopes of these relationships between different preparations. In one preparation, a 50% increase in spike duration was correlated with a fivefold increase in the size of VSI-to-VFN EPSCs (Fig. 7C, dark triangles), whereas in other preparations, only a doubling in EPSC amplitude was observed (Fig. 7C, solid circles). Thus, there appears to be individual variability when spike width is translated to synaptic strength.
To determine whether the individual variation in the relationship between synaptic strength and spike width was an artifact of the wash-in related to differing degrees of penetration of 4-AP into the neuropil, we examined steady-state responses to different concentrations of 4-AP. 4-AP caused a dose-dependent increase in both VSI spike duration and VSI-to-VFN EPSC amplitude (Fig. 7Di,Dii). The VSI action potential increased significantly by 37.9 ± 3.7 and 145 ± 33% in 0.1 and 1.0 mm 4-AP. Concurrent with the increase in action potential duration, the amplitude of the VSI-evoked EPSC increased significantly by 159 ± 53 and 457 ± 55% in 0.1 and 1.0 mm 4-AP. Plotting the mean spike EPSC amplitude versus the mean spike width for the two concentrations of 4-AP tested yielded a somewhat linear relationship (Fig. 7E, large gray squares with a dashed line); however, when the values for each preparation were plotted separately, then a large degree of variability was again observed. Thus, as with the wash-in experiment, the increase in EPSC amplitude was linearly related to the increase in spike duration within each preparation, but there was a multiplicative factor between preparations that determined the extent to which spike broadening affected synaptic strength.
Computer simulation of VSI
The experimental results suggest that enhancement of IA may lead to spike narrowing in the synaptic terminals that decreases the strength of the VSI-to-VFN synapse. To determine whether the small enhancement in IA produced by 5-HT could be sufficient to cause spike narrowing, we constructed a conductance-based simulation of VSI using the A-current model described in Materials and Methods. The parameters of the A-current model (Table 1) reproduced the observed A-current properties of VSI (Fig. 8A). The parameters of the other conductances in the simulation were chosen to reproduce the spike shape and action potential firing pattern of VSI (Fig. 8B).
To test the effect of IA on spike shape, we varied ḡA in our model neuron from zero to two times the normal value, with all other parameters kept constant (Fig. 8C). The broadened spike waveform in the simulation has a low-voltage shoulder wider than that usually observed in the spikes of real VSIs (compare Figs. 2C, 3C; also see Fig. 2D). Therefore, to increase the fidelity of the comparison with experimental results, we measured spike duration at two different heights (50 and 25%) (Fig. 8C, inset). We found that lowering ḡA caused spike broadening, thereby mimicking the effect of 4-AP. When ḡA was reduced to zero, spike half-width increased by 91.2% at 50% spike height and by 147.0% at 25% spike height. By comparison, 4-AP (2 mm) caused more spike broadening by an average increase of 153%, with less of a decrease in gA by −75.1% (Fig. 8C, gray squares) (compare Figs. 3D, 5C).
In the model, we increased ḡA to mimic the effect of 5-HT. The model prediction matched the experimentally observed range: the simulations predicted that to produce a 6.6% reduction in spike duration (similar to what we observed in 5-HT), ḡA would need to increase by ∼14.8% (Fig. 8C). In voltage-clamp experiments, we observed that 5-HT increased IA by 14.6% (compare Fig. 5C). Thus, the model prediction is in the range of the experimental data and suggests that this degree of IA enhancement could account for the observed degree of spike narrowing.
Dynamic clamp of IA in VSI
The computer simulation relied on a number of assumptions that may not match the physiological conditions. First, the simulation is a single-compartment model that assumes uniform current and receptor densities. Second, although IA was modeled to accurately reflect the dynamics of the A-current in VSI, the other currents and their densities were fit simply to make the firing properties of the model resemble those of VSI. To more accurately test the effect of varying gA on spike shape in the actual ion channel environment of VSI, we introduced an artificial A-current into a real VSI using the dynamic clamp (Sharp et al., 1993; Prinz et al., 2004).
Dynamic clamp of A-current in VSI with parameters from our voltage-clamp studies (see Materials and Methods) altered spike shape in a manner that resembled the computer simulation. In control saline, we observed that subtracting A-current by reducing ḡA in the dynamic-clamp system caused spike broadening, whereas adding artificial A-current by increasing ḡA caused spike narrowing (n = 6) (Fig. 9A).
Figure 9.
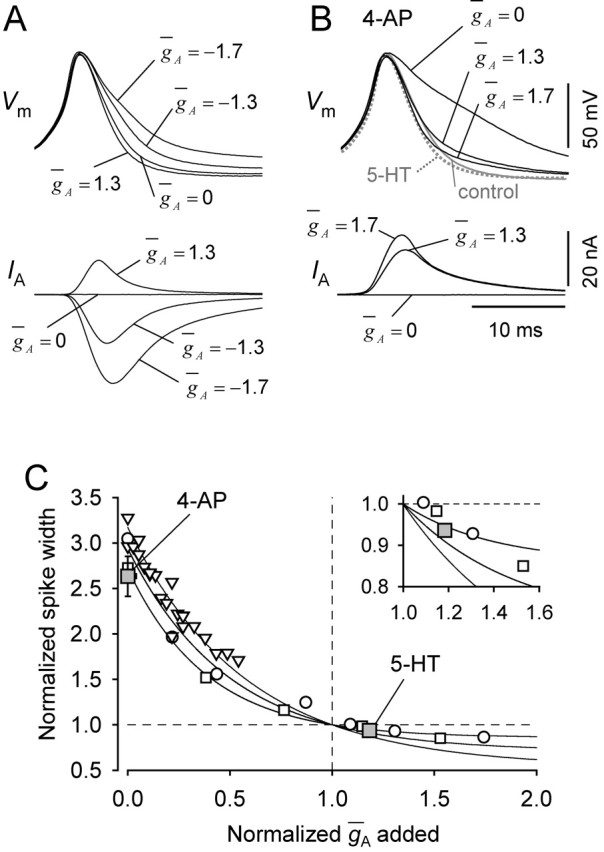
Dynamic clamp of VSI. A, Spike width can be changed by the dynamic-clamp introduction of IA. Dynamic-clamp insertion of gA (ḡA = 1.3 μS) reduced the width of the action potential, whereas subtraction of gA (ḡA = −1.3 and −1.7 μS) increased the spike duration. B, Spike broadening by 4-AP was reversed by dynamic-clamp introduction of IA. Gray trace shows the VSI action potential in control saline. 4-AP (2 mm) caused spike broadening (ḡA = 0). Dynamic-clamp insertion of gA (ḡA = 1.3 μS) restored the width of the action potential to its control value. Increasing ḡA further to 1.7 μS narrowed the spike to the level seen in 5-HT (dotted gray trace). C, Relationships of spike half-width to normalized gA as determined with dynamic clamp in 2 mm 4-AP (open symbols). ḡA values in each experiment were normalized such that a single exponential regression line crossed the normalized spike width of 1. Spike half-width and the 4-AP-sensitive conductance recorded from real VSIs in the presence of 2 mm 4-AP or 10 μm 5-HT were also normalized and plotted with gray squares with error bars (mean ± SEM) (compare Figs. 3D, 5C). In one preparation, we failed to inject enough current to restore control spike width (triangles). Inset, The same graph having enlarged y-axis with limited range of x-axis.
We then tested whether introduction of artificial A-current into VSI could reverse the effects of 4-AP on spike duration (Fig. 9B). In the presence of 2 mm 4-AP, which reduced the amount of A-current to 9.4% of control in this preparation, the VSI spike was broadened to ∼170% of the control spike half-width (Fig. 9B). Adding artificial A-current by dynamic clamp reduced the action potential duration to close to the control value when ḡA in dynamic clamp was set to 1.3 μS. A further increase of ḡA in dynamic clamp to 1.7 μS reproduced spike narrowing to the level of 5-HT-induced spike narrowing (6.6%). Thus, in this preparation (Fig. 9B), it took a 30% increase in the baseline A-current to cause the same degree of spike narrowing as with 5-HT, which produces, on average, a 14.6% increase in gA. Similar results were obtained in three preparations.
The dynamic clamp may overestimate the magnitude of the conductance needed to rescue the A-current blocked by 4-AP. Dynamic clamp inserts the artificial conductance only in the soma, but the remainder of the neuron still has a broadened action potential. This was further reflected in the slight difference in magnitude of the afterhyperpolarization compared with the spikes evoked in control saline and in 5-HT (Fig. 9B). Nevertheless, the relationship between normalized spike width and gA change by means of the dynamic clamp (Fig. 9C) resembled the one obtained with 4-AP wash-in (Fig. 7B) and the simulation (Fig. 8C), further supporting the hypothesis that increases in A-current conductance can account for the decrease in spike shape caused by 5-HT.
Discussion
Neuromodulatory actions often are thought to have tonic, gain-setting functions. In contrast, the discovery of STDN demonstrated that neuromodulatory actions also can convey temporal information (Sakurai and Katz, 2003b). This study examined the mechanisms by which a single occurrence of synaptically released 5-HT produces time-varying neuromodulatory actions. The results from this study suggest that 5-HT released from the DSIs has two separate effects: a short-lasting spike duration-independent enhancement of VSI-to-VFN synaptic strength and a longer-lasting enhancement of IA, which narrows the VSI spike and therefore may lead to depression of the VSI-to-VFN synapse. Thus, the time-varying effects of STDN are likely caused by the different durations of two independent intracellular signaling events triggered by 5-HT.
The mechanism underlying the depression phase
Our conclusions about the mechanism underlying the depression phase are based on indirect evidence. Although the A-channel blocker 4-AP eliminated the depression phase of STDN, in our soma recordings we did not observe VSI spike narrowing or IA enhancement in response to DSI stimulation. The inability to observe spike narrowing in response to DSI stimulation is presumably because of the electrotonic distance between the soma, where the recordings were made, and the synaptic terminals, where DSI probably contacts VSI (Fig. 1Ai). It is not surprising that the actively propagated action potentials recorded from the soma are not affected by modulatory actions in this distant region. Similarly, altering spike shape in the soma with dynamic clamp did not affect synaptic strength in the terminals (data not shown), again indicating a large electrotonic distance.
The distribution of ion channels and 5-HT receptors also affects the magnitude of responses observed with soma recordings. A-current channels are likely to be distributed all over the cell because 4-AP has a large effect on the amplitude of IA, on spike shape, and on synaptic strength; however, 5-HT receptors are probably not as abundant on the soma as near the terminals. Although 5-HT receptors are known to be distributed widely over molluscan neurons (Drummond et al., 1980; Kadan and Hartig, 1988), studies in Tritonia and other molluscs have shown that soma responses to 5-HT can be smaller than neuropil responses (Sun et al., 1996; Popova and Katz, 1998, 2002). Thus, although bath-applied 5-HT may have activated receptors closer to the soma than the DSI synaptic inputs, the full effect of 5-HT may not be apparent in the soma. Therefore, the 14.6% change in IA and the 6.6% decrease in spike duration observed in the soma in response to bath-applied 5-HT may be underestimates of the changes that occur at the synaptic terminals in response to either bath-applied 5-HT or DSI stimulation.
Nevertheless, evidence suggests that DSI stimulation leads to spike narrowing at the terminals. It has been shown previously that DSIs are serotonergic (Katz et al., 1994; McClellan et al., 1994; Sudlow et al., 1998) and that 5-HT released from the DSIs is responsible for both the potentiation and depression phases of STDN (Sakurai and Katz, 2003b). We showed here that bath-applied 5-HT caused spike narrowing and an enhancement of IA in VSI. Both effects of 5-HT were blocked by 4-AP. The role of IA in altering spike shape was further supported by 4-AP wash-in experiments as well as a conductance-based simulation and dynamic-clamp experiments. Together with the observation that 4-AP blocks the depression phase of STDN, the most parsimonious explanation for the depression phase is that DSI stimulation causes spike narrowing at the terminals by enhancing the A-current conductance, although there may be other more complex explanations.
Despite many studies that have shown 5-HT-induced synaptic potentiation via spike broadening (Abrams et al., 1984; Baxter and Byrne, 1989, 1990; Goldsmith and Abrams, 1992; Ghirardi et al., 1992; Hochner and Kandel, 1992; Byrne and Kandel, 1996), relatively few studies have demonstrated 5-HT-induced spike narrowing as a mechanism for decreasing synaptic strength (but see Holz et al., 1986; Piomelli et al., 1987; Rosen et al., 1989; Satterlie et al., 2000), yet there are numerous examples of heterosynaptic depression mediated by modulation of ionic conductances in the presynaptic neuron. For example, in grasshopper sensorimotor synapse, the synaptic depression is caused by a reduction of the amplitude and duration of presynaptic spikes (Niven and Burrows, 2003). In prefrontal cortex, 5-HT downmodulates dendritic synaptic plasticity by depressing rapidly inactivating Na+ currents (Carr et al., 2003). Similarly, in rat hippocampal pyramidal neurons, 5-HT directly reduces calcium influx (Sandler and Ross, 1999). Distinct dose-dependent actions of 5-HT on multiple voltage-gated conductances have been reported in dorsal root ganglion cells in bullfrog (Holz et al., 1986).
Two phases of STDN are pharmacologically separable
The previous study suggested that two independent mechanisms underlie the DSI-evoked STDN of the VSI-to-VFN synapse (Sakurai and Katz, 2003b). The results from the current study definitively show that the two phases of STDN are produced by pharmacologically separable actions of 5-HT. First, the depression phase of STDN, but not the potentiation phase, was eliminated by 4-AP (Fig. 2). Second, blocking of IA with 4-AP enhanced the 5-HT-induced potentiation by reducing conflicting simultaneous depression; thus the variability of the effect of bath-applied 5-HT was removed by 4-AP (Fig. 3E,F).
Although 4-AP increased the extent of potentiation produced by bath-applied 5-HT (Fig. 3E,F), it did not enhance the potentiation phase of STDN produced by DSI stimulation (Fig. 2). If DSI stimulation causes a simultaneous increase in synaptic strength and a decrease in spike duration, then it would be expected that blocking IA with 4-AP would cause an increase in both the amplitude and duration of the potentiation phase of STDN. There are two possible explanations for the lack of an effect of 4-AP on DSI-evoked potentiation. One is that there is a ceiling effect to the potentiation: broadening the spike increases the synaptic output so much that DSI stimulation cannot multiply it further. Moreover, DSI-evoked potentiation (70–100% increase at peak) (Figs. 1C, 2B, 6B) was stronger in the control saline and thus may be closer to the maximal value than 5-HT-induced potentiation (∼30%). Alternatively, the two phases might not arise simultaneously; the onset of the depression phase might be delayed with respect to the potentiation phase of STDN. This could be tested if there were a means to block just the potentiation phase, which there currently is not.
The mechanism underlying the potentiation phase is still unknown. The fact that broadening the spike with 4-AP did not decrease the extent of DSI-evoked potentiation (Fig. 2) indicates that the mechanism of potentiation is independent from spike duration, and thus it may not likely be dependent on changes in calcium influx through the plasma membrane. It is remarkable that increasing the synaptic output 10-fold with 4-AP did not diminish the ability of 5-HT or DSI stimulation to cause further potentiation (Figs. 2, 3).
Multiple functions for the A-current
Getting (1983b) showed previously that VSI has a characteristic voltage-dependent transient outward current similar to the A-current in pleural neurons 2 and 3 described by Thompson (1977). He suggested that the major functional role of this current is to cause a delayed firing of VSI on excitation, which may play an essential role in generating the alternating bursting pattern during the swim motor program. Here we show that A-current also plays a major role in determining spike shape and hence synaptic output. Our finding that DSI stimulation modulates A-current in VSI is likely to have implications for the pattern-generating mechanism itself.
In conclusion, our results suggest that a serotonergic neuron intrinsic to the Tritonia swim CPG evokes neuromodulatory effects that change over time because of the different decay rates of opposite actions: a rapidly decaying spike duration-independent enhancement of synaptic strength and a more slowly decaying enhancement of IA, which leads to spike narrowing and hence to a decrease in synaptic strength. These results highlight the importance of studying the dynamic actions of neuromodulatory neurons rather than the static application of applied neuromodulatory substances. They also illustrate how a single neuromodulatory signal may provide temporal information to other neurons.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant R01-NS035371 and by a Research Program Enhancement grant from Georgia State University to P.S.K. R.J.B. and N.R.D. were supported by National Science Foundation Grant DBI-9987074 and NIH Grant R01-RR20115. We thank the anonymous reviewers for their helpful suggestions.
References
- Abrams TW, Castellucci VF, Camardo JS, Kandel ER, Lloyd PE (1984). Two endogenous neuropeptides modulate the gill and siphon withdrawal reflex in Aplysia by presynaptic facilitation involving cAMP-dependent closure of a serotonin-sensitive potassium channel. Proc Natl Acad Sci USA 81:7956–7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter DA, Byrne JH (1989). Serotonergic modulation of two potassium currents in the pleural sensory neurons of Aplysia. J Neurophysiol 62:665–679. [DOI] [PubMed] [Google Scholar]
- Baxter DA, Byrne JH (1990). Differential effects of cAMP and serotonin on membrane current, action-potential duration, and excitability in somata of pleural sensory neurons of Aplysia. J Neurophysiol 64:978–990. [DOI] [PubMed] [Google Scholar]
- Baxter DA, Canavier CC, Clark JW Jr, Byrne JH (1999). Computational model of the serotonergic modulation of sensory neurons in Aplysia. J Neurophysiol 82:2914–2935. [DOI] [PubMed] [Google Scholar]
- Buchholtz F, Golowasch J, Epstein IR, Marder E (1992). Mathematical model of an identified stomatogastric ganglion neuron. J Neurophysiol 67:332–340. [DOI] [PubMed] [Google Scholar]
- Byrne JH, Kandel ER (1996). Presynaptic facilitation revisited: state and time dependence. J Neurosci 16:425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese RL (1998). Cellular, synaptic, network, and modulatory mechanisms involved in rhythm generation. Curr Opin Neurobiol 8:710–717. [DOI] [PubMed] [Google Scholar]
- Carr DB, Cooper DC, Ulrich SL, Spruston N, Surmeier DJ (2002). Serotonin receptor activation inhibits sodium current and dendritic excitability in prefrontal cortex via a protein kinase C-dependent mechanism. J Neurosci 22:6846–6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JA, Stevens CF (1971). Voltage clamp studies of a transient outward membrane current in gastropod neural somata. J Physiol (Lond) 213:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darghouth NR, Sakurai A, Butera RJ, Katz PS (2004). Computer simulations and dynamic clamp experiments support a role for a 4-AP-sensitive current in serotonin-induced spike-narrowing leading to synaptic depression. Soc Neurosci Abstr 30:420.2. [Google Scholar]
- Drummond AH, Bucher F, Levitan IB (1980). Distribution of serotonin and dopamine receptors in Aplysia tissues: analysis by [3H]LSD binding and adenylate cyclase stimulation. Brain Res 184:163–177. [DOI] [PubMed] [Google Scholar]
- Frankenhaeuser B, Hodgkin AL (1957). The action of calcium on the electrical properties of squid axons. J Physiol (Lond) 137:218–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getting PA (1981). Mechanisms of pattern generation underlying swimming in Tritonia. I. Neuronal network formed by monosynaptic connections. J Neurophysiol 46:65–79. [DOI] [PubMed] [Google Scholar]
- Getting PA (1983a). Mechanisms of pattern generation underlying swimming in Tritonia. II. Network reconstruction. J Neurophysiol 49:1017–1035. [DOI] [PubMed] [Google Scholar]
- Getting PA (1983b). Mechanisms of pattern generation underlying swimming in Tritonia. III. Intrinsic and synaptic mechanisms for delayed excitation. J Neurophysiol 49:1036–1050. [DOI] [PubMed] [Google Scholar]
- Getting PA (1989). Emerging principles governing the operation of neural networks. Annu Rev Neurosci 12:185–204. [DOI] [PubMed] [Google Scholar]
- Getting PA, Dekin MS (1985). Mechanisms of pattern generation underlying swimming in Tritonia. IV. Gating of central pattern generator. J Neurophysiol 53:466–480. [DOI] [PubMed] [Google Scholar]
- Getting PA, Lennard PR, Hume RI (1980). Central pattern generator mediating swimming in Tritonia. I. Identification and synaptic interactions. J Neurophysiol 44:151–164. [DOI] [PubMed] [Google Scholar]
- Ghirardi M, Braha O, Hochner B, Montarolo PG, Kandel ER, Dale N (1992). Roles of PKA and PKC in facilitation of evoked and spontaneous transmitter release at depressed and nondepressed synapses in Aplysia sensory neurons. Neuron 9:479–489. [DOI] [PubMed] [Google Scholar]
- Gilly WF, Armstrong CM (1982a). Slowing of sodium channel opening kinetics in squid axon by extracellular zinc. J Gen Physiol 79:935–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilly WF, Armstrong CM (1982b). Divalent cations and the activation kinetics of potassium channels in squid giant axons. J Gen Physiol 79:965–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith BA, Abrams TW (1992). cAMP modulates multiple K+ currents, increasing spike duration and excitability in Aplysia sensory neurons. Proc Natl Acad Sci USA 89:11481–11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman AL, Mirolli M (1972). The passive electrical properties of the membrane of a molluscan neurone. J Physiol (Lond) 227:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahin R, Campbell DT (1983). Simple shifts in the voltage dependence of sodium channel gating caused by divalent cations. J Gen Physiol 82:785–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines ML (1998). The neurosimulator NEURON. In: Methods in neuronal modeling: from synapses to networks (Koch C, Segev I, eds) , pp. 121–136. Cambridge, MA: MIT.
- Hochner B, Kandel ER (1992). Modulation of a transient K+ current in the pleural sensory neurons of Aplysia by serotonin and cAMP: implications for spike broadening. Proc Natl Acad Sci USA 89:11476–11480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol (Lond) 117:500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GG, Shefner SA, Anderson EG (1986). Serotonin decreases the duration of action potentials recorded from tetraethylammonium-treated bullfrog dorsal root ganglion cells. J Neurosci 6:620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA (1992). Simulation of the currents involved in rhythmic oscillations in thalamic relay neurons. J Neurophysiol 68:1373–1383. [DOI] [PubMed] [Google Scholar]
- Kadan MJ, Hartig PR (1988). Autoradiographic localization and characterization of [125I]lysergic acid diethylamide binding to serotonin receptors in Aplysia. Neuroscience 24:1089–1102. [DOI] [PubMed] [Google Scholar]
- Katz PS (1998). Neuromodulation intrinsic to the central pattern generator for escape swimming in Tritonia. Ann NY Acad Sci 860:181–188. [DOI] [PubMed] [Google Scholar]
- Katz PS, Getting PA, Frost WN (1994). Dynamic neuromodulation of synaptic strength intrinsic to a central pattern generator circuit. Nature 367:729–731. [DOI] [PubMed] [Google Scholar]
- Kiehn O, Katz PS (1999). Making circuits dance: neuromodulation of motor systems. In: Beyond neurotransmission: neuromodulation and its importance for information processing (Katz PS, ed.) , pp. 275–317. Oxford: Oxford UP.
- Lynn-Bullock CP, Clemens S, Sakurai A, Katz PS (2002). Functional anatomy of the escape swim CPG in Tritonia. Soc Neurosci Abstr 28:67.25. [Google Scholar]
- Marder E, Bucher D (2001). Central pattern generators and the control of rhythmic movements. Curr Biol 11:R986–R996. [DOI] [PubMed] [Google Scholar]
- Marder E, Calabrese RL (1996). Principles of rhythmic motor pattern generation. Physiol Rev 76:687–717. [DOI] [PubMed] [Google Scholar]
- McClellan AD, Brown GD, Getting PA (1994). Modulation of swimming in Tritonia: excitatory and inhibitory effects of serotonin. J Comp Physiol [A] 174:257–266. [DOI] [PubMed] [Google Scholar]
- Mercer AR (1999). Changing the way we perceive things: sensory systems modulation. In: Beyond neurotransmission: neuromodulation and its importance for information processing (Katz PS, ed.) , pp. 198–240. Oxford: Oxford UP.
- Mirolli M, Talbott SR (1972). The geometrical factors determining the electrotonic properties of a molluscan neurone. J Physiol (Lond) 227:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Kim DJ, Delay RJ, Roper SD (1996). Neuromodulation of transduction and signal processing in the end organs of taste. Chem Senses 21:353–365. [DOI] [PubMed] [Google Scholar]
- Niven JE, Burrows M (2003). Spike width reduction modifies the dynamics of short-term depression at a central synapse in the locust. J Neurosci 23:7461–7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasztor VM, Bush BM (1989). Primary afferent responses of a crustacean mechanoreceptor are modulated by proctolin, octopamine, and serotonin. J Neurobiol 20:234–254. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Volterra A, Dale N, Siegelbaum SA, Kandel ER, Schwartz JH, Belardetti F (1987). Lipoxygenase metabolites of arachidonic acid as second messengers for presynaptic inhibition of Aplysia sensory cells. Nature 328:38–43. [DOI] [PubMed] [Google Scholar]
- Popova LB, Katz PS (1998). Serotonin responses of acutely isolated identified neurons from the Tritonia swim CPG. Soc Neurosci Abstr 24:358. [Google Scholar]
- Popova LB, Katz PS (2002). The effects of 5-HT on acutely isolated serotonergic Tritonia swim CPG neurons. Soc Neurosci Abstr 28:270.12. [Google Scholar]
- Prinz AA, Abbott LF, Marder E (2004). The dynamic clamp comes of age. Trends Neurosci 27:218–224. [DOI] [PubMed] [Google Scholar]
- Raikov I, Preyer A, Butera RJ (2004). MRCI: a flexible real-time dynamic clamp system for electrophysiology experiments. J Neurosci Methods 132:109–123. [DOI] [PubMed] [Google Scholar]
- Rosen SC, Susswein AJ, Cropper EC, Weiss KR, Kupfermann I (1989). Selective modulation of spike duration by serotonin and the neuropeptides, FMRFamide, SCPB, buccalin and myomodulin in different classes of mechanoafferent neurons in the cerebral ganglion of Aplysia. J Neurosci 9:390–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai A, Katz PS (2003a). 4-AP and IBMX selectively block the depression phase of a biphasic neuromodulation of synaptic strength by serotonergic neurons in the Tritonia swim CPG. Soc Neurosci Abstr 29:168.18. [Google Scholar]
- Sakurai A, Katz PS (2003b). Spike timing-dependent serotonergic neuromodulation of synaptic strength intrinsic to a central pattern generator circuit. J Neurosci 23:10745–10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler VM, Ross WN (1999). Serotonin modulates spike backpropagation and associated [Ca2+]i changes in the apical dendrites of hippocampal CA1 pyramidal neurons. J Neurophysiol 81:216–224. [DOI] [PubMed] [Google Scholar]
- Satterlie RA, Norekian TP, Pirtle TJ (2000). Serotonin-induced spike narrowing in a locomotor pattern generator permits increases in cycle frequency during accelerations. J Neurophysiol 83:2163–2170. [DOI] [PubMed] [Google Scholar]
- Serrano EE, Getting PA (1989). Diversity of the transient outward potassium current in somata of identified molluscan neurons. J Neurosci 9:4021–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AA, O’Neil MB, Abbott LF, Marder E (1993). Dynamic clamp: computer-generated conductances in real neurons. J Neurophysiol 69:992–995. [DOI] [PubMed] [Google Scholar]
- Sudlow LC, Jing J, Moroz LL, Gillette R (1998). Serotonin immunoreactivity in the central nervous system of the marine molluscs Pleurobranchaea californica and Tritonia diomedea. J Comp Neurol 395:466–480. [PubMed] [Google Scholar]
- Sun ZY, Kauderer B, Schacher S (1996). Differential distribution of functional receptors for neuromodulators evoking short-term heterosynaptic plasticity in Aplysia sensory neurons. J Neurosci 16:7540–7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SH (1977). Three pharmacologically distinct potassium channels in molluscan neurones. J Physiol (Lond) 265:465–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willows AO, Dorsett DA, Hoyle G (1973). The neuronal basis of behavior in Tritonia. I. Functional organization of the central nervous system. J Neurobiol 4:207–237. [DOI] [PubMed] [Google Scholar]