

Two hallmark features of neurodegenerative diseases are the accumulation of protein aggregates and degeneration of a select population of neurons. The underlying mechanism(s) of cell death in neurodegenerative diseases are debated intensely, and presumably, delineating the molecular pathways responsible for neuronal dysfunction and death will lead to therapeutic interventions for these devastating diseases. Apoptosis is an evolutionary conserved mechanism of cell suicide, and evidence for this form of death is abundant in neurodegenerative diseases, as is evidence for necrotic and autophagic cell death (Yuan et al., 2003). The role of apoptotic cell death in the pathogenesis of amyotrophic lateral sclerosis (ALS), which is associated with the misfolding of a key antioxidant enzyme, superoxide dismutase 1 (SOD1), and loss of motor neurons (Bruijn et al., 2004) has generated great interest.
In a recent paper in The Journal of Neuroscience, Gould et al. (2006) demonstrate that deletion of Bax (Bcl-2-associated X protein), a proapoptotic multidomain Bcl-2 (B-cell lymphoma protein 2) family member, abrogates neuronal cell death and delays disease onset but fails to alter disease duration in ALS. The authors used the well characterized human mutant SOD1(G93A) transgenic mice, which display a robust paralytic phenotype (Gurney et al., 1994). When crossed into a Bax knock-out (KO) background, SOD1 mutants survived significantly longer than controls [Gould et al., 2006, their Fig. 1A (http://www.jneurosci.org/cgi/content/full/26/34/8774#F1)]. Also, this rescue was paralleled by a delay in the decline in motor performance [Gould et al., 2006, their Fig. 1B (http://www.jneurosci.org/cgi/content/full/26/34/8774#F1)]. However, the duration of the disease, as defined by the time between disease onset and end stage, remained unchanged by Bax deletion similar to the results from Bcl-2 overexpression in SOD1 mutant mice, which causes a similar delay in disease (Kostic et al., 1997). Importantly, the authors observed a 50% reduction in the number of motor neurons (MNs) within the spinal cord in end-stage Bax wild-type (WT)/SOD1 mutants compared with controls but no loss in Bax KO/SOD1 mutants, demonstrating that Bax completely rescues MN cell death [Gould et al., their Fig. 2 (http://www.jneurosci.org/cgi/content/full/26/34/8774#F2)]. Because Bax deficiency is sufficient to protect MNs from mutant SOD1, Bax-mediated cell death may be the primary mechanism of cell death in this model of ALS. Furthermore, this suggests that Bak (Bcl-2-antagonist/killer 1), which is functionally similar to Bax, may not compensate for Bax deletion in MNs, at least in terms of the execution of cell death. Interestingly, the MNs from both mutants revealed a similar decrease in somal area, suggesting that neuronal atrophy is unaffected by Bax deletion.
A crucial observation made by Gould and colleagues was a marked delay in the onset of denervation at the neuromuscular junctions (NMJs) in Bax KO/SOD1 mice compared with Bax WT/SOD1 mice (Fig. 1) [Gould et al., 2006, their Fig. 3E (http://www.jneurosci.org/cgi/content/full/26/34/8774#F3)]. This denervation coincided with another disease marker for ALS, mitochondrial vacuolization within MN presynaptic terminals, which was delayed by Bax deletion [Gould et al., 2006, their Fig. 7 (http://www.jneurosci.org/cgi/content/full/26/34/8774#F7)]. Furthermore, Bax deletion delayed the loss of myelinated axons in the spinal cord and target tissue [Gould et al., 2006, their Table 2 (http://www.jneurosci.org/cgi/content/full/26/34/8774#T1)]. By end stage, however, mitochondrial vacuolization and loss in innervated synapses and percentage loss of myelinated axons were similar in Bax WT/SOD1 and Bax KO/ SOD1 mutants [Gould et al., 2006, their Table 1 (http://www.jneurosci.org/cgi/content/full/26/34/8774#T1), Figs. 3B,E (http://www.jneurosci.org/cgi/content/full/26/34/8774#F3), 4 B (http://www.jneurosci.org/cgi/content/full/26/34/8774#F4), 7 (http://www.jneurosci.org/cgi/content/full/26/34/8774#F7)], suggesting that a Bax-independent pathway may mediate these degenerative processes. Strikingly, accumulation of SOD1 aggregates in MNs occurred earlier in Bax WT/SOD1 mice than in Bax KO/SOD1 mice [Gould et al., 2006, their Fig. 5C (http://www.jneurosci.org/cgi/content/full/26/34/8774#F5)]. Because SOD1 aggregation was associated with denervation, the authors suggest that withdrawal of axons promotes protein accumulation. However, it remains unclear whether Bax is directly involved in these processes. Finally, a subtle delay in astrogliosis, a surrogate marker of neurodegeneration, was observed in Bax KO/SOD1 mice relative to controls [Gould et al., 2006, their Fig. 6 (http://www.jneurosci.org/cgi/content/full/26/34/8774#F6)]. Neuromuscular denervation precedes SOD1 aggregation and gliosis, suggesting that axonal retraction from target tissue is the primary insult in this model of ALS. These observations further suggest that the delayed disease onset and prolonged survival of Bax KO/SOD1 mice results from delayed denervation. Because deletion of Bax only delays but does not rescue MN denervation, there are clearly other factors responsible for the retraction of synapses from the NMJ. These results clearly demonstrate that neuromuscular denervation is a key pathological event in ALS rather than MN cell death.
Figure 1.
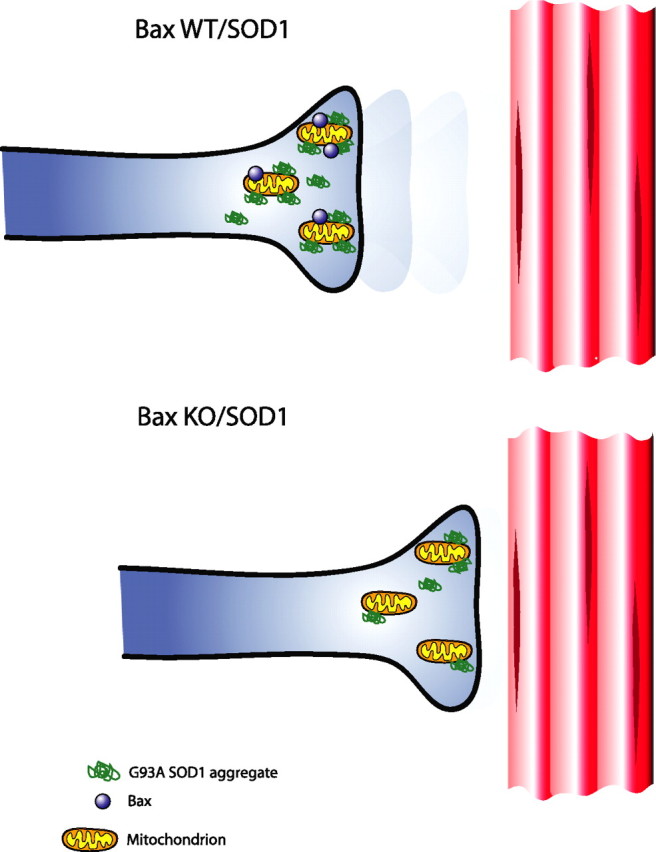
Bax deletion delays denervation of neuromuscular junctions. A neuromuscular junction in a Bax WT/SOD1 mutant is presented in the top. Note the retraction of the synapse from the muscle, whereas in Bax KO/SOD1 mutant (bottom), the muscle remains innervated. In addition, there are fewer SOD1 protein aggregates in the Bax KO/SOD1 mutant at this time point.
The molecular mechanism(s) by which Bax affects axonal retraction remains a mystery. Given reports regarding a biochemical (Pasinelli et al., 2004) and genetic (Kostic et al., 1997) interaction between Bcl-2 and SOD1 along with the results described above, it is tempting to suggest a simple model for mutant SOD1 toxicity. In this model, mutant SOD1 would sequester Bcl-2, freeing Bax from inhibition and thereby promoting mitochondrial permeabilization. This model would explain the partial rescues of the disease phenotype during Bcl-2 overexpression or Bax deletion. However, Gould et al. fail to detect such an interaction by immunoprecipitation [Gould et al., 2006, their Fig. 1E–G (http://www.jneurosci.org/cgi/content/full/26/34/8774#F1)], in contrast to what has been reported (Pasinelli et al., 2004).
In summary, Bax deletion led to expected and unexpected results. Bax deletion protected MNs against cell death, consistent with what has been observed in several other cell death paradigms, but also led to a delay in the onset of denervation, protein aggregation, gliosis, and mitochondrial vacuolation. According to the authors, this suggests a novel nonapoptotic function for Bax in all of these processes. Several key questions remain: How does Bax promote denervation, SOD1 aggregation, gliosis, and mitochondrial vacuolation? (Fig. 1). Likely, Bax is not directly involved in all of these processes. With regard to the suggested nonapoptotic role of Bax in denervation, why was the delay so moderate? What other components of the cell death machinery are upstream or downstream of Bax in ALS? Interestingly, a dominant-negative mutant of caspase-1 delayed disease in the SOD1(G93A) transgenic mice, but deletion of caspase-11, a critical activator of caspase-1, did not alter the phenotype in this same ALS model (Kang et al., 2003 and references therein). It is likely that multiple pathways affect denervation of NMJs in ALS.
There is a growing consensus that neuronal dysfunction, which is of critical importance for the phenotypes observed in neurodegenerative disease, long precedes neuronal death. Future studies will determine whether Bax plays a broad role in the death and dysfunction of neurons in a range of neurodegenerative diseases. In the case of prion diseases, which are very distinct protein misfolding disorders from ALS, Bax deletion in a transgenic model of inherited prion disease completely rescued cell loss without modulating the disease phenotype (Chiesa et al., 2005). However, in the case of infectious prion disease, Bax deletion had no consequences on phenotype or pathology (Coulpier et al., 2006). Thus, the role(s) of Bax in neuronal dysfunction and/or death may be dependent on the disease context. It is very intriguing that Bax may have a role in both the execution of cell death and cellular dysfunction. Future studies may tease apart these seemingly discrete functions and possibly broaden our understanding of “apoptotic” proteins in roles beyond cell death.
Footnotes
We are grateful to Tom DiCesare for assistance with graphics, James Shorter for critical comments, and Susan Lindquist (A.D.S.) and Junying Yuan (C.H.Y.) for mentorship.
Editor's Note: These short reviews of a recent paper in the Journal, written exclusively by graduate students or postdoctoral fellows, are intended to mimic the journal clubs that exist in your own departments or institutions. For more information on the format and purpose of the Journal Club, please see http://www.jneurosci.org/misc/ifa_features.shtml.
References
- Bruijn et al., 2004.Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Chiesa et al., 2005.Chiesa R, Piccardo P, Dossena S, Nowoslawski L, Roth KA, Ghetti B, Harris DA. Bax deletion prevents neuronal loss but not neurological symptoms in a transgenic model of inherited prion disease. Proc Natl Acad Sci USA. 2005;102:238–243. doi: 10.1073/pnas.0406173102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulpier et al., 2006.Coulpier M, Messiaen S, Hamel R, Fernandez de Marco M, Lilin T, Eloit M. Bax deletion does not protect neurons from BSE-induced death. Neurobiol Dis. 2006;23:603–611. doi: 10.1016/j.nbd.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Gould et al., 2006.Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney et al., 1994.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Kang et al., 2003.Kang SJ, Sanchez I, Jing N, Yuan J. Dissociation between neurodegeneration and caspase-11-mediated activation of caspase-1 and caspase-3 in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2003;23:5455–5460. doi: 10.1523/JNEUROSCI.23-13-05455.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic et al., 1997.Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–562. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- Pasinelli et al., 2004.Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Yuan et al., 2003.Yuan J, Lipinski M, Degterev A. Diversity in the mechanisms of neuronal cell death. Neuron. 2003;40:401–413. doi: 10.1016/s0896-6273(03)00601-9. [DOI] [PubMed] [Google Scholar]