

Abstract
An emerging area of research has documented a novel role for the plasminogen activation system in the regulation of neurotransmitter release. Prohormones, secreted by cells within the sympathoadrenal system, are processed by plasmin to bioactive peptides that feed back to inhibit secretagogue-stimulated release. Catecholaminergic cells of the sympathoadrenal system are prototypic prohormone-secreting cells. Processing of prohormones by plasmin is enhanced in the presence of catecholaminergic cells, and the enhancement requires binding of plasmin(ogen) to cellular receptors. Consequently, modulation of the local cellular fibrinolytic system of catecholaminergic cells results in substantial changes in catecholamine release. However, mechanisms for enhancing prohormone processing and cell-surface molecules mediating the enhancement on catecholaminergic cells have not been investigated. Here we show that plasminogen activation was enhanced >6.5-fold on catecholaminergic cells. Carboxypeptidase B treatment decreased cell-dependent plasminogen activation by ∼90%, suggesting that the binding of plasminogen to proteins exposing C-terminal lysines on the cell surface is required to promote plasminogen activation. We identified catecholaminergic plasminogen receptors required for enhancing plasminogen activation, using a novel strategy combining targeted specific proteolysis using carboxypeptidase B with a proteomics approach using two-dimensional gel electrophoresis, radioligand blotting, and tandem mass spectrometry. Two major plasminogen-binding proteins that exposed C-terminal lysines on the cell surface contained amino acid sequences corresponding to β/γ-actin. An anti-actin monoclonal antibody inhibited cell-dependent plasminogen activation and also enhanced nicotine-dependent catecholamine release. Our results suggest that cell-surface-expressed forms of actin bind plasminogen, thereby promoting plasminogen activation and increased prohormone processing leading to inhibition of neurotransmitter release.
Keywords: chromaffin cell, plasminogen, release, actin, nicotinic, catecholamine
Introduction
An emerging area of research has demonstrated a novel role for the serine protease plasmin as a prohormone-processing protease in the neuroendocrine system (Parmer et al., 2000; Hoover-Plow et al., 2001; Jiang et al., 2001; Colombo et al., 2002; Q. Jiang et al., 2002; Pang et al., 2004; N. Wang et al., 2004). For example, prohormones, secreted by cells within the sympathoadrenal system, are processed by plasmin to bioactive peptides that feed back to inhibit secretagogue-stimulated catecholamine release (Parmer et al., 2000; Jiang et al., 2001; Q. Jiang et al., 2002). Catecholaminergic cells of the sympathoadrenal system, including chromaffin cells of the adrenal medulla and sympathetic neurons, are prototypic prohormone-secreting cells that concomitantly secrete the plasminogen activator tissue plasminogen activator (t-PA), in response to specific secretagogue stimulation (Gualandris et al., 1996; Parmer et al., 1997). The processing of prohormones by plasmin is markedly enhanced when plasminogen is bound to the catecholaminergic cell surface (Parmer et al., 2000; Miles et al., 2002). Consequently, positive and negative modulation of the local cellular fibrinolytic system of catecholaminergic cells results in substantial changes in catecholamine release (Parmer et al., 2000). The mechanisms for enhancing prohormone processing and cell-surface molecules mediating the enhancement on the catecholaminergic cell surface have not been elucidated. Therefore, in the present study we investigated the ability of plasminogen binding sites on catecholaminergic cells to promote plasminogen activation as a mechanism to enhance local plasmin production and regulate neurotransmitter release. We found that plasminogen activation was markedly stimulated on the catecholaminergic cell surface via a specific interaction with plasminogen binding sites.
Catecholaminergic cells, including PC12 pheochromocytoma cells and bovine chromaffin cells, have a high capacity for plasminogen (Parmer et al., 2000; Miles et al., 2002). Therefore, no single molecule can account for the entire plasminogen binding capacity of these cells. Therefore, we focused specifically on identification of the plasminogen binding sites that stimulate plasminogen activation. Carboxypeptidase B (CpB) treatment decreased cell-dependent plasminogen activation by ∼90%, suggesting that the binding of plasminogen to proteins exposing C-terminal lysines on the cell surface is required to promote plasminogen activation. Therefore, we used a novel strategy, combining targeted specific proteolysis with CpB with a proteomics approach using two-dimensional gel electrophoresis (2D-PAGE), radioligand blotting, and tandem mass spectrometry and identified cell-surface forms of actin as major plasminogen receptors responsible for stimulation of plasminogen activation, leading to prohormone processing and inhibition of catecholamine release by catecholaminergic cells. The results of our study define a key cell-surface-dependent mechanism underlying the ability of neurosecretory cells to locally promote plasminogen activation for enhancement of prohormone processing and modulation of neurotransmitter release.
Materials and Methods
Proteins and peptides.
Glu-plasminogen was purified from fresh human blood as described previously (Deutsch and Mertz, 1970; Parmer et al., 2000). Peptides were synthesized and purified as in our previous studies (Beebe et al., 1989; Jiang et al., 2001).
Cells.
PC12 cells derived from a rat pheochromocytoma (Greene and Tischler, 1976) were obtained from Dr. D. Schubert (Salk Institute, La Jolla, CA) and were grown as described in DMEM supplemented with 5% fetal calf serum, 10% horse serum, 100 U/ml penicillin G, and 100 μg/ml streptomycin at 37°C, 6% CO2 (Parmer et al., 1993, 1997, 2000; Jiang et al., 2001).
Bovine chromaffin cells were isolated from bovine adrenal glands as described previously (Parmer et al., 1997, 2000; Miles et al., 2002). The cells were cultured in minimal essential medium containing 1% nonessential amino acids, 1% l-glutamine, 10% fetal calf serum, 1% amphotericin B, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Before CpB treatment, subconfluent, adherent PC12 cells that had been cultured for 48 h without a change of media were harvested by rinsing flasks twice with PBS at 4°C and detached with 5 mm EDTA/PBS at 37°C for 5 min. The cells were resuspended in 20 mm HEPES (HBSS) at a final concentration of 3 × 107 cells/ml, and CpB (Boehringer Mannheim, Indianapolis, IN) was added to a final concentration of 100 U/ml. The cells were incubated for 30 min at 37°C with gentle agitation every 5 min. Control cells were incubated with an equivalent volume of PBS.
Before subcellular fractionation, cells were washed three times in HBSS and resuspended at 3 × 107 cells/ml in 20 mm HEPES, pH 7.2, containing the protease inhibitors 2 μm leupeptin (Calbiochem, La Jolla, CA), 1.5 μm pepstatin A (Calbiochem), 50 kIU/ml Trasylol, 8 μm 2-guanidinoethylmercaptosuccinic acid (Calbiochem), and 2 mm PMSF. The cells were chilled on ice for 5 min and sheared in a 7 ml Dounce homogenizer (Kontes, Vineland, NJ). After homogenization, an equivalent volume of 20 mm HEPES containing 0.5 m sucrose, 10 mm MgCl2, 0.1 m KCl, 2 mm CaCl2, and the protease inhibitors above was added to the cell homogenate. Cell debris was pelleted by centrifugation at 500 × g, and the supernatant was then centrifuged at 100,000 × g for 1 h. The membrane pellet was washed by centrifugation three times with 20 mm HEPES containing 0.25 m sucrose, 5 mm MgCl2, 0.2 m KCl, 1 mm CaCl2, and the protease inhibitors above.
Ligand binding assays.
Ligand binding assays were performed as described previously (Parmer et al., 2000) with PC12 cells in suspension (0.5–1.0 × 107 cells/ml) in HBSS containing 0.1% bovine serum albumin (BSA) in a volume of 200 μl in 1.5 ml polypropylene tubes with 0.2 μm 125I-plasminogen. Bound and free ligand were separated by layering three 50 μl aliquots from each reaction mixture over 300 μl of 20% sucrose, centrifuging for 2 min, and cutting off the tube tips. Nonspecific binding was determined as counts bound in the presence of 0.2 m ε-aminocaproic acid (EACA), and specific binding was determined by subtracting nonspecific binding from total binding.
Plasminogen activation assays.
For plasminogen activation assays, cells were preincubated with 2.7 μm glu-plasminogen at 37°C for 30 min. Then 20 nm single-chain recombinant human t-PA (Genentech, South San Francisco, CA) was added. Plasmin activity (expressed as OD 405 nm) was measured after 6 min by diluting the reaction mixture 1:10 into S-2251 (DiaPharma Group, Franklin, OH) to a final concentration of 1 mm and monitoring absorbance at 405 nm as described previously (Felez et al., 1996).
Fluorescence-activated cell-sorting analysis.
Subconfluent, adherent PC12 cells that had been cultured for 48 h without a change of medium were harvested by rinsing flasks twice with PBS at 4°C and detached with 5 mm EDTA/PBS at 37°C for 5 min. All fluorescence-activated cell-sorting (FACS) analyses were performed as described previously (Ranson et al., 1998). Briefly, for the detection of cell-surface actin on viable PC12 cells and viable bovine adrenal chromaffin cells, indirect immunofluorescence staining and dual-color FACS analyses were performed. [Bovine adrenal chromaffin cells were identified with an anti-CD56 antibody (Exalpha Biological, Watertown, MA) and gated after plotting forward scatter versus side scatter as described previously (Muench et al., 2003).] Cells (2 × 105) were incubated with 60 μg/ml of either an anti-actin monoclonal antibody (clone 4, IgG1K; Chemicon, Temecula, CA) or an irrelevant IgG1K isotype control, MOPC-21C (Sigma, St. Louis, MO), for 30 min in binding buffer (HBSS containing 0.1% BSA) at 4°C. The cells were washed three times with 200 μl of binding buffer and incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (1:50 dilution of stock in binding buffer) for 30 min at 4°C in the dark. The cells were washed again, resuspended in 200 μl of binding buffer containing the non-vital dye propidium iodide (PI) at 5 μg/ml, and immediately analyzed by dual-color FACS as described above. Populations of cells were gated according to the fluorescence intensity of PI staining. The population of cells with low cell-associated PI fluorescence intensity (cells that excluded PI) were defined as viable cells, whereas the population of cells with high PI fluorescence intensity (inclusion of PI) were defined as non-viable.
Quantitative flow cytometry.
Quantitative flow cytometric equilibrium binding of an FITC-conjugated anti-actin Fab fragment (Sigma) to the cells was performed as described previously (Waller et al., 2001). Briefly, the output from the flow cytometer was standardized into mean equivalent standard fluorescence units (MESF) using beads impregnated with different MESF units of FITC as described previously (Waller et al., 2001). The fluorescence intensity change of the FITC-conjugated anti-actin Fab fragment conformational change (Q = DIf/DIo, where Ii and If are the initial and final fluorescence intensities of FITC–anti-actin, respectively) was determined using a F500 fluorometric plate reader.
Two-dimensional gel electrophoresis.
Denaturing first-dimension isoelectric focusing (IEF) was performed in a vertical slab format. The IEF gel mixture contained 2% Triton X-100 and 9 m urea with 2.0% Bio-Lytes (Bio-Rad, Hercules, CA), pH 6–8, in a 4% polyacrylamide gel with piperazine diacrylamide (Bio-Rad) as a cross-linker. Either 100 μg of membrane proteins or 10 μg of cytoplasmic proteins in 0.25% SDS, 2% Triton X-100, 9 m urea, and 2% ampholines was loaded onto the IEF gels and focused under constant voltage using a stepped-voltage gradient from 50 to 250 V for a total of 4000 V-hours, with the current never exceeding 15 mA. The IEF gels were fixed in 12% trichloroacetic acid and washed six times with 50 ml of H2O. Individual lanes were excised from the gels and soaked in reduced sample buffer to resolubilize the proteins. Each lane was placed on the second dimension gel and overlaid with molten 0.5% agarose in 120 mm Tris, pH 6.8. Second-dimension SDS-PAGE was performed according to the method of Laemmli (1970).
Ligand blotting.
Samples were subjected to 2D-PAGE and transferred to Immobilon P (Millipore, Bedford, MA) at 100 mA for 18 h in either 25 mm Tris-HCl containing 192 mm glycine and 10% methanol or 10 mm CAPS (3-(cyclohexylamino)propanesulfonic acid) buffer, pH 11, with 10% methanol. Blots were blocked for 2 h in 2% BSA in PBS containing 0.1% Tween 20 (PBS-BSA) and incubated with 50 nm 125I-plasminogen in PBS-BSA for 2 h at 22°C. The blots were washed seven times with PBS-BSA containing 0.5 m NaCl, dried, and subjected to autoradiography. Control blots were incubated with 50 nm 125I-plasminogen in the presence of 0.1 m EACA. Autoradiography was performed using BioMax MS film (Eastman Kodak, Rochester, NY). Autoradiograms were digitized and analyzed by two-dimensional spot densitometry (Alpha Innotech, San Leandro, CA) to determine differences in spot density.
Secretagogue-stimulated catecholamine release.
Chromaffin cell catecholamine secretion was determined as described previously (Parmer et al., 1993, 1997, 2000; Jiang et al., 2001). Briefly, PC12 cells were labeled for 2 h with [3H]norepinephrine (PerkinElmer Life Sciences, Boston, MA) at 1 μCi/ml in cell culture medium, washed twice with release buffer (150 mm NaCl, 5 mm KCl, 2 mm CaCl2, and 10 mm HEPES, pH 7.0), and incubated at 37°C for 30 min in release buffer in either the presence or absence of nicotine. After aspirating the release buffer, the cells were harvested and lysed in release buffer containing 0.1% Triton X-100. [3H]norepinephrine content of release buffer, and cell lysates were determined by liquid-scintillation counting. The percentage of release was calculated as percentage of secretion [amount released/(amount released + amount in cell lysate)], and results are expressed as net release (percentage of secretagogue-stimulated release − percentage of basal release).
Protein determinations.
Protein concentrations were determined by the BCA protein assay (Bio-Rad) using BSA as a standard.
Statistics.
Data are presented as means ± SEM. Results were analyzed by ANOVA, followed by Student–Newman–Keuls post hoc tests for multiple comparisons.
Results
Cellular augmentation of plasminogen activation on catecholaminergic cells
Prohormone processing by the serine protease plasmin is enhanced in the presence of catecholaminergic cells (Parmer et al., 2000). Therefore, as a mechanism to promote processing, we tested whether plasminogen activation is enhanced when plasminogen is bound to PC12 pheochromocytoma cells. In the presence of cells, plasminogen activation was markedly increased in a cell concentration-dependent manner, reaching a plateau at 1.67 × 105 cells/ml (Fig. 1A). At this saturating cell density, the enhancement caused by the presence of cells was 6.5-fold. PC12 cells synthesize and release t-PA in response to secretagogue stimulation (Gualandris et al., 1996; Parmer et al., 1997). Furthermore, the plasminogen activator urokinase and its receptor uPAR are present in these cells (Pittman et al., 1989; Fowler et al., 1998; Herschman et al., 2000). Therefore, we tested whether endogenous cellular plasminogen activators could contribute to the cell-dependent plasminogen activation. The rate of plasminogen activation in the presence of 3.3 × 105 cells/ml plus plasminogen without the addition of exogenous t-PA was only 13% of that in the presence of cells plus plasminogen plus t-PA. These data were consistent with the effect of the cells in enhancement of plasminogen activation, being primarily attributable to a cell-surface-dependent mechanism.
Figure 1.
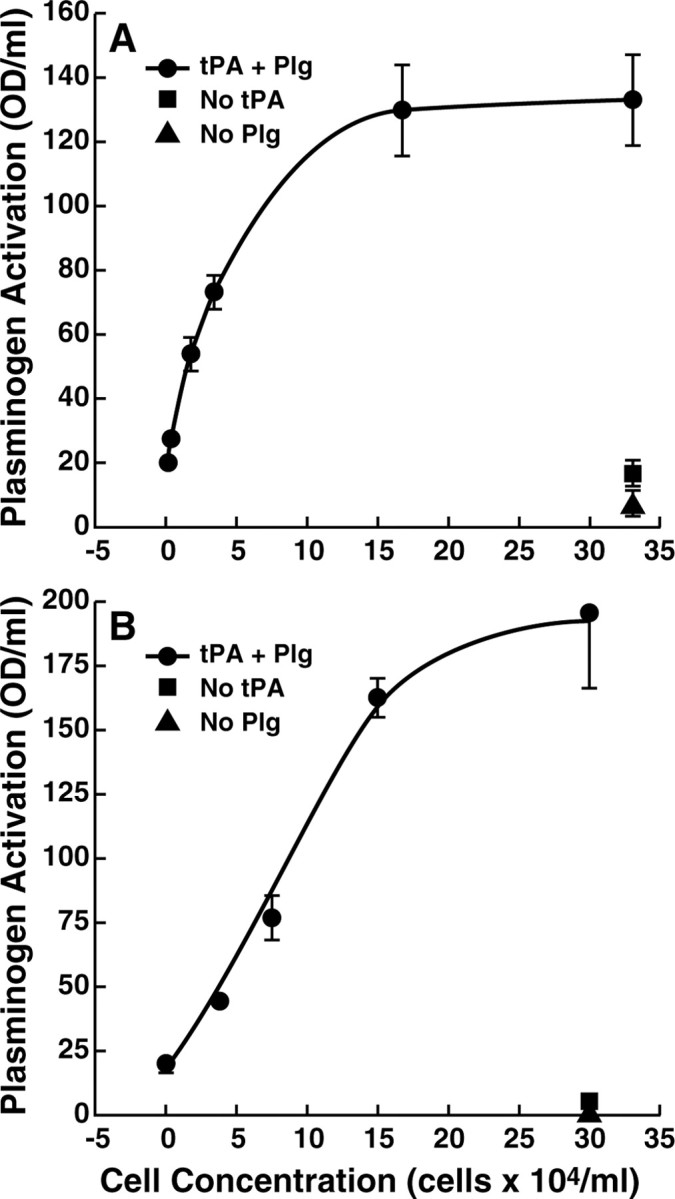
Cell-dependent plasminogen activation. PC12 cells (A) or bovine adrenal chromaffin cells (B) at increasing concentrations were preincubated with 2.7 μm plasminogen (Plg) for 30 min. Then, 20 nm t-PA was added, and plasminogen activation was measured as cleavage of the tripeptide substrate S2251 (1 mm) after 6 min (circles). The squares represent cells without the addition of t-PA. The triangles represent cells without the addition of plasminogen.
We performed similar cell-dependent plasminogen activation assays using primary bovine adrenal chromaffin cells that also express plasminogen binding sites (Miles et al., 2002). Bovine chromaffin cells markedly enhanced t-PA-dependent plasminogen activation in a dose-dependent manner, approaching a plateau above 1.5 × 105 cells/ml (Fig. 1B). At a saturating cell density of 3 × 105 cells/ml, the enhancement caused by the presence of the bovine chromaffin cells was ∼10-fold. As with the PC12 cells, the enhancement was not attributable to secretion of t-PA by the cells, because plasminogen activation in the presence of cells plus plasminogen only was not detected.
Role of C-terminal lysines in plasminogen binding and activation on the catecholaminergic cell surface
The interactions of plasminogen with substrates and regulatory molecules are dependent on lysine binding sites within the disulfide-bonded kringle structures of plasminogen, and therefore these interactions are inhibited in the presence of lysine and lysine analogs, such as EACA (for review, see Castellino and McCance, 1997). The interaction of plasminogen with catecholaminergic cells is also blocked by EACA (Parmer et al., 2000; Miles et al., 2002), suggesting that proteins with C-terminal lysines, exposed on the extracellular face of the cell membrane, might serve as plasminogen binding sites. To explore the relationship between plasminogen binding and stimulation of plasminogen activation by the cells, we examined the role of proteins with C-terminal lysines in the interactions of the cells with plasminogen. Intact cells were treated with increasing concentrations of CpB, which has a preference for cleavage at the carboxyl sides of lysyl and arginyl residues. A dose-dependent decrease in plasminogen binding was observed, reaching a plateau at ∼70% inhibition in the presence of 100 U/ml CpB (Fig. 2A). Interestingly, ∼30% of the plasminogen binding sites were not susceptible to CpB treatment, suggesting that this subpopulation of sites could bind plasminogen, but not in an orientation that would stimulate plasminogen activation.
Figure 2.

Effect of treatment of PC12 cells with CpB on the interaction with plasminogen. PC12 cells (1 × 107/ml) were incubated with increasing concentrations of CpB for 30 min at 37°C. The cells were washed six times with 1 ml of HBSS/0.1% BSA, and specific binding of 0.2 μm 125I-plasminogen (A) and plasminogen activation (B) were assessed as described in Materials and Methods. In B, the no-cell control is the plasminogen activation reaction performed in the absence of cells. We have shown previously that the number of molecules of plasminogen-bound/PC12 cells at 37°C is not decreased at 4°C (Parmer et al., 2000).
To determine whether the CpB-sensitive population of plasminogen-binding proteins was responsible for stimulating plasminogen activation on the cell surface, we examined whether CpB treatment of the PC12 cells affected the ability of the cells to promote plasminogen activation. CpB treatment decreased the stimulating effect of the cells on plasminogen activation in a dose-dependent manner, approaching a plateau at 88% inhibition of cell-dependent stimulation (Fig. 2B). This result suggests that the plasminogen receptors that are sensitive to CpB [i.e., proteins exposing C-terminal lysines (with the conformation of EACA) on the cell surface] are primarily responsible for the promotion of plasminogen activation by these cells. Although CpB also can remove C-terminal arginyl residues, the results are most consistent with an effect on a C-terminal lysyl residue because EACA interacts with plaminogen with an affinity in the range of the affinity of plasminogen for the cells (Violand et al., 1975; Markus et al., 1978; Parmer et al., 2000), whereas the affinity of arginine for plasminogen is ∼10-fold lower (Violand et al., 1975).
Identification of CpB-sensitive cell-surface plasminogen-binding proteins
We sought to identify the CpB-sensitive plasminogen-binding protein(s) on the surfaces of the PC12 cells as the major plasminogen receptors responsible for stimulating plasminogen activation on these cells. We wanted to identify only proteins that exposed C-terminal lysines on the cell surface and to exclude proteins present exclusively in the cytoplasm, proteins on the inner surface of the cell membrane, or inaccessible proteins on the cell surface. Therefore, intact cells were treated with 100 U/ml CpB, followed by washing. Then, cell membrane and cytoplasmic fractions were subsequently prepared, subjected to 2D-PAGE, and transferred to polyvinylidene difluoride membranes, and ligand blotted with 125I-plasminogen. Several plasminogen-binding proteins were present in the cell membrane preparations (Fig. 3A). [The binding of 125I-plasminogen was specific because no plasminogen binding spots were detected in the presence of 100 mm EACA (data not shown).] Two major spots, one with an Mrapp of 45.1 thousand and a pI of 5.27 (spot 1) and the other with an Mrapp of 34.7 thousand and a pI of 5.43 (spot 2), exhibited a marked reduction in plasminogen binding capacity after treatment of the intact cells with CpB. The susceptibility to treatment of intact cells with CpB suggested that these proteins exposed C-terminal lysines on the cell surface (Fig. 3, compare A, B). The extent of reduction in plasminogen binding was 89% for spot 1 and 71% for spot 2, based on laser densitometry of the gels. In controls for the method, plasminogen-binding proteins were present also in the cytoplasmic fractions but retained activity after CpB treatment of the intact cells, indicating that cytoplasmic proteins were not accessible to treatment of intact cells by CpB (Fig. 3, compare C, D). Hence, only proteins on the cell surface were accessible to CpB. A major advantage of using the criterion of CpB sensitivity of proteins on the surfaces of intact cells is that even if minor amounts of cytoplasm are present in the subsequently prepared membrane fraction, cytoplasmic contaminants are not identified as proteins exposing C-terminal lysines on the cell surface, because proteins in the cytoplasm are not processed by CpB before membrane fractionation.
Figure 3.

Broad pH range analysis and identification of plasminogen-binding proteins that expose C-terminal lysines on the PC12 cell surface. Intact PC12 cells were incubated either with 100 U/ml CpB (B, D) or buffer (A, C) for 30 min at 37°C before fractionation into membrane or cytosolic fractions. 2D-PAGE was performed on 100 μg of membrane proteins (A, B) or 10 μg of cytoplasmic proteins (C, D), followed by ligand blotting with 125I-plasminogen. In specificity controls, the binding of 125I-plasminogen in the ligand blots was specific because no plasminogen binding spots were detected in the presence of 0.1 m EACA.
We purified spot 1 and spot 2 directly from the two-dimensional gels. One hundred micrograms per lane of PC12 membrane fractions were subjected to 2D-PAGE, and replicate gels were either ligand blotted with 125I-plasminogen or stained with colloidal Coomassie. Two replicate colloidal Coomassie-stained gels were aligned with the 125I-plasminogen ligand blot. Spot 1 and spot 2 were excised, eluted from the gels, and digested with trypsin. A capillary reverse-phase chromatograph coupled to the electrospray ionization source of a Finnigan LCQ Quadrupole Ion Trap Mass Spectrometer was used to obtain 10 peptide sequences comprising a total of 128 unique amino acid sequences from spot 1 and 7 peptide sequences comprising a total of 79 unique amino acid residues from the tryptic digest of spot 2 (Table 1). The exact peptides (with one exception of an I for V75 substitution in spot 2) are present in the amino acid sequences of both rat β-actin and rat γ-actin. (Rat β- and γ-actin are identical, with the exception of a substitution of E2E3E4 in γ-actin for D2D3D4 in β-actin.) We did not obtain sequences corresponding to the extreme N terminus of either protein from the tryptic digests of spot 1 and spot 2. Therefore, we could not distinguish between the γ and β isoforms. Our data were also consistent with processing of native β/γ-actin to spot 2 by removal of both N- and C-terminal peptides because we did not obtain any sequences from the spot 2 digestion that were N terminal to D184; we did, however, obtain a peptide with an amino acid sequence corresponding to amino acids 360–372 of the β- and γ-actin sequences and thus lacking only the three most C-terminal amino acids. The absence of these three amino acids, together with the CpB sensititivy of spot 2, is consistent only with either actin residue K373 or R372 as the C terminus and thus consistent with exposure of either K373 or R372 on the cell surface. Likewise, sequencing of spot 1 also yielded a peptide corresponding to amino acids 360–372 of the β- and γ-actin sequences and therefore is also consistent only with exposure of either K373 or R372 on the cell surface. The results are most consistent with cell-surface exposure of K373 (with the structure of EACA) because EACA interacts with plaminogen with an affinity, in the range of the affinity of plasminogen for the cells (Violand et al., 1975; Markus et al., 1978; Parmer et al., 2000), whereas the affinity of arginine for plasminogen is ∼10-fold lower (Violand et al., 1975).
Table 1.
Tandem mass spectrometry peptide sequences of tryptic digests of spot 1 and spot 2
| Spot 1 | Spot 2 |
|---|---|
| (K) 51DSYVGDEAQSK61 | (R) 184DLTDYLMK191 |
| (K) 69YPIEHGIITNWDMMEK82 | (R) 197GYSFTTTAER206 |
| (K) 85IWHHTFYNELR95 | (K) 239SYELPDGQVITIGNER254 |
| (R) 96VAPEEHPVLLTEAPLNPK113 | (R) 291KDLYANTVLSGGTTMYPGIADR322 |
| (R) 197GYSFTTTAER206 | (K) 292DLYANTVLSGGTTMYPGIADR322 |
| (K) 239SYELPDGQVITIGNER254 | (K) 316EITALAPSTMK326 |
| (R) 291KDLYANTVLSGGTTMYPGIADR312 | (K) 360QEYDESGPSIVHR372 |
| (N) 297TVLSGGTTMYPGIADR313 | |
| (K) 316EITALAPSTMK326 | |
| (K) 360QEYDESGPSIVHR372 |
Because all isoforms of actin perform major intracellular functions (Kabsch and Vandekerckhove, 1992), we used FACS analysis with an anti-actin monoclonal antibody as a second method to verify the cell-surface expression of actin. When viable cells were incubated with a fluoresceinated Fab fragment of an anti-actin monoclonal antibody, the mean fluorescence intensity of the cell population was increased twofold over the isotype control, demonstrating the presence of actin on the cell surface (Fig. 4). FACS data from the viable populations of cells that were treated with different concentrations of FITC-conjugated anti-actin Fab were used to derive the apparent Kd and Bmax of the interaction of the antibody with the cells by fitting these data (after subtracting the fluorescence of the FITC-conjugated MOPC21C isotype control) to the single-site binding equation [LR] = ([L]Bmax)/([L] + Kd). In quantitative flow cytometry with the FITC-labeled monoclonal anti-actin Fab fragment, a Bmax of 9.8 × 104 molecules of actin was determined per cell. The apparent Kd for the interaction of the anti-actin Fab fragment with cell-surface actin was 645 nm.
Figure 4.

Expression of cell-surface actin on viable PC12 cells. A histogram plot of viable PC12 cells (i.e., the PI-negative population) incubated with an anti-actin antibody is shown. PC12 cells were incubated with 0.4 μm of either an anti-actin-specific monoclonal antibody (right trace) or an irrelevant isotype control antibody (left trace), washed, and incubated with an FITC-labeled secondary antibody. The labeled cells were resuspended in binding buffer containing PI and analyzed by dual-color flow cytometry. FACS analysis of actin expression on viable PC12 cells is shown.
Actin expression on the surfaces of primary adrenal chromaffin cells was also evaluated. Primary adrenal chromaffin cells were identified by CD56 expression and plotting forward scatter versus side scatter as described previously (Muench et al., 2003). Viable primary bovine adrenal chromaffin cells bound actin specifically (mean fluorescence intensity obtained with the fluoresceinated Fab fragment of an anti-actin monoclonal was twofold greater than that of the isotype control). Furthermore, FITC-plasminogen binding to the primary bovine adrenal chromaffin cells was decreased by 22 ± 1% (n = 3; p = 0.001) in the presence of the anti-actin monoclonal antibody (400 nm). Correspondingly, in the presence of the monoclonal antibody, cell-surface-dependent plasminogen activation was decreased by 23 ± 1% (n = 3; p < 0.001).
Role of cell-surface actin in catecholamine secretion
Because localization of plasminogen on catecholaminergic cells results in processing of chromogranin A (CgA) (released from catecholamine storage vesicles after secretagogue stimulation) to bioactive peptides that inhibit secretagogue-stimulated catecholamine release (Parmer et al., 2000; Jiang et al., 2001), we tested whether antibodies against actin could modulate secretagogue-stimulated catecholamine release from catecholaminergic cells. Accordingly, PC12 cells were preloaded with [3H]norepinephrine and stimulated with the chromaffin cell secretagogue nicotine in the presence or absence of an anti-actin monoclonal antibody or the isotype (IgG1κ) control (MOPC-21C). In the presence of the anti-actin antibody, norepinephrine release in response to nicotine stimulation was enhanced in a dose-dependent manner, with release increased by 40% compared with the isotype control (MOPC-21C) at an antibody concentration of 400 nm (Fig. 5). Consistent with these results, when plasminogen activation assays were performed (as in Fig. 1) in the presence of the anti-actin monoclonal antibody, plasminogen activation was decreased by 31.4 ± 0.3% (n = 3; p < 0.001) compared with the isotype control. These results are consistent with decreased plasminogen activation and, consequently, decreased production of inhibitory peptides. We have shown previously that plasmin processes the prototypical prohormone CgA to a specific peptide [ARAYGFRGPGPQLR, corresponding to human CgA-(360–373)] that inhibits nicotinic-mediated catecholamine release (Jiang et al., 2001; Q. Jiang et al., 2002). Consistent with the foregoing results, in the presence of 10 μm of the synthetic peptide ARAYGFRGPGPQLR, the stimulatory effect of the anti-actin antibody was markedly inhibited (by 73.9 ± 6.1%; n = 3; p < 0.001). In control experiments, the presence of the reverse peptide RLQPGPGRFGYARA had no effect on the stimulatory effect of the anti-actin antibody.
Figure 5.

Effect of an anti-actin monoclonal antibody on catecholamine release. PC12 cells were preincubated with either an anti-actin monoclonal antibody (■) or the isotype control (MOPC-21C; □) for 30 min and treated with 60 μm nicotine or untreated at 37°C for 15 min, and catecholamine release was measured as described in Materials and Methods. Results are mean ± SEM; n = 3 for each experimental group. **p < 0.01 for the anti-actin antibody compared with corresponding values for the MOPC-21C isotype control.
Discussion
Processing of secreted prohormones is a recently demonstrated function of the enzyme plasmin (Parmer et al., 2000; Hoover-Plow et al., 2001; Jiang et al., 2001; Colombo et al., 2002; Q. Jiang et al., 2002; Pang et al., 2004; N. Wang et al., 2004). Notably, plasmin processes the prototypical prohormone CgA to a specific peptide [human CgA-(360–373)] that inhibits nicotinic-mediated catecholamine release (Parmer et al., 2000; Jiang et al., 2001). Previously, we found that plasminogen binding sites on chromaffin cells markedly stimulate plasmin processing of CgA (Parmer et al., 2000). These interactions in the environment of catecholaminergic cells represent a novel autocrine/paracrine system that has a substantial impact on catecholamine secretion and may represent a broad paradigm for neurosecretory processing throughout the neuroendocrine system. Therefore, in this study, we investigated the mechanism by which catecholaminergic cells enhance prohormone processing and identified plasminogen receptors responsible for stimulation of plasminogen activation on these cells. We found that (1) plasminogen activation was markedly stimulated when plasminogen was bound to catecholaminergic cells, (2) proteins exposing C-terminal lysines on the cell surface were responsible for ∼90% of the ability of the cells to stimulate plasminogen activation, (3) two isoforms of β/γ-actin provided major plasminogen binding sites responsible for stimulation of plasminogen activation on these cells, (4) in the presence of specific anti-actin antibody catecholaminergic cell-dependent plasminogen activation was markedly decreased, and (5) treatment of catecholaminergic cells with anti-actin antibody resulted in marked enhancement of secretagogue-stimulated catecholamine release. Together, these studies suggest the presence of cell-surface forms of actin that provide major plasminogen binding sites and promote plasminogen activation on catecholaminergic cells, resulting in production of inhibitory peptides that modulate catecholamine release during stimulation with secretagogues.
In a previous study, we found that processing of CgA is markedly enhanced in the presence of catecholaminergic cells and is dependent on the specific interaction of plasminogen with the cells (Parmer et al., 2000). In the current study, our data show that the major effect of the catecholaminergic cells was to promote plasminogen activation. The enhancement in activation required the interaction of plasminogen with cell-surface proteins exposing C-terminal lysines, because ∼90% of the cell-dependent stimulation was lost after treatment of the cells with CpB. Of plasminogen binding sites on the cells [i.e., ∼4 × 105 high-affinity sites (Parmer et al., 2000)], 70% were accessible to CpB treatment of intact cells. Using quantitative FACS analysis, we determined 9.8 × 104 molecules of surface-associated actin per cell. Thus, cell-surface actin represents ∼35% of the CpB susceptible cell-surface plasminogen binding sites. Furthermore, we found that an anti-actin antibody blocked 31.4% of cell-dependent stimulation of plasminogen activation. Thus, although plasminogen is expected to bind to other proteins exposing C-terminal lysines on the cell surface, cell-surface actin appears to be responsible for a major effect on cell-dependent plasminogen activation and, consequently, CgA processing to produce peptides that inhibit secretagogue-stimulated catecholamine release.
In the current study, we have used a novel method to define plasminogen receptors that is designed to exclusively identify molecules that expose a C-terminal lysine on the cell surface and thus stimulate plasminogen activation (Hawley et al., 2000, 2001). In this method, intact cells are treated with CpB, followed by preparation of membrane fractions. A comparison of 125I-plasminogen ligand blots of untreated with CpB-treated membranes reveals the plasminogen-binding proteins that expose C-terminal lysines on the cell surface. This method eliminates from consideration any plasminogen binding proteins on the inner surface of the cell membrane, inaccessible proteins on the cell surface, or proteins present exclusively in the cytoplasmic compartment. Using this method, we identified two major plasminogen-binding proteins that expose C-terminal lysines on the PC12 cell surface. The two proteins were eluted from gels, digested with trypsin, analyzed by mass spectrometry, and identified as isoforms of β/γ-actin. Our data were also consistent with loss of an ∼10,000 fragment at the N terminus of full-length actin (spot 1), resulting in the 34.7 thousand actin form (spot 2). The Mrapp of spot 2 at 34.7 thousand is consistent with proteolytic processing within the protease sensitive actin subdomain 2 (Jacobson and Rosenbusch, 1976; Mornet et al., 1981; Muhlrad et al., 2004).
Our data suggested that the plasminogen-binding proteins identified as rat β/γ-actin were present on the cell surface and accessible to CpB. The cDNA sequences of these actin isoforms do not encode C-terminal lysines. Therefore, processing at the C terminus of actin is necessary to expose the C-terminal lysine. Our mass spectrometric sequencing results are consistent with exposure of either actin residue, K373 or R372, on the cell surface after proteolytic processing. The results are most consistent with cell-surface exposure of K373 because EACA (with an equivalent structure to a C-terminal lysine in a protein) interacts with plasminogen with an affinity in the range of the affinity of plasminogen for the cells (Violand et al., 1975; Markus et al., 1978; Parmer et al., 2000), whereas the affinity of arginine for plasminogen is ∼10-fold lower than that of EACA (Violand et al., 1975).
Our results suggest a major extracellular function of native β/γ-actin and a processed form of β/γ-actin on the catecholaminergic cell surface, based on the ability to bind plasminogen and stimulate plasminogen activation, leading to inhibitory effects on catecholamine release. As an independent approach, we demonstrated the presence of actin on the catecholaminergic cell surface using FACS analyses. Several reports suggest the presence of a cell-surface form of actin on other cell types (Owen et al., 1978; Bachvaroff et al., 1980; Sanders and Craig, 1983; Moroianu et al., 1993; Dudani and Ganz, 1996; Andronicos and Ranson, 2001; Dudani et al., 2005). Actin mediates autoproteolysis of plasminogen to angiostatin on cancer cells (H. Wang et al., 2004, 2006). The cDNA sequence of actin does not encode a classical signal sequence. Nonetheless, actin is present in the circulation of healthy subjects (Thorstensson et al., 1982; Emerson et al., 1983; Mejean et al., 1987) and release of actin from viable cultured myoblasts has been demonstrated (Rubenstein et al., 1982), consistent with direct translocation of actin to the cell membrane or release of actin, followed by its membrane localization, although the mechanisms are not well understood. It is well known that a number of proteins lacking cleavable signal sequences are expressed extracellularly (Muesch et al., 1990).
Our results suggest that cell-surface forms of actin bind plasminogen and stimulate plasminogen activation on the catecholaminergic cell surface. The Kd for the interaction of plasminogen with purified actin is ∼70–140 nm (Dudani and Ganz, 1996; Wang et al., 2006). These values are consistent with the high-affinity interaction of plasminogen that we have determined previously on PC12 cells (Kd = 77 nm) (Parmer et al., 2000). Furthermore, actin enhances plasminogen activation by t-PA (Lind and Smith, 1991, 1993). In the current study, we found that a specific monoclonal antibody against actin inhibited cell-dependent plasminogen activation. Furthermore, in the presence of anti-actin antibody, nicotine-dependent stimulation of catecholamine release was enhanced, consistent with blockade of plasminogen activation and decreased formation of bioregulatory inhibitory peptides.
The results of our study define a key cell-surface-dependent mechanism underlying the ability of catecholaminergic cells to promote plasminogen activation and hence stimulate local autocrine/paracrine prohormone processing (see working model in supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Expression of binding sites for plasminogen (Parmer et al., 2000; Miles et al., 2002) and t-PA (Pittman et al., 1989; Parmer et al., 2000), together with the demonstration of trafficking of t-PA to catecholamine storage vesicles (Parmer et al., 1997; Parmer and Miles, 1998), suggests the presence of a local catecholaminergic cell plasminogen/t-PA system that regulates cell-associated neuroendocrine prohormone processing that, in turn, may play a key role in the regulation of neurotransmitter release. Of note, molecules of the plasminogen activation pathway are present in a variety of neuroendocrine sites, including the cerebral cortex (Sappino et al., 1993), cerebellum (Sappino et al., 1993; Friedman and Seeds, 1995; Zhang et al., 2002), hippocampus (Qian et al., 1993; Sappino et al., 1993; Tsirka et al., 1997a; Baranes et al., 1998; Salles and Strickland, 2002; Zhang et al., 2002), adrenal medulla (Parmer et al., 1997; Zhang et al., 2002), and in peripheral sympathetic neurons (X. Jiang et al., 2002; Hao et al., 2006). Hence, these results may suggest an important system and mechanism for the regulation of neurosecretory and catecholaminergic pathways in both central and peripheral nervous systems, with implications, for example, for t-PA/plasminogen-dependent processes such as long-term potentiation, learning, and memory (Qian et al., 1993; Carmeliet et al., 1994; Frey et al., 1996; Huang et al., 1996; Baranes et al., 1998; Seeds et al., 2003; Pang et al., 2004), and excitotoxin-induced neuronal injury in the CNS (Tsirka et al., 1996, 1997a,b; Chen-Liu and Strickland, 1997), as well as for the regulation of key systemic cardiovascular and metabolic homeostatic physiological responses governed by sympathoadrenal and sympathoneural activity (Parmer et al., 2000; Jiang et al., 2001; Q. Jiang et al., 2002; X. Jiang et al., 2002). Our current results, identifying specific, profibrinolytic plasminogen binding sites on catecholaminergic cells, thus may suggest a broad paradigm for regulating neurotransmitter secretion within the neuroendocrine system.
Footnotes
This work was supported by National Institutes of Health Grants HL-45934 and HL-38272 (L.A.M.) and HL-50398 (R.J.P.) and by the Department of Veterans Affairs (R.J.P.). Blood drawing was performed in the General Clinical Research Center of the Scripps Clinic, supported by National Institutes of Health Grant M01 RR00833. We thank Dr. Stephen B. Hawley for helpful discussions and Derek Hacke and Mike Green for excellent technical assistance. This is publication number 14609-CB from The Scripps Research Institute.
References
- Andronicos and Ranson, 2001.Andronicos NM, Ranson M. The topology of plasminogen binding and activation on the surface of human breast cancer cells. Br J Cancer. 2001;85:909–916. doi: 10.1054/bjoc.2001.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachvaroff et al., 1980.Bachvaroff RJ, Miller F, Rapaport FT. Appearance of cyoskeletal components on the surface of leukemia cells and of lymphocytes transformed by mitogens and Epstein-Barr virus. J Immunol. 1980;131:370–377. doi: 10.1073/pnas.77.8.4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranes et al., 1998.Baranes D, Lederfein D, Huang YY, Chen M, Bailey CH, Kandel ER. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 1998;21:813–825. doi: 10.1016/s0896-6273(00)80597-8. [DOI] [PubMed] [Google Scholar]
- Beebe et al., 1989.Beebe DP, Miles LA, Plow EF. A linear amino acid sequence involved in the interaction of t-PA with its endothelial cell receptor. Blood. 1989;74:2034–2037. [PubMed] [Google Scholar]
- Carmeliet et al., 1994.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, Van den Oord JJ, Collen D, Mulligan RC. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- Castellino and McCance, 1997.Castellino FJ, McCance SG. The kringle domains of human plasminogen. Ciba Found Symp. 1997;212:46–60. doi: 10.1002/9780470515457.ch4. [DOI] [PubMed] [Google Scholar]
- Chen-Liu and Strickland, 1997.Chen-Liu LW, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Colombo et al., 2002.Colombo B, Longhi R, Marinzi C, Magni F, Cattoretti G, Yoo S-H, Curnis F, Corti A. Cleavage of chromogranin A N-terminal domain by plasmin provides a new mechanism for regulating cell adhesion. J Biol Chem. 2002;277:45911–45919. doi: 10.1074/jbc.M202637200. [DOI] [PubMed] [Google Scholar]
- Deutsch and Mertz, 1970.Deutsch DG, Mertz ET. Plasminogen: Purification from human plasma by affinity chromatography. Science. 1970;170:1995–1996. doi: 10.1126/science.170.3962.1095. [DOI] [PubMed] [Google Scholar]
- Dudani and Ganz, 1996.Dudani AK, Ganz PR. Endothelial cell surface actin serves as a binding site for plasminogen, tissue plasminogen activator and lipoprotein(a) Br J Haematol. 1996;95:168–178. doi: 10.1046/j.1365-2141.1996.7482367.x. [DOI] [PubMed] [Google Scholar]
- Dudani et al., 2005.Dudani AK, Ben Tchavtchavadze M, Porter S, Tackaberry E. Angiostatin and plasminogen share binding to endothelial cell surface actin. Biochem Cell Biol. 2005;83:28–35. doi: 10.1139/o04-109. [DOI] [PubMed] [Google Scholar]
- Emerson et al., 1983.Emerson DL, Arnaud P, Galbraith RM. Evidence of increased Gc:actin complexes in pregnant serum: a possible result of trophoblast embolism. Am J Reprod Immunol. 1983;4:185–189. doi: 10.1111/j.1600-0897.1983.tb00276.x. [DOI] [PubMed] [Google Scholar]
- Felez et al., 1996.Felez J, Miles LA, Fabregas P, Jardi M, Plow EF, Lijnen RJ. Characterization of cellular binding sites and interactive regions within reactants required for enhancement of plasminogen activation by tPA on the surface of leukocytic cells. Thromb Haemost. 1996;76:577–584. [PubMed] [Google Scholar]
- Fowler et al., 1998.Fowler B, Mackman N, Parmer RJ, Miles LA. Binding of human single chain urokinase to Chinese hamster ovary cells and cloning of hamster u-PAR. Thromb Haemost. 1998;80:148–154. [PubMed] [Google Scholar]
- Frey et al., 1996.Frey U, Muller M, Kuhl D. A different form of long-lasting potentiation revealed in tissue plasminogen activator mutant mice. J Neurosci. 1996;16:2057–2063. doi: 10.1523/JNEUROSCI.16-06-02057.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman and Seeds, 1995.Friedman GC, Seeds NW. Tissue plasminogen activator mRNA expression in granule neurons coincides with their migration in the developing cerebellum. J Comp Neurol. 1995;360:658–670. doi: 10.1002/cne.903600410. [DOI] [PubMed] [Google Scholar]
- Greene and Tischler, 1976.Greene LA, Tischler AS. Establishment of a noradrenergic clonal cell line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualandris et al., 1996.Gualandris A, Jones TE, Strickland S, Tsirka SE. Membrane depolarization induces calcium-dependent secretion of tissue plasminogen activator. J Neurol Sci. 1996;16:2220–2225. doi: 10.1523/JNEUROSCI.16-07-02220.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao et al., 2006.Hao Z, Guo C, Jiang X, Krueger S, Pietri T, Dufour S, Cone RE, O'Rourke J. New transgenic evidence for a system of sympathetic axons able to express tissue plasminogen activator (t-PA) within arterial/arteriolar walls. Blood. 2006;108:200–202. doi: 10.1182/blood-2005-12-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley et al., 2000.Hawley SB, Green MA, Miles LA. Discriminating between cell surface and intracellular plasminogen-binding proteins: heterogeneity in profibrinolytic plasminogen-binding proteins on monocytoid cells. Thromb Haemost. 2000;84:882–890. [PubMed] [Google Scholar]
- Hawley et al., 2001.Hawley SB, Tamura T, Miles LA. Purification, cloning, and characterization of a profibrinolytic plasminogen-binding protein, TIP49a. J Biol Chem. 2001;276:179–186. doi: 10.1074/jbc.M004919200. [DOI] [PubMed] [Google Scholar]
- Herschman et al., 2000.Herschman HR, Ferguson GD, Feldman JD, Farias-Eisner R, Vician L. Searching for depolarization-induced genes that modulate synaptic plasticity and neurotrophin-induced genes that mediate neuronal differentiation. Neurochem Res. 2000;25:591–602. doi: 10.1023/a:1007546600535. [DOI] [PubMed] [Google Scholar]
- Hoover-Plow et al., 2001.Hoover-Plow J, Skomorovska-Prokvolit O, Welsh S. Selective behaviors altered in plasminogen-deficient mice are reconstituted with intracerebroventricular injection of plasminogen. Brain Res. 2001;898:256–264. doi: 10.1016/s0006-8993(01)02191-6. [DOI] [PubMed] [Google Scholar]
- Huang et al., 1996.Huang YY, Bach ME, Lipp HP, Zhuo M, Wolfer DP, Hawkins RD, Schoonjans L, Kandel ER, Godfraind JM, Mulligan R, Collen D, Carmeliet P. Mice lacking the gene encoding tissue-type plasminogen activator show a selective interference with late-phase long-term potentiation in both Schaffer collateral and mossy fiber pathways. Proc Natl Acad Sci USA. 1996;93:8699–8704. doi: 10.1073/pnas.93.16.8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson and Rosenbusch, 1976.Jacobson GR, Rosenbusch JP. ATP binding to a protease-resistant core of actin. Proc Natl Acad Sci USA. 1976;73:2742–2746. doi: 10.1073/pnas.73.8.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang et al., 2001.Jiang Q, Taupenot L, Mahata SK, Mahata M, O'Connor DT, Miles LA, Parmer RJ. Proteolytic cleavage of chromogranin A (CgA) by plasmin. Selective liberation of a specific bioactive CgA fragment that regulates catecholamine release. J Biol Chem. 2001;276:25022–25029. doi: 10.1074/jbc.M101545200. [DOI] [PubMed] [Google Scholar]
- Jiang et al., 2002.Jiang Q, Yasothornsrikul S, Taupenot L, Miles LA, Parmer RJ. The local chromaffin cell plasminogen/plasmin system and the regulation of catecholamine secretion. Ann NY Acad Sci. 2002;971:445–449. doi: 10.1111/j.1749-6632.2002.tb04506.x. [DOI] [PubMed] [Google Scholar]
- Jiang et al., 2002.Jiang X, Wang Y, Hand AR, Gillies C, Cone RE, Kirk J, O'Rourke J. Storage and release of tissue plasminogen activator by sympathetic axons in resistance vessel walls. Microvasc Res. 2002;64:438–447. doi: 10.1006/mvre.2002.2441. [DOI] [PubMed] [Google Scholar]
- Kabsch and Vandekerckhove, 1992.Kabsch W, Vandekerckhove J. Structure and function of actin. Annu Rev Biophys Biomol Struct. 1992;21:49–76. doi: 10.1146/annurev.bb.21.060192.000405. [DOI] [PubMed] [Google Scholar]
- Laemmli, 1970.Laemmli UK. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lind and Smith, 1991.Lind SE, Smith CJ. Actin accelerates plasmin generation by tissue plasminogen activator. J Biol Chem. 1991;266:17673–17678. [PubMed] [Google Scholar]
- Lind and Smith, 1993.Lind SE, Smith CJ. Actin stimulates plasmin generation by tissue and urokinase-type plasminogen activators. Arch Biochem Biophys. 1993;307:138–145. doi: 10.1006/abbi.1993.1572. [DOI] [PubMed] [Google Scholar]
- Markus et al., 1978.Markus G, De Pasquale JL, Wissler FC. Quantitative determination of the binding of epsilon-aminocaproic acid to native plasminogen. J Biol Chem. 1978;253:727–732. [PubMed] [Google Scholar]
- Mejean et al., 1987.Mejean C, Roustan C, Benyamin Y. Anti-actin antibodies. Detection and quantitation of total and skeletal muscle actin in human plasma using a competitive ELISA. J Immunol Methods. 1987;99:129–135. doi: 10.1016/0022-1759(87)90041-x. [DOI] [PubMed] [Google Scholar]
- Miles et al., 2002.Miles LA, Hawley SB, Parmer RJ. Chromaffin cell plasminogen receptors. Ann NY Acad Sci. 2002;971:454–459. doi: 10.1111/j.1749-6632.2002.tb04508.x. [DOI] [PubMed] [Google Scholar]
- Mornet et al., 1981.Mornet D, Bertrand R, Pantel P, Audemard E, Kassab R. Structure of the actin-myosin interface. Nature. 1981;292:301–306. doi: 10.1038/292301a0. [DOI] [PubMed] [Google Scholar]
- Moroianu et al., 1993.Moroianu J, Fett JW, Riordan JF, Vallee BL. Actin is a surface component of calf pulmonary artery endothelial cells in culture. Proc Natl Acad Sci USA. 1993;90:3815–3819. doi: 10.1073/pnas.90.9.3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muench et al., 2003.Muench MO, Ratcliffe JV, Nakanishi M, Ishimoto H, Jaffe RB. Isolation of definitive zone and chromaffin cells based upon expression of CD56 (neural cell adhesion molecule) in the human fetal adrenal gland. J Clin Endocrinol Metab. 2003;88:3921–3930. doi: 10.1210/jc.2003-030154. [DOI] [PubMed] [Google Scholar]
- Muesch et al., 1990.Muesch A, Hartmann E, Rohde K, Rubartelli A, Sitia R, Rapoport TA. A novel pathway for secretory proteins? Trends Biochem Sci. 1990;15:86–88. doi: 10.1016/0968-0004(90)90186-f. [DOI] [PubMed] [Google Scholar]
- Muhlrad et al., 2004.Muhlrad A, Kudryashov D, Michael PY, Bobkov AA, Almo SC, Reisler E. Cofilin induced conformational changes in F-actin expose subdomain 2 to proteolysis. J Mol Biol. 2004;342:1559–1567. doi: 10.1016/j.jmb.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Owen et al., 1978.Owen MJ, Auger J, Barber BH, Edwards AJ, Walsh FS, Crumpton MJ. Actin may be present on the lymphocyte surface. Proc Natl Acad Sci USA. 1978;75:4484–4488. doi: 10.1073/pnas.75.9.4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang et al., 2004.Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Parmer and Miles, 1998.Parmer RJ, Miles LA. Targeting of tissue plasminogen activator to the regulated pathway of secretion. Trends Cardiovasc Med. 1998;8:306–312. doi: 10.1016/s1050-1738(98)00025-5. [DOI] [PubMed] [Google Scholar]
- Parmer et al., 1993.Parmer RJ, Xi X-P, Wu H-J, Helman LJ, Petz LN. Secretory Protein Traffic. Chromogranin A contains a dominant signal for the regulated pathway. J Clin Invest. 1993;92:1042–1054. doi: 10.1172/JCI116609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmer et al., 1997.Parmer RJ, Mahata M, Mahata S, Sebald MT, O'Connor DT, Miles LA. Tissue plasminogen activator (t-PA) is targeted to the regulated secretory pathway: Catecholamine storage vesicles as a reservoir for the rapid release of t-PA. J Biol Chem. 1997;272:1976–1982. doi: 10.1074/jbc.272.3.1976. [DOI] [PubMed] [Google Scholar]
- Parmer et al., 2000.Parmer RJ, Mahata M, Gong Y, Mahata S, Jiang Q, O'Connor DT, Xi X-P, Miles LA. Processing of chromogranin A by plasmin provides a novel mechanism for regulating catecholamine secretioin. J Clin Invest. 2000;106:907–915. doi: 10.1172/JCI7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman et al., 1989.Pittman RN, Ivins JK, Buettner HM. Neuronal plasminogen activators: Cell surface binding sites and involvement in neurite outgrowth. J Neurosci. 1989;9:4269–4286. doi: 10.1523/JNEUROSCI.09-12-04269.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian et al., 1993.Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-type plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- Ranson et al., 1998.Ranson M, Andronicos NM, O'Mullane MJ, Baker MS. Increased plasminogen binding is associated with metastatic breast cancer cells: differential expression of plasminogen binding proteins. Br J Cancer. 1998;77:1586–1597. doi: 10.1038/bjc.1998.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein et al., 1982.Rubenstein P, Ruppert T, Sandra A. Selective isoactin release from cultured embryonic skeletal muscle cells. J Cell Biol. 1982;92:164–169. doi: 10.1083/jcb.92.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salles and Strickland, 2002.Salles FJ, Strickland S. Localization and regulation of the tissue plasminogen activator-plasmin system in the hippocampus. J Neurosci. 2002;22:2125–2134. doi: 10.1523/JNEUROSCI.22-06-02125.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders and Craig, 1983.Sanders SK, Craig SW. A lymphocyte cell surface molecule that is antigenically related to actin. J Immunol. 1983;131:370–377. [PubMed] [Google Scholar]
- Sappino et al., 1993.Sappino A-P, Madani R, huarte J, Belin D, Kiss JZ, Wohlwend A, Vassalli J-D. Extracellular proteolysis in the adult murine brain. J Clin Invest. 1993;92:679–685. doi: 10.1172/JCI116637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeds et al., 2003.Seeds NW, Basham ME, Ferguson JE. Absence of tissue plasminogen activator gene or activity impairs mouse cerebellar motor learning. J Neurosci. 2003;23:7368–7375. doi: 10.1523/JNEUROSCI.23-19-07368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorstensson et al., 1982.Thorstensson R, Utter G, Norberg R. Further characterization of the Ca2+-dependent F-actin-depolymerizing protein of human serum. Eur J Biochem. 1982;126:11–16. doi: 10.1111/j.1432-1033.1982.tb06738.x. [DOI] [PubMed] [Google Scholar]
- Tsirka et al., 1996.Tsirka SE, Rogove AD, Strickland S. Neuronal cell death and tPA. Nature. 1996;384:123–124. doi: 10.1038/384123b0. [DOI] [PubMed] [Google Scholar]
- Tsirka et al., 1997a.Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997a;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirka et al., 1997b.Tsirka SE, Bugge TH, Degen JL, Strickland S. Neuronal death in the central nervous system demonstrates a non-fibrin substrate for plasmin. Proc Natl Acad Sci USA. 1997b;94:9779–9781. doi: 10.1073/pnas.94.18.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violand et al., 1975.Violand BN, Sodetz JM, Castellino FJ. The effect of epsilon-amino caproic acid on the gross conformation of plasminogen and plasmin. Arch Biochem Biophys. 1975;170:300–305. doi: 10.1016/0003-9861(75)90121-6. [DOI] [PubMed] [Google Scholar]
- Waller et al., 2001.Waller A, Pipkorn D, Sutton KL, Linderman JJ, Omann GM. Validation of flow cytometric competitive binding protocols and characterization of fluorescently labeled ligands. Cytometry. 2001;45:102–114. doi: 10.1002/1097-0320(20011001)45:2<102::aid-cyto1152>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Wang et al., 2004.Wang H, Schultz R, Hong J, Cundiff DL, Jiang K, Soff GA. Cell surface-dependent generation of angiostatin 4.5. Cancer Res. 2004;64:162–168. doi: 10.1158/0008-5472.can-03-1862. [DOI] [PubMed] [Google Scholar]
- Wang et al., 2006.Wang H, Doll JA, Jiang K, Cundiff DL, Czarnecki JS, Wilson M, Ridge KM, Soff GA. Differential binding of plasminogen, plasmin, and angiostatin4.5 to cell surface beta-actin: implications for cancer-mediated angiogenesis. Cancer Res. 2006;66:7211–7215. doi: 10.1158/0008-5472.CAN-05-4331. [DOI] [PubMed] [Google Scholar]
- Wang et al., 2004.Wang N, Zhang L, Miles L, Hoover-Plow J. Plasminogen regulates pro-opiomelanocortin processing. J Thromb Haemost. 2004;2:785–796. doi: 10.1111/j.1538-7836.2004.00694.x. [DOI] [PubMed] [Google Scholar]
- Zhang et al., 2002.Zhang L, Seiffert D, Fowler BJ, Jenkins GR, Thinnes TC, Loskutoff DJ, Parmer RJ, Miles LA. Plasminogen has a broad extrahepatic distribution. Thromb Haemost. 2002;87:493–501. [PubMed] [Google Scholar]