

Abstract
Multiple sclerosis (MS) is pathologically characterized by inflammatory demyelination and neuronal injury. Although phagocytosis of myelin debris by microglia and macrophages in acute MS lesions is well documented, its pathophysiological significance is unclear. Using real-time quantitative PCR, flow cytometry, ELISA, and reactive oxygen species (ROS) measurement assays, we demonstrated that phagocytosis of myelin modulates activation of microglial cells prestimulated by interferon-γ (IFN-γ) or a combination of IFN-γ and lipopolysaccharide with a biphasic temporal pattern, i.e., enhanced production of proinflammatory mediators during the first phase (≤6 h), followed by suppression during the second (6–24 h) phase. In this second phase, myelin phagocytosis leads to an enhanced release of prostaglandin E2 and ROS in microglia, whereas the production of anti-inflammatory cytokines (particularly interleukin-10) remains unchanged. Suppression of inflammatory microglial activation by myelin phagocytosis was reversed by treatment with superoxide dismutase and catalase, by inhibition of the NADPH–oxidase complex, or by specific knockdown of the NADPH–oxidase-required adaptor p47–phagocyte oxidase (PHOX). Furthermore, we observed that myelin phagocytosis destabilized tumor necrosis factor-α and interferon-induced protein-10 mRNA through an adenine–uridine-rich elements-involved mechanism, which was reversed by blocking the function of NADPH–oxidase complex. We conclude that phagocytosis of myelin suppresses microglial inflammatory activities via enhancement of p47-PHOX-mediated ROS generation. These results suggest that intervention in ROS generation could represent a novel therapeutic strategy to reduce neuroinflammation in MS.
Keywords: multiple sclerosis, inflammation, myelin, phagocytosis, reactive oxygen species, Ncf1
Introduction
Multiple sclerosis (MS) is characterized by disseminated demyelination and the presence of activated T cells, microglia, and macrophages (Sospedra and Martin, 2005). Phagocytosis of myelin by activated microglia/macrophages is well documented in MS and its animal model experimental autoimmune encephalomyelitis (EAE) (Tanaka et al., 1975; Epstein et al., 1983; Bauer et al., 1994; Williams et al., 1994; Mosley and Cuzner, 1996; van der Laan et al., 1996). However, the effects of myelin phagocytosis on microglia/macrophages-mediated inflammatory responses remain controversial. Several in vitro studies showed that myelin phagocytosis by microglia and macrophages triggers release of proinflammatory cytokines and nitric oxide (Williams et al., 1994; Mosley and Cuzner, 1996; van der Laan et al., 1996), suggesting that phagocytosis of myelin could enhance neuroinflammation. In contrast, Boven's group (Boven et al., 2006) reported an anti-inflammatory effect of myelin phagocytosis. At present, the mechanisms by which myelin phagocytosis could affect activation of mononuclear phagocytes are unknown.
In both MS and EAE, myelin phagocytosis has been shown to induce the generation of reactive oxygen species (ROS) (Williams et al., 1994; van der Goes et al., 1998), but the significance of ROS on regulation of inflammation is still unclear. Recently, mice with experimental autoimmune arthritis and EAE were reported to suffer a more severe disease course when they carried mutations in the neutrophil cytosolic factor 1 (Ncf1) gene, which encodes p47–phagocyte oxidase (PHOX), a key adaptor of NADPH–oxidase (Hultqvist et al., 2004). Together with the previous work that demonstrated that a defect of the p47–PHOX protein decreases ROS generation in NADPH–oxidase-expressing cells (Olofsson et al., 2003), these findings strongly suggest that p47–PHOX-mediated oxidative burst has the potential to suppress inflammatory responses.
In our study, we demonstrate that myelin phagocytosis induces the generation of ROS via p47–PHOX and that, by this mechanism, a marked downregulation of microglial inflammatory responses occurs.
Materials and Methods
Mouse myelin isolation and labeling.
Myelin was isolated from the nervous tissue of 6-month-old mice using sucrose (0.32 and 0.85 m) density gradient centrifugation at 75,000 × g, as described previously (Norton and Poduslo, 1973). The myelin protein content was determined using the Bradford method with bovine serum albumin as the standard. Endotoxin concentrations of myelin preparations were below the detection limit (<0.01 U/ml) as determined by the Limulus amebocyte lysate assay system (Cambrex, Verviers, Belgium) according to the instructions of the manufacturer. For labeling, myelin was first dissolved in 0.1 m NaHCO3–Na2CO3 buffer at 1 mg/ml protein concentration and then labeled using a cyanine 3 (Cy3) mono-reactive dye pack (Amersham Biosciences, Freiburg, Germany) according to the instructions of the manufacturer.
Culture of primary microglial cells and the microglial cell line BV-2 cells.
Primary microglial cells were prepared from brains of postnatal day 3–5 C57BL/6J mice. Briefly, meninges were mechanically removed, and the cells were cultured in DMEM (Invitrogen, Eggenstein, Germany) supplemented with 10% fetal calf serum (PAN Biotech, Aidenbach, Germany) under a humidified atmosphere of 10% CO2 at 37°C for at least 14 d. To collect microglial cells, microglia–astrocyte cocultures were shaken on a rotary shaker (220 rpm) for 2 h, and microglial cells were obtained from the supernatant. Flow cytometric analysis routinely showed the microglial cell preparation to consist of >95% CD11b+ cells.
BV-2 cells, derived from an immortalized microglial cell line (Blasi et al., 1990) (kindly provided by Prof. K. Frei, University Hospital Zürich, Zürich, Switzerland), were maintained in the same microglial culture medium.
Flow cytometric analysis of primary microglial phagocytosis of labeled myelin.
Primary microglia cultured in a 24-well plate at a density of 3 × 105 cells per well were treated with Cy3-labeled mouse myelin (2 μg/ml) for 1, 2, 6, and 24 h. In parallel, some cells were pretreated with 5 μm cytochalasin D (Sigma, St. Louis, MO) for 1 h and then treated with 2 μg/ml myelin containing 5 μm cytochalasin D for 1 h. Microglia were then washed with PBS and detached from the plate with trypsin–EDTA (Invitrogen, Eggenstein, Germany), and the mean fluorescence intensity of Cy3-labeled myelin was immediately determined by flow cytometry.
Laser scanning confocal microscopy analysis of myelin internalization by primary microglia.
Primary microglial cells grown on chamber slides (Nunc, Wiesbaden, Germany) were treated with 2 μg/ml Cy3-labeled myelin for 2 h in microglial culture medium at 37°C and 10% CO2. The cells were fixed in 4% paraformaldehyde and then permeabilized with 0.2% Triton X-100. After blocking with 10% normal goat serum, microglial cells were incubated overnight at 4°C with rat anti-mouse lysosome-associated membrane protein-2 (LAMP-2) monoclonal antibody (obtained from ABL-93 hybridoma cells culture; Development Studies Hybridoma Bank, Department of Biological Sciences, University of Iowa, Iowa City, IA) (Chen et al., 1985). After rinsing, slides were incubated for 1 h at room temperature with Cy5-conjugated goat anti-rat IgG (Jackson ImmunoResearch, West Grove, PA). The slides were mounted with Mowiol and kept at 4°C in the dark until analysis under a laser scanning confocal microscope (LSC; Leica, Heidelberg, Germany). Images of microglia treated with isotype control primary antibodies were used to assess background fluorescence.
Reverse transcription and quantitative PCR for analysis of inflammatory gene transcripts after myelin phagocytosis in primary microglia.
Primary microglia were seeded in 24-well plates at a density of 3.5 × 105 cells per well. All cells were pretreated with 100 U/ml mouse interferon-γ (IFN-γ) (R & D Systems, Wiesbaden, Germany) for 24 h. Cells were then split into four groups. One group of cells were incubated in the presence of IFN-γ for another 24 h with (1) myelin at 5, 10, or 50 μg/ml and (2) 1 μl/ml FluoSpheres fluorescent microspheres (1 × 107 polystyrene microspheres/ml, 1 μm diameter; Invitrogen, Leiden, The Netherlands) or 100 μg/ml zymosan A from Saccharomyces cerevisiae (Sigma) as a positive control of phagocytosis. The second group of cells were treated with 10 μg/ml myelin in the presence of IFN-γ for 1, 3, 6, 24, and 48 h. The third group were cotreated in the presence of IFN-γ for 24 h with 10 μg/ml myelin and a scavenger of extracellular ROS [either 500 U/ml superoxide dismutase (SOD) or 1000 U/ml catalase (both from Sigma)] or 5 mm apocynin (Sigma), an inhibitor of assembly of NADPH–oxidase complex. If apocynin was used, it was administrated 1 h before exposure of the cells to myelin. The fourth group were treated in the presence of IFN-γ for 24 h with 50 or 200 μm H2O2 (Sigma) as ROS. Cells in this group were not exposed to myelin.
Total RNA was isolated from primary microglia after treatments described above by the RNEasy Mini Kit (Qiagen, Hilden, Germany). First-strand cDNA was synthesized by priming total RNA with hexamer random primers (Roche Molecular Biochemicals, Mannheim, Germany) and using Superscript III reverse transcriptase according to the instructions of the manufacturer (Invitrogen, Karlsruhe, Germany). For quantification, quantitative real-time (RT)-PCR was performed using the Applied Biosystems (Foster City, CA) GeneAmp 5700 Sequence Detection System or 7500 Fast real-time PCR system. The following oligonucleotides (Invitrogen, Karlsruhe, Germany) were used for PCR amplification: Ncf1, sense, 5′-GCC CAA AGA TGG CAA GAA TA-3′ and antisense, 5′-TGT TCC CGA ACT CTT CTC GT-3′; prostaglandin E (PGE2) synthase 1 (Ptges1), sense, 5′-GAG TTT TCA CGT TCC GGT GT-3′ and antisense, 5′-GGT AGG CTG TCA GCT CAA GG-3′; and Ptges2, sense, 5′-ACT TCC ACT CCC TGC CCT AT-3′ and antisense, 5′-GTT GCA AGC TGT CTC CTT CC-3′. Previously described primers were used for amplification of tumor necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS), interleukin-1β (IL-1β), transformation growth factor-β1 (TGF-β1), IL-10, macrophage inflammatory protein-1α (MIP-1α), monocyte chemoattractant protein-1 (MCP-1), and interferon-induced protein-10 (IP-10) (Olson and Miller, 2004; Liu et al., 2005; Takahashi et al., 2005).
The amount of double-stranded PCR product synthesized in each cycle was measured using SYBR green I dye. Threshold cycle (Ct) values for each test gene from the replicate PCRs was normalized to the Ct values for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control from the same cDNA preparations. The ratio of transcription of each gene was calculated as 2(ΔCt), where ΔCt is the difference Ct (test gene) − Ct (GAPDH).
Myelin challenge and ELISA detection of inflammatory mediators released by primary microglia.
Primary microglia (1 × 105/well in 48-well plates) were prestimulated with 100 U/ml IFN-γ or a combination of IFN-γ and 10 ng/ml lipopolysaccharide (LPS) from Escherichia coli (Sigma) for 24 h and then incubated in the presence of the prestimulator for an additional 24 h with 5, 10, or 50 μg/ml myelin, 100 μg/ml zymosan A, and 1 μl/ml FluoSpheres fluorescent microspheres. To analyze the effects of ROS on release of inflammatory cytokines and chemokines, preactivated microglial cells were treated with (1) myelin in the presence of SOD, catalase, or apocynin, or (2) H2O2 as described in the last section. Supernatants were collected for detection of TNF-α, IL-1β, IL-10, IP-10, and PGE2 by ELISA (ELISA kits for TNF-α, IL-1β, IP-10, and PGE2 detection were from R & D Systems; IL-10 ELISA kit was from PharMingen, Heidelberg, Germany).
To assess possible absorption of cytokines by myelin in the culture media, we incubated 1 ng/ml recombinant mouse TNF-α and 100 U/ml IFN-γ with 0, 5, 10, and 50 μg/ml myelin in a cell-free culture system for 24 h. We removed the myelin by centrifugation and quantified the levels of TNF-α and IFN-γ in the myelin-free medium with ELISA (IFN-γ ELISA kit was from PharMingen).
Assessment of reactive oxygen species produced by primary microglia.
The production of ROS in primary microglia treated with myelin was determined by measuring the reduction changes of water-soluble tetrazolium salt (WST-1) (Roche Diagnostics, Mannheim, Germany) caused by SOD (Zhang et al., 2005). Microglia were seeded at a density of 4 × 104/well on a 96-well plate (BD Falcon, Bedford, MA) in culture medium and incubated with 100 U/ml IFN-γ for 24 h. Microglial cells were then incubated in the presence of IFN-γ for 24 h with (1) myelin at 5, 10, or 50 μg/ml, (2) 1 μl/ml FluoSpheres fluorescent microspheres, or (3) 100 μg/ml zymosan A or with 10 μg/ml myelin for 1, 2, 3, 6, 24, and 48 h. As a control to confirm NADPH–oxidase activity in ROS production, an aliquot of IFN-γ-activated microglial cells were cotreated with myelin and 5 mm apocynin for another 24 h (apocynin was administrated 1 h before exposure to myelin). WST-1 was then added to the treated microglia (100 μl/well, 1:10 dilution in culture medium) with or without SOD (500 U/ml), and cells were incubated for 1 h. The amount of ROS production was determined as the increase in absorbance at 450 nm and expressed as percentage of the control cultures without myelin treatment.
Intracellular generation of ROS was measured using the luminol chemiluminescence assay as described previously (Dahlgren and Karlsson, 1999). In brief, primary microglia cultured in 96-well black plate (BD Falcon) (4 × 104 cells per well) treated as described above were mixed with luminol (100 μm; Sigma), SOD (500 U/ml), and catalase (2000 U/ml) in HBSS with 0.5 mm CaCl2 and 1 mm MgCl2. The production of light over time (relative fluorescence units per seconds) was recorded by a luminometer (MicroBeta TriLux with software version 4.7; PerkinElmer, Turku, Finland) for 1 s. As a control of NADPH–oxidase activity, an aliquot of IFN-γ-activated microglial cells were cotreated with 5 mm apocynin.
Knockdown of p47–PHOX in BV-2 cells and analysis of regulation of inflammatory mediators after myelin phagocytosis.
The following Stealth small interfering RNA (siRNA) targeting mouse Ncf1 sequence was synthesized by Invitrogen (Karlsruhe, Germany): 5′-GCACUAUGUGUACAUGUUCCUGGUU-3′. The sequence of control Stealth siRNA was 5′-GCAGUAUGUACAUGUCCUGUCUGUU-3′. BV-2 cells were plated on a 24-well plate (1 × 105/well) and transfected with double-stranded siRNA using Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany) according to the protocol of the manufacturer. Twenty-four hours after transfection, the culture medium was replaced by fresh medium containing 100 U/ml IFN-γ or a combination of 100 U/ml IFN-γ and 10 ng/ml LPS as indicated in Results. The cells were cultured for an additional 24 h. Cells were then treated with 5, 10, or 50 μg/ml myelin for 24 h in the presence of the prestimulator. Thereafter, culture supernatant was collected for determination of TNF-α, IL-1β, IL-10, and IP-10 levels by ELISA, and total RNA was isolated to quantify transcripts of Ncf1, TNF-α, iNOS, IL-1β, TGF-β1, IL-10, MIP-1α, MCP-1, and IP-10 using real-time quantitative PCR (detailed procedures were described above).
Protein expression level of p47–PHOX was measured by flow cytometry after intracellular staining of p47–PHOX with 4 μg/ml mouse monoclonal antibody against p47–PHOX (Santa Cruz Biotechnology, Heidelberg, Germany), followed by staining with 1 μg/ml Alexa488-conjugated donkey anti-mouse IgG (Invitrogen, Leiden, The Netherlands) according to the previously published protocol (Liu et al., 2005). Extracellular ROS levels were determined by measuring SOD-inhibitible reduction of WST-1.
Analysis of TNF-α and IP-10 mRNA stability in IFN-γ-preactivated primary microglia after myelin phagocytosis.
Primary microglia (1 × 105/well in 48-well plates) were prestimulated with 100 U/ml IFN-γ for 24 h and then incubated with 10 μg/ml myelin in the presence of IFN-γ for an additional 3 h. Thereafter, the 10 μg/ml actinomycin D (Sigma) was added to the culture medium. After 0, 10, 20, 30, 60, and 120 min, total RNA was isolated from the cells. The amount of TNF-α and IP-10 transcripts was measured with real-time quantitative PCR as described in the previous section. To analyze the effects of ROS on stability of TNF-α and IP-10 mRNA, apocynin at a final concentration of 5 mm was added to culture system 1 h before myelin treatment.
Analysis of adenine–uridine-rich elements-regulated stability of TNF-α and IP-10 mRNA in p47–PHOX-knocked down BV-2 cells after myelin phagocytosis.
The 3′ untranslated region (3′ UTR) containing adenine–uridine-rich elements (AREs) of TNF-α, IP-10, or β-actin was cloned by RT-PCR using total RNA from IFN-γ-activated microglia according to the previous publication (Sun and Ding, 2006). The following oligonucleotides (Invitrogen, Karlsruhe, Germany) containing XbaI or FseI restriction sites (underlined) were used for PCR amplification: TNF-α, sense, 5′-GCTCTAGAGCAGTCCGGGCAGGTCTACTTT-3′ and antisense, 5′-TTTGGCCGGCCAAATGTAGGGCAATTACAGTCACG-3′; IP-10, sense, 5′-GCTCTAGAGCCTGGAGTGAAGCCACG-3′ and antisense, 5′-TTTGGCCGGCCAAACATTTTGGGTCTCTTACTAC-3′; and β-actin, sense, 5′-GCTCTAGAGCGCGGACTGTTACTGAG-3′ and antisense, 5′-TTTGGCCGGCCAAAGTTTGTGTAAGGTAAGGTGTG-3′. The PCR products were cleaved by XbaI and FseI and ligated into pGL3-basic vectors (Promega, Madison, WI) downstream to a luciferase reporter gene. Then, the cytomegalovirus (CMV) promoter from pLenti6/V5-D-TOPO (Invitrogen, Karlsruhe, Germany) was inserted into pGL3 between KpnI and XhoI.
BV-2 cells (2 × 105/well in 24-well plate) were transfected with 0.15 μg of luciferase reporter genes carrying various 3′ UTRs together with 0.05 μg of pcDNA3.1/His/LacZ (Invitrogen, Karlsruhe, Germany) as an internal transfection control and 10 pmol of double-stranded siRNA targeting mouse Ncf1 sequence or a nonsense sequence using 1.5 μl of Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany) according to the protocol of the manufacturer. Six hours after transfection, the culture medium was replaced by fresh medium containing 100 U/ml IFN-γ. The cells were cultured for an additional 18 h before treatment with 10 μg/ml myelin for 24 h in the presence of IFN-γ. Luciferase and β-galactosidase activity was then assayed with Bright-Glo Luciferase Assay System and β-galactosidase Enzyme Assay Systems (both from Promega).
Statistics.
Data in figures and tables are presented as mean ± SEM. One-way ANOVA followed by Tukey's HSD or Tamhane's T2 post hoc test (dependent on the result of Levene's test to determine the equality of variances) was used for multiple comparisons. Two-independent-samples t test was used to compare means for two groups of cases. All statistical analysis was performed on SPSS 11.0 for Windows (SPSS, Chicago, IL). Statistical significance was set at p < 0.05.
Results
Microglia readily phagocytose myelin
Microglia/macrophages are the key effector cells that remove damaged myelin sheaths by phagocytosing myelin debris. To confirm the biological activity of our cell preparations, we incubated primary microglia with 2 μg/ml Cy3-labeled myelin for 1, 2, 6, and 24 h and measured myelin internalization over time by flow cytometry (Fig. 1A). Internalization was detectable 1 h after myelin administration and increased along with the incubation time. After 24 h, the fluorescence intensity of Cy3-labeled myelin decreased possibly attributable to release of Cy3 from the degraded protein in internalized myelin. Treatment with cytochalasin D, a known inhibitor of phagocytosis (Mimura and Asano, 1976), blocked the internalization of myelin within 1 h.
Figure 1.
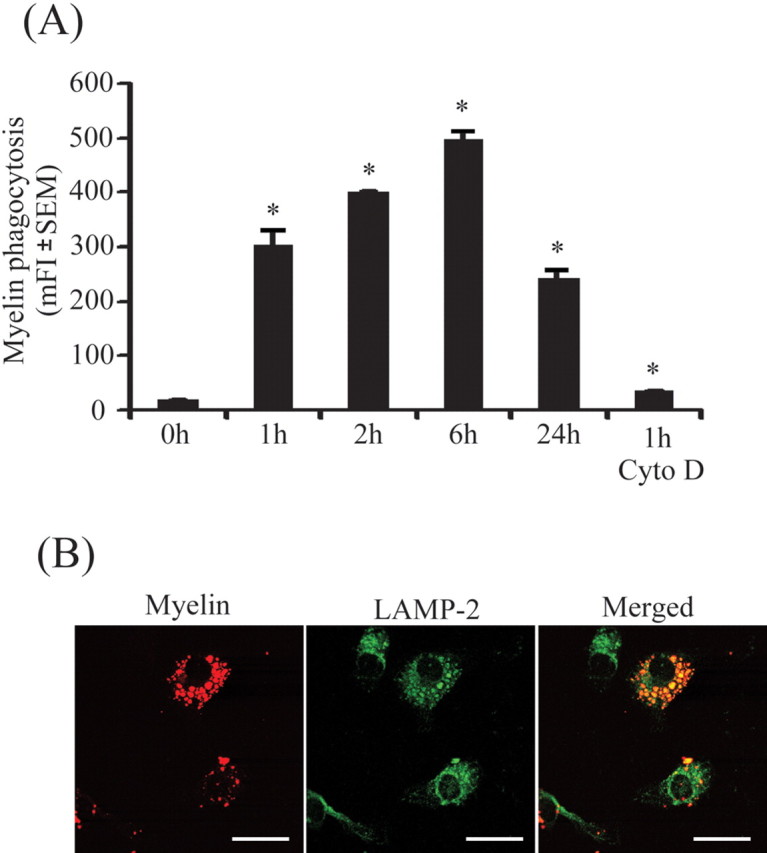
Microglia readily phagocytose myelin. Primary microglia were plated on 24-well plates and treated with Cy3-labeled myelin for 0, 1, 2, 6, and 24 h. A, The mean fluorescence intensity (mFI) of Cy3 in microglia was analyzed by flow cytometry. Microglial internalization reached its maximum ∼6 h after myelin administration. Myelin internalization was blocked by 5 μm cytochalasin D (Cyto D) as showed by mean fluorescence intensity of Cy3 after 1 h (all of the experiments were independently repeated 3 times with the same trends; one-way ANOVA vs that at zero incubation time point, *p < 0.05; n = 3 per group). Under confocal microscopy, myelin (shown in red) colocalized (shown in yellow) with LAMP-2, a specific marker of lysosomes (shown in green), within 2 h after myelin administration. Scale bars, 10 μm.
To confirm internalization within lysosomes, we performed fluorescence labeling of the cell preparation with the antibody against LAMP-2 and observed the cells under confocal microscopy. The internalized myelin colocalized with LAMP-2-stained lysosomes at 2 h after addition of Cy3-labeled myelin (Fig. 1B).
Myelin phagocytosis regulates transcription and secretion of inflammatory mediators in IFN-γ-activated primary microglia
IFN-γ is produced by auto-antigen-reactive T cells and has a pivotal role in the initiation of inflammation in MS. We asked whether myelin phagocytosis could regulate IFN-γ-activated inflammation in microglial cells. After 24 h, myelin phagocytosis resulted in a dose-dependent downregulation of transcripts of proinflammatory cytokines and chemokines TNF-α, iNOS, IL-1β, MIP-1α, and IP-10 (Fig. 2A,B,D) (supplemental Table 1, available at www.jneurosci.org as supplemental material). In contrast, transcription levels of the anti-inflammatory cytokines TGF-β and IL-10 were not downregulated, and IL-10 gene transcription was even enhanced by addition of myelin (Fig. 2C) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Treatment of IFN-γ-activated microglia with 10 μg/ml myelin for various intervals resulted in a transient increase in transcription of TNF-α, IL-1β, IL-10, and IP-10 during the first phase (≤6 h), followed by a rapid and marked decrease in transcription during the second phase (6–24 h) (Fig. 2). A similar temporal pattern was seen in secreted TNF-α and IP-10 proteins as determined by ELISA (Fig. 2A,D).
Figure 2.

Myelin phagocytosis downregulates transcription and secretion of proinflammatory mediators in IFN-γ-activated microglia. Primary microglia were activated by IFN-γ and then treated in the presence of IFN-γ with (1) myelin at 0, 5, 10, and 50 μg/ml for 24 h, or (2) 10 μg/ml myelin for 0, 1, 3, 6, 24, and 48 h. Total RNA was isolated, and transcripts of TNF-α (A), IL-1β (B), IL-10 (C), and IP-10 (D) were measured using real-time quantitative PCR. Release of TNF-α and IP-10 in the culture medium was detected with ELISA. After 24 h, transcription of TNF-α (A), IL-1β (B), or IP-10 (D) and secretion of TNF-α (A) or IP-10 (D) were dose-dependently downregulated by myelin phagocytosis. In contrast, transcription of IL-10 (C) was slightly enhanced by myelin. After treatment with 10 μg/ml myelin, transcription of TNF-α (A), IL-1β (B), IL-10 (C), or IP-10 (D) and secretion of TNF-α (A) or IP-10 (D) were initially (≤6 h) upregulated and later (24 h and thereafter) downregulated. Beads and zymosan A were used as positive controls of phagocytosis. After 24 h, beads did not significantly affect inflammatory response except by downregulating IL-1β (B); zymosan A strongly increased production of TNF-α (A) and IL-1β (B) (all of the experiments were independently repeated at least 3 times with the same trends; one-way ANOVA vs IFN-γ-activated control, *p < 0.05, #p > 0.05; n = 3–10 per group).
As controls, microglia were incubated with beads and zymosan A instead of myelin. Beads did not significantly affect transcription of TNF-α, IL-10, and IP-10 (independent-samples t test, p values >0.05; n = 3 per group) (Fig. 2) except downregulating IL-1β (p = 0.03). Zymosan A strongly stimulated transcription and release of TNF-α and IL-1β (change of the TNF-α/GAPDH ratio from 0.17 ± 0.02 to 0.56 ± 0.09 and the IL-1β/GAPDH ratio from 1.01 ± 0.20 × 10−2 to 5.57 ± 0.52 × 10−2; independent-samples t test, p < 0.05; n = 3 per group) (Fig. 2A,B).
To investigate the role of phagocytosis on transcriptional modulation, we treated IFN-γ-preactivated microglia with 50 μg/ml myelin in the presence of 5 μm cytochalasin D for 24 h. We observed that cytochalasin D strongly reversed myelin-induced transcriptional suppression of TNF-α and IP-10 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Comparison of the Ct values of GAPDH from all samples with or without myelin treatment in each experiment showed no significant changes in GAPDH transcription after myelin treatment (data not shown).
To exclude whether downregulation of proinflammatory gene transcription and protein secretion after myelin internalization could be attributable to a possible absorption of released cytokines by myelin in the culture media, we incubated recombinant mouse TNF-α and IFN-γ with myelin in the cell-free system and then quantified TNF-α and IFN-γ in the myelin-free medium with ELISA. We observed no reduction of TNF-α or IFN-γ concentrations after myelin incubation (data not shown).
Myelin phagocytosis decreases release of TNF-α, IL-1β, IP-10, and IL-10 from IFN-γ and LPS-activated primary cultured microglia
To confirm the regulatory effects of myelin phagocytosis on release of inflammatory mediators on a protein level, we activated primary cultured microglia with a combination of IFN-γ and LPS and then treated cells with myelin for 24 h at different concentrations (5, 10, and 50 μg/ml). As seen in Figure 3, myelin challenge significantly reduced release of TNF-α, IL-1β, and IL-10 in a dose-dependent manner (one-way ANOVA, p values <0.05); IP-10 secretion was also significantly reduced (one-way ANOVA, p value <0.05), although not in a concentration-dependent manner. Incubation with beads did not affect release of cytokines except IP-10, which was downregulated. Zymosan A significantly increased release of TNF-α and IL-1β and decreased release of IP-10 (independent-samples t test, p values <0.05). The myelin-mediated suppression of inflammatory mediators release was also observed in mouse peritoneal macrophages and human monocytes after 24 h of incubation with 10 μg/ml myelin (data not shown).
Figure 3.

Myelin phagocytosis decreases release of inflammatory mediators from IFN-γ- and LPS-activated microglia. Primary microglia preactivated by a combination of IFN-γ and LPS were treated with myelin at 0, 5, 10, and 50 μg/ml in the presence of preactivators for 24 h. A–C, Release of cytokines into the supernatant was determined by ELISA. The release of TNF-α (A), IL-1β (B), and IL-10 (C) was downregulated by myelin in a dose-dependent manner. D, IP-10 secretion was significantly reduced but not in a dose-dependent manner (all of the experiments were independently repeated at least 3 times with the same trends; one-way ANOVA vs control activated by IFN-γ plus LPS, *p < 0.05, #p > 0.05; n = 3–8 per group).
To further investigate the suppressing effects of engulfed myelin on inflammatory mediator release, we first treated primary microglia with myelin for 24 h and then washed the non-engulfed myelin away and replaced the culture medium with fresh medium containing 10 ng/ml LPS for an additional 6 h. We observed that higher concentrations (200 μg/ml) of myelin were needed to suppress the TNF-α release (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), which suggests that it is rather the process of phagocytosing myelin than the internalized myelin itself that mediates inflammatory suppression.
Myelin phagocytosis increases release of PGE2 from IFN-γ-activated primary microglia
The basal level of the inflammatory marker PGE2 in IFN-γ-activated microglia was 4.66 ± 0.11 ng/ml, as determined by ELISA. After treatment with 10 and 50 μg/ml myelin, concentrations of released PGE2 were significantly increased from the basal level to 6.45 ± 0.53 and 7.53 ± 0.64 ng/ml, respectively (mean ± SEM; one-way ANOVA, p value <0.05; n = 5 per group) (Fig. 4A). Myelin phagocytosis did not affect the transcription of constitutively expressed Ptges2 in IFN-γ-activated primary microglia (Fig. 4C), but the inducible Ptges1 was significantly reduced (one-way ANOVA, p values <0.05) (Fig. 4B).
Figure 4.
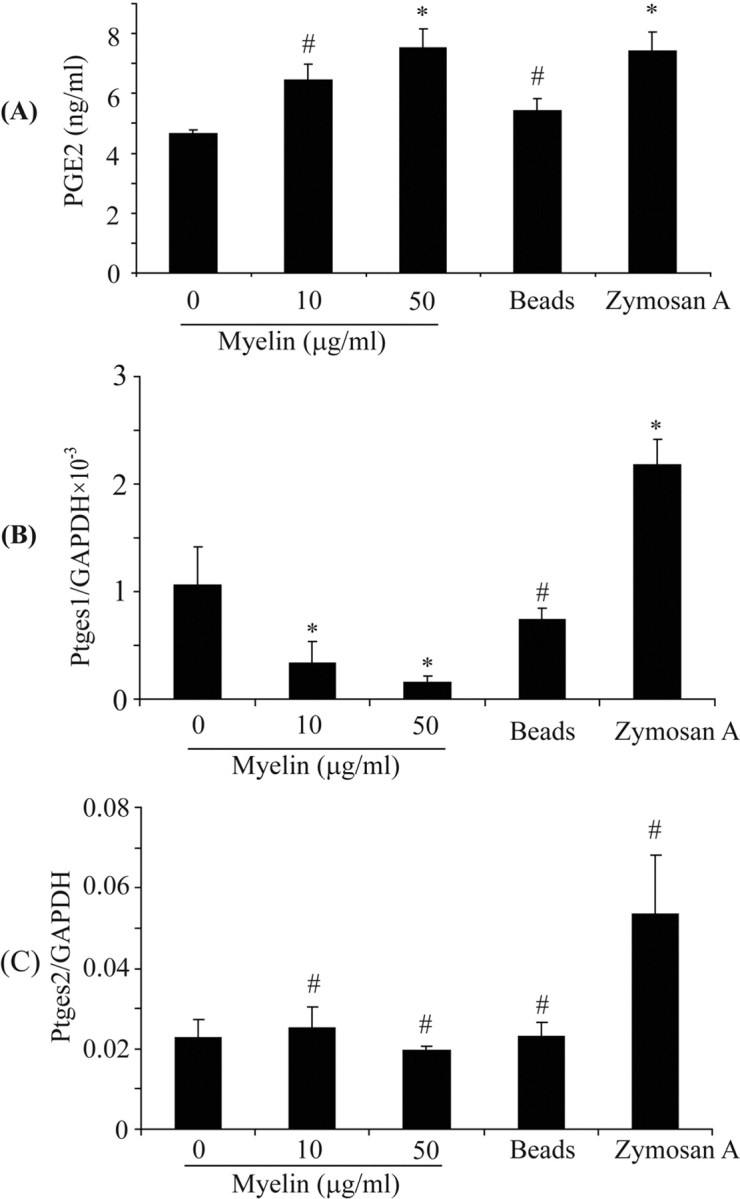
Myelin phagocytosis induces production of PGE2. IFN-γ-preactivated primary microglia were treated with myelin at 0, 10, and 50 μg/ml in the presence of IFN-γ for 24 h. Release of PGE2 as well as gene transcription of Ptges1 and Ptges2 were determined by ELISA and real-time PCR, respectively. A, The release of PGE2 detected by ELISA was significantly enhanced in relation to the myelin concentrations. B, C, The transcription of Ptges1 was downregulated by myelin in a dose-dependent manner (B) and that of Ptges2 was not significantly changed (C) (all of the experiments were independently repeated at least 4 times with the same trends; one-way ANOVA vs IFN-γ-activated control, *p < 0.05, #p > 0.05; n = 4–5 per group).
Myelin phagocytosis induces production of superoxide anion in IFN-γ-activated primary microglial cells
Internalization of myelin through phagocytosis induces a respiratory burst in microglia. To assess the effects of myelin phagocytosis on IFN-γ-activated primary microglial cells, we measured the transcription of Ncf1, a gene encoding p47–PHOX that is an essential component of the phagocyte NADPH–oxidase and quantified ROS levels. As seen in Figure 5, microglial phagocytosis of myelin did not affect Ncf1 transcription (Fig. 5A) but clearly enhanced microglial production of extracellular and intracellular ROS. After treatment of IFN-γ-activated microglia with myelin at 5, 10, and 50 μg/ml for 24 h, the extracellular ROS generation was 147.26 ± 14.00, 161.87 ± 7.21, and 214.36 ± 17.52%, respectively, compared with the basal level (Fig. 5B,C). Both intracellular and extracellular ROS generation could be inhibited by 5 mm apocynin (Fig. 5B). When treated with beads or zymosan A, microglia released significantly less ROS than did microglia treated with myelin (Fig. 5B).
Figure 5.
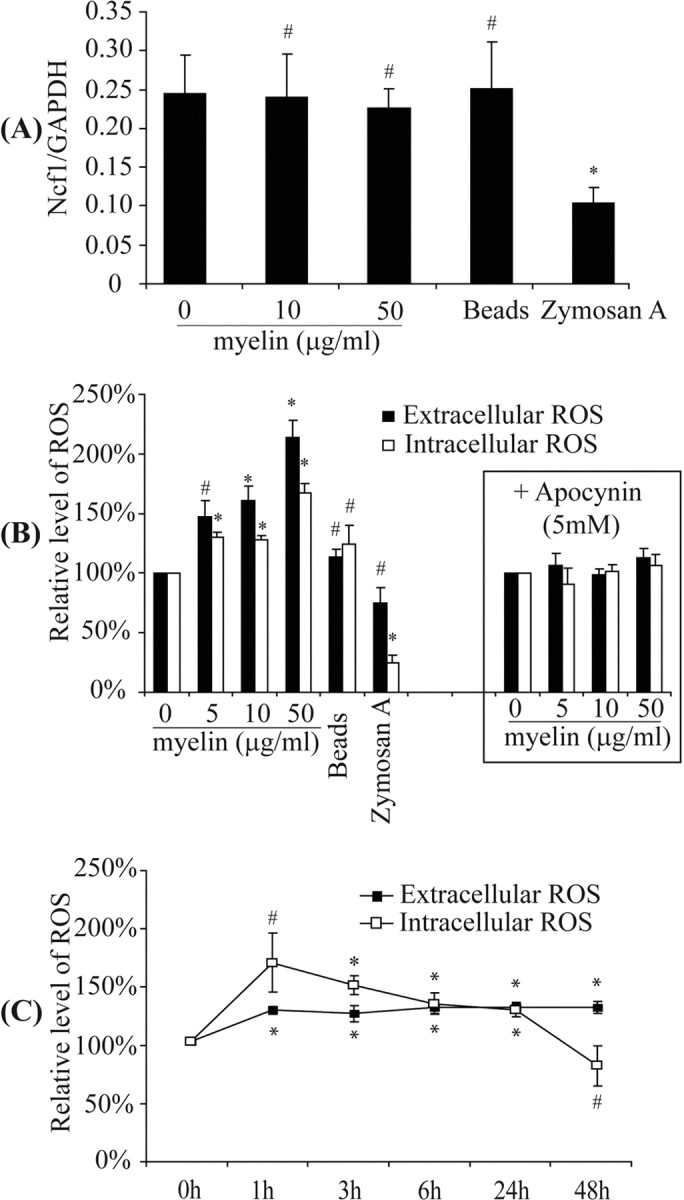
Myelin phagocytosis induces production of reactive oxygen species. IFN-γ-preactivated primary microglia were treated in the presence of IFN-γ with myelin at 0, 5, 10, and 50 μg/ml for 24 h or with 10 μg/ml myelin for 0, 1, 3, 6, 24, and 48 h. A, After 24 h, the transcription of Ncf1, encoding p47–PHOX, the main adaptor of NADPH–oxidase complex, was not changed. B, The extracellular and intracellular ROS were significantly enhanced relative to the concentration of myelin used as determined by measuring SOD-inhibitible reduction of WST-1 and luminol chemiluminescence assay, respectively. Apocynin at 5 mm was able to block the production of ROS caused by treatment of myelin. C, Production of both extracellular and intracellular ROS caused by phagocytosis of 10 μg/ml myelin was maintained at significantly higher level for >24 h (all of the experiments were independently repeated at least 4 times with the same trends; one-way ANOVA vs control without myelin treatment, *p < 0.05, #p > 0.05; n = 4–6 per group).
Reactive oxygen species mediate downregulation of inflammatory activation
We next asked whether ROS could modulate inflammatory gene transcription and secretion of cytokines and chemokines. We cotreated IFN-γ-activated primary microglia with myelin and SOD or catalase for 24 h. Treatment with SOD or catalase partially reversed the transcriptional downregulation of TNF-α, iNOS, and IP-10 but did not affect transcription of IL-1β and MIP-1α (Fig. 6A) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Similarly, cotreatment with SOD or catalase also reversed the myelin-triggered downregulation of TNF-α and IP-10 secretion (Fig. 6B). Limulus amebocyte lysate assay of SOD and catalase confirmed the absence of endotoxin contamination.
Figure 6.
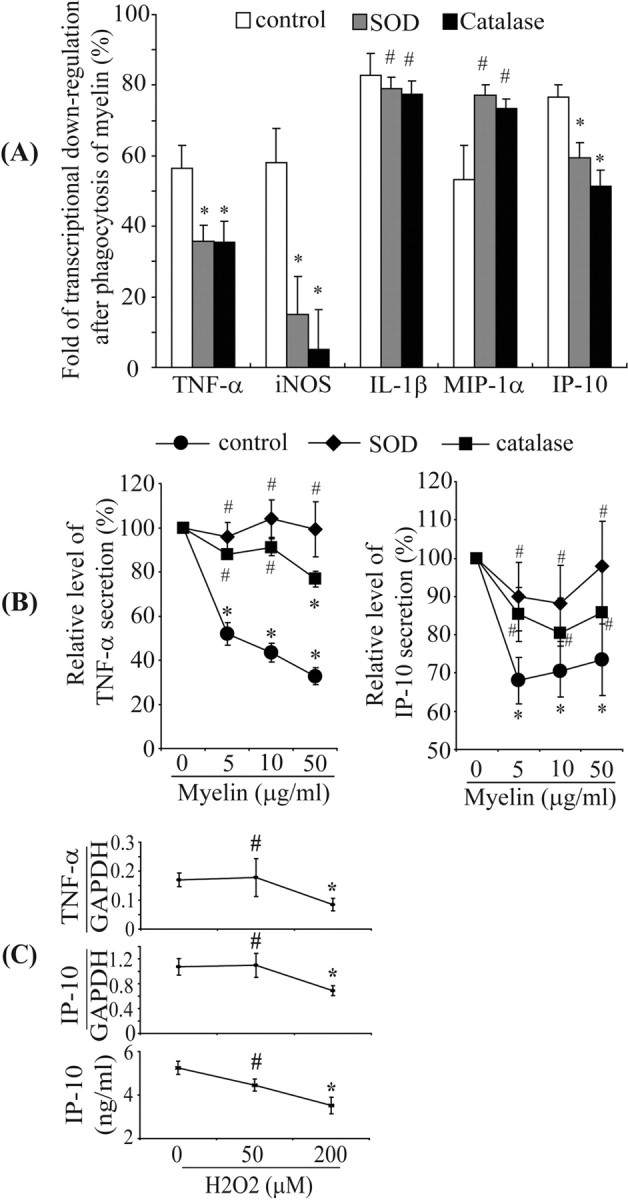
Reactive oxygen species are involved in downregulation of inflammatory activities. IFN-γ-preactivated primary microglia were treated with IFN-γ and myelin at 0, 5, 10, and 50 μg/ml in the presence or absence of 500 U/ml SOD or 1000 U/ml catalase for 24 h. Total RNA was isolated, and transcripts of inflammatory genes were measured using real-time quantitative PCR. Release of TNF-α and IP-10 in the culture medium was detected with ELISA. A, After 24 h, the folds of transcriptional downregulation of TNF-α, iNOS, and IP-10 were significantly reduced by cotreatment of SOD or catalase, but the downregulation of IL-1β or MIP-1α transcription was not altered (all of the experiments were independently repeated at least 5 times with the same trends; two-independent-samples t test vs control without cotreatment, *p < 0.05, #p > 0.05; n = 5–10 per group). B, Secretion of TNF-α or IP-10 was dose-dependently downregulated by myelin phagocytosis, which could be reversed by SOD and catalase (all of the experiments were independently repeated 4 times with the same trends; one-way ANOVA vs control without myelin treatment, *p < 0.05, #p > 0.05; n = 4 per group). C, When IFN-γ-activated primary microglia were directly treated with 50 and 200 μm H2O2 for 24 h, the transcription of TNF-α or IP-10 and secretion of IP-10 were downregulated in a dose-dependent manner (all of the experiments were independently repeated 4 times with the same trends; one-way ANOVA vs control without H2O2 treatment, *p < 0.05, #p > 0.05; n = 4–5 per group).
Treatment of IFN-γ-activated microglia with 200 μm H2O2 significantly downregulated TNF-α and IP-10 transcription and IP-10 release (one-way ANOVA, all p values <0.05) (Fig. 6C). However, IL-1β transcription was not reduced. After treatment of 200 μm H2O2, the transcripts were 9.46 ± 4.00 × 10−3 compared with basal levels, 5.00 ± 1.69 × 10−3 (mean ± SEM; independent-samples t test, p value >0.05; n = 4 per group). Based on MTT assay measurements and determinations of the release of lactate dehydrogenase, we excluded that treatment of IFN-γ-activated microglia with 50 and 200 μm H2O2 affected microglial viability (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Inhibiting assembly of NADPH–oxidase complex reverses the downregulation of transcription and secretion of inflammatory mediators induced by myelin phagocytosis
We cotreated IFN-γ-activated primary microglia with 10 μg/ml myelin and apocynin, a specific inhibitor of the NADPH–oxidase complex (Van der Worm et al., 2001), for 24 h. Apocynin treatment reversed the transcriptional downregulation of TNF-α, iNOS, IL-1β, MIP-1α, and IP-10 by myelin phagocytosis (Fig. 7A) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Similarly, apocynin treatment also reversed the reduced release of TNF-α, from IFN-γ-activated primary microglia (Fig. 7B). Administration of apocynin during myelin treatments of microglia activated by a combination of IFN-γ and LPS reversed the suppression of release of TNF-α, IL-1β, IL-10, and IP-10 (Fig. 7C–F), but apocynin treatment did not change the beads- and zymosan A-induced modulation of cytokine secretion from IFN-γ and LPS-activated microglia (data not shown). This suggests that myelin-mediated effects on the inflammatory responses of IFN-γ and LPS preactivated microglia are specific.
Figure 7.

Inhibition of NADPH–oxidase reverses downregulation of microglial transcription and secretion of inflammatory mediators by myelin phagocytosis. A–F, Primary microglia were preactivated by IFN-γ (A, B) or a combination of IFN-γ and LPS (C–F) and then treated with myelin (0, 5, 10, or 50 μg/ml) and the preactivator in the presence or absence of 5 mm apocynin for 24 h. Transcripts and secretion of inflammatory mediators were measured by quantitative PCR and ELISA, respectively. In the absence of apocynin treatment, transcription of TNF-α and IP-10 (A) and secretion of TNF-α (B) in IFN-γ-activated microglia were significantly downregulated; apocynin treatment reversed the downregulation of TNF-α and IP-10. Similarly, in IFN-γ plus LPS-activated microglia, release of TNF-α (C), IL-1β (D), IL-10 (E), and IP-10 (F) were significantly downregulated by myelin treatments; apocynin treatment reversed the downregulation of cytokines and IP-10 (all of the experiments were independently repeated at least 3 times with the same trends; one-way ANOVA vs control without myelin treatment, *p < 0.05, #p > 0.05; n = 3–8 per group).
Knockdown of Ncf1 reverses suppression of transcription and secretion of inflammatory mediators by myelin phagocytosis
To confirm the role of p47–PHOX in myelin phagocytosis-induced suppression of microglial activation, we specifically knocked down p47–PHOX expression by transfecting BV-2 cells with siRNA directed against gene Ncf1. Transfection with Ncf1 knockdown siRNA reduced the transcription of Ncf1 and expression of p47–PHOX, which, in turn, resulted in reduced levels of extracellular ROS after myelin phagocytosis (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
After preactivation of siRNA-transfected BV-2 cells with IFN-γ or a combination of IFN-γ and LPS, we administrated myelin at different concentrations (5, 10, and 50 μg/ml) for 24 h. Myelin phagocytosis did not downregulate transcription of TNF-α, iNOS, IL-1β, and IP-10 in IFN-γ-activated BV-2 cells transfected with the Ncf1 knockdown siRNA but did cause downregulation of the transcripts in IFN-γ-activated BV-2 cells transfected with control siRNA (Fig. 8A) (supplemental Table 2, available at www.jneurosci.org as supplemental material). Similarly, in BV-2 cells activated by a combination of LPS and IFN-γ, transfection with the Ncf1 knockdown siRNA prevented the myelin phagocytosis-induced downregulation of TNF-α, IL-1β, IL-10, and IP-10 secretion that was observed in cells transfected with control siRNA (Fig. 8B–E).
Figure 8.

Knockdown of p47–PHOX reverses downregulation of transcription and secretion of inflammatory mediators by myelin phagocytosis in siRNA-transfected BV-2 cells. BV-2 cells were transfected with control siRNA (ct siRNA) or siRNA targeting mouse Ncf1 (Ncf1 siRNA), activated by IFN-γ and then administrated with 10 μg/ml myelin in the presence of IFN-γ for 24 h. Transcripts of TNF-α and IP-10 were measured by quantitative PCR. As seen in A, in ct siRNA-transfected cells, TNF-α and IP-10 were significantly downregulated during challenge with 10 μg/ml myelin, but after transfection with Ncf1 knockdown siRNA, no decreased transcription of TNF-α and IP-10 was observed (all of the experiments were independently repeated 3 times with the same trends; two-independent-samples t test vs control without myelin treatment, *p < 0.05, #p > 0.05; n = 3 per group). To detect secretion of inflammatory mediators, transfected BV-2 cells were activated by IFN-γ and LPS and then administrated with 0, 5, 10, and 50 μg/ml myelin in the presence of preactivators for 24 h. Secretions were determined by ELISA. As seen in the B–E, myelin phagocytosis significantly reduced release of TNF-α, IL-1β, IL-10, and IP-10 from ct siRNA-transfected BV-2 cells but not from Ncf1 siRNA-transfected BV-2 cells (all of the experiments were independently repeated at least 3 times with the same trends; one-way ANOVA vs control without myelin treatment, *p < 0.05, #p > 0.05; n = 3–8 per group).
Myelin phagocytosis impairs adenine–uridine-rich elements-related stability of TNF-α and IP-10 mRNA in microglial or BV-2 cells
We continued to investigate the potential mechanisms of myelin phagocytosis-induced suppression of microglial activation. We treated IFN-γ-activated primary cultured microglia with 10 μg/ml myelin for 3 h because we demonstrated that, within this time, the myelin phagocytosis and ROS release were substantially induced and the transcription and secretion of specific inflammatory mediators were maximal. Thereafter, we added 10 μg/ml actinomycin D to block de novo mRNA synthesis. We measured IP-10 and TNF-α transcripts by quantitative RT-PCR at the time of addition of actinomycin D and at subsequent time points. The lifetime of both TNF-α and IP-10 transcripts was reduced by the treatment of myelin, which could be reversed by cotreatment of apocynin (Fig. 9A,B).
Figure 9.
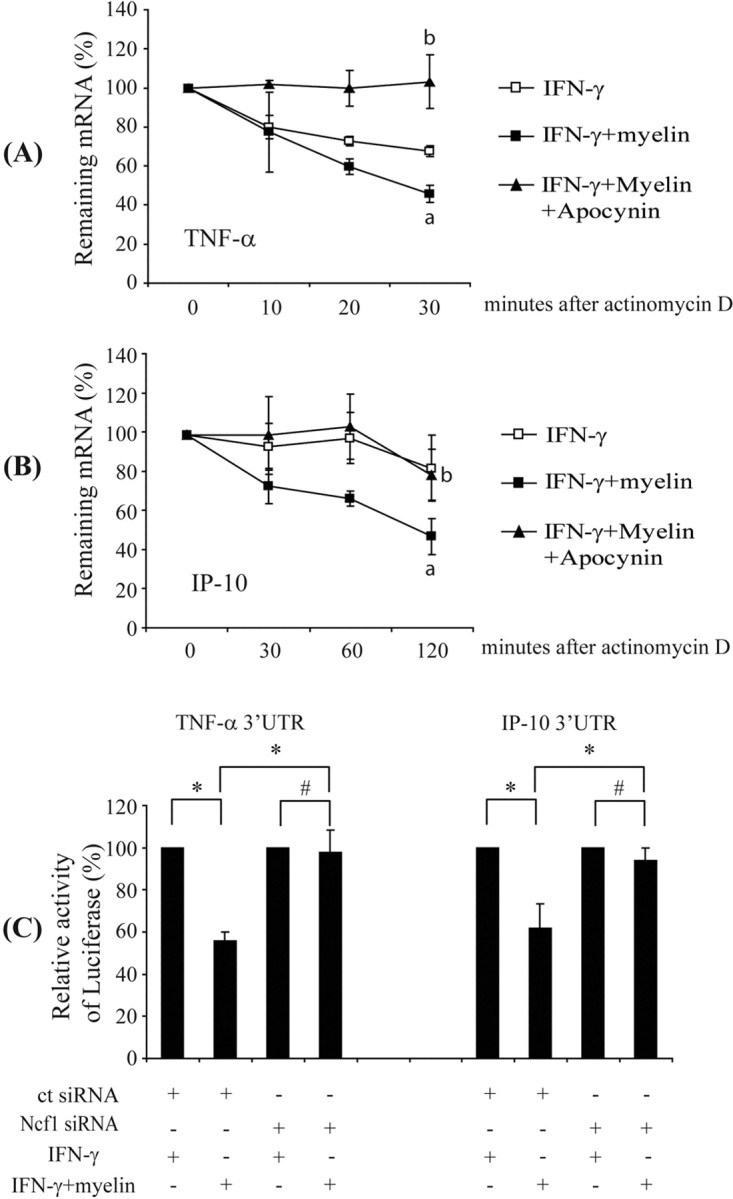
Myelin phagocytosis impairs TNF-α and IP-10 mRNA stability in microglia through an AREs-involved mechanism. IFN-γ-preactivated primary microglia were treated with 10 μg/ml myelin in the presence or absence of 5 mm apocynin for 3 h before incubation with 10 μg/ml actinomycin D. TNF-α and IP-10 transcripts were measured by quantitative RT-PCR. Lifetime of transcripts were determined by plotting of the percentage of remaining mRNA versus time after treatment with actinomycin D. A, B, Lifetime of TNF-α and IP-10 transcripts after treatment with myelin and IFN-γ were significantly shortened compared with treatment with only IFN-γ. Apocynin treatment could prolong the lifetime of TNF-α and IP-10 transcripts (all of the experiments were independently repeated 4 times with the same trends; two-independent-samples t test, ap < 0.05 vs IFN-γ treatment; bp < 0.05 vs cotreatment of IFN-γ and myelin; n = 4 per group). BV-2 cells were cotransfected with 0.15 μg of luciferase reporter genes carrying various 3′ UTR of gene encoding TNF-α or IP-10, 0.05 μg of pcDNA3.1/His/LacZ, and 10 pmol of double-stranded siRNA targeting mouse Ncf1 sequence (Ncf1 siRNA) or a nonsense sequence (ct siRNA). Reporter activity in cell lysates of BV-2 cells was measured after IFN-γ preactivation and myelin treatment. The ratio of luciferase activity to β-galactosidase activity was calculated. C, Reporter activity of BV-2 cells transfected with siRNA targeting a nonsense sequence but not with siRNA targeting Ncf1 was reduced by myelin treatment (all of the experiments were independently repeated 4 times with the same trends; two-independent-samples t test, *p < 0.05, #p > 0.05; n = 4 per group).
AREs in 3′ UTR regulate the decay of TNF-α, IP-10, and other cytokine and chemokine transcripts (Sun and Ding, 2006). AREs from unstable transcripts can destabilize reporter gene mRNAs to whose 3′ UTRs they are appended (Jing et al., 2005; Sun and Ding, 2006). To determine whether AREs could be involved in myelin phagocytosis-mediated transcript destabilization, we generated CMV promoter-driven luciferase reporter constructs containing 3′ UTRs derived from genes encoding IP-10, TNF-α, or β-actin and transfected BV-2 cells. Myelin phagocytosis reduced the luciferase reporter activity of the cells cotransfected with the reporter constructs containing ARETNF-α or AREIP-10 and siRNA targeting a nonsense sequence (Fig. 9C) but did not change the luciferase activity of the cells cotransfected with the reporter constructs containing ARETNF-α or AREIP-10 and siRNA targeting Ncf1. The reported activity of the construct containing the β-actin 3′ UTR after myelin treatment was not altered by siRNA transfection (data not shown).
Discussion
MS lesions contain a large number of proinflammatory products from activated microglia and macrophages that are highly toxic to myelin sheets, oligodendrocytes, and neurons (Selmaj and Raine, 1988; Selmaj et al., 1991; Probert et al., 1995; Merrill and Benveniste, 1996; Raivich and Banati, 2004; Sospedra and Martin, 2005). We have shown in our cell culture systems that phagocytosis of myelin affects microglial inflammatory activity: after an initial (first hours) increase in transcription and secretion of inflammatory microglial products, consistent with previous reports on proinflammatory effects of myelin phagocytosis (Williams et al., 1994; Mosley and Cuzner, 1996; van der Laan et al., 1996), phagocytosis of myelin, however, strongly suppressed microglial inflammatory activation at a late phase as demonstrated by the pronounced downregulated production of inflammatory cytokines and chemokines. These results are consistent with findings of an anti-inflammatory phenotype of myelin-laden mononuclear phagocytes in histological studies (Boven et al., 2006).
As a major mechanism for that inflammatory suppressive effect of myelin phagocytosis, we identified the release of ROS via p47–PHOX. Thus, we observed that blocking the assembly of the NADPH–oxidase complex or knocking down p47–PHOX expression reverses suppression of inflammatory activation by myelin phagocytosis.
Activated microglia exert quite different functions, including production of inflammatory cytokines, chemokines, and reactive superoxide ions, as well as phagocytotic activities. Based on our results, it is likely that there is a functionally relevant crosstalk between those different inflammatory events, e.g., secretion of cytokines/chemokines and phagocytosis, which is mediated by ROS synthesized through the assembly of NADPH–oxidase. In the context of the autoimmune attack in MS, ROS generation induced by phagocytosis of myelin debris may represent a negative feedback loop to self-limit cytokine-driven neuroinflammation. This view correlates with previous observations that the genetic defect of p47–PHOX enhances autoimmune encephalomyelitis and arthritis in animal models (Hultqvist et al., 2004) and leads to chronic non-infectious inflammation in chronic granulomatous disease (Morgenstern et al., 1997). Additional support for this hypothesis comes from a recently published study showing that p47–PHOX-mediated ROS negatively regulate expression of IL-8 and IL-1β in human neutrophils (Lekstrom-Himes et al., 2005).
However, the mechanism by which ROS modulate inflammation appears to be more complex. In our study, ROS generation caused by myelin phagocytosis suppressed both myeloid differentiation factor 88 (MyD88)-dependent (e.g., LPS-induced) and MyD88-independent (e.g., IFN-γ-induced) inflammatory responses. Further investigating the underlying mechanisms, we observed that myelin phagocytosis leads to ROS-dependent accelerated decay of TNF-α and IP-10 mRNA via their AREs in 3′ UTR. Therefore, the myelin-induced downregulation of different inflammatory mediators activated by different signal pathways could be explained by the gene structure of 3′ UTR. We then focused on extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK), and c-Jun N-terminal kinase (JNK) activation as upstream signal pathways because they have been shown to impair mRNA stability in pheochromocytoma cells, cardiomyocytes, or fibroblasts (Akiyama et al., 2005; Vassilopoulos and Papazafiri, 2005; DiCamillo et al., 2006; Glaser et al., 2006; Zhou et al., 2006). We observed that treatments with inhibitors of ERK and JNK pathways reversed myelin phagocytosis-mediated reduction of TNF-α or IP-10 transcription and secretion (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). We thus expect that JNK and ERK activation could be involved in mediating ROS-induced mRNA destabilization, although additional experiments that directly prove ROS-induced MAPK phosphorylation, which subsequently could impair mRNA stability, are still necessary.
Compared with the pronounced suppression of proinflammatory activities, the anti-inflammatory activities of myelin-treated microglia were relatively unchanged. Thus, expression of IL-10 and TGF-β1 was not downregulated by myelin phagocytosis in our study. However, myelin phagocytosis-induced alteration of IL-10 expression appeared to depend on the state of cellular preactivation because expression was increased in IFN-γ-activated but not in LPS plus IFN-γ-activated microglial cells. The anti-inflammatory cytokine IL-10 was also transcribed in the same time-dependent manner as the proinflammatory cytokines. Similarly, release of PGE2, a potential anti-inflammatory molecule (Hilkens et al., 1995; Kalinski et al., 1997; Harizi et al., 2002; Vassiliou et al., 2003), was increased in myelin-phagocytosing microglial cells. However, myelin phagocytosis did not upregulate transcription of the PGE2 syntheses, suggesting that stimulation of PGE2 release occurs at a posttranslational level. Therefore, it is possible that myelin phagocytosis inhibits overall microglial inflammatory activities.
Interestingly, myelin phagocytosis may also regulate secretion of inflammatory molecules. After myelin phagocytosis, TNF-α and IP-10 was immediately released within an interval too short for protein synthesis, and IP-10 transcription and secretion were modulated in different myelin dose-related manners. Furthermore, IL-1β secretion but not its transcription was strongly attenuated by knockdown of Ncf1. Because of the multiple sources of ROS within the cell and the broad and pleiotropic targets of ROS (Lambeth, 2004), the detailed mechanisms for this myelin phagocytosis-mediated inflammatory suppression need additional investigation.
In summary, this study demonstrates that p47–PHOX-mediated production of ROS after myelin phagocytosis suppresses microglial inflammatory activation. ROS generation could function as a negative feedback loop to self-limit neuroinflammation, once endogenous materials are cleared after an immune attack. Intervention in ROS generation could possibly represent a novel therapeutic target to attenuate neuroinflammation in MS patients.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Research Centre for Molecular Physiology of the Brain in Göttingen (K.F.), the “Deutsch Stiftung” (K.F.), and Homburger Forschungsförderungsprogramm Grant HOMFOR2006 (Y.L.). The Neuroimmunology Group at the European Neuroscience Institute Göttingen was supported by the University of Göttingen, the Hertie-Foundation (Institute for Multiple Sclerosis Research), and the Deutsche Forschungsgemeinschaft. The Neural Regeneration Group at the University of Bonn Life and Brain Center is supported by the Hertie-Foundation and Walter-und-Ilse-Rose-Foundation. The monoclonal antibody (ABL-93) developed by J. T. August was obtained from the Development Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biological Sciences (Iowa City, IA). We thank Dr. Stefan Graeber and Dr. Stefan Jung for their helpful comments.
References
- Akiyama et al., 2005.Akiyama N, Shimma N, Takashiro Y, Hatori Y, Hirabayashi T, Horie S, Saito T, Murayama T. Decrease in cytosolic phospholipase A2alpha mRNA levels by reactive oxygen species via MAP kinase pathways in PC12 cells: effects of dopaminergic neurotoxins. Cell Signal. 2005;17:597–604. doi: 10.1016/j.cellsig.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Bauer et al., 1994.Bauer J, Sminia T, Wouterlood FG, Dijkstra CD. Phagocytic activity of macrophages and microglial cells during the course of acute and chronic relapsing experimental autoimmune encephalomyelitis. J Neurosci Res. 1994;38:365–375. doi: 10.1002/jnr.490380402. [DOI] [PubMed] [Google Scholar]
- Blasi et al., 1990.Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Boven et al., 2006.Boven LA, Van Meurs M, Van Zwam M, Wierenga-Wolf A, Hintzen RQ, Boot RG, Aerts JM, Amor S, Nieuwenhuis EE, Laman JD. Myelin-laden macrophages are anti-inflammatory, consistent with foam cells in multiple sclerosis. Brain. 2006;129:517–526. doi: 10.1093/brain/awh707. [DOI] [PubMed] [Google Scholar]
- Chen et al., 1985.Chen JW, Murphy TL, Willingham MC, Pastan I, August JT. Identification of two lysosome membrane glycoproteins. J Cell Biol. 1985;101:85–95. doi: 10.1083/jcb.101.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren and Karlsson, 1999.Dahlgren C, Karlsson A. Respiratory burst in human neutrophils. J Immunol Methods. 1999;232:3–14. doi: 10.1016/s0022-1759(99)00146-5. [DOI] [PubMed] [Google Scholar]
- DiCamillo et al., 2006.DiCamillo SJ, Yang S, Panchenko MV, Toselli PA, Naggar EF, Rich CB, Stone PJ, Nugent MA, Panchenko MP. Neutrophil elastase-initiated EGFR/MEK/ERK signaling counteracts stabilizing effect of autocrine TGF-beta on tropoelastin mRNA in lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2006;291:L232–L243. doi: 10.1152/ajplung.00530.2005. [DOI] [PubMed] [Google Scholar]
- Epstein et al., 1983.Epstein LG, Prineas JW, Raine CS. Attachment of myelin to coated pits on macrophages in experimental allergic encephalomyelitis. J Neurol Sci. 1983;61:341–348. doi: 10.1016/0022-510x(83)90167-3. [DOI] [PubMed] [Google Scholar]
- Glaser et al., 2006.Glaser ND, Lukyanenko YO, Wang Y, Wilson GM, Rogers TB. JNK activation decreases PP2A regulatory subunit B56alpha expression and mRNA stability and increases AUF1 expression in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2006;291:H1183–H1192. doi: 10.1152/ajpheart.01162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harizi et al., 2002.Harizi H, Juzan M, Pitard V, Moreau JF, Gualde N. Cyclooxygenase-2-issued prostaglandin E2 enhances the production of endogenous IL-10, which down-regulates dendritic cell functions. J Immunol. 2002;168:2255–2263. doi: 10.4049/jimmunol.168.5.2255. [DOI] [PubMed] [Google Scholar]
- Hilkens et al., 1995.Hilkens CM, Vermeulen H, van Neerven RJ, Snijdewint FG, Wierenga EA, Kapsenberg ML. Differential modulation of T helper type 1 (Th1) and T helper type 2 (Th2) cytokine secretion by prostaglandin E2 critically depends on interleukin-2. Eur J Immunol. 1995;25:59–63. doi: 10.1002/eji.1830250112. [DOI] [PubMed] [Google Scholar]
- Hultqvist et al., 2004.Hultqvist M, Olofsson P, Holmberg J, Baeckstroem BT, Tordsson J, Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci USA. 2004;101:12646–12651. doi: 10.1073/pnas.0403831101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing et al., 2005.Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–634. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- Kalinski et al., 1997.Kalinski P, Hilkens CM, Snijders A, Snijdewint FG, Kapsenberg ML. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J Immunol. 1997;159:28–35. [PubMed] [Google Scholar]
- Lambeth, 2004.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Lekstrom-Himes et al., 2005.Lekstrom-Himes JA, Kuhns DB, Alvord WG, Gallin JI. Inhibition of human neutrophil IL-8 production by hydrogen peroxide and dysregulation in chronic granulomatous disease. J Immunol. 2005;174:411–417. doi: 10.4049/jimmunol.174.1.411. [DOI] [PubMed] [Google Scholar]
- Liu et al., 2005.Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, Heine H, Penke B, Neumann H, Fassbender K. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain. 2005;128:1778–1789. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- Merrill and Benveniste, 1996.Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- Mimura and Asano, 1976.Mimura N, Asano A. Synergistic effect of colchicine and cytochalasin D on phagocytosis by peritoneal macrophages. Nature. 1976;261:319–321. doi: 10.1038/261319a0. [DOI] [PubMed] [Google Scholar]
- Morgenstern et al., 1997.Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, Dinauer MC. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J Exp Med. 1997;185:207–218. doi: 10.1084/jem.185.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley and Cuzner, 1996.Mosley K, Cuzner ML. Receptor-mediated phagocytosis of myelin by macrophages and microglia: effect of opsonization and receptor blocking agents. Neurochem Res. 1996;21:481–487. doi: 10.1007/BF02527713. [DOI] [PubMed] [Google Scholar]
- Norton and Poduslo, 1973.Norton WT, Poduslo SE. Myelination in rat brain: method of myelin isolation. J Neurochem. 1973;21:749–757. doi: 10.1111/j.1471-4159.1973.tb07519.x. [DOI] [PubMed] [Google Scholar]
- Olofsson et al., 2003.Olofsson P, Holmberg J, Tordsson J, Lu S, Akerstrom B, Holmdahl R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet. 2003;33:25–32. doi: 10.1038/ng1058. [DOI] [PubMed] [Google Scholar]
- Olson and Miller, 2004.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Probert et al., 1995.Probert L, Akassoglou K, Pasparakis M, Kontogeorgos G, Kollias G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc Natl Acad Sci USA. 1995;92:11294–11298. doi: 10.1073/pnas.92.24.11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich and Banati, 2004.Raivich G, Banati R. Brain microglia and blood-derived macrophages: molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res Rev. 2004;46:261–281. doi: 10.1016/j.brainresrev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Selmaj et al., 1991.Selmaj K, Raine CS, Farooq M, Norton WT, Brosnan CF. Cytokine cytotoxicity against oligodendrocytes. Apoptosis induced by lymphotoxin. J Immunol. 1991;147:1522–1529. [PubMed] [Google Scholar]
- Selmaj and Raine, 1988.Selmaj KW, Raine CS. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol. 1988;23:339–346. doi: 10.1002/ana.410230405. [DOI] [PubMed] [Google Scholar]
- Sospedra and Martin, 2005.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Sun and Ding, 2006.Sun D, Ding AH. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375–381. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- Takahashi et al., 2005.Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka et al., 1975.Tanaka R, Iwasaki Y, Koprowski H. Ultrastructural studies of perivascular cuffing cells in multiple sclerosis brain. Am J Pathol. 1975;81:467–478. [PMC free article] [PubMed] [Google Scholar]
- van der Goes et al., 1998.van der Goes A, Brouwer J, Hoekstra K, Roos D, van den Berg TK, Dijkstra CD. Reactive oxygen species are required for the phagocytosis of myelin by macrophages. J Neuroimmunol. 1998;92:67–75. doi: 10.1016/s0165-5728(98)00175-1. [DOI] [PubMed] [Google Scholar]
- van der Laan et al., 1996.van der Laan LJ, Ruuls SR, Weber KS, Lodder IJ, Dopp EA, Dijkstra CD. Macrophage phagocytosis of myelin in vitro determined by flow cytometry: phagocytosis is mediated by CR3 and induces production of tumor necrosis factor-alpha and nitric oxide. J Neuroimmunol. 1996;70:145–152. doi: 10.1016/s0165-5728(96)00110-5. [DOI] [PubMed] [Google Scholar]
- Van der Worm et al., 2001.Van der Worm E, Beukelman CJ, Van der Berg AJ, Kroes BH, Labadie RP, Van Dijk H. Effects of methoxylation of apocynin and analogs on the inhibition of reactive oxygen species production by stimulated human neutrophils. Eur J Pharmacol. 2001;433:225–230. doi: 10.1016/s0014-2999(01)01516-3. [DOI] [PubMed] [Google Scholar]
- Vassiliou et al., 2003.Vassiliou E, Jing H, Ganea D. Prostaglandin E2 inhibits TNF production in murine bone marrow-derived dendritic cells. Cell Immunol. 2003;223:120–132. doi: 10.1016/s0008-8749(03)00158-8. [DOI] [PubMed] [Google Scholar]
- Vassilopoulos and Papazafiri, 2005.Vassilopoulos A, Papazafiri P. Attenuation of oxidative stress in HL-1 cardiomyocytes improves mitochondrial function and stabilizes Hif-1alpha. Free Radic Res. 2005;39:1273–1284. doi: 10.1080/10715760500166685. [DOI] [PubMed] [Google Scholar]
- Williams et al., 1994.Williams K, Ulvestad E, Waage A, Antel JP, McLaurin J. Activation of adult human derived microglia by myelin phagocytosis in vitro. J Neurosci Res. 1994;38:433–443. doi: 10.1002/jnr.490380409. [DOI] [PubMed] [Google Scholar]
- Zhang et al., 2005.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J. Aggregated a-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- Zhou et al., 2006.Zhou C, Ziegler C, Birder LA, Stewart AF, Levitan ES. Angiotensin II and stretch activate NADPH–oxidase to destabilize cardiac Kv4.3 channel mRNA. Circ Res. 2006;98:1040–1047. doi: 10.1161/01.RES.0000218989.52072.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]