

Abstract
Mints/X11s are adaptor proteins composed of three isoforms: neuron-specific Mints 1 and 2, and the ubiquitously expressed Mint 3. We have now analyzed constitutive and conditional knock-out mice for all three Mints/X11s. We found that ∼80% of mice lacking both neuron-specific Mint isoforms (Mints 1 and 2) die at birth, whereas mice lacking any other combination of Mint isoforms survive normally. The ∼20% surviving Mint 1/2 double knock-out mice exhibit a decrease in weight and deficits in motor behaviors. Hippocampal slice electrophysiology uncovered a decline in spontaneous neurotransmitter release, lowered synaptic strength, and enhanced paired-pulse facilitation in Mint-deficient mice, suggesting a decreased presynaptic release probability. Acute ablation of Mint expression in cultured neurons from conditional Mint 1/2/3 triple knock-in mice also revealed a decline in spontaneous release, confirming that deletion of Mints impair presynaptic function. Quantitation of synaptic proteins showed that acute deletion of Mints caused a selective increase in Munc18-1 and Fe65 proteins, and overexpression of Munc18-1 in wild-type neurons also produced a decrease in spontaneous release, suggesting that the interaction of Mints with Munc18-1 may contribute to the presynaptic phenotype observed in Mint-deficient mice. Our studies thus indicate that Mints are important regulators of presynaptic neurotransmitter release that are essential for mouse survival.
Keywords: Mint, X11, knock-out, neurotransmitter release, survival, adaptor proteins
Introduction
Vertebrates contain three Mint/X11 proteins: Mint 1/X11α, Mint 2/X11β, and Mint 3/X11γ (Duclos et al., 1993; Duclos and Koenig, 1995; Okamoto and Südhof, 1997, 1998). Mints are composed of variable isoform-specific N-terminal, and conserved C-terminal sequences composed of one phosphotyrosine-binding (PTB) domain and two postsynaptic density-95/Discs large/Zona occludens-1 (PDZ) domains (Okamoto and Südhof, 1997, 1998). The N-terminal sequence of Mint 1 (but not of Mints 2 and 3) binds to calcium/calmodulin-dependent serine kinase-related protein (CASK), a PDZ-domain protein that belongs to the membrane-associated guanylate kinase family and was identified because it in turn binds to neurexins, which are presumptive synaptic cell-adhesion molecules (Hata et al., 1996). CASK also binds to another PDZ-domain protein called mammalian LIN-7/vertebrate homolog of LIN-7 (Veli/MALS), forming an evolutionarily conserved tripartite complex composed of Mint 1, CASK, and Velis (Borg et al., 1998, 1999; Butz et al., 1998; Kaech et al., 1998). In Caenorhabditis elegans, the Mint 1/CASK/Veli complex is essential for vulva development (Kaech et al., 1998; Whitfield et al., 1999), and Mint 1 is separately required for trafficking of AMPA- but not NMDA-type glutamate receptors to synapses (Rongo et al., 1998). Although only Mint 1 binds to CASK, both neuron-specific Mints (Mints 1 and 2) but not Mint 3 bind to the synaptic vesicle fusion protein Munc18-1 via an N-terminal sequence (Okamoto and Südhof, 1997). Moreover, via their PTB domains all three Mints bind to the cytoplasmic tail of the amyloid-β precursor protein (APP) and alter its proteolytic processing, which is centrally involved in the pathogenesis of Alzheimer's disease (Borg et al., 1996; McLoughlin and Miller, 1996; Sastre et al., 1998; Biederer et al., 2002). Finally, via their PDZ domains, Mints bind in vitro to multiple proteins such as presenilins (Lau et al., 2000; Biederer et al., 2002), neurexins (Biederer and Südhof, 2000), calcium channels (Maximov et al., 1999), and the kinesin motor protein KIF17 (Setou et al., 2000).
Various functional hypotheses about Mints have emerged (see the >30 papers cited in supplemental Table 1, available at www.jneurosci.org as supplemental material). However, in vertebrates, only Mint 1 was examined genetically, and little is known about the in vivo functions of vertebrate Mints (Mori et al., 2002; Ho et al., 2003). Deletion of Mint 1 does not alter survival and brain development but selectively impairs inhibitory synaptic transmission (Ho et al., 2003), similar to the short-term synaptic plasticity changes observed in Veli/MALS knock-out (KO) mice (Olsen et al., 2005). Mints 1, 2, and 3 are coexpressed in neurons throughout the brain; interestingly, Mint 1 is particularly abundant in inhibitory interneurons (Ho et al., 2003), suggesting that the limited phenotype in inhibitory neurons observed in Mint 1 KO mice is attributable to functional redundancy between Mints. Thus, to better define the essential role of Mints, we have now generated and analyzed mutant mice lacking multiple Mints as constitutive and conditional KOs. Our data demonstrate that Mints are essential for mouse survival and that their deletion produces a presynaptic functional deficit.
Materials and Methods
Generation of Mint KO mice.
The strategies used to generate conditional and constitutive Mint KO mice are depicted in Figure 1. Mint 1 mutant mice were described previously (Ho et al., 2003). In the Mint 2 targeting vector, we flanked the exon encoding residues 368–416 of the PTB domain with loxP sites. Of 444 clones analyzed, 11 were targeted correctly, and 2 of these clones were used to generate mice containing the Mint 2 knock-in. Genotyping for Mint 2 was performed using PCR with oligonucleotide primers AH0183 and AH0184 (5′-GGTAGCTGTGTGTTCAGG-3′ and 5′-GGCTAGATCACTCTGCAG-3′, respectively) for the wild-type (product size, 400 bp) and mutant alleles (product size, 440 bp). In the Mint 3 targeting vector, we flanked exon 3 of the Mint 3 gene encoding residues 201–249 of the PTB domain with loxP sites. Of 531 clones analyzed, 24 were targeted correctly, and 4 of these clones were used to generate mice containing the Mint 3 knock-in. Genotyping for Mint 3 was performed using PCR with oligonucleotide primers AH0178 and AH0182 (5′-GGATCTTGTGCATGCTAAGCC-3′ and 5′-GGACATGCGCTGGGTGGGCTG-3′, respectively) for the wild-type allele (product size, 505 bp). For Mint 3 mutant allele, oligonucleotide primers AH0182 and AH0052 were used (5′-GGACATGCGCTGGGTGGGCTG-3′ and 5′-GGCGCCTACCGGTGGATGTGGAATGTGTGC0–3′, respectively) with product size at 400 bp. We generated mice lacking individual Mint proteins or all possible combinations of Mint family members by intercrossing the individual lines. Null phenotype was also confirmed by Western blotting of brain homogenates with antibodies to Mint 1, 2, and 3.
Figure 1.
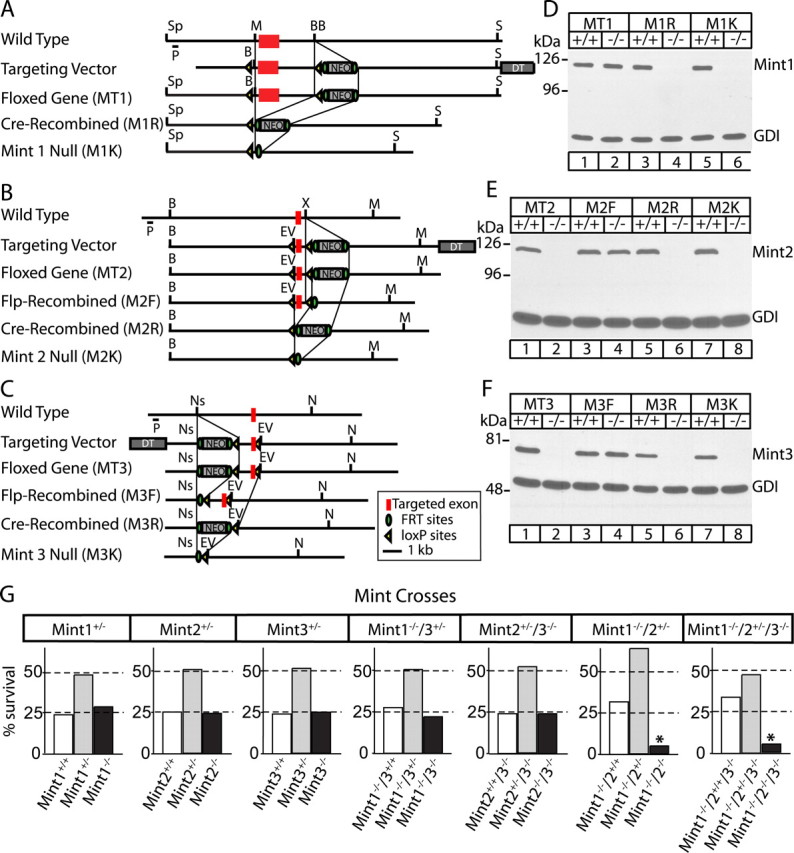
Generation and survival of Mint 1, 2, and 3 KO mice. A–C, Homologous recombination strategy for Mint 1, 2, and 3, respectively. The targeted exon (red box) was flanked by loxP sites (yellow arrowheads) to allow conditional removal of exon, a NEO for positive selection surrounded by FRT sites (green circles), and a diphtheria toxin (DT) was attached for negative selection. D–F, Immunoblot analysis of brain proteins from wild-type (+/+) and homozygous (−/−) mice. A–D, Homozygous floxed Mint 1 mutants (MT1) showed normal Mint 1 protein expression in the presence of NEO resistance cassette but were abolished when we crossed these mice with transgenic mice that express cre recombinase in the male germline (M1R). B, C, E, F, For homozygous floxed Mint 2 and 3 mutants (MT2 and MT3, respectively), the presence of the NEO resistance cassette abolished protein expression before floxed exon was excised. However, when NEO was excised by crossing with mice expressing FRT recombinase in the germline, both Mints 2 and 3 expressions returned as expected (M2F, M3F, respectively). In addition, Mints 2 and 3 protein expression were abolished when exon was removed by crossing with mice that express cre recombinase (M2R, M3R, respectively). In addition, we have created Mint KO lines in which protein expression was abolished with excision of both the targeted exon and NEO cassette (M1K, M2K, and M3K). G, Genotype analysis of offspring obtained from single Mint 1, 2, or 3 heterozygous matings show normal Mendelian ratio of genotypes. Genotype analysis of offspring obtained from Mint 1−/−/3+/− or Mint 2+/−/3−/− matings revealed normal Mendelian ratio of genotypes. However, interbreeding of Mint 1−/−/2+/− animals resulted in a Mendelian ratio of 5% instead of the expected 25% in double Mint 1/2 offsprings. Triple KO of all three Mints showed a Mendelian ratio similar to Mint 1/2 double KO mice. Sp, Spe; P, probe; M, Mfe1; BB, BstB1; B, BamH1; S, Sal1; EV, EcoRV; X, Xho1; Ns, Nsi1; N, Not1.
Morphological, quantitative protein analyses, and force-plate actometry.
Brains were removed from newborn pups and directly fixed with 4% fresh paraformaldehyde overnight at 4°C and cryoprotected with 30% sucrose in PBS, while adult mice (6–8 weeks of age) were anesthetized and perfusion fixed as described previously (Ho et al., 2003). Sections were stained with either hematoxylin and eosin or Nissl. For immunohistochemistry, sections were permeabilized with 0.5% Triton X-100, blocked with 2% goat serum/0.1% Triton X-100, and incubated with primary antibodies overnight at 4°C. This was followed by incubation with horseradish peroxidase-coupled secondary antibodies and developed in 3,3′-diaminobenzidine tetrahydrochloride with nickel chloride as a metal enhancement and analyzed by standard light microscopy. To determine presynaptic density, sections were incubated with monoclonal antibody against synaptophysin (Millipore, Billerica, MA), followed by incubation with Alexa Fluor 633 goat anti-mouse IgG (Invitrogen, Eugene, OR). Sections were transferred onto SuperFrost slides and mounted under glass coverslips with Vectashield with DAPI (Vector Laboratories, Burlingame, CA). Sections of the CA1 subfields of the hippocampus were then imaged with a laser-scanning confocal microscope (Leica Microsystems, Wetzlar, Germany) at 63× and magnified fivefold. Confocal images of synaptophysin-immunoreactive presynaptic terminals (dissector grid, 152.9 μm2) were obtained, and additional images were captured at the same x and y coordinates at a greater depth (1.0 μm for synapses). The number of immunolabeled objects was obtained by ImageJ analysis. For protein quantifications, brain tissue from three adult littermate mice per genotype was isolated and homogenized in PBS, 10 mm EDTA, 1 mm PMSF, and proteinase inhibitors. Brain proteins (40 μg) were separated by SDS-PAGE, and immunoblotting was performed with standard methods as described previously (Rosahl et al., 1995). Radioisotope 125I-labeled secondary antibodies were used for quantitative analyses followed by PhosphorImager (Molecular Dynamics, Sunnyvale, CA) detection with GDP dissociation inhibitor or vasolin-containing protein as internal controls. For protein quantifications in neuronal cultures, high-density neocortical cultures at 14 d in vitro (DIV) were harvested by washing neurons with cold PBS, followed by the addition of SDS sample buffer. All data shown are means ± SEMs. Statistical significance was determined by the Student's t test. For force-plate analyses, adult littermate mice per genotype performed on a force plate (28 × 28 cm) for 6 min as described previously (Fowler et al., 2001). Ataxia index is calculated as the area over the distance traveled on the force plate.
Primary neuronal cultures from Mint KOs.
Dissociated high-density hippocampal and neocortical cultures were prepared from newborn mice as described previously (Kavalali et al., 1999), and analyzed at 13–15 DIV.
Lentiviral infection.
Production of recombinant lentiviruses is achieved by transfecting human embryonic kidney 293T cells with three plasmids using FuGENE reagent. VSVg and CMVΔ8.9 are plasmids encoding the elements essential for packing of viral particles. pFUGW (or its derivates) is a shuttle vector encoding the gene of interest and containing recombination arms for incorporating into mammalian genome. For our purposes, we generated a pFUGW plasmid containing the enhanced green fluorescent protein (EGFP) with nuclear localization signal (NLS) carrying cre recombinase sequence (EGFP-NLS-CRE) with ubiquitin promoter. For control, we generated a pFUGW plasmid containing only EGFP-NLS (Cre mutant). For overexpression of Munc18-1 in wild-type neurons, we generated a pFUGW plasmid containing Munc18-1 with GFP or GFP alone. Viruses were harvested 48 h after transfection by collecting the medium from transfected cells, and 0.45 μm filter was used to remove cellular debris. Neurons were infected with 500 μl of cell medium to each 24-well of high-density neurons at 4 DIV. Neurons were infected for 48 h for expression, and media were exchanged back to normal growth media and sustained until 13–15 DIV for biochemical and electrophysiological analyses.
Electrophysiological analyses.
Synaptic transmission was recorded from pyramidal cells in 13–15 DIV cultures using whole-cell voltage-clamp techniques. Recordings were acquired with an Axopatch 700B amplifier and Clampex 9.0 software (Molecular Devices, Sunnyvale, CA). Synaptic recordings were filtered at 1 kHz and sampled at 5 kHz. Electrode solution for the pipette contained the following (in mm): 105 Cs-MeSo3, 10 CsCl, 5 NaCl, 10 HEPES, 0.6 EGTA, 20 TEA-Cl, 4 Mg-ATP, 0.3 Na3GTP, and 10 lidocaine N-ethyl bromide, pH 7.35 (300 mOsm). Spontaneous event recordings were performed in the presence of 1 μm tetrodotoxin (TTX). For recording miniature EPSCs (mEPSCs), 50 μm picrotoxin was added to the bath. For miniature IPSCs (mIPSCs), 10 μm CNQX and 50 μm d-2-amino-5-phosphonovaleric acid (d-APV) were added to block glutamatergic transmission. Hypertonic sucrose solution was prepared by the addition of 500 mm sucrose to the modified Tyrode solution and applied by gravity perfusion for measuring evoked sucrose response. To measure 10 Hz stimulation, we used 25 action potentials.
Hippocampal slice electrophysiology.
Synaptic responses were recorded from transverse hippocampal slices (0.4 mm) of 4- to 6-week-old mice. Slices were maintained and perfused in external solution containing the following (in mm): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1 NaH2PO4, 26.2 NaHCO3, and 11 d-glucose saturated with 95% O2/5% CO2, pH 7.4. Slices were equilibrated at 28–30°C for 1.5 h before transfer to a recording chamber and superfused at 2 ml/min with external solution warmed to 28–30°C. Extracellular field and whole-cell voltage-clamp recordings were obtained from CA1 pyramidal cells. The composition of the patch solution contained the following (in mm): 117.5 CsMeSO4, 10 HEPES, 10 TEACl, 15.5 CsCl, 8 NaCl, 1 MgCl, 0.3 NaGTP, 4 MgATP, 0.25 EGTA, 314 5 QX (295–305 mOsm, pH adjusted to 7.3 with CsOH). Synaptic responses to paired-pulse stimuli, synaptic AMPA/NMDA ratios, and 14 Hz induced synaptic depression were obtained as described previously (Ho et al., 2003). mEPSCs were recorded in the presence of 50 μm d-APV and 1 μm TTX. mIPSCs were recorded at a holding potential of +10 mV in the presence of 50 μm d-APV, 10 μm NBQX, 5 μm CGP-55845 and 1 μm TTX. Dendritic field EPSPs (fEPSPs) were recorded in the CA1 region with a patch electrode filled with external solution. Basal fEPSPs were evoked in the presence of 50 μm picrotoxin at 0.05 Hz with a monopolar stimulating electrode positioned in the stratum radiatum. Input–output curves were constructed by averaging 10 consecutive fEPSPs evoked at each stimulus strength, and the amplitude of the presynaptic volley was plotted against the corresponding slope of the fEPSP. Whole cell and field recordings were filtered at 2 and 1 kHz, respectively, and digitized at 5 kHz with an analog-to-digital board (National Instruments, Austin, TX) driven by custom acquisition software designed to run on IGOR Pro (WaveMetrics, Lake Oswego, OR). mEPSCs and mIPSCs were analyzed with Synaptosoft (Decatur, GA) software using a minimum of 200 events. Statistical significance of data were determined using a Student's t test or a Kolmogorov–Smirnov nonparametric test with a level of significance of p < 0.05.
Results
Generation of KO mice lacking all three Mint isoforms
Mint 2 and 3 KO mice were produced using a strategy similar to that previously applied for Mint 1 KO mice (Ho et al., 2003). Briefly, genomic clones containing the 5′ ends of the murine Mint 2 and 3 genes were isolated and characterized and then used to construct targeting vectors for homologous recombination experiments in embryonic stem cells (Rosahl et al., 1995). The targeting strategy was designed to generate conditional KO mice by flanking a 5′ coding exon with loxP sites such that expression of Mints can be inactivated by excision of exons with cre recombinase (Fig. 1 A–C). In addition, a neomycin resistance gene cassette (NEO) was introduced for positive selection and was flanked by FLP recognition target (FRT) sites so that it can be excised from the targeted gene because the NEO cassette can occasionally suppress transcription from a targeted gene. Finally, a cassette encoding diphtheria toxin was placed at the end of one of the homologous arms for negative selection (Fig. 1 A–C).
Using the targeting vectors in homologous recombination experiments in embryonic stem cells, we generated mice containing mutant Mints 2 and 3 genes and systematically analyzed these mice together with previously produced mice containing a mutant Mint 1 gene (Ho et al., 2003). We first examined Mint expression in mutant mice that still contain the floxed exon and the NEO cassette (referred to as MT1, MT2, and MT3 for Mints 1, 2, and 3, respectively). In contrast to Mint 1 mutant mice (MT1) that fully express Mint 1 (Ho et al., 2003), Mints 2 and 3 mutant mice (MT2 and MT3, respectively) containing the NEO cassette did not express Mints 2 or 3, suggesting that the NEO cassette may block expression (Fig. 1 D–F). We then excised the NEO cassette by crossing the Mints 2 and 3 mutant mice with mice expressing FRT recombinase in the germ line. In the resulting mice (referred to as M2F and M3F) (Fig. 1 A–C), Mint 2 and 3 expression was restored (Fig. 1 E,F). When we subsequently deleted the floxed exon by crossing mice expressing cre recombinase in the germline with Mint mutant mice that either retain the NEO cassette (resulting in M1R, M2R, and M3R lines) or lack the NEO cassette (resulting in M1K, M2K, and M3K lines) (Fig. 1 A–C), expression of the respective Mint proteins was abolished (Fig. 1 D–F). Immunoblotting of the KO mice with antibodies to the N-terminal sequences of Mints failed to reveal any remaining N-terminal fragments (data not shown). Thus, our homologous recombination experiments produced truly conditional and constitutive KO mice for all three Mint/X11 proteins.
Neuronal Mint proteins are essential for survival and are functionally redundant
To determine whether Mints are essential for mouse survival, we systematically examined the survival of mice that carry constitutive deletions of various individual Mints, of two Mints in any combination, and of all three Mints.
All single Mint KO mice were viable and fertile with a normal Mendelian ratio of genotypes in the offspring (Fig. 1 G). Although Mint 1 KO mice exhibit a significant decrease in size (Ho et al., 2003), Mints 2 and 3 KO mice did not display weight changes or overt abnormalities (data not shown). To obtain Mint double KO mice, we bred mice deficient for Mint 1 and heterozygous for Mint 2 or 3 with each other (for Mints 1/2 and 1/3 double KO mice), or mice deficient for Mint 3 and heterozygous for Mint 2 (for Mint 2/3 double KO mice). Mint 1/2 double KO and Mint 1/2/3 triple KO mice exhibited a severe survival deficit, with only ∼20% of mice surviving postnatally for each genotype. In contrast, Mint 1/3 and 2/3 double KO mice survived normally (Fig. 1 G). Together these data demonstrate that deletion of neuronal Mints 1 and 2 causes redundant lethality that is independent of Mint 3, and exhibits an incomplete penetrance.
Deletion of Mints 1 and 2 could be lethal either during embryonic development or within the first postnatal weeks. To test this, we analyzed the genotypes of offsprings from crossings of Mint 1 homozygous/Mint 2 heterozygous KO mice immediately after birth. We observed a normal Mendelian ratio of genotypes in the offspring (0.25, 0.50, 0.25 for wild-type/heterozygous/homozygous, respectively; n = 263), indicating that Mint 1/2 double KO mice perish after birth. Newborn Mint 1/2 double KO mice were visually indistinguishable from their littermates and displayed no major alterations in breathing, movements, or reaction to stimuli for several hours after birth. Later, however, most Mint 1/2 double and Mint 1/2/3 triple KO mice (except for those that survived) had no milk in their stomachs, appeared progressively weaker within 12 h, and died within 24 h presumably of dehydration.
We next investigated the effect of Mint deletions by analyzing the brains of newborn littermate Mint 1 KO mice that either contain or lack Mint 2 (Fig. 2). Mint 1 KO brains were used as controls in these experiments because previous studies showed that these KO mice exhibit no significant developmental changes or major impairments in brain structure (Ho et al., 2003). CASK binds to Mint 1 (Butz et al., 1998), and CASK KO mice are similar to Mint 1/2 double KO mice in that they also die within 12 h after birth and lack milk in their stomachs (T. C. Südhof, unpublished observation). Because in the case of the CASK KO mice this phenotype is associated with a cleft palate and Mint 1 binds to CASK, we paid particular attention in the morphological analyses to the possibility of a cleft palate. However, we detected no obvious alterations in brain morphology or body structures in newborn Mint 1/2 double KO mice compared with Mint 1 single KO littermates, and we especially observed no cleft palate (Fig. 2 A,B). Moreover, we detected no major developmental and structural abnormalities such as changes in the formation or layering of the cortex, cerebellum, hippocampus, and olfactory bulb (Fig. 2 C). Immunocytochemistry revealed a normal distribution of Mint 2 staining in the CA3 region of the hippocampus in the brains of newborn single Mint 1 KO mice but, as expected, failed to detect Mint 2 staining in the brains from Mint 1/2 double KO mice (Fig. 2 D). Furthermore, immunocytochemistry experiments for APP showed that APP was similarly localized in neurons of the CA3 region of the hippocampus in Mint 1 single and Mint 1/2 double KO mice (Fig. 2 E). Moreover, quantitations of 18 neuronal proteins failed to reveal significant changes (i.e., changes of >20%) in newborn Mint 1/2 double KO mice compared with Mint 1 single KO mice (supplemental Table 2, available at www.jneurosci.org as supplemental material). Viewed together, these data indicate that newborn Mint 1/2 double KO mice do not perish because of major developmental abnormalities or significant changes in brain structure or composition, suggesting that deletion of Mints causes a functional rather than a structural change in the brain.
Figure 2.
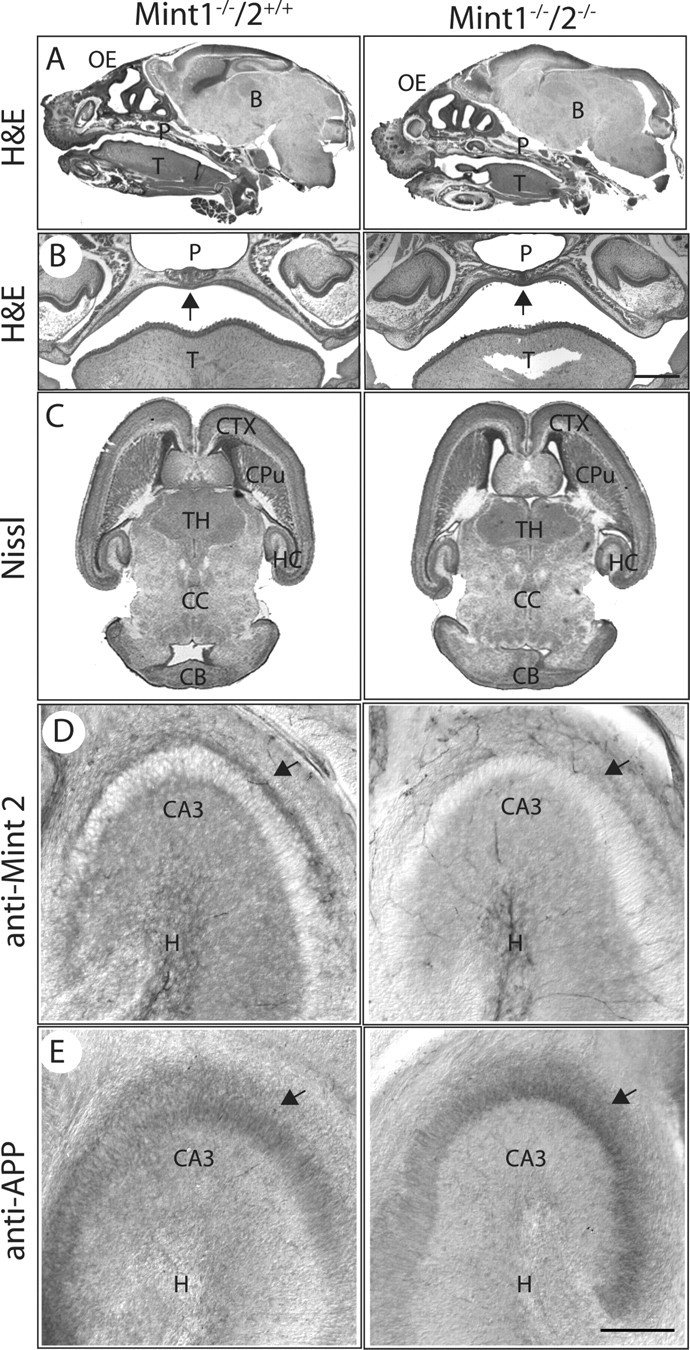
Histological examination of newborn Mint mutant mice. A, B, Mint 1/2 double KO mice revealed normal brain development and showed no obvious palate abnormalities by hematoxylin and eosin (H&E) stain compared with Mint 1 single KO littermates. C, Nissl-stained brain sections showed normal development and formation of the cortex, caudate–putamen, thalamus, and hippocampus in Mint 1 single and Mint 1/2 double KO mice. D, Single deletion of Mint 1 stained with Mint 2 revealed normal distribution of expression in CA3 region of the hippocampus, and as expected, all Mint 2 staining was abolished in Mint 1/2 double KO mice. E, Immunostaining of APP showed normal and distinct localization at the CA3 region of the hippocampus in Mint 1 KO mice, and this was indistinguishable in Mint 1/2 double KOs. OE, Olfactory epithelium; B, brain; P, palate; T, tongue; CTX, cerebral cortex; CPu, caudate–putamen; TH, thalamus; HC, hippocampus; CC, central canal; CB, cerebellum; H, hilus. Scale bars: B, 150 μm; (in E) D, E, 50 nm.
To expand these conclusions, we examined protein localizations and levels in surviving adult Mint 1/2 double knock-out mice. Nissl-stained brain sections from 6- to 8-week-old surviving Mint 1/2 double KO mice failed to uncover structural abnormalities or changes in brain architecture, including the cerebellum, despite the fact that Mint 1/2 double KO mice exhibited movement dysfunction (Fig. 3 A). As expected, all Mint 2 immunostaining was abolished, as shown in the CA3 region of the hippocampus and molecular layer of the cerebellum in adult Mint 1/2 double KO mice compared with single Mint 1 KO littermate brains (Fig. 3 B,C). Immunostaining for APP and for the synaptic protein Rab3A demonstrated that the distinct and typical localization of these proteins in the soma and synapses of hippocampal neurons, respectively, was not altered in brains lacking either Mint 1 alone or both Mints 1 and 2 (Fig. 3 D,E). This result suggests that the overall structure and distribution of these proteins in the mutant brains are normal. Consistent with this conclusion, measurements of the levels of synaptic proteins failed to reveal major changes in the adult mutant brains (supplemental Table 3, available at www.jneurosci.org as supplemental material).
Figure 3.
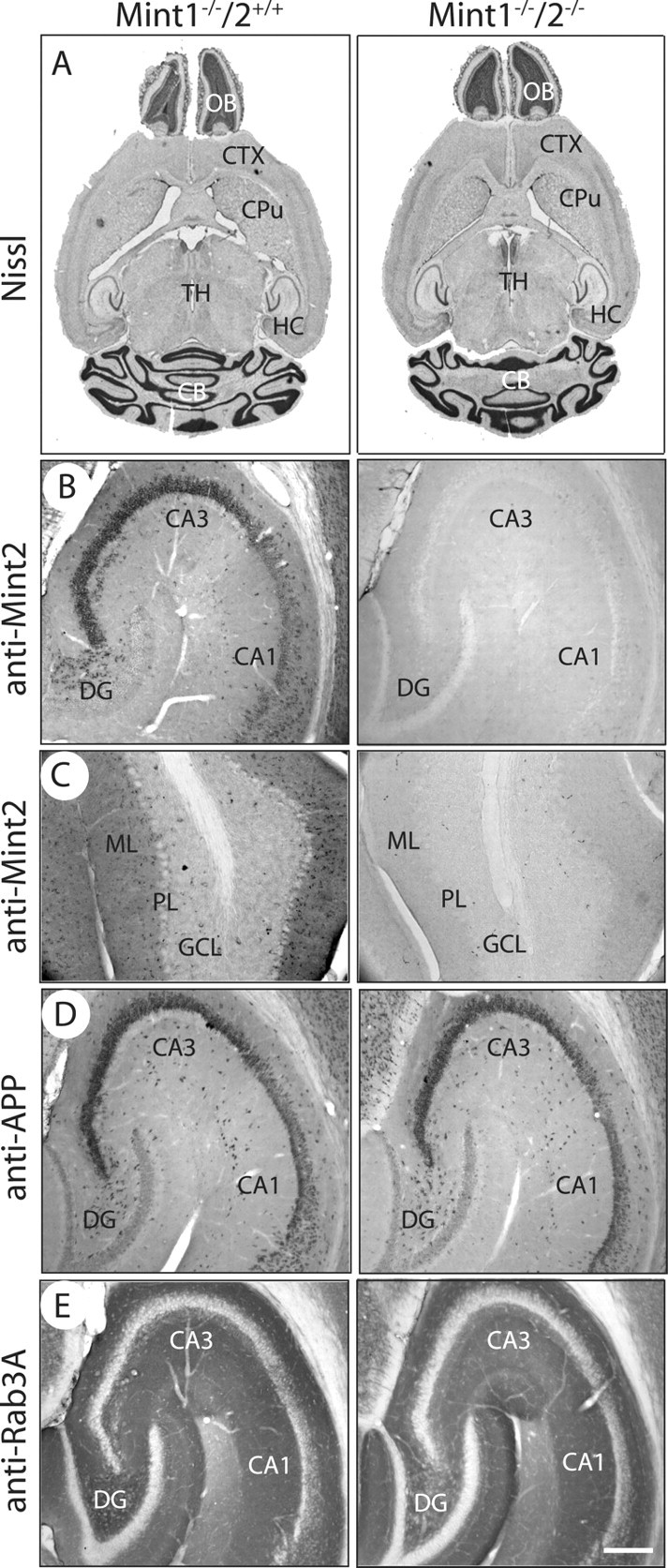
Overall brain structure and morphology of adult Mint KOs. A, Nissl brain sections of Mint 1−/−/2+/+ (left) and Mint 1−/−/2−/− double KO mice (right) at 6–8 weeks of age showed normal brain architecture. CTX, Cerebral cortex; CPu, caudate–putamen; TH, thalamus; OB, olfactory bulb; HC, hippocampus; CB, cerebellum. B, C, Mint 2 immunostaining was abolished in Mint 1−/−/2−/− double KO mice (right) as expected in the hippocampus (B) and cerebellum (C) compared with Mint 1−/−/2+/+ littermate control mice. DG, Dentate gyrus; ML, molecular layer; PL, Purkinje cell layer; GCL, granule cell layer. D, E, Immunostaining for APP (D) and Rab3A (E) displayed normal and distinct localization in the hippocampus in Mint 1−/−/2+/+ and Mint 1−/−/2−/− double KO littermate mice. Scale bar: (in E) B–E, 100 μm.
Surviving Mint 1/2 double KO mice are behaviorally abnormal
The ∼20% Mint 1/2 double KO mice that survive postnatally were significantly smaller than littermate Mint 1 single KO control mice (Fig. 4 A). Because Mint 1 KO mice but not Mint 2 KO mice are smaller than wild-type mice (Ho et al., 2003) (data not shown), the additional Mint 2 deletion on top of the Mint 1 deletion further impairs the normal weight gain of developing female and male mice (Fig. 4 B). However, different from many other KO mice with functional changes in synapses (Rosahl et al., 1995; Janz et al., 1999), no seizures were evident in Mint 1/2 double KO mice.
Figure 4.
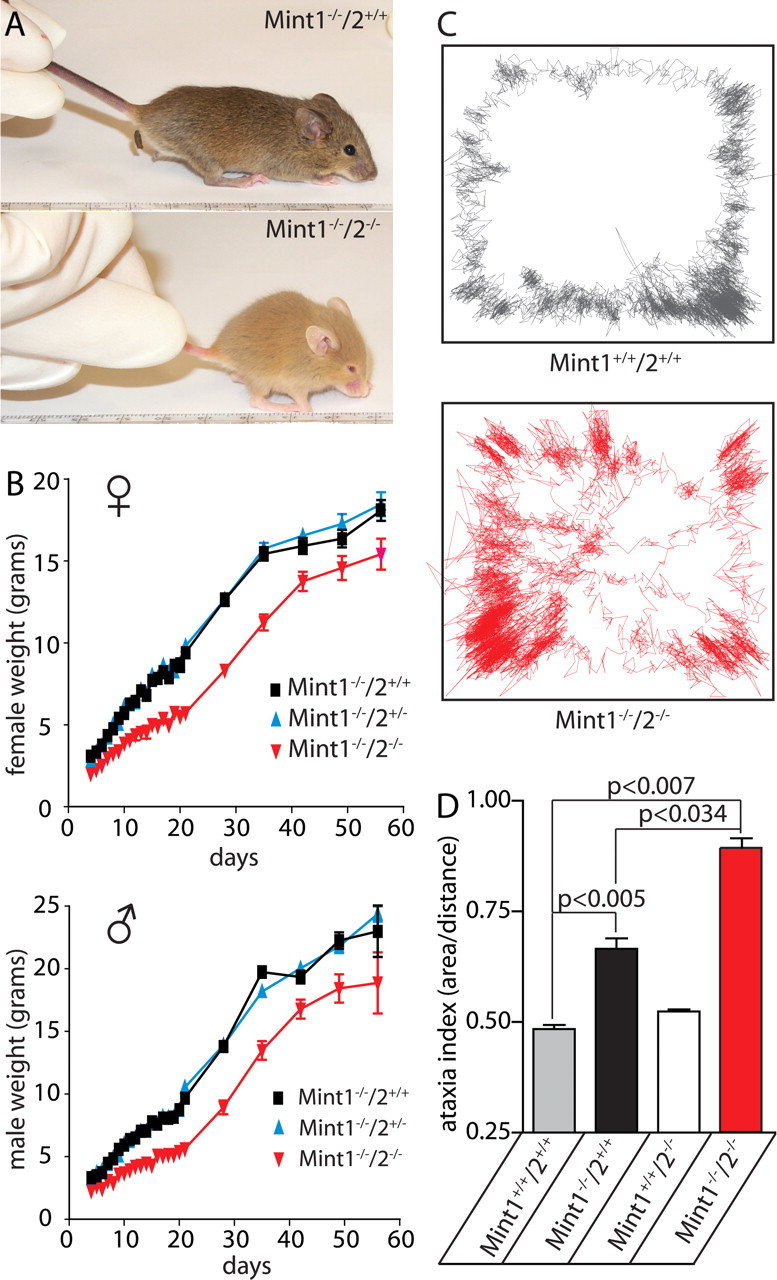
Abnormal weight and motor behavior of adult Mint 1/2 double KO mice. A, Photograph of Mint 1 single KO and Mint 1/2 double KO mice at 21 d of age. B, Plot of average weight of wild-type, heterozygous, and homozygous Mint 2 KO mice in a Mint 1 KO background as a function of age for littermate female (♀) and male mice (♂). Data shown represent means ± SEMs. C, Locomotor sample traces of wild-type (Mint 1+/+/2+/+; gray) and Mint 1−/−/2−/− double KO mice (red), which display the position of the mouse as it moves within the force plate as represented by the box within 6 min. D, Force plate analysis revealed a significant increase in ataxia indices (area covered in 6 min/net distance traveled) of Mint 1/2 double KO mice compared with wild type. Single Mint 1 KO mice exhibited a slight increase, whereas single Mint 2 KO mice displayed normal locomotive movements. Data shown represent means ± SEMs.
Behavioral testing revealed that Mint 1/2 double KO mice exhibit normal righting reflexes in a lateral recumbancy test (data not shown) but move abnormally. To measure this quantitatively, we evaluated 6- to 8-week-old mice on a force-plate actometer, which tracks the movements of a mouse on a square plate suspended on four corner sensors (Fowler et al., 2001) (Fig. 4 C,D). In the force-plate experiments, movements are quantitated as the ataxia index, which measures the area over distance traveled. Compared with wild-type littermate mice, Mint 1 but not Mint 2 single KO mice exhibited a small increase in the ataxia index, whereas deletion of both Mint 1 and 2 caused a major increase in the ataxia index (Fig. 4 C,D). Thus, both in terms of weight and movement, deletion of Mint 2 alone did not produce a phenotype but aggravated the Mint 1 deletion phenotype.
Adult Mint 1/2 double KO mice exhibit presynaptic deficits
To analyze the effect of the deletion of Mints on synaptic transmission, we recorded field EPSPs from CA1 pyramidal cells in acute hippocampal slices from surviving adult (4–6 weeks of age) Mint 1/2 double KO mice. Because in previous studies we had analyzed Mint 1 single KO mice with this approach compared with littermate wild-type control mice (Ho et al., 2003), we now examined Mint 1/2 double KO mice compared with littermate Mint 1 single KO mice. Input–output curves, the slopes of which represents the relationship between the size of the presynaptic fiber volley (input) and the resulting field EPSPs (output), revealed that Mint 1/2 double KO mice exhibited a significant decrease in the input–output relationship compared with Mint 1 single KO mice, thus strongly suggesting a decrease in basal synaptic strength (Fig. 5 A). In addition, we found a similar decrease in basal synaptic strength in Mint 1/2/3 triple KO mice; thus, deletion of Mint 3 in addition to Mint 1/2 double KO did not cause a major additional synaptic phenotype (Fig. 5 B).
Figure 5.
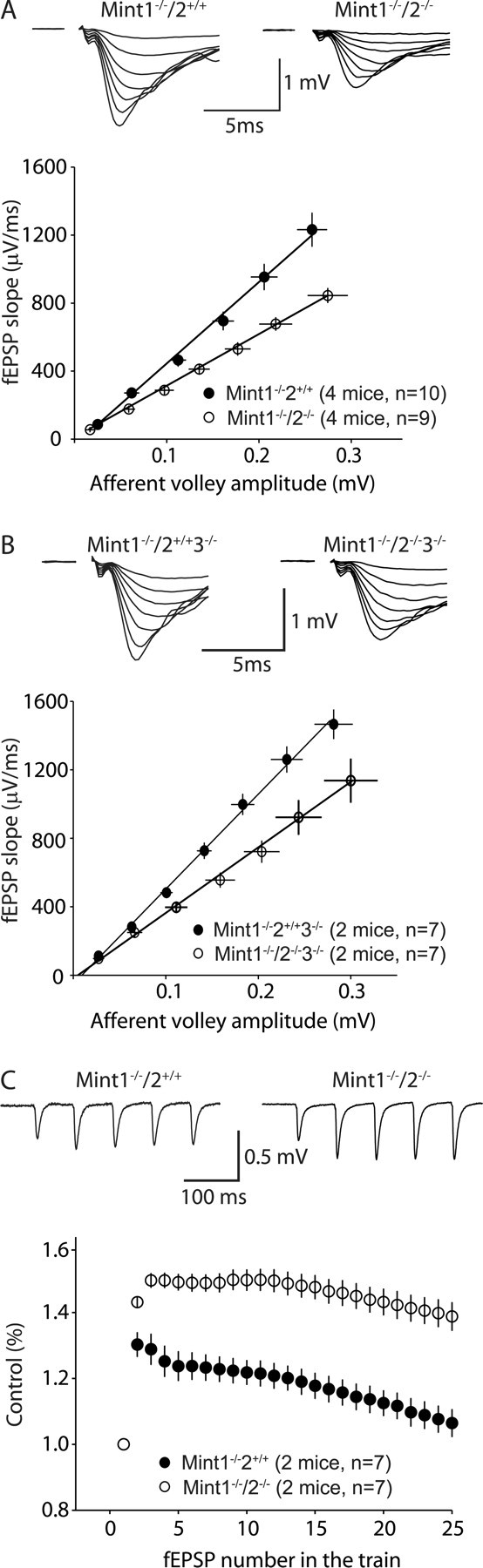
Synaptic transmission deficits at Schaffer collateral/CA1 pyramidal cell synapses of Mint 1/2 double KO mice. A, Synaptic strength analysis using input–output measurements. The fEPSP slope is plotted as a function of the afferent volley amplitude (in millivolts). Linear fit slopes are significantly different between Mint 1−/−/2−/− double KO mice, which displayed a decrease in the magnitude of synaptic responses compared with Mint 1−/−/2+/+ single KO littermate controls. B, Input–output measurements illustrating Mint 1−/−2−/−3−/− triple KO mice showed a similar decrease in the magnitude of synaptic responses compared with Mint 1−/−2−/− double KO mice shown in A. C, Response to 14 Hz stimulus trains. Control synaptic responses (Mint 1−/−/2+/+) exhibited initial facilitation that slowly decreased, in contrast to Mint 1−/−/2−/− double KO mice, which displayed extended facilitation.
Several observations suggest that the changes in input–output curves are attributable to impairments in presynaptic function in Mint-deficient mice. Repetitive stimulation of excitatory synapses by 14 Hz trains of action potentials revealed a typical profile of facilitation followed by depression in Mint 1 single KO control slices. In Mint 1/2 double KO mice, in contrast, repetitive stimulation produced a much larger degree of facilitation, suggesting that the probability of neurotransmitter release is decreased (Fig. 5 C). We next monitored the relative size of synaptic responses to two closely spaced stimuli as a function of the interstimulus interval (paired-pulse facilitaton and depression). Paired-pulse responses are inversely correlated with the probability of neurotransmitter release. Consistent with the notion that excitatory synapses have a relatively low, and inhibitory synapses a relatively high, average release probability, excitatory responses exhibited facilitation, and inhibitory synapses exhibited depression at short interstimulus intervals in Mint wild-type and KO mice. Deletion of Mints 1 and 2 caused a significant increase in paired-pulse facilitation at excitatory synapses, in agreement with the increased facilitation in the 14 Hz stimulus train (Fig. 6 A). In contrast, no differences were observed on inhibitory synaptic responses (Fig. 6 B), suggesting that the additional deletion of Mint 2 did not cause a further impairment of inhibitory synaptic transmission beyond that observed in the Mint 1 single KO mice (Ho et al., 2003).
Figure 6.

Excitatory and inhibitory synaptic responses to paired-pulse stimulation of Mint 1/2 double KO mice. A, Mint 1−/−/2−/− double KO mice display an enhanced increase in paired-pulse facilitation compared with Mint 1−/−/2+/+ single KO mice. A, Bottom, Ratio of the paired-pulse facilitation values between Mint 1−/−/2−/− to Mint 1−/−/2+/+ KO mice at excitatory synapses. B, Paired-pulse depression of inhibitory synaptic currents did not exhibit any difference between Mint 1−/−/2+/+ single and Mint 1−/−/2−/− double KO mice. B, Bottom, Ratio of paired-pulse responses at inhibitory synapses between Mint 1−/−/2−/− to Mint 1−/−/2+/+ KO mice. ISI, Interstimulus interval.
To further examine the effect of the deletion of Mints on synaptic transmission, we performed whole-cell recordings to monitor spontaneous mEPSCs and mIPSCs. No changes in the amplitude or shape of mEPSCs or mIPSCs were observed, suggesting that the Mint 1/2 double KO does not alter the amount of neurotransmitter released from a single synaptic vesicle or the number and sensitivity of postsynaptic receptors (Fig. 7 A,B). However, we observed a more than twofold decrease in the frequency of mEPSCs, but not of mIPSCs (Fig. 7 C,D), suggesting a change in the presynaptic release apparatus. These results provide further evidence that additional deletion of Mint 2 on top of Mint 1 caused a defect in glutamate release but had no additional effect on the impairment of GABA release caused by deletion of Mint 1 alone. To examine whether the presynaptic deficits we observed electrophysiologically are associated with morphological synaptic changes, we performed a quantitative analysis of the density of presynaptic terminals in the CA1 subfield of the hippocampus. We stained cryosections from surviving adult (4–6 weeks of age) Mint 1/2 double KO mice and control mice with an antibody to the presynaptic marker synaptophysin and measured the density of terminals by confocal imaging and image processing (Fig. 7 E,F). We observed no significant difference in the densities of synaptophysin-immunoreactive presynaptic terminals between wild-type, Mint 1 single, and Mint 1/2 double KO mice that might account for the synaptic deficits observed in Mint 1/2 double KO mice (Fig. 7 G), suggesting that the phenotype of the KO mice cannot be accounted for by a large change in synapse numbers.
Figure 7.
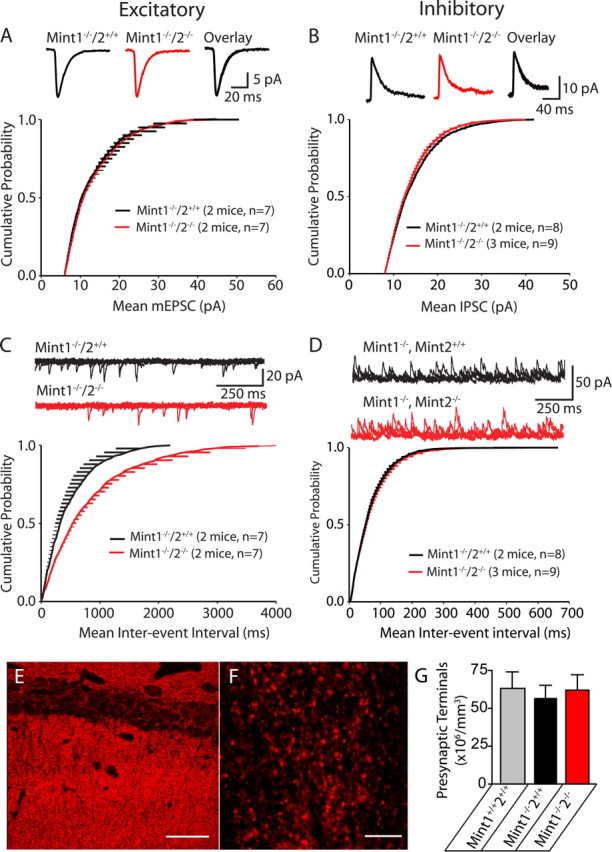
Deletion of Mints 1 and 2 decreases spontaneous release at excitatory, but not inhibitory synapses onto CA1 pyramidal neurons. A–B, Cumulative probability plot of mEPSC (A) and mIPSC (B) amplitudes for Mint 1−/−/2+/+ and Mint 1−/−/2−/− KO neurons did not show any significant differences between the genotypes. C, Meanwhile, there was a statistically significant decline in the cumulative probability plot of mEPSC frequency in Mint 1−/−/2−/− double KO compared with Mint 1−/−/2+/+ mice (*p = 0.0024). D, No changes were detected in the cumulative probability plot of mIPSC frequency in Mint 1−/−/2−/− double KO compare with Mint 1−/−/2+/+ mice. E, F, Representative confocal image of the CA1 region of the hippocampus immunostained for synaptophysin. Scale bars: E, 47.62 μm; F, 9.52 μm. G, Quantitative assessment of presynaptic terminals in the CA1 subfields of the hippocampus (n = 3 mice per group and genotype).
Acute deletion of Mint proteins by lentiviral cre infection of cultured neurons
A potential problem in the analysis of the adult surviving Mint 1/2 double KO mice (Figs. 3 –7) is that unknown compensatory processes operating prenatally or postnatally may alter the phenotype. Understanding such processes would be interesting in its own right because they could potentially provide insight into the function of Mints, but they cannot be pursued without an idea of whether such compensatory processes actually exist and what their direction may be. Because we produced the Mint KO mice as conditional deletions, we can potentially address this problem by acutely deleting Mint expression postnatally using cre recombinase. Such an approach provides the added advantage that it makes it possible to delete all three Mints at once, allowing a direct comparison of neurons with the same genetic background and developmental history that either contain or lack all three Mint proteins.
To achieve an acute Mint KO, we established a mouse line that is homozygous for the floxed mutant alleles of all three Mint genes. The homozygous triple knock-in mice are viable and fertile and exhibit no overt morbidity, abnormalities, or survival impairment, consistent with the notion that Mint proteins in these mice are fully functional. We then cultured neurons from newborn homozygous Mint triple knock-in mice and infected them with lentiviruses at 4 DIV for 48 h. The lentiviruses used either express a functional cre recombinase GFP fusion protein or a mutant cre recombinase GFP fusion protein as a negative control. Infected neurons were cultured until 13–15 DIV and then analyzed biochemically, morphologically, and electrophysiologically.
Immunoblotting of infected cultured neurons expressing mutant cre recombinase showed normal expression of all three Mint proteins, whereas infected neurons expressing active cre recombinase lacked all three Mint proteins (Fig. 8 A). This result was confirmed by PCR analyses (data not shown). As assessed by immunocytochemical staining for synapsin and MAP2 (microtubule-associated protein 2), Mint-deficient neurons appeared to be healthy and exhibited an apparently normal morphology (Fig. 8 B and data not shown). Electron microscopy revealed normal synaptic structure of presynaptic nerve terminals with clustered synaptic vesicles at the active zone and a filamentous postsynaptic density in Mint-deficient neurons (Fig. 8 C).
Figure 8.
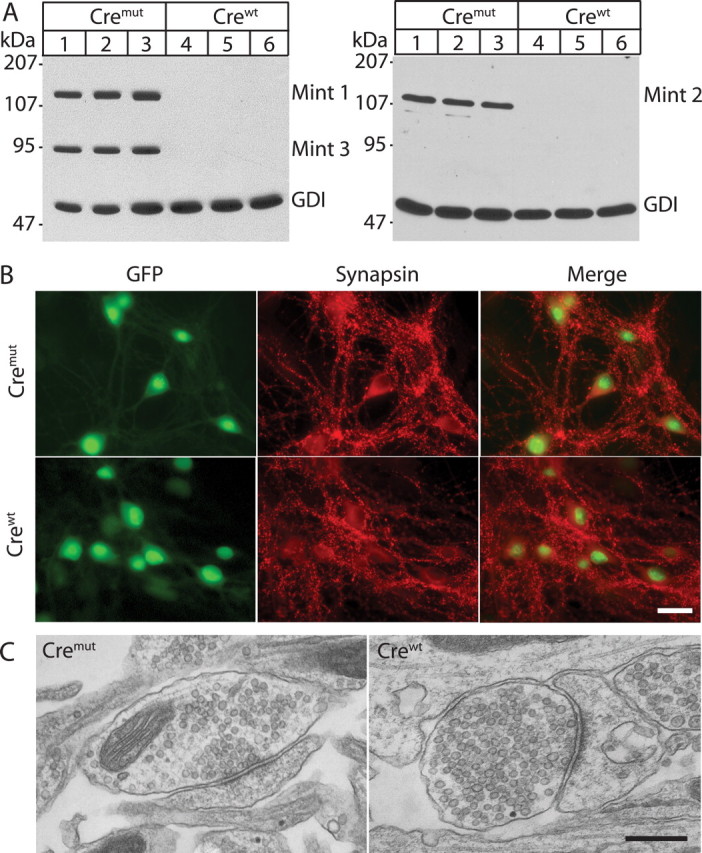
Acute deletion of all three Mint proteins in neuronal cultures infected with lentiviral cre recombinase (crewt) or cremut (vector carrying deletion of cre recombinase). Neurons were cultured from Mint triple-floxed newborn pups and infected with either lentiviral crewt or cremut at 4 DIV for 48 h. Lysates were collected at 14 DIV and subjected for SDS-PAGE and immunoblot for all Mint 1, 2, and 3 with GDP dissociation inhibitor (GDI), a ubiquitously expressed protein used as internal loading control. A, Neuronal cultures infected with cremut virus showed normal expression of all three Mint proteins, whereas lentiviral infection with crewt efficiently deleted all three Mint protein expressions. B, Neuronal cultures transfected with lentiviral cremut or crewt displayed normal synaptic staining with the synaptic marker synapsin. Both lentiviral vectors contain nuclear localization signal, and a GFP tag that labels cell nuclei of infected neurons exclusively. C, Synaptic structure of neuronal cultures transfected with lentiviral cremut and crewt showed normal presynaptic nerve terminal clustered with synaptic vesicles at the active zone with a filamentous postsynaptic density. Scale bars: B, 30 μm; C, 400 nm.
Quantitative immunoblotting uncovered dramatic changes in a subset of neuronal proteins (Fig. 9). Specifically, four changes were observed: a large increase (27–51%) in members of the Fe65 adaptor protein family, a similarly significant increase in the levels of Munc18-1 (33%; note that Munc18-1 interacts with Mints 1 and 2) (Okamoto et al., 1997) and of the AMPA-type glutamate receptor GluR1 (27%; note that AMPA-type glutamate receptor trafficking is altered in C. elegans Mint 1/LIN-10 mutants) (Rongo et al., 1998), and a smaller but still significant decrease (14%) in the cell-surface receptor low-density lipoprotein receptor-related protein (LRP). Several proteins that are thought to interact with Mints were not changed, in particular APP. The levels of proteins that might be functionally related to Mints, such as CASK, Velis, and NMDA-type glutamate receptors were also unchanged. The increase in Fe65 expression was specific because the levels of another adaptor protein called Disabled that also contain a PTB domain with APP-binding activity did not change in Mint KO mice. These results strongly suggest that Mint proteins function by binding to Munc18-1 and to Fe65 adaptor proteins but do not exclude an additional functional relation of Mints to APP (which binds to both Fe65-like adaptors and to Mints).
Figure 9.

Effect of Mint-deficient neurons on protein level expression. Levels of selected brain proteins in Mint-deficient neurons (crewt; black bars) and control (cremut; gray bars) at 13–15 DIV. As expected, we efficiently abolished all three Mint expressions in neurons that were treated with crewt compared with cremut control neurons. We found a selective increase in Fe65 family of adaptor proteins (Fe65, Fe65L1, and Fe65L2), postsynaptic receptor GluR1, and synaptic protein Munc18-1. Meanwhile, a small significant decrease in LRP protein was observed in Mint-deficient neurons. APOE, Apolipoprotein E; NR1, NMDA receptor subunit 1; SNAP25, soluble N-ethylmaleimide-sensitive factor attached protein 25.
Acute deletion of Mints impairs presynaptic function
To test whether acute deletion of all Mints impairs synaptic transmission, we performed whole-cell recordings in cultured neurons from triple Mint knock-in mice in which Mint expression was acutely ablated. The cultured neurons were infected with lentivirus that expresses either cre recombinase or a mutant cre recombinase as a control, and synaptic events were monitored by patch-clamp recordings. We first measured spontaneous miniature currents of excitatory and inhibitory synapses of Mint-deficient neurons. Mint-deficient neurons showed an ∼30% reduction in “mini” frequency in both excitatory and inhibitory synapses compared with control neurons that were infected with cre mutant (Fig. 10 A,B). However, mini amplitudes were not significantly different between Mint-deficient neurons and control neurons, suggesting that acute deletion of Mints alters the presynaptic release machinery but is unlikely to impair postsynaptic receptors (Fig. 10 A,B).
Figure 10.
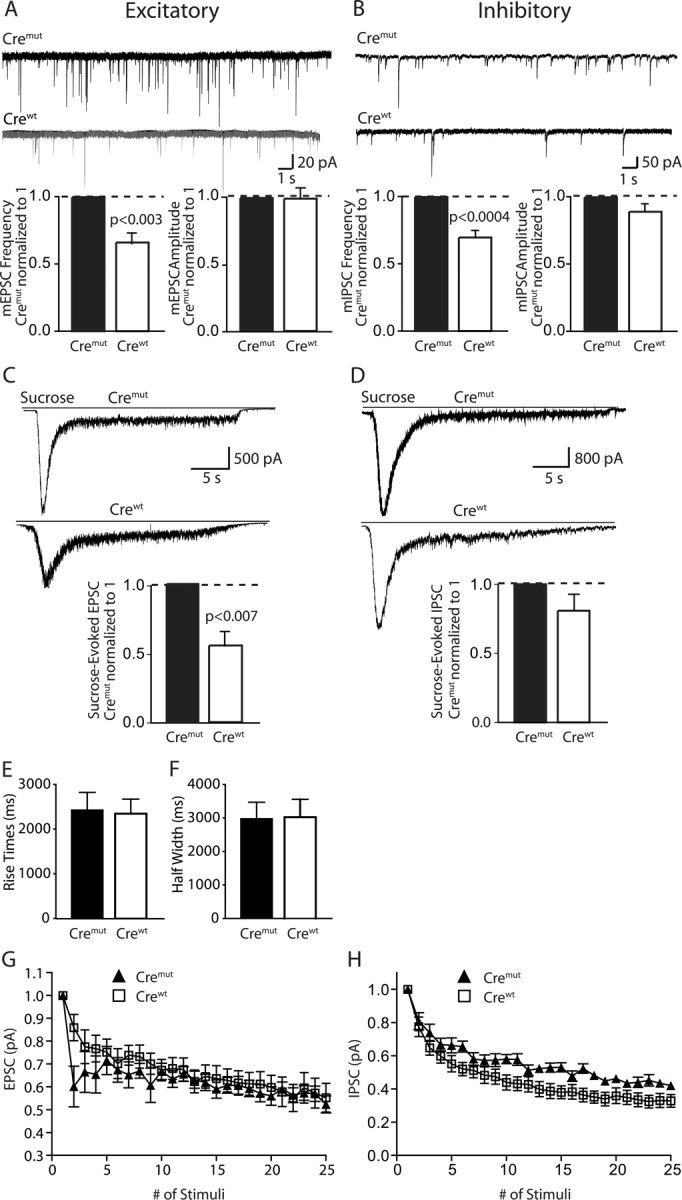
Altered synaptic responses in Mint-deficient neurons. A, B, Sample traces showing miniature excitatory and inhibitory recordings of cremut and crewt at 13–15 DIV. Bar graphs of the quantification revealed a significant decrease in miniature frequency but not amplitude in neurons transfected with crewt that abolished all three Mint proteins. C, D, Sample traces showing sucrose-evoked excitatory and inhibitory recordings of cremut and crewt at 13–15 DIV. Bar graphs showed a significant decrease in sucrose-evoked response in excitatory but not inhibitory synapses of Mint deficient neurons. Data shown represent absolute mean EPSP and IPSP amplitudes. E, F, Plots of 10–90% rise times and half-width showed no changes. G, H, Traces of synaptic depression responses recorded from excitatory and inhibitory neurons, respectively, during a 10 Hz stimulation.
We then examined whether the decrease in the frequency of spontaneous synaptic events was caused by a change in the size of the readily-releasable pool of vesicles in Mint-deficient neurons by measuring the synaptic response to hypertonic sucrose application (Rosenmund and Stevens, 1996) (Fig. 10 C,D). We found a substantial, highly significant decrease in sucrose-evoked excitatory postsynaptic currents (∼45%), and a much smaller, not statistically significant decrease in sucrose-evoked inhibitory postsynaptic currents in Mint-deficient neurons compared with control neurons (Fig. 10 C,D). In these experiments, we applied sucrose by gravity perfusion that may be variable and thereby contribute to differences in the size of the evoked currents. To control for this possible artifact, we plotted the 10–90% rise times and half-width of sucrose-induced responses in floxed control and Mint-deficient neurons (Fig. 10 E,F). We observed no changes in either of the plots, suggesting that the experiments were internally controlled and that the differences we observed between excitatory and inhibitory sucrose-evoked responses are accurate. Viewed together, these results indicate that in excitatory synapses, deletion of Mints induces a similar decrease in spontaneous miniature frequency and in the number of vesicles in the readily-releasable pool, whereas in inhibitory synapses, no such correlation is present. Consistent with this conclusion, we found that use-dependent depression was initially increased in the mutant excitatory synapses during repetitive 10 Hz stimulation, but not in inhibitory synapses (Fig. 10 G,H).
Is it possible that the increase in Munc18-1 observed in Mint-deficient neurons contributes to the synaptic phenotype of these neurons, a possibility that is raised by the fact that Munc18-1 acts in neurotransmitter release (reviewed in Südhof, 2004)? To test this possiblity, we overexpressed a Munc18–GFP fusion protein in cultured wild-type neurons, using overexpression of GFP alone as a control, and then measured spontaneous release in excitatory and inhibitory synapses in these neurons. In both excitatory and inhibitory synapses, Munc18-1 overexpression suppressed the mini frequency ∼40% compared with control neurons overexpressing GFP alone (Fig. 11 A,B). As observed before with the Mint deletions, Munc18-1 overexpression did not alter mini amplitudes, suggesting that the increase in Munc18-1 expression may contribute to the selective presynaptic deficits we observed in Mint-deficient neurons.
Figure 11.

Munc18-1 overexpression decreases spontaneous miniature frequency in excitatory and inhibitory synapses. A, B, Sample traces showing miniature excitatory and inhibitory recordings of GFP and GFP–Munc18 at 13–15 DIV. Bar graphs showed a significant decrease in miniature frequency but not amplitude in both excitatory and inhibitory synapses in Munc18-1 overexpressed neurons compared with GFP alone.
Discussion
The present study was designed to gain insight into the elusive functions of Mint/X11 proteins. The evolutionary conservation, multiple protein interactions, and dramatic in vitro effects mediated by Mints [as documented in >50 papers (see supplemental Table 1, available at www.jneurosci.org as supplemental material)] suggest that they are important. Moreover, careful analyses in C. elegans mutants lacking the single Mint 1/X11 homolog revealed an important developmental role for Mints. Despite this wealth of information, however, little progress has been made in understanding the functions of Mints in vertebrates. As a first step to address this question, we have now performed a systematic genetic study in which all three vertebrate Mint genes were targeted in mice. We analyzed both constitutive and conditional KOs and examined the mutant mice for changes in neuronal development, brain composition, and synaptic function. Our results allow five principal conclusions.
First, the genetic analysis of the various mouse Mint mutants revealed that Mints 1 and 2 are functionally redundant and are important for mouse survival. Whereas single deletions of either Mint 1 or 2 had no effect on survival, the double deletion of both Mints 1 and 2 severely impaired survival (Fig. 1). In addition, double deletion of Mints 1 and 2 increased the growth and movement impairments observed in mice lacking only Mint 1 (Fig. 4). Since Mints 1 and 2 are exclusively expressed in brain, Mints must perform an essential brain function. In contrast to the deletions of Mints 1 and 2, lack of Mint 3 had no effect on any measured parameter. This result by no means implies that Mint 3 does not have an essential function, but suggests that its function is not as penetrating as that of Mints 1 and 2.
Second, although Mints are essential for survival, they do not appear to perform an essential developmental role as observed for the C. elegans Mint homolog Lin-10. We cannot exclude discrete changes in development, for example in axonal pathfinding, but drastic changes were clearly absent even in Mint 1/2/3 triple KO mice.
Third, Mints 1 and 2 perform a critical synaptic function that manifests as changes in the presynaptic release of neurotransmitters in Mint KO mice. In the current study, we analyzed two very different preparations electrophysiologically: acute hippocampal slices from the ∼20% surviving adult Mint 1/2 double KO mice compared with littermate Mint 1 single KO mice and cultured neurons from newborn Mint triple knock-in mice in which the deletion of Mints was induced acutely by lentiviral expression of cre recombinase. Both of the preparations exhibited electrophysiological changes consistent with impairments in presynaptic neurotransmitter release (Figs. 5, 6, 7, 10). Furthermore, the presynaptic deficits we observe electrophysiologically were not caused by a decrease in synaptic density (Fig. 7), suggesting that the synaptic deficit we observe is most likely attributable to a decrease in synaptic vesicle release probability. One difference between the two preparations was that in acute triple KO mice, both excitatory and inhibitory minis were abnormal, whereas in chronic Mint 1/2 double KO mice, only excitatory minis were changed. This difference is probably attributable to the fact that in the acute KO in cultured neurons, we compared Mint 1/2/3 triple KO neurons with wild-type neurons, whereas in the chronic KO analysis, we compared Mint 1/2 double KO mice with Mint 1 single KO mice. Since Mint 1 KO mice exhibit an impairment in the regulation of GABA release at inhibitory synapses (Ho et al., 2003), we would not notice such an impairment in a comparison of Mint 1/2 double KO mice with Mint 1 single KO mice. Mints 1 and 2 clearly perform redundant functions, but Mint 1 is dominant in inhibitory synapses because it is highly enriched in inhibitory interneurons. In our previous analysis (Ho et al., 2003), we observed increased paired-pulse depression at inhibitory synapses in slices from Mint 1 KO mice, consistent with an increased release probability, whereas the current data analyzing neurons that lack multiple Mints suggest a decreased release probability both in slices and in cultured neurons. This observation suggests that Mints 1 and 2 may at least partly differ functionally, but other alternative explanations could also account for this observation (e.g., a protein dosage effect or changes in network activity).
Fourth, the redundancy of Mints 1 and 2 in terms of survival (Fig. 1), behavior (Fig. 4), and excitatory synaptic transmission (Figs. 5, 6, 7, 10) suggests a common function. Because only Mint 1 but not Mints 2 or 3 bind to CASK and participate in the tripartite CASK/Mint/Veli complex (Butz et al., 1998; Ho et al., 2003), this function has to be independent of CASK, similar to the previously described function of C. elegans Mint homolog lin-10 in glutamate receptor trafficking (Rongo et al., 1998). An interesting candidate for mediating the presynaptic function of Mints 1 and 2 is their common binding to Munc18-1, a membrane-fusion protein at the synapse (Hata et al., 1993). A role for Munc18-1 binding in the presynaptic function of Mints would also be consistent with the lack of Munc18-1 binding by Mint 3, the lack of functional redundancy in terms of survival between Mints 1 and 2 on the one hand, and Mint 3 on the other. The observation that Munc18-1 exhibits a significant increase (33%) in neurons in which Mints were acutely deleted strongly supports the physiological importance of Munc18-1 binding to Mints. This increase indicates that the previously observed in vitro interaction of Mints with Munc18-1 (Okamoto and Südhof, 1997, 1998) operates in vivo and suggests that this interaction may be responsible for the observed changes in presynaptic function (Figs. 5, 6, 7, 10). This view was supported by overexpression of Munc18-1 in neuronal cultures in which we were able to induce a similar decrease in miniature frequency in both excitatory and inhibitory synapses (Fig. 11). Although the overall evidence is not conclusive, viewed together these results strongly suggest that Munc18-1 binding to Mints is a major regulator of presynaptic neurotransmitter release.
Finally, our data indicate that Mints are functionally related to the Fe65 adaptor family of proteins. In the acutely Mint-deleted neurons, the levels of Fe65 and its homologs increased up to 50% (Fig. 9). Mints and Fe65 proteins both contain a PTB domain that binds to the cytoplasmic NPXY motif of APP, and both have been shown to modulate APP processing in transfected cells (Borg et al., 1996; McLoughlin and Miller, 1996). It is thus conceivable that Mints and Fe65 proteins are partially redundant and that the increase in Fe65 expression compensates in part for the lack of Mint proteins, possibly in a function that is related to their common APP binding. The fact that we did not observe a change in APP levels in the Mint KO mice supports the notion that Mint function with respect to APP may be functionally redundant with Fe65 proteins, or that the effect does not manifest under the conditions analyzed here. If there is redundancy, it can only be partial because both Mint and Fe65 KO mice exhibit major phenotypes on their own, and these phenotypes display no similarity (this study and Guenette et al., 2006). In addition to Fe65 changes, we observed a small change in LRP protein expression. LRP is a large transmembrane glycoprotein with multiple binding properties and has been shown to modulate several Alzheimer's disease-related proteins, including α2-macroglobulin, apolipoprotein E, and APP (Herz et al., 1990; Kang et al., 2000; Herz and Strickland, 2001). Interestingly, LRP-mediated regulation of APP processing requires its interaction with the adaptor protein Fe65, thus providing a functional linker between LRP and APP (Pietrzik et al., 2004). Because it was previously shown that overexpression of Fe65L1 results in decreased steady-state levels of LRP (Guenette et al., 2002), this correlates well with the simultaneous increase in Fe65 proteins and decrease in LRP protein levels in our Mint-deficient neurons (Fig. 9). Thus, the aggregate data are consistent with the notion that Mints and Fe65 perform a related function by binding to APP and/or LRP and other cell-surface receptors (Borg et al., 1996; McLoughlin and Miller, 1996; Trommsdorff et al., 1998; Gotthardt et al., 2000). This is a selective effect because Disabled, another adaptor protein that binds to both APP and LRP (Trommsdorff et al., 1998), did not change in the acutely deleted Mint-deficient neurons. A possible scenario is that Mints and Fe65 perform multiple functions in the presynaptic, and possibly postsynaptic, organization of synapses that are either redundant and/or modulatory and that among others operates via Munc18-1 binding. Future experiments will have to clarify these questions.
In addition to providing an initial insight into the functions of Mint proteins, our study is to our knowledge the first to directly compare the effects of a chronic constitutive deletion of a protein in adult animals with that of an acute deletion in cultured neurons. We demonstrate that it is feasible to delete three genes at once in cultured neurons by lentiviral expression of cre recombinase, opening up new avenues to analyzing KO mice in which multiple genes are manipulated. This feasibility would not necessarily have been expected considering the fact that six alleles have to be acutely recombined in order for this experiment to work and suggests that similar deletions should be possible in adult animals by local injection of lentiviruses. Our results show that chronic versus acute deletions of Mints gives similar but not identical results, despite the comparable presynaptic impairment observed electrophysiologically. Specifically, we did not detect in the constitutively mutant adult brains that lack Mints 1 and 2 the same changes in protein expression that were detected in acutely Mint-deleted cultured neurons (Fig. 9, supplemental Tables 2, 3, available at www.jneurosci.org as supplemental material). This observation is consistent with the notion that constitutive deletions elicit compensatory changes, which make interpretation of KO results more difficult but also, at least potentially, more interesting if the compensatory processes can be identified.
Footnotes
This work was supported by National Institute of Health Grant R37-MH52804-06 (T.C.S.) and National Institutes of Health Fellowship F32-AG05844 (A.H.). We are grateful to I. Kornblum, A. Roth, and E. Borowicz for excellent technical assistance and N. Hamlin and J. Mitchell for animal care. We thank Drs. E. Kavalali, F. Deak, and M. Khvochtchev for their help and U. Beffert and J. Herz for antibodies.
References
- Biederer and Südhof, 2000.Biederer T, Südhof TC. Mints as adaptors: direct binding to neurexins and recruitment of munc18. J Biol Chem. 2000;275:39803–39806. doi: 10.1074/jbc.C000656200. [DOI] [PubMed] [Google Scholar]
- Biederer et al., 2002.Biederer T, Cao X, Südhof TC, Liu X. Regulation of APP-dependent transcription complexes by Mint/X11s: Differential functions of mint isoforms. J Neurosci. 2002;22:7340–7351. doi: 10.1523/JNEUROSCI.22-17-07340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg et al., 1996.Borg JP, Ooi J, Levy E, Margolis B. The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol. 1996;16:6229–6241. doi: 10.1128/mcb.16.11.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg et al., 1998.Borg JP, Straight SW, Kaech SM, de Taddeo-Borg M, Kroon DE, Karnak D, Turner RS, Kim SK, Margolis B. Identification of an evolutionarily conserved heterotrimeric protein complex involved in protein targeting. J Biol Chem. 1998;273:31633–31636. doi: 10.1074/jbc.273.48.31633. [DOI] [PubMed] [Google Scholar]
- Borg et al., 1999.Borg JP, Lopez-Figueroa MO, de Taddeo-Borg M, Kroon DE, Turner RS, Watson SJ, Margolis B. Molecular analysis of the X11-mLin-2/CASK complex in brain. J Neurosci. 1999;19:1307–1316. doi: 10.1523/JNEUROSCI.19-04-01307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butz et al., 1998.Butz S, Okamoto M, Südhof TC. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell. 1998;94:773–782. doi: 10.1016/s0092-8674(00)81736-5. [DOI] [PubMed] [Google Scholar]
- Duclos and Koenig, 1995.Duclos F, Koenig M. Comparison of primary structure of a neuron-specific protein, X11, between human and mouse. Mamm Genome. 1995;6:57–58. doi: 10.1007/BF00350899. [DOI] [PubMed] [Google Scholar]
- Duclos et al., 1993.Duclos F, Boschert U, Sirugo G, Mandel JL, Hen R, Koenig M. Gene in the region of the Friedreich ataxia locus encodes a putative transmembrane protein expressed in the nervous system. Proc Natl Acad Sci USA. 1993;90:109–113. doi: 10.1073/pnas.90.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler et al., 2001.Fowler SC, Birkestrand BR, Chen R, Moss SJ, Vorontsova E, Wang G, Zarcone TJ. A force-plate actometer for quantitating rodent behaviors: illustrative data on locomotion, rotation, spatial patterning, stereotypies, and tremor. J Neurosci Methods. 2001;107:107–124. doi: 10.1016/s0165-0270(01)00359-4. [DOI] [PubMed] [Google Scholar]
- Gotthardt et al., 2000.Gotthardt M, Trommsdorff M, Nevitt MF, Shelton J, Richardson JA, Stockinger W, Nimpf J, Herz J. Interactions of the low density lipoprotein receptor gene family with cytosolic adaptor and scaffold proteins suggest diverse biological functions in cellular communication and signal transduction. J Biol Chem. 2000;275:25616–25624. doi: 10.1074/jbc.M000955200. [DOI] [PubMed] [Google Scholar]
- Guenette et al., 2002.Guenette SY, Chang Y, Hyman BT, Tanzi RE, Rebeck GW. Low-density lipoprotein receptor-related protein levels and endocytic function are reduced by overexpression of the FE65 adaptor protein, FE65L1. J Neurochem. 2002;82:755–762. doi: 10.1046/j.1471-4159.2002.01009.x. [DOI] [PubMed] [Google Scholar]
- Guenette et al., 2006.Guenette S, Chang Y, Hiesberger T, Richardson JA, Eckman CB, Eckman EA, Hammer RE, Herz J. Essential roles for the FE65 amyloid precursor protein interacting proteins in brain development. EMBO J. 2006;25:420–431. doi: 10.1038/sj.emboj.7600926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata et al., 1993.Hata Y, Slaughter CA, Südhof TC. Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature. 1993;366:347–351. doi: 10.1038/366347a0. [DOI] [PubMed] [Google Scholar]
- Hata et al., 1996.Hata Y, Butz S, Südhof TC. CASK: a novel dlg/PSD95 homolog with an N-terminal CaM kinase domain identified by interaction with neurexins. J Neurosci. 1996;16:2488–2494. doi: 10.1523/JNEUROSCI.16-08-02488.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz and Strickland, 2001.Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779–784. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz et al., 1990.Herz J, Kowal RC, Ho YK, Brown MS, Goldstein JL. Low density lipoprotein receptor-related protein mediates endocytosis of monoclonal antibodies in cultured cells and rabbit liver. J Biol Chem. 1990;265:21355–21362. [PubMed] [Google Scholar]
- Ho et al., 2003.Ho A, Morishita W, Hammer RE, Malenka RC, Südhof TC. A role for Mints in transmitter release: Mint 1 knockout mice exhibit impaired GABAergic synaptic transmission. Proc Natl Acad Sci USA. 2003;100:1409–1414. doi: 10.1073/pnas.252774899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz et al., 1999.Janz R, Goda Y, Geppert M, Missler M, Südhof TC. SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron. 1999;24:1003–1016. doi: 10.1016/s0896-6273(00)81046-6. [DOI] [PubMed] [Google Scholar]
- Kaech et al., 1998.Kaech SM, Whitfield CW, Kim SK. The LIN-2/LIN-7/LIN-10 complex mediates basolateral membrane localization of the C. elegans EGF receptor LET-23 in vulval epithelial cells. Cell. 1998;94:761–771. doi: 10.1016/s0092-8674(00)81735-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang et al., 2000.Kang DE, Pietrzik CU, Baum L, Chevallier N, Merriam DE, Kounnas MZ, Wagner SL, Troncoso JC, Kawas CH, Katzman R, Koo EH. Modulation of amyloid beta-protein clearance and Alzheimer's disease susceptibility by the LDL receptor-related protein pathway. J Clin Invest. 2000;106:1159–1166. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali et al., 1999.Kavalali ET, Klingauf J, Tsien RW. Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proc Natl Acad Sci USA. 1999;96:12893–12900. doi: 10.1073/pnas.96.22.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau et al., 2000.Lau KF, McLoughlin DM, Standen C, Miller CC. X11 alpha and X11 beta interact with presenilin-1 via their PDZ domains. Mol Cell Neurosci. 2000;16:557–565. doi: 10.1006/mcne.2000.0898. [DOI] [PubMed] [Google Scholar]
- Maximov et al., 1999.Maximov A, Südhof TC, Bezprozvanny I. Association of neuronal calcium channels with modular adaptor protein. J Biol Chem. 1999;274:24453–24456. doi: 10.1074/jbc.274.35.24453. [DOI] [PubMed] [Google Scholar]
- McLoughlin and Miller, 1996.McLoughlin DM, Miller CC. The intracellular cytoplasmic domain of the Alzheimer's disease amyloid precursor protein interacts with phosphotyrosine-binding domain proteins in the yeast two-hybrid system. FEBS Lett. 1996;397:197–200. doi: 10.1016/s0014-5793(96)01128-3. [DOI] [PubMed] [Google Scholar]
- Mori et al., 2002.Mori A, Okuyama K, Horie M, Taniguchi Y, Wadatsu T, Nishino N, Shimada Y, Miyazawa N, Takeda S, Niimi M, Kyushiki H, Kondo M, Mitsumoto Y. Alteration of methamphetamine-induced striatal dopamine release in mint-1 knockout mice. Neurosci Res. 2002;43:251–257. doi: 10.1016/s0168-0102(02)00041-x. [DOI] [PubMed] [Google Scholar]
- Okamoto and Südhof, 1997.Okamoto M, Südhof TC. Mints, Munc18-interacting proteins in synaptic vesicle exocytosis. J Biol Chem. 1997;272:31459–31464. doi: 10.1074/jbc.272.50.31459. [DOI] [PubMed] [Google Scholar]
- Okamoto and Südhof, 1998.Okamoto M, Südhof TC. Mint 3: a ubiquitous mint isoform that does not bind to munc18-1 or 2. Eur J Cell Biol. 1998;77:161–165. doi: 10.1016/S0171-9335(98)80103-9. [DOI] [PubMed] [Google Scholar]
- Olsen et al., 2005.Olsen O, Moore KA, Fukata M, Kazuta T, Trinidad JC, Kauer FW, Streuli M, Misawa H, Burlingame AL, Nicoll RA, Bredt DS. Neurotransmitter release regulated by a MALS-liprin-alpha presynaptic complex. J Cell Biol. 2005;170:1127–1134. doi: 10.1083/jcb.200503011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzik et al., 2004.Pietrzik CU, Il-Sang Y, Jaeger S, Busse T, Weggen S, Koo EH. FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J Neurosci. 2004;24:4259–4265. doi: 10.1523/JNEUROSCI.5451-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongo et al., 1998.Rongo C, Whitfield CW, Rodal A, Kim SK, Kaplan JM. LIN-10 is a shared component of the polarized protein localization pathways in neurons and epithelia. Cell. 1998;94:751–759. doi: 10.1016/s0092-8674(00)81734-1. [DOI] [PubMed] [Google Scholar]
- Rosahl et al., 1995.Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Rosenmund and Stevens, 1996.Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Sastre et al., 1998.Sastre M, Turner RS, Levy E. X11 interaction with beta-amyloid precursor protein modulates its cellular stabilization and reduces amyloid beta-protein secretion. 1998;273:22351–22357. doi: 10.1074/jbc.273.35.22351. [DOI] [PubMed] [Google Scholar]
- Setou et al., 2000.Setou M, Nakagawa T, Seog DH, Hirokawa N. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science. 2000;288:1796–1802. doi: 10.1126/science.288.5472.1796. [DOI] [PubMed] [Google Scholar]
- Südhof, 2004.Südhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Trommsdorff et al., 1998.Trommsdorff M, Borg JP, Margolis B, Herz J. Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem. 1998;273:33445–33560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- Whitfield et al., 1999.Whitfield CW, Benard C, Barnes T, Hekimi S, Kim SK. Basolateral localization of the Caenorhabditis elegans epidermal growth factor receptor in epithelial cells by the PDZ protein LIN-10. Mol Biol Cell. 1999;10:2087–2100. doi: 10.1091/mbc.10.6.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]