

Abstract
Multiple cues, including growth factors and circuit activity, signal to regulate the initiation and growth of mammalian dendrites. In this study, we have asked how these environmental cues regulate dendrite formation, and in particular, whether dendrite initiation and growth requires integrin-linked kinase (ILK) or its downstream effector, glycogen synthase kinase-3β (GSK-3β). In cultured sympathetic neurons, NGF and neuronal depolarization activated ILK and promoted dendrite initiation and growth, and inhibition of ILK (either pharmacologically, with a dominant-negative form of ILK, or by genetic knockdown) reduced depolarization-induced dendrite formation. In sympathetic neurons, ILK phosphorylated and inhibited GSK-3β, and inhibition of GSK-3β (either pharmacologically, with dominant-negative GSK-3β, or by genetic knockdown) caused robust dendrite initiation. GSK-3β inhibition also caused dendrite initiation in cultured cortical neurons and growth of hippocampal neurons in slice cultures. GSK-3β functioned downstream of ILK to regulate dendrite formation, because inhibition of GSK-3β promoted dendrite initiation even when ILK was simultaneously inhibited. Moreover, GSK-3β promoted dendrite formation in sympathetic neurons by regulating the activity of a key dendrite formation effector, the MAP (microtubule-associated protein) kinase kinase (MEK)–extracellular signal-regulated protein kinase (ERK) pathway. Specifically, inhibition of GSK-3β led to increased ERK phosphorylation, and inhibition of MEK completely blocked the effects of GSK-3β inhibition on dendrite initiation and growth. Thus, the ILK–GSK-3β pathway plays a key role in regulating dendrite formation in developing mammalian neurons.
Keywords: sympathetic neurons, neuronal activity, MEK, cortical neurons, hippocampal neurons, neuronal growth, neuronal development, MAP kinase
Introduction
The development and maintenance of functional neuronal circuitry requires the regulated initiation and growth of a dendritic arbor that can receive and process synaptic information. Although significant progress has been made in identifying extrinsic cues that regulate dendritic morphogenesis, the underlying signaling mechanisms are just now being elucidated. Two cues that are known to play a key role in regulating dendrite formation in multiple populations of mammalian neurons are neurotrophins and neuronal activity (Miller and Kaplan, 2003). For example, sympathetic neurons in vivo require NGF and appropriate connectivity to promote genesis of their dendritic arbor (Snider, 1988; Ruit et al., 1990; Ruit and Snider, 1991), and neonatal sympathetic neurons cultured in NGF require coincident neuronal activity for both the formation and maintenance of dendrites (Vaillant et al., 2002). What are the signaling mechanisms that allow extrinsic cues such as these to promote dendrite initiation and growth? One set of signaling proteins essential for dendrite growth is key regulators of the cytoskeleton such as members of the Rho GTPase family (Van Aelst and Cline, 2004). In addition, signaling proteins known to be downstream of growth factor receptors and calcium influx, such as the microtubule-associated protein (MAP) kinase (MAPK) kinase (MEK)–extracellular signal-regulated protein kinase (ERK) pathway, calcium/calmodulin-dependent protein kinase II (CaMKII), and a Wnt-2–β-catenin pathway are also important regulators of dendritic development (Miller and Kaplan, 2003; Yu and Malenka, 2003).
Integrin-linked kinase (ILK) is one signaling protein that has recently been implicated in axonal growth (Mills et al., 2003) but is not known to play a role in dendrite morphogenesis. ILK is a serine–threonine kinase that is activated in a phosphatidylinositol 3 (PI3)-kinase-dependent manner (Delcommenne et al., 1998) and that functions downstream of β-integrins and growth factor receptors to regulate cell growth, differentiation, and migration (Dedhar et al., 1999). The physiological importance of this signaling protein has recently been demonstrated genetically; mice lacking ILK are embryonic lethal (Sakai et al., 2003), and neural precursor-specific deletion of ILK perturbs cortical lamination (Niewmierzycka et al., 2005). Although ILK has several known downstream targets, only one of these, glycogen synthase kinase-3β (GSK-3β), is currently known to mediate the effects of ILK during NGF-mediated axon growth (Arevalo and Chao, 2005). GSK-3β is a serine/threonine kinase implicated in a variety of cellular functions (Doble and Woodgett, 2003; Arevalo and Chao, 2005), including glycogen synthesis, microtubule dynamics, and regulation of Wnt-mediated gene expression. ILK phosphorylates and inactivates GSK-3β (Delcommenne et al., 1998), preventing GSK-3β from phosphorylating its downstream targets, which include β-catenin (Hinoi et al., 2000), kinesin light chain (Morfini et al., 2002), and a number of microtubule-associated proteins such as Tau, MAP2, and adenomatous polyposis coli (APC) (Sanchez et al., 2000; Alvarez et al., 2002; Zhou et al., 2004). GSK-3β also regulates neuronal polarity (Jiang et al., 2005) and axonal growth and branching in hippocampal neurons by phosphorylating the tubulin-binding protein collapsin response mediator protein-2 (CRMP-2) (Yoshimura et al., 2005).
Here, we have asked whether an ILK–GSK-3β pathway is an important mediator of dendritogenesis, as it is for axogenesis. We demonstrate that this pathway is activated in response to cues that promote dendrite formation, such as neurotrophins and neuronal depolarization, and that it plays a key role in regulating both the initiation and growth of dendrites in multiple populations of developing mammalian neurons.
Materials and Methods
Primary neuron cultures.
Superior cervical ganglia (SCGs) were dissected from postnatal day 1 (P1) Sprague Dawley rats, neurons were cultured onto collagen-coated four-well chamber slides (20,000 cells per well) and established for 5 d in 50 ng/ml NGF as per Vaillant et al. (2002). Neurons were switched into 10 ng/ml NGF plus 50 mm KCl for 3–5 d to induce dendrite formation. For low-density SCG cultures, ganglia were dissected and cultured at a density of 10,000 cells per well of four-well chamber slides and then treated as described above. Primary cortical neuron cultures were cultured from embryonic day 16 (E16) to E17 CD-1 mouse or E17–E18 rat embryos as described previously (Lee et al., 2004). To eliminate non-neuronal cells, 7 μm cytosine arabinoside was added to the medium after 3 d. Treatments with kinase inhibitors or DMSO were done at 5 d in vitro for 3 d. The following inhibitors were used (in μm): 40 QLT0254; 20 kenpaullone (Calbiochem, La Jolla, CA), and 1 alsterpaullone (Calbiochem), 50 [2-(2′amino-3′-methoxyphenyl)-oxanaphthalen-4-one] (PD98059; Calbiochem).
Hippocampal slice cultures and biolistic transfections.
Hippocampi from P7 rats were dissected into cold medium containing 50% Medium 199 (Invitrogen, Carlsbad, CA), 25% HBSS (Invitrogen), 25% horse serum (Invitrogen), 6.5 mg/ml glucose, 2 mg/ml sodium bicarbonate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.002% Fungizone (Invitrogen), and immediately placed onto a McIlwain tissue chopper. Slices were cut at a thickness of 350 μm, transferred onto a Millicell-CM 0.4 μm membrane, and allowed to establish themselves for a minimum of 5–7 d. Then, slices were transfected with an enhanced green fluorescent protein (EGFP) expression plasmid (Clontech, Mountain View, CA) using the Helios Gene Gun (Bio-Rad, Hercules, CA) and Gold-Coat cartridges. Helium (150–200 psi) was used to deliver the DNA.
Adenoviral infections, transfections, and siRNA knockdown in sympathetic neurons.
Established sympathetic neurons were switched overnight to medium containing 10 ng/ml NGF, 50 mm KCl, and replication-defective recombinant adenovirus at a multiplicity of infection of 50–100. Cultures were then washed free of the adenovirus and grown for 3 d in 10 ng/ml NGF or 10 ng/ml NGF plus 50 mm KCl. Recombinant adenoviruses used were dominant-negative ILK (dnILK) and wild-type ILK (wtILK), both expressing EGFP bicistronically (Kumar et al., 2004). An EGFP-expressing recombinant adenovirus (Pozniak et al., 2000) was used as a control.
Sympathetic neurons were transfected on the day of plating using Lipofectamine 2000 (Invitrogen). Transfection was performed at a 3:1 ratio of plasmid DNA encoding the gene of interest relative to an EGFP-expressing plasmid. The DNA–Lipofectamine 2000 complexes were prepared in Opti-MEM (Invitrogen) according to the manufacturer's guidelines. To optimize the transfection rate, Ultraculture medium was supplemented with 50 mm KCl. Twenty-four hours after transfection, cells were switched to Ultraculture medium containing 50 ng/ml NGF but no KCl and established for 5 d. Recombinant plasmids expressing dominant-negative GSK-3β (dnGSK-3β) and constitutively activated ILK were hemagglutinin (HA)- and V5-His-tagged, respectively. Transfection with an EGFP expression plasmid (Clontech) was used as control.
For the ILK small interfering RNA (siRNA) transfection, two duplex siRNAs against the rat ILK gene were used that specifically targeted the kinase domain, ILK siRNA1, and the pH domain, ILK siRNA2. siRNAs targeted to the homologous sequences in human ILK have been used previously to silence the human ILK gene (Troussard et al., 2003; Edwards et al., 2006). These two duplex siRNAs were designed and purchased from Dharmacon (Lafayette, CO). They were as follows: for ILK siRNA1, 5′-UGUAAAGUUUUCUUUCCAGUGdTdT-3′ (sense) and 5′-CACUGGAAAGAAAACUUUACAdTdT-3′ (antisense); and for ILK siRNA2, 5′-CCUGGCAAAGCUCAACGAGAAdTdT-3′ (sense) and 5′-UUCUCGUUGAGCUUUGCCAGGdTdT-3′ (antisense). For the GSK-3β siRNA transfection, specific pooled siRNAs against rat GSK-3β were purchased from Dharmacon. A nonspecific sequence was used as the siRNA negative control (Qiagen, Valencia, CA). The target sequence for this siRNA control was AATTCTCCGAACGTGTCACGT and does not represent any gene accession number. The siRNA duplex sequence for control siRNA was sense, UUCUCCGAACGUGUCACGUdTdT; antisense, ACGUGACAGGUUCGGAGAAdTdT. Sympathetic neurons were transfected with siRNAs on the day of plating using Lipofectamine 2000. Neurons were transfected overnight with 60 pmol of siRNA, maintained for 5 d in Ultraculture medium containing 50 ng/ml NGF, and then switched for 3 d into 10 ng/ml NGF with or without 50 mm KCl, QLT0254, kenpaullone, or DMSO. PC12 6-24 cells were maintained in DMEM (BioWhittaker, Verviers, Belgium) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mm glutamine, 10% horse serum (Invitrogen), and 5% bovine calf serum (Hyclone, Logan, UT). Cells were transfected overnight with 200 pmol of siRNA using Lipofectamine 2000, according to the manufacturer's instructions and as we have described previously (Lin et al., 2006), and then lysed 48 h after being transfected.
Immunocytochemistry.
Immunocytochemistry was performed as described by Vaillant et al. (2002). The following primary antibodies were used: mouse anti-high-molecular-weight (HMW)-MAP2 (1:500; Sigma-Aldrich, St. Louis, MO), mouse anti-β-tubulin (1:1000; Covance, Princeton, NJ), rabbit anti-ILK (1:500; Millipore, Billerica, MA), mouse anti-GSK-3β (1:500; BD Biosciences Pharmingen, San Diego, CA), rabbit anti-GFP (1:400; Millipore), rabbit anti-V5 (1:1000; Sigma-Aldrich), and rabbit anti-HA (1:400; Covance). Antibody binding was detected with the conjugated secondary antibodies goat anti-mouse Alexa 555 (1:1000) or goat anti-rabbit Alexa 488 (1:1000). Hoechst staining was performed by incubating the cells with 1 μg/ml Hoechst.
Neuronal imaging and analysis.
Dendrites were analyzed either by measuring HMW-MAP2-positive processes, as we have described previously (Vaillant et al., 2002), or by measuring dendrites morphologically by phase-contrast microscopy in low-density cultures. For the MAP2 measurements, random HMW-MAP2-labeled fields were acquired. The lengths of all of the MAP2-positive processes were then measured by tracing the MAP2-labeled processes from the perimeter of the cell body to the farthest extent of the labeled process using Northern Eclipse software (Empix, Mississauga, Ontario, Canada). In each experiment, at least 150–500 neurons were measured for each condition. These values were pooled for each treatment and divided by the total number of cells measured (including those with no detectable MAP2-positive process) to give a mean dendrite length per neuron ± SEM. The number of neurons with no detectable MAP2-positive process was also determined. These are shown as percentages of the total number of neurons for individual experiments. For the distribution histograms of single experiments, all MAP2-positive processes were binned by length and plotted; these same values were used to determine a mean dendrite length per neuron in that particular experiment. For morphological measurements, random HMW-MAP2-labeled fields were acquired from low-density sympathetic neuron cultures. The same fields were also acquired in phase-contrast. Dendrites were then distinguished by their morphology and by their expression of MAP2. The length of these dendrites was measured under phase-contrast microscopy by tracing the processes from the perimeter of the cell body to the farthest extent of the process using Northern Eclipse software. The length of the same dendrites, as indicated by HMW-MAP2 immunoreactivity, was also measured. In each experiment, ∼25–50 neurons were measured for each condition. Statistics were performed with the paired Student's t test. To determine survival, neurons were Hoechst stained, and the percentage with fragmented nuclei was determined.
For Sholl analysis, images of EGFP-positive neurons in hippocampal slices were taken live just before (day 0) and 3 d after (day 3) treatment on a Zeiss (Thornwood, NY) Axiovert 200M inverted microscope. Digital images were analyzed using Northern Eclipse software. Sholl analysis was used to measure the growth of dendrites of healthy, nonstellate neurons. To provide a measure of the rate of growth, the same neurons were photographed on days 0 and 3, and the extent of growth and branching at these two time points was determined by Sholl analysis. These values were then subtracted from each other, and the mean difference was calculated. Statistics were performed using SigmaStat 2.03 (SPSS, Chicago, IL).
Western blot analysis.
Western blot analysis of sympathetic neuron lysates was performed as described previously (Vaillant et al., 1999, 2002). Antibodies used were as follows: rabbit anti-phosphorylated GSK-3β (pGSK-3β) (Ser9; 1:1000; Cell Signaling Technology, Beverly, MA) and mouse anti-GSK-3β (1:5000; BD Biosciences Pharmingen). Other antibodies used were as follows: rabbit anti-phosphorylated Akt (pAkt; S473; 1:1000; Cell Signaling Technology), rabbit anti-phosphorylated ERK (pERK; 1:1000; Promega, Madison, WI), rabbit anti-ILK (1:1000; Cell Signaling Technology), mouse anti-phosphorylated Tau (pTau) (1:50; kind gift from Dr. P. Davies, Albert Einstein College of Medicine, Bronx, NY), rabbit anti-Akt (1:1000; Cell Signaling Technology), rabbit anti-ERK1 (C-16; 1:10,000; Santa Cruz Biotechnology, Santa Cruz, CA), and mouse anti-Tau (1:1000; Millipore). After washing, blots were incubated with HRP-conjugated goat anti-mouse (1:5000; Millipore) or goat anti-rabbit (1:5000) secondary antibodies, and protein was detected using the ECL chemiluminescent kit (GE Healthcare, Little Chalfont, UK).
Results
Depolarization increases GSK-3β phosphorylation during activity-dependent dendrite formation
We have previously reported that neuronal activity, either KCl-mediated depolarization or patterned electrical field stimulation, is required to induce dendrite formation in cultured neonatal sympathetic neurons (Vaillant et al., 2002). When neonatal sympathetic neurons were plated on collagen in 50 ng/ml NGF, they grew an extensive tubulin-positive axonal network but extended no or rudimentary dendrites, as indicated by immunostaining for the dendrite-specific HMW-MAP2 (Fig. 1A). However, when neurons were depolarized in 50 mm KCl, this caused dendrite formation that continued for at least 5 d, as indicated by measuring the total length of MAP2-positive processes per neuron (Fig. 1A–D). We have shown previously that this KCl-mediated dendrite formation is dependent on both L-type Ca2+ channels and CaMKII (Vaillant et al., 2002). Thus, depolarization causes both dendrite initiation and growth in developing sympathetic neurons.
Figure 1.
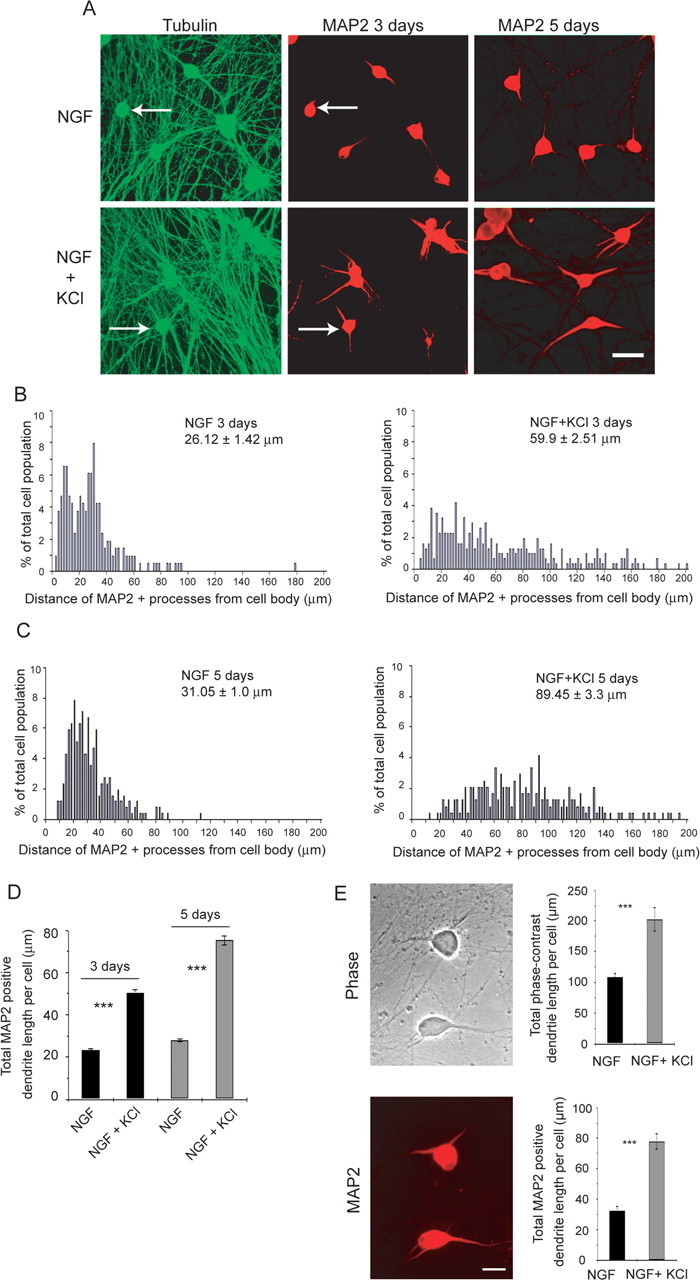
Dendrite growth is regulated by both neurotrophins and neuronal depolarization. A, Double-label immunocytochemistry for total β-tubulin (green), which is present in all processes, and dendrite-specific HMW-MAP2 (red) in neonatal sympathetic neurons grown in 10 ng/ml NGF with or without 50 mm KCl for 3 d (left and center) or for 5 d (right). The left and center panels show the same fields, and arrows indicate the same neurons. Scale bar, 50 μm. B–D, Quantification of dendritic arbor growth and complexity in sympathetic neuron cultures grown in NGF with and without KCl-mediated depolarization for 3 d (B, D) or 5 d (C, D). Mean total dendrite length per cell (D) is calculated from the pooled data from three independent experiments, with 150 to 500 neurons per experiment. Error bars indicate SEM; ***p < 0.001 (Student's t test). Distribution histograms (B, C) represent the distribution of total MAP2-positive dendrite lengths per neuron from a single representative experiment of three that were performed. The data were normalized so that the total number of dendrites equaled 100%, and bin sizes are 2 μm. Neurons with no detectable MAP2-positive processes were not included in these histograms. Numbers indicate the mean ± SEM of the measurements represented in the histogram. E, Phase-contrast image (top) and HMW-MAP2 immunostaining (bottom) of sympathetic neurons grown at low density in 10 ng/ml NGF with or without 50 mm KCl for 3 d. Both panels show the same field. Scale bar, 50 μm. Quantitation of the mean total length of phase-positive dendrites and of MAP2-positive processes is shown on the right. Similar results were obtained in two independent experiments. Error bars indicate SEM; ***p < 0.001 (Student's t test).
To confirm that measurements of HMW-MAP2-positive processes reflected dendrite initiation and growth, we also performed similar experiments measuring dendrites directly by phase-contrast microscopy (Fig. 1E). Sympathetic neurons were cultured at low density in 50 ng/ml NGF for 5 d, depolarized in 10 ng/ml NGF plus 50 mm KCl for an additional 3 d, and then immunostained for HMW-MAP2. Analysis of the length of MAP2-positive processes revealed that, under these low-density conditions, basal dendrite formation in NGF alone was somewhat higher but that, as seen at higher density, coincident KCl-mediated depolarization significantly increased the length of MAP2-positive processes (Fig. 1E). Analysis of the same cultures by phase microscopy confirmed that HMW-MAP2 immunoreactivity was limited to cell bodies and morphologically recognizable dendrites and demonstrated that although the absolute length of phase-positive dendrites was greater than the corresponding MAP2-positive process length, the magnitude of the increase in dendrite length induced by depolarization was similar with both (Fig. 1E). Thus, measurement of MAP2-positive processes is a valid reflection of alterations in dendrite initiation and growth in sympathetic neurons.
ILK is a PI3-kinase substrate that has been implicated in axonal growth. To ask whether ILK might regulate dendrite initiation and/or growth, we first determined whether it was activated by neurotrophins and/or neuronal depolarization by assaying phosphorylation of its substrate, GSK-3β (Delcommenne et al., 1998). Sympathetic neurons were established for 5 d in NGF, washed, and stimulated with NGF, KCl, or NGF plus KCl for 15 min. Western blot analysis of lysates with an antibody for phosphorylated GSK-3β revealed that NGF and KCl both stimulated GSK-3β phosphorylation, with maximal activation seen in NGF plus KCl (Fig. 2A). We then asked whether ILK was present in dendrites; sympathetic neurons were established, treated with NGF plus KCl for 3 d, and double labeled for HMW-MAP2 and ILK. This analysis revealed that ILK was present in cell bodies and processes, including MAP2-positive dendrites (Fig. 2B) and MAP2-negative axons (data not shown).
Figure 2.

ILK is localized to sympathetic neuron dendrites and is regulated by both neurotrophins and neuronal depolarization. A, Western blot analysis for phosphorylated GSK-3β in sympathetic neurons stimulated for 15 min with medium alone or with medium plus 10 ng/ml NGF, 50 mm KCl, or NGF plus KCl. The same blot was reprobed for total GSK-3β as a loading control. Similar results were obtained in three independent experiments. Both of these antibodies react with two GSK-3β-specific bands (Jiang et al., 2005; Yoshimura et al., 2005). B, Double-label immunocytochemistry for HMW-MAP2 (MAP2; green; left) and ILK (red; center; right, merge of the two) in sympathetic neurons grown in NGF plus KCl for 3 d. Scale bar, 25 μm. C–E, Western blot analysis for pGSK-3β (C) or pAkt or pERK (D, E) in sympathetic neurons pretreated for 1 h with QLT0254 (254) and stimulated for 15 min with 10 ng/ml NGF, 50 mm KCl, or NGF plus KCl. As controls, cultures were treated with DMSO alone or stimulated in the absence of DMSO or inhibitors. Blots were reprobed for total GSK-3β (C) or total Akt or ERK (D, E) as loading controls. E shows short and long exposures of the same pAkt blot. Note that QLT0254 suppresses GSK-3β, but not Akt or ERK phosphorylation. F, Quantification of the percentage of surviving sympathetic neurons in cultures treated with QLT0254 for 3 d in the presence of NGF plus KCl, as monitored by counting intact, uncondensed Hoechst-positive nuclei. As controls, cultures were treated with DMSO alone or left untreated. Error bars indicate SEM.
ILK is necessary for depolarization-induced dendrite initiation and growth
Although depolarization induces phosphorylation of GSK-3β, there are a number of GSK-3β kinases in addition to ILK. We therefore asked whether ILK was required for the observed increase in GSK-3β phosphorylation, using the specific pharmacological ILK inhibitor QLT0254 (formerly KP-392; Mills et al., 2003; Yau et al., 2005). Neurons were established, pretreated for 1 h with or without the ILK inhibitor, and stimulated for 15 min with NGF plus KCl. Western blot analysis demonstrated that QLT0254 decreased the NGF-plus-KCl-induced stimulation of GSK-3β (Fig. 2C) but had no effect on the phosphorylation of the signaling kinases Akt or ERK (Fig. 2D,E), demonstrating its efficacy and specificity. We also ensured that ILK inhibition did not affect neuronal survival; established neurons were treated for 3 d with NGF plus KCl with or without QLT0254. Hoechst staining of neuronal nuclei revealed that neurons survived equally well in all conditions (Fig. 2F).
We then used QLT0254 to ask whether ILK regulates dendrite formation. Neurons were established and treated for 3 d with NGF plus KCl with or without this inhibitor. Immunostaining for HMW-MAP2 (Fig. 3A) revealed that ILK inhibition almost completely inhibited depolarization-induced dendrite formation (Fig. 3B–D). In particular, at 3 d in NGF plus KCl, total MAP2-positive dendrite length was increased twofold to threefold, and this growth was completely blocked by QLT0254 (Fig. 3B,C). Moreover, dendrite initiation was also inhibited, as demonstrated by considering the percentage of neurons in the population with no dendrites at 3 d; QLT0254 increased the number of neurons without dendrites in NGF alone and inhibited the depolarization-induced decrease in neurons lacking dendrites (Fig. 3D).
Figure 3.

Inhibition of ILK activity reduces dendrite initiation and growth. A, Immunocytochemistry for HMW-MAP2 in sympathetic neurons cultured for 3 d in NGF plus KCl with or without QLT0254 (254). As controls, cultures were treated with DMSO (center) or maintained in NGF plus DMSO (left). Representative images from one of three independent experiments are shown. Scale bar, 50 μm. B–D, Quantitative analysis of cultures similar to those shown in A. B, Distribution histograms representing total MAP2-positive dendrite lengths per neuron for cultures treated with NGF versus NGF plus KCl with or without QLT0254 for 3 d. Results derive from a single representative experiment of three that were performed. The data were normalized so that the total number of dendrites equaled 100%, and bin sizes are 2 μm. Neurons with no detectable MAP2-positive processes were not included in these histograms. Numbers indicate the mean ± SEM of the measurements represented in the histogram. C, Mean total MAP2-positive dendrite length per cell calculated from pooled data deriving from three independent experiments. In each experiment, 200–300 neurons were measured, and all neurons, including those with no dendrites, were included in the calculations. Error bars indicate SEM; ***p < 0.001 (Student's t test). D, The number of neurons with no dendrites in two independent experiments similar to those shown in A, represented as a percentage of the total number of neurons measured. Exp., Experiment. E, F, Mean total dendrite length and number of dendrites per cell in sympathetic neurons grown at low density in 10 ng/ml NGF with or without 50 mm KCl plus or minus QLT0254 for 3 d and then measured by HMW-MAP2 immunoreactivity (E) or phase-contrast microscopy (F). Results in E and F derive from a single representative experiment of two that were performed. Error bars indicate SEM; **p < 0.01; ***p < 0.001 (Student's t test).
To ensure that the observed changes in MAP2-positive processes reflected alterations in the number and length of dendrites, we performed similar experiments, analyzing low-density cultures by phase-contrast microscopy as well as HMW-MAP2 immunoreactivity. In these low-density cultures, QLT0254 led to a small but significant decrease in the length and number of MAP2-positive processes in the presence of NGF alone and, as seen in the high-density cultures, almost completely blocked the increased dendrite formation observed in NGF plus KCl (Fig. 3E). Analysis of the same cultures by phase-contrast microscopy revealed that although the length of morphologically recognizable dendrites was greater than the MAP2-positive process length, the effects of inhibiting ILK were similar (Fig. 3F). Moreover, both the number of dendrites per cell and the changes in dendrite number elicited by QLT0254 were similar whether we measured phase-contrast-positive dendrites or MAP2-positive processes (Fig. 3E,F). Thus, reducing ILK activity with the pharmacological inhibitor QLT0254 blocked the enhanced dendrite initiation and growth induced by sympathetic neuron depolarization.
To confirm these pharmacological studies, we used both dominant-negative and genetic knockdown approaches. Initially, we transduced sympathetic neurons with a previously described dnILK (Kumar et al., 2004) using a replication-defective adenovirus that also expressed EGFP. As a control, we used an adenovirus expressing EGFP only in the same adenoviral backbone. Initially, we confirmed that dnILK was expressed in transduced sympathetic neurons by performing Western blot analysis with an antibody to ILK (Fig. 4A). We then asked whether dnILK inhibited depolarization-induced dendrite initiation and growth; established neurons were infected overnight and then maintained in NGF plus KCl for 3 d. As seen with pharmacological inhibition, neuronal survival was unaffected by expression of dnILK (data not shown). Double-label immunocytochemistry for HMW-MAP2 and for EGFP (Fig. 4B) revealed that dnILK reduced the length of dendrites observed in NGF plus KCl, as measured by MAP2 immunoreactivity (Fig. 4C,D). Dominant-negative ILK also inhibited depolarization-induced dendrite initiation; the percentage of neurons with no dendrites increased approximately threefold in neurons transduced with dnILK relative to those transduced with EGFP (Fig. 4E). In contrast to dnILK, wtILK expression had no effect on dendrite initiation or growth (data not shown), arguing that it is ILK activity, and not levels, that are important.
Figure 4.

ILK is essential for depolarization-induced sympathetic neuron dendrite growth but not for maintenance of preformed sympathetic neuron dendrites. A, Western blot analysis for ILK in sympathetic neurons transduced with recombinant adenoviruses expressing EGFP or EGFP plus dnILK. B, Double-label immunocytochemistry for EGFP (green) and HMW-MAP2 (red) in sympathetic neurons transduced with adenoviruses expressing EGFP or EGFP plus dnILK and cultured in NGF plus KCl for 3 d. Representative images from one of three independent experiments are shown. Each set of left and right panels shows the same field, and arrows indicate transduced neurons. Scale bar, 50 μm. C–E, Quantitative analysis of cultures similar to those shown in B, in which neurons were transduced with EGFP alone or EGFP plus dnILK. C, Mean total MAP2-positive dendrite length per cell calculated from pooled data deriving from three independent experiments. In each experiment, 90–120 neurons were measured, and all neurons, including those with no dendrites, were included in the calculations. Error bars indicate SEM; ***p < 0.001 (Student's t test). D, Distribution histograms representing total MAP2-positive dendrite lengths per neuron for cultures transduced with EGFP or dnILK for 3 d. Results derive from a single representative experiment of three that were quantitated. The data were normalized so that the total number of dendrites equaled 100%, and bin sizes are 2 μm. Neurons with no detectable MAP2-positive processes were not included in these histograms. Numbers indicate the mean ± SEM of the measurements represented in the histogram. E, The number of neurons with no dendrites in two independent experiments similar to those shown in D, represented as a percentage of the total number of neurons measured. Exp., Experiment. F, Western blot analysis for ILK in rat PC12 6-24 cells transfected for 48 h with ILK siRNA1, ILK siRNA2, or a control siRNA. The same blot was reprobed for total ERK1 as a loading control. G, ILK immunocytochemistry in sympathetic neurons transfected with ILK siRNA1 or control siRNA and cultured in NGF plus KCl for 3 d. The arrow indicates a neuron expressing ILK, and the arrowheads indicate neurons that do not express ILK at levels above background. Scale bar, 50 μm. H, Mean total MAP2-positive dendrite length per cell calculated from pooled data deriving from cells transfected with ILK siRNA1, ILK siRNA2, or control siRNA. A total of 70–100 neurons were measured. Similar results were obtained with both ILK siRNAs independently. Error bars indicate SEM; ***p < 0.001 (Student's t test). I, Quantitative analysis of sympathetic neuron cultures that were established for 5 d in NGF, switched to NGF plus KCl for 3 d (day 3), and then maintained in NGF plus KCl plus or minus QLT0254 for an additional 2 d (day 5). In some controls, neurons were treated for the final 2 d with DMSO alone, and in others, neurons were maintained in NGF alone. Total MAP2-positive dendrite lengths per neuron in each condition were derived from one of three independent experiments, in each of which 200–600 neurons were measured per condition. Error bars are SEM; ***p < 0.001 (Student's t test).
We then performed a similar series of studies, using siRNA to knock down ILK in sympathetic neurons. To do this, we used two siRNA sequences (ILK siRNA1 and siRNA2) that have previously been shown to be effective in targeting ILK (Troussard et al., 2003; Edwards et al., 2006). To confirm their efficacy, we transfected these siRNAs, or a control siRNA, into PC12 6-24 cells, lysed the cells 48 h after transfection, and monitored ILK levels by Western blots. This analysis revealed that total ILK levels were significantly reduced relative to PC12 cells transfected with the control siRNA (Fig. 4F), despite the fact that only ∼40–60% of the cells are transfected under these conditions. We then performed similar transfections with sympathetic neurons; cultures were transfected with ILK siRNA1 or siRNA2 or with control siRNA on the day of plating, established for 5 d in NGF, and then switched to NGF plus KCl for 3 d. Immunocytochemistry for ILK revealed that in sympathetic neurons transduced with the control siRNA, all of the neurons expressed similar, easily detectable levels of ILK (Fig. 4G). In contrast, in cultures transduced with one of the two ILK siRNAs, although most of the neurons expressed easily detectable levels of ILK, ∼23% did not express ILK at levels above background (ILK-low neurons) (Fig. 4G, arrowheads). We then quantitated dendrite formation in these ILK-low neurons relative to neurons in sister cultures treated with the control siRNA. This analysis demonstrated that genetic knockdown of ILK significantly reduced dendrite length in sympathetic neurons grown in NGF plus KCl (Fig. 4H) and that the magnitude of this decrease was similar to that observed with dnILK (Fig. 4C,H). These results, together with the dominant-negative and pharmacological data, suggest that ILK activity is essential for dendrite formation in sympathetic neurons.
ILK activity is not required for maintenance of sympathetic neuron dendrites
We have previously shown that activity is required for both growth and maintenance of sympathetic dendrites (Vaillant et al., 2002). We therefore asked whether ILK was also necessary for dendrite maintenance; neurons were established in NGF, switched to NGF plus KCl for 3 d, and then maintained in NGF plus KCl with or without QLT0254 for 2 additional days (5 d total in NGF plus KCl). Immunostaining for HMW-MAP2 at 3 and 5 d revealed that depolarized dendrites continued to grow over this time period in terms of total length per neuron, whereas neurons maintained in NGF for this length of time maintained only rudimentary MAP2-positive processes (Fig. 4I). Inhibition of ILK activity with QLT0254 blocked the new dendrite growth seen between days 3 and 5 in NGF plus KCl but had no effect on preexistent dendrites (Fig. 4I). Thus, ILK activity is essential for growth of sympathetic neuron dendrites but is not apparently required for their maintenance once established.
GSK-3β, an ILK target, regulates dendrite initiation and growth in sympathetic neurons
One target of ILK implicated in the establishment of neuronal polarity and axonal growth is GSK-3β. To ask whether GSK-3β was localized to dendrites, we established sympathetic neuron cultures, treated them with NGF plus KCl for 3 d, and performed immunocytochemistry for HMW-MAP2 and GSK-3β. This analysis demonstrated that GSK-3β was expressed in cell bodies and processes, including MAP2-positive dendrites (Fig. 5A) and MAP2-negative axons (data not shown). We therefore asked whether GSK-3β was important for dendrite development using two potent GSK-3β inhibitors, kenpaullone and alsterpaullone (Schultz et al., 1999; Leost et al., 2000; Bain et al., 2003). Initially, we confirmed that these inhibitors reduced GSK-3β activity in sympathetic neurons by assaying phosphorylation of the GSK-3β target Tau; neurons were stimulated with NGF plus KCl with or without one of the inhibitors for 15 min or 3 d. Western blots revealed that the inhibitors reduced phosphorylated Tau levels in both the short- and long-term experiments (Fig. 5B). We also confirmed that inhibition of GSK-3β using these pharmacological inhibitors had no effect on neuronal survival (Fig. 5C).
Figure 5.

GSK-3β inhibition is sufficient to promote dendrite initiation in sympathetic neurons. A, Double-label immunocytochemistry for HMW-MAP2 (red; left) and GSK-3β (green; center; right, merge of the two) in sympathetic neurons grown in NGF plus KCl for 3 d. Scale bar, 25 μm. B, Western blot analysis for the GSK-3β substrate pTau, in sympathetic neurons established in NGF and stimulated for 15 min or 3 d in NGF plus KCl with or without kenpaullone, alsterpaullone, or DMSO. Blots were reprobed for total Tau levels as a loading control. C, Quantification of the percentage of surviving sympathetic neurons in cultures treated with kenpaullone (Ken) for 3 d in the presence of NGF or NGF plus KCl, as monitored by counting intact, uncondensed Hoechst-positive nuclei. As controls, cultures were treated with DMSO alone. Error bars indicate SEM. D, Representative photomicrographs of sympathetic neurons cultured for 3 d in NGF or NGF plus KCl with or without kenpaullone or DMSO and immunostained for HMW-MAP2. Scale bar, 50 μm. E–G, Quantitative analysis of cultures similar to those shown in D. E, The mean number of dendrites per neuron was calculated by determining the total number of HMW-MAP2 processes and dividing by the total number of neurons measured. Results are pooled from three independent experiments, in each of which 100–300 neurons were measured per condition. F, The number of neurons with no dendrites in two independent experiments similar to those shown in D, represented as a percentage of the total number of neurons measured. Exp., Experiment. G, Mean total MAP2-positive dendrite length per cell calculated from pooled data deriving from three independent experiments. In each experiment, 100–300 neurons were measured, and all neurons, including those with no dendrites, were included in the calculations. Error bars indicate SEM; ***p < 0.001 (Student's t test). H, I, Mean total dendrite length and number of dendrites per cell in sympathetic neurons grown at low density in 10 ng/ml NGF with or without 50 mm KCl plus or minus kenpaullone for 3 d and then measured by HMW-MAP2 immunoreactivity (H) or phase-contrast microscopy (I). Results in H and I derive from a single representative experiment of three that were performed. Error bars indicate SEM; *p < 0.05; ***p < 0.001 (Student's t test).
Having established the efficacy of these inhibitors, we asked whether GSK-3β activity regulated dendrite formation. Neurons were established and then maintained for 3 d in NGF or NGF plus KCl with or without kenpaullone. Because phosphorylation of GSK-3β by ILK inhibits its activity, we predicted that GSK-3β inhibition would promote dendrite initiation and/or growth. As predicted, immunocytochemistry for HMW-MAP2 revealed that GSK-3β inhibition increased the number of dendrites per neuron (Fig. 5D). In NGF alone, the number of MAP2-positive processes was increased by kenpaullone to the same level as it was by depolarization (Fig. 5E), whereas the number of neurons without dendrites decreased (Fig. 5F). In NGF plus KCl, GSK-3β inhibition increased primary dendrite number by approximately twofold (Fig. 5E) and robustly reduced the number of neurons without dendrites (Fig. 5F). GSK-3β inhibition also increased total dendritic growth (Fig. 5G), primarily because of its effects on dendrite initiation. Similar results were obtained with the second pharmacological inhibitor, alsterpaullone (data not shown). The enhanced dendrite initiation was not simply a result of increased cell size, because kenpaullone increased soma size by only 6.5 and 12% in NGF and NGF plus KCl conditions, respectively.
We also performed similar experiments with low-density sympathetic neuron cultures, analyzing them by both phase microscopy and HMW-MAP2 immunocytochemistry. Both approaches provided similar results with regard to dendrite initiation; inhibition of GSK-3β caused a small but significant increase in the number of dendrites in NGF alone and an approximately twofold increase in dendrite number in NGF plus KCl (Fig. 5H,I). Moreover, although the absolute length of dendrites measured by phase contrast was greater than that measured by MAP2 immunoreactivity, in both cases kenpaullone caused an increase in dendrite length of approximately the same magnitude (Fig. 5H,I). Thus, pharmacological inhibition of GSK-3β led to increased dendrite initiation and growth.
We next confirmed that GSK-3β regulated dendrite initiation using both dominant-negative and genetic knockdown approaches. Initially, we transduced sympathetic neurons with a previously defined, HA-tagged dnGSK-3β (Liu et al., 2004). Sympathetic neurons were cotransfected with plasmids encoding EGFP and dnGSK-3β on the day of plating, established for 5 d, and then switched to NGF plus KCl for an additional 3 d. We confirmed that neurons were cotransfected by performing immunocytochemistry for EGFP and the HA tag on dnGSK-3β (data not shown) and then analyzed dendrite growth. Immunostaining for HMW-MAP2 (Fig. 6A) confirmed that GSK-3β inhibition enhanced dendrite initiation; the number of dendrites per neuron was increased (Fig. 6B), and the number of neurons without dendrites was greatly decreased (Fig. 6C). This increased initiation was also reflected in an enhancement of total dendritic length (Fig. 6D).
Figure 6.

GSK-3β inhibition promotes dendrite initiation and growth in multiple neuronal populations. A, Representative photomicrographs of sympathetic neurons cotransfected with EGFP and dnGSK-3β and immunostained for EGFP (left) and HMW-MAP2 (right) after 3 d in NGF plus KCl. Arrows indicate transfected neurons and arrowheads indicate nontransfected neurons. Scale bar, 50 μm. B–D, Quantitative analysis of cultures similar to that shown in A for number of dendrites per neuron (B), number of neurons with no dendrites (C), and total MAP2-positive dendrite length per neuron (D). Exp., Experiment. Measurements and calculations are similar to those shown in Figure 5E–G. Results in B and D are pooled from two independent experiments, in which 76 EGFP- and 109 dnGSK-transfected neurons were measured, whereas C shows the results from these two experiments graphed independently. Error bars are SEM; ***p < 0.001 (Student's t test). E, Western blot analysis for GSK-3β in rat PC12 6-24 cells transfected for 48 h with GSK-3β siRNA or with control siRNA. The same blot was reprobed for total ERK1 as a loading control. F, GSK-3β immunocytochemistry in sympathetic neurons transfected with GSK-3β siRNA or control siRNA and cultured in NGF plus KCl for 3 d. The arrow indicates a neuron expressing GSK-3β, and the arrowhead indicates a neuron that does not express GSK-3β at levels above background. Scale bar, 50 μm. G, H, Number of dendrites per cell (G) and mean total MAP2-positive dendrite length per cell (H) of cultures similar to those shown in F. A total of 40–50 neurons were measured for each condition. Results derive from a single representative experiment of three that were quantitated. Error bars indicate SEM; **p < 0.01; ***p < 0.001 (Student's t test). I, Representative photomicrographs of cortical neurons treated for 3 d with kenpaullone (Ken; bottom) or DMSO (top) and immunostained for HMW-MAP2. Scale bar, 50 μm. J, Quantitative analysis of cultures similar to those shown in E, measuring the number of primary dendrites per neuron. Results are representative data from one of three experiments, in each of which 100–250 neurons were measured. ***p < 0.001 (Student's t test). K, Representative photomicrographs of EGFP-transfected hippocampal neurons in slice cultures treated for 3 d with DMSO or kenpaullone. Scale bar, 50 μm. L, Sholl analysis of transfected, EGFP-expressing neurons in hippocampal slice cultures similar to those shown in K. *p < 0.05; **p < 0.01; ***p < 0.001; all values not indicated are significant to ***p < 0.001 (Student's t test). Results are representative data from one of three experiments, in each of which 21–63 neurons were measured.
As a second approach, we used a pool of GSK-3β siRNAs to knock down GSK-3β levels in sympathetic neurons. To do this, we first confirmed that these siRNAs targeted GSK-3β in rat cells; PC12 6-24 cells were transfected with either the pool of GSK-3β siRNAs or a control siRNA. Forty-eight hours after transfection, cells were lysed, and GSK-3β levels were analyzed by Western blot. This analysis revealed that the pooled siRNAs significantly reduced GSK-3β levels (Fig. 6E). We then performed similar transfections with sympathetic neurons; SCGs were transfected with GSK-3β siRNA or control siRNA on the day of plating, established for 5 d in NGF, and then switched to NGF plus KCl for 3 d. Immunocytochemistry for GSK-3β revealed that in sympathetic neurons transduced with the control siRNA, all of the neurons expressed similar, easily detectable levels of GSK-3β (Fig. 6F). In contrast, in cultures transduced with the GSK-3β-pooled siRNAs, although most of the neurons expressed easily detectable levels of GSK-3β, ∼32% did not express GSK-3β at levels above background (GSK-low neurons) (Fig. 6F, arrowhead). We then quantitated dendrite formation in these GSK-low neurons relative to neurons in sister cultures treated with the control siRNA. This analysis demonstrated that genetic knockdown of GSK-3β significantly increased the number and length of sympathetic neuron dendrites in NGF plus KCl (Fig. 6G,H) and that the magnitude of these increases was similar to that seen with the dnGSK-3β (Fig. 6B,D). Thus, activated GSK-3β is a negative regulator of dendrite initiation and growth, and when GSK-3β is phosphorylated and inactivated by ILK, this negative regulation is suppressed.
GSK-3β regulates dendrite growth and initiation in isolated cortical neurons and in hippocampal neurons in slice cultures
To determine whether GSK-3β is a general negative regulator of dendrite growth, we analyzed cultured cortical neurons. Immunocytochemistry and image analysis revealed that inhibition of GSK-3β with the pharmacological inhibitor kenpaullone for 3 d significantly increased the number of primary MAP2-positive cortical dendrites relative to DMSO-treated controls (Fig. 6I,J), results similar to those observed in sympathetic neurons. We then asked whether GSK-3β played a similar role in polarized neurons within an intact synaptic system by examining hippocampal neurons in slice cultures. Hippocampi were dissected from P7–P9 rats, sliced at 350 μm, cultured for 5–7 d, and then biolistically transfected with a plasmid encoding EGFP. This led to the selective labeling of a subpopulation of pyramidal and nonpyramidal neurons of the CA1, CA3, and dentate gyrus regions. Two days later, slice cultures were treated with kenpaullone for 3 d (ending on the equivalent of P17–P20) to inhibit GSK-3β activity, and images of live EGFP-positive neurons were captured (Fig. 6K). Sholl analysis revealed that the kenpaullone-treated hippocampal neurons had increased dendritic length and branching relative to DMSO-treated slice cultures from the same hippocampi (Fig. 6L). Moreover, a direct measurement of the number of dendrites per hippocampal neuron revealed that GSK-3β inhibition promoted dendrite initiation; kenpaullone-treated hippocampal neurons had a mean of 6.28 ± 0.3 processes, whereas controls had a mean of 5.05 ± 0.2 (p < 0.01; n = 64 and n = 39 for kenpaullone- and DMSO-treated neurons, respectively). Thus, GSK-3β is a negative regulator of dendrite initiation and growth in multiple neuronal populations, including those within intact networks.
ILK regulates dendrite initiation and growth via GSK-3β
Because our biochemical data indicated that GSK-3β is a downstream target of ILK in neurons (Fig. 2C), we asked whether ILK mediates its effects on dendrite formation via GSK-3β, by determining whether GSK-3β inhibition was sufficient to override the effects of ILK inhibition. Established sympathetic neurons were treated for 3 d with NGF plus KCl with or without ILK inhibitor QLT0254 and/or the GSK-3β inhibitor kenpaullone. Immunocytochemistry for HMW-MAP2 demonstrated that GSK-3β inhibition promoted dendrite formation whether or not ILK was coincidently inhibited. Specifically, for neurons treated with kenpaullone and QLT0254, dendrite number and total length were robustly increased in both NGF and NGF plus KCl (Fig. 7A,B,D), and the number of neurons without dendrites was reduced to 5% or less (Fig. 7C), as seen with kenpaullone alone (Fig. 5D–I). Thus, suppressing GSK-3β activity rescues the effects of inhibiting ILK, arguing that ILK signals via GSK-3β to regulate dendrite initiation and growth.
Figure 7.

GSK-3β regulates dendrite initiation and growth downstream of ILK. A, Representative photomicrographs of sympathetic neurons cultured in NGF or NGF plus KCl with or without QLT0254 (254) and kenpaullone (Ken) for 3 d and then immunostained for HMW-MAP2. Controls were treated with DMSO. Scale bar, 50 μm. B–D, Quantitative analysis of cultures similar to those shown in A. B, The mean number of dendrites per neuron was calculated by determining the total number of HMW-MAP2 processes and dividing by the total number of neurons measured. Results are pooled from three independent experiments, in each of which 100–400 neurons were measured per condition. C, The number of neurons with no dendrites in two independent experiments similar to those shown in A, represented as a percentage of the total number of neurons measured. Exp., Experiment. D, Mean total MAP2-positive dendrite length per cell calculated from pooled data deriving from three independent experiments. In each experiment, 100–400 neurons were measured, and all neurons, including those with no dendrites, were included in the calculations. Error bars indicate SEM; ***p < 0.001 (Student's t test).
GSK-3β inhibits dendrite initiation and growth by negatively regulating ERK activity
The MEK–ERK pathway has previously been shown to be essential for activity-dependent dendrite formation in sympathetic (Vaillant et al., 2002), cortical, and hippocampal neurons (Wu et al., 2001; Redmond et al., 2002), and GSK-3β has been shown to negatively regulate ERK activity (Wang et al., 2006). We therefore asked whether GSK-3β decreased dendrite formation by inhibiting ERK. Initially, we confirmed our previous results that PD98059, a pharmacological MEK inhibitor, blocked ERK phosphorylation in sympathetic neurons (Fig. 8A) and inhibited KCl-mediated formation of HMW-MAP2-positive dendrites (Fig. 8B,C) (Vaillant et al., 2002). We then asked whether GSK-3β inhibited ERK phosphorylation in neurons as it does in non-neuronal cells (Wang et al., 2006); sympathetic neurons were established, pretreated for 1 h with kenpaullone or PD98059, and then stimulated for 15 min with NGF plus KCl. Western blot analysis revealed that depolarization-mediated ERK activation was increased by the coincident inhibition of GSK-3β with kenpaullone (Fig. 8A). Conversely, Western blot analysis for phosphorylated Tau indicated that MEK inhibition had no effect on GSK-3β activity under these same conditions (Fig. 8A). Thus, GSK-3β normally inhibits ERK activation in sympathetic neurons. We then asked whether blockade of ERK activity could reverse the enhanced dendrite formation caused by GSK-3β inhibition; sympathetic neurons were established and treated for 3 d with NGF plus KCl with PD98059 and/or kenpaullone. Immunocytochemical analysis for HMW-MAP2 revealed that the enhanced dendrite initiation and growth observed after GSK-3β inhibition was completely blocked by PD98059 (Fig. 8B–D), indicating that ERK was downstream of GSK-3β with regard to dendrite formation. These results suggest that under basal conditions, GSK-3β inhibits ERK, thereby inhibiting dendrite formation, and depolarization causes ILK-mediated GSK-3β phosphorylation and inhibition, ultimately leading to increased ERK activity and enhanced dendrite formation.
Figure 8.

GSK-3β negatively regulates ERK activity, and ERK is downstream of GSK-3β during dendrite formation. A, Western blot analysis for pERK and pTau in sympathetic neurons pretreated for 1 h with kenpaullone (Ken) and/or PD98059 (PD) and stimulated for 15 min with NGF plus KCl. Controls were pretreated with DMSO or were maintained in media. Blots were reprobed for total ERK and Tau levels as loading controls. Note that GSK-3β inhibition increases pERK. B, Representative photomicrographs of sympathetic neurons cultured for 3 d in NGF plus KCl with kenpaullone and/or PD98059 and then immunostained for HMW-MAP2. Controls were treated with DMSO. Scale bar, 50 μm. C, D, Quantitative analysis of cultures similar to those shown in B, measuring the mean total MAP2-positive dendrite length per neuron (C) and the mean number of dendrites per neuron (D). Results are representative data from one of three experiments, in each of which 200–600 neurons were measured for each condition. Error bars are SEM; ***p < 0.001 (Student's t test). Note that MEK inhibition suppresses dendrite formation caused by GSK-3β inhibition.
Discussion
The data presented here report a novel role for the ILK–GSK-3β signaling pathway in regulating dendrite initiation and growth and support four major conclusions. First, our results demonstrate that ILK is regulated by cues that promote dendrite formation, such as neurotrophins and depolarization, and that ILK is essential for dendrite growth in cultured sympathetic neurons. Second, ILK regulates the phosphorylation and activity of GSK-3β, which on its own is crucial in modulating dendrite initiation. Indeed, inhibition of GSK-3β activity increased dendrite initiation in cultured sympathetic and cortical neurons and in hippocampal neurons within intact hippocampal networks. Third, GSK-3β acts downstream of ILK, because ILK inhibition suppressed GSK-3β phosphorylation, and GSK-3β suppression rescued the effects of ILK inhibition, consistent with ILK negatively regulating GSK-3β. Finally, GSK-3β mediates its effects at least partially via the MEK–ERK pathway, because GSK-3β regulates ERK activation in sympathetic neurons, and inhibition of MEK is sufficient to repress the enhanced dendrite initiation observed when GSK-3β is inhibited. Thus, our data indicate that ILK signals via GSK-3β to regulate neuronal dendrite formation.
Although many signals have been identified that control dendrite growth and maturation, particularly for spines, few have been identified that regulate initiation (Miller and Kaplan, 2003). In this study, the use of sympathetic neurons that elaborate no or rudimentary dendrites in the absence of an activity stimulus enabled us to specifically study dendrite initiation. Manipulation of this system using pharmacological, dominant-negative, and genetic knockdown approaches led to the conclusion that suppression of ILK activity decreased, whereas suppression of GSK-3β activity promoted, dendrite initiation in these neurons. Moreover, our experiments demonstrated that ILK inhibition suppressed GSK-3β phosphorylation and that GSK-3β suppression was dominant to ILK suppression, indicating that GSK-3β functions downstream of ILK in dendrite initiation. Our results also indicate that high GSK-3β activity normally suppresses dendrite initiation and does so much more potently than it suppresses axon initiation, suggesting different GSK-3β thresholds for these two activities.
How does neuronal depolarization lead to ILK activation? Depolarization of sympathetic neurons stimulates PI3-kinase activity (Vaillant et al., 1999), which is required to activate ILK (Hannigan et al., 2005). Ca2+ influx can also regulate the intracellular localization and cytoskeletal interactions of ILK (Vespa et al., 2003), which can alter both ILK activity and access to its substrates that are involved in dendrite plasticity such as β1-integrin (Hannigan et al., 2005; Shi and Ethell, 2006). However, ILK is also activated by NGF alone, and NGF in our cultures stimulates axon but not dendrite growth (Fig. 1) (Vaillant et al., 2002). Dendrite initiation requires an additional depolarization stimulus, or additional exogenous inactivation of GSK-3β, and the relatively modest increase in ILK activation seen in response to KCl-mediated depolarization may not be sufficient to explain the “all or nothing” effect that depolarization has on dendrite initiation and growth. In this regard, it is possible that ILK is not the only activity-dependent signal that suppresses GSK-3β in sympathetic neurons. For example, a number of kinases and kinase signaling pathways that regulate dendrite morphology are activated by neurotrophins and activity and inactivate GSK-3β, including Akt, MAPK/p90S6 kinase, cAMP-activated protein kinase (PKA), and protein kinase C (PKC) (Doble and Woodgett, 2003). Of particular interest is the finding that activity can stimulate dendrite growth through a CaMK-mediated enhancement of Wnt-2 transcription and secretion, which in turn leads to GSK-3β inactivation (Le Floch et al., 2005).
What are the targets of GSK-3β activity that regulate dendrite initiation and growth? Our data indicate that one of these targets is the MEK–ERK signaling pathway, which we and others have previously shown to be required for activity-dependent dendrite growth (Wu et al., 2001; Vaillant et al., 2002; Wayman et al., 2006). We show here that GSK-3β decreases ERK activation in sympathetic neurons, findings similar to those in transformed cells (Wang et al., 2006), and that inhibition of MEK completely suppresses the enhanced dendrite initiation observed when GSK-3β activity is blocked. Thus, depolarization-mediated suppression of GSK-3β activity leads to increased activation of the MEK–ERK pathway, a key regulator of dendrite formation. Interestingly, suppression of ILK activity in sympathetic neurons was not sufficient to inhibit ERK phosphorylation, indicating that signals other than ILK must converge onto GSK-3β to regulate ERK. These might include CaMKII (Miller and Kaplan, 2003) and/or PKC (Wang et al., 2006), the latter of which is known to regulate ERK activity, in part through GSK-3β (Wang et al., 2006). In addition to ERK1/2, GSK-3β has been reported to regulate axonal growth and transport by modulating the phosphorylation of a number of other targets, including Tau (Brownlees et al., 1997; Alvarez et al., 2002), APC (Zhou et al., 2004), CRMP-2 (Yoshimura et al., 2005), kinesin light chain (Morfini et al., 2002), and cytoskeletal proteins such as MAP1B (Trivedi et al., 2005) and MAP2 (Sanchez et al., 1998, 2000). We are currently systematically examining the phosphorylation of each of these proteins in neurons treated with ILK and GSK-3β inhibitors.
Recent reports indicate that GSK-3β is important for the initial establishment of neuronal polarity and that GSK-3β inhibition caused hippocampal neurons to make axons at the expense of dendrites (Jiang et al., 2005; Yoshimura et al., 2005). The data reported here are quite different; GSK-3β inhibition by pharmacological, dominant-negative, and genetic knockdown approaches markedly stimulated dendrite formation. These different outcomes for GSK-3β inhibition are likely caused by differences in developmental stage of the neurons that were examined. Jiang et al. (2005) and Yoshimura et al. (2005) studied cultured E18 hippocampal neurons, at a stage when these neurons were as yet unpolarized. In contrast, we used postnatal sympathetic neurons, hippocampal neurons in postnatal slice cultures, and cultured cortical neurons, all of which were polarized neurons with well established axons. Additional support for the idea that GSK-3β suppression leads to enhanced dendrite formation comes from a recent study (Kwon et al., 2006), in which Pten (phosphatase and tensin homolog) was genetically ablated in subsets of forebrain postmitotic neurons in vivo; these neurons exhibited hypertrophic and ectopic dendrites, a phenotype that was accompanied by inactivation of GSK-3β. Thus, it is likely that GSK-3β plays important but distinct roles during the establishment of neuronal polarity versus later morphological development.
Although our data indicate an important role for an ILK–GSK-3β pathway in dendrite initiation, this does not exclude the involvement of other pathways. In fact, our data demonstrating that the effects of GSK-3β are dependent on the MEK–ERK pathway suggest that the latter is a convergence point that is responsible for integrating a number of pathways activated during neuronal depolarization. For example, we (Vaillant et al., 1999, 2002) and others (Farnsworth et al., 1995; Finkbeiner and Greenberg, 1996) have previously demonstrated that depolarization leads to direct activation of CaMKII and Ras and that this leads to enhanced MEK–ERK signaling. As a second example, Wayman et al. (2006) recently showed that NMDA-receptor mediated Ca2+ influx led to CaMKI-dependent activation of the Ras–MEK–ERK pathway. Thus, it is likely that neuronal depolarization activates multiple signals that converge on the MEK–ERK pathway, including the ILK–GSK-3β pathway that we report here. A number of signaling cascades in addition to GSK-3βb and the MEK–ERK pathway have also been implicated in dendrite formation, including PI3-kinase, Akt, and mTor (mammalian target of rapamycin) (Dijkhuizen and Ghosh, 2005; Jaworski et al., 2005; Kumar et al., 2005), JNK1 (c-Jun N-terminal protein kinase 1) (Bjorkblom et al., 2005), and the GTP-binding proteins Cdc42 and Rac1 (Li et al., 2000; Tolias et al., 2005). Interestingly, some of these latter proteins regulate ILK, such as PI3-kinase (Delcommenne et al., 1998), or are substrates of ILK activity, such as Cdc42 and Rac1 (Filipenko et al., 2005), indicating that there is likely to be significant cross talk among these different activity-dependent pathways.
Because the dendritic arbor is the main site to receive and process synaptic inputs, the regulation of dendrite initiation and growth is fundamental not just to neural development but to the ongoing remodeling that is seen in the adult nervous system. In this regard, our data indicate that GSK-3β is a major negative regulator of dendrite initiation. We therefore suggest that extracellular cues such as growth factors and activity converge via ILK and potentially other upstream GSK-3β regulators to phosphorylate and suppress GSK-3β and thus promote dendrite initiation and growth. Because these pathways are present within dendrites, this provides a mechanism whereby positive cues can locally promote dendrite formation. Interestingly, one prediction of such a model is that the loss of positive cues, such as a local decrease in calcium influx, would cause derepression of GSK-3β and lead to local destabilization and even retraction of dendrites. Thus, GSK-3β may act as a key integration point in both the developmental establishment and ongoing remodeling of the local dendritic arbor.
Footnotes
S.N. is supported by a postdoctoral fellowship from the Canadian Heart and Stroke Foundation. K.J.P. is supported by a studentship from the Ontario Student Opportunities Trust Fund, Hospital for Sick Children Foundation. This work was supported by research grants from the Canadian Institute of Health Research (D.R.K., F.D.M.). D.R.K. and F.D.M. are Canada Research Chairs. We thank Drs. Patrizia Zanassi and Monica Wetzel for assistance with the cortical cultures, Dr. Nao Kobayashi for help with hippocampal slice cultures, and Jean-Francois Lavoie for statistical analysis. The recombinant plasmid expressing dnGSK-3β was a kind gift from Dr. J. Woodgett, the anti-pTau antibody was a kind gift of Dr. P. Davies, and the ILK inhibitor QLT0254 was provided by QLT (Vancouver, British Columbia, Canada).
References
- Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J. Regulation of tau phosphorylation and protection against beta-amyloid-induced neurodegeneration by lithium. Possible implications for Alzheimer's disease. Bipolar Disord. 2002;4:153–165. doi: 10.1034/j.1399-5618.2002.01150.x. [DOI] [PubMed] [Google Scholar]
- Arevalo JC, Chao MV. Axonal growth: where neurotrophins meet Wnts. Curr Opin Cell Biol. 2005;17:112–115. doi: 10.1016/j.ceb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkblom B, Ostman N, Hongisto V, Komarovski V, Filen JJ, Nyman TA, Kallunki T, Courtney MJ, Coffey ET. Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J Neurosci. 2005;25:6350–6361. doi: 10.1523/JNEUROSCI.1517-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlees J, Irving NG, Brion JP, Gibb BJ, Wagner U, Woodgett J, Miller CC. Tau phosphorylation in transgenic mice expressing glycogen synthase kinase-3beta transgenes. NeuroReport. 1997;8:3251–3255. doi: 10.1097/00001756-199710200-00013. [DOI] [PubMed] [Google Scholar]
- Dedhar S, Williams B, Hannigan G. Integrin-linked kinase (ILK): a regulator of integrin and growth-factor signalling. Trends Cell Biol. 1999;9:319–323. doi: 10.1016/s0962-8924(99)01612-8. [DOI] [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkhuizen PA, Ghosh A. BDNF regulates primary dendrite formation in cortical neurons via the PI3-kinase and MAP kinase signaling pathways. J Neurobiol. 2005;62:278–288. doi: 10.1002/neu.20100. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LA, Verreault M, Thiessen B, Dragowska WH, Hu Y, Yeung JH, Dedhar S, Bally MB. Combined inhibition of the phosphatidylinositol 3-kinase/Akt and Ras/mitogen-activated protein kinase pathways results in synergistic effects in glioblastoma cells. Mol Cancer Ther. 2006;5:645–654. doi: 10.1158/1535-7163.MCT-05-0099. [DOI] [PubMed] [Google Scholar]
- Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature. 1995;376:524–527. doi: 10.1038/376524a0. [DOI] [PubMed] [Google Scholar]
- Filipenko NR, Attwell S, Roskelley C, Dedhar S. Integrin-linked kinase activity regulates Rac- and Cdc42-mediated actin cytoskeleton reorganization via alpha-PIX. Oncogene. 2005;24:5837–5849. doi: 10.1038/sj.onc.1208737. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Greenberg ME. Ca(2+)-dependent routes to Ras: mechanisms for neuronal survival, differentiation, and plasticity? Neuron. 1996;16:233–236. doi: 10.1016/s0896-6273(00)80040-9. [DOI] [PubMed] [Google Scholar]
- Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer. 2005;5:51–63. doi: 10.1038/nrc1524. [DOI] [PubMed] [Google Scholar]
- Hinoi T, Yamamoto H, Kishida M, Takada S, Kishida S, Kikuchi A. Complex formation of adenomatous polyposis coli gene product and axin facilitates glycogen synthase kinase-3 beta-dependent phosphorylation of beta-catenin and down-regulates beta-catenin. J Biol Chem. 2000;275:34399–34406. doi: 10.1074/jbc.M003997200. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Kumar AS, Naruszewicz I, Wang P, Leung-Hagesteijn C, Hannigan GE. ILKAP regulates ILK signaling and inhibits anchorage-independent growth. Oncogene. 2004;23:3454–3461. doi: 10.1038/sj.onc.1207473. [DOI] [PubMed] [Google Scholar]
- Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras–PI3K–Akt–mTOR and Ras–MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Floch N, Rivat C, De Wever O, Bruyneel E, Mareel M, Dale T, Gespach C. The proinvasive activity of Wnt-2 is mediated through a noncanonical Wnt pathway coupled to GSK-3beta and c-Jun/AP-1 signaling. FASEB J. 2005;19:144–146. doi: 10.1096/fj.04-2373fje. [DOI] [PubMed] [Google Scholar]
- Lee AF, Ho DK, Zanassi P, Walsh GS, Kaplan DR, Miller FD. Evidence that DeltaNp73 promotes neuronal survival by p53-dependent and p53-independent mechanisms. J Neurosci. 2004;24:9174–9184. doi: 10.1523/JNEUROSCI.1588-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leost M, Schultz C, Link A, Wu YZ, Biernat J, Mandelkow EM, Bibb JA, Snyder GL, Greengard P, Zaharevitz DW, Gussio R, Senderowicz AM, Sausville EA, Kunick C, Meijer L. Paullones are potent inhibitors of glycogen synthase kinase-3beta and cyclin-dependent kinase 5/p25. Eur J Biochem. 2000;267:5983–5994. doi: 10.1046/j.1432-1327.2000.01673.x. [DOI] [PubMed] [Google Scholar]
- Li Z, Van Aelst L, Cline HT. Rho GTPases regulate distinct aspects of dendritic arbor growth in Xenopus central neurons in vivo. Nat Neurosci. 2000;3:217–225. doi: 10.1038/72920. [DOI] [PubMed] [Google Scholar]
- Lin DC, Quevedo C, Brewer NE, Bell A, Testa J, Grimes ML, Miller FD, Kaplan DR. APPL1 associates with TrkA and GIPC1 and is required for nerve growth factor-mediated signal transduction. Mol Cell Biol. 2006;26:8928–8941. doi: 10.1128/MCB.00228-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Yu S, Hasegawa Y, Lapushin R, Xu HJ, Woodgett JR, Mills GB, Fang X. Glycogen synthase kinase 3beta is a negative regulator of growth factor-induced activation of the c-Jun N-terminal kinase. J Biol Chem. 2004;279:51075–51081. doi: 10.1074/jbc.M408607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR. Signaling mechanisms underlying dendrite formation. Curr Opin Neurobiol. 2003;13:391–398. doi: 10.1016/s0959-4388(03)00072-2. [DOI] [PubMed] [Google Scholar]
- Mills J, Digicaylioglu M, Legg AT, Young CE, Young SS, Barr AM, Fletcher L, O'Connor TP, Dedhar S. Role of integrin-linked kinase in nerve growth factor-stimulated neurite outgrowth. J Neurosci. 2003;23:1638–1648. doi: 10.1523/JNEUROSCI.23-05-01638.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewmierzycka A, Mills J, St-Arnaud R, Dedhar S, Reichardt LF. Integrin-linked kinase deletion from mouse cortex results in cortical lamination defects resembling cobblestone lissencephaly. J Neurosci. 2005;25:7022–7031. doi: 10.1523/JNEUROSCI.1695-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304–306. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron. 2002;34:999–1010. doi: 10.1016/s0896-6273(02)00737-7. [DOI] [PubMed] [Google Scholar]
- Ruit KG, Snider WD. Administration or deprivation of nerve growth factor during development permanently alters neuronal geometry. J Comp Neurol. 1991;314:106–113. doi: 10.1002/cne.903140110. [DOI] [PubMed] [Google Scholar]
- Ruit KG, Osborne PA, Schmidt RE, Johnson EM, Jr, Snider WD. Nerve growth factor regulates sympathetic ganglion cell morphology and survival in the adult mouse. J Neurosci. 1990;10:2412–2419. doi: 10.1523/JNEUROSCI.10-07-02412.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T, Li S, Docheva D, Grashoff C, Sakai K, Kostka G, Braun A, Pfeifer A, Yurchenco PD, Fassler R. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 2003;17:926–940. doi: 10.1101/gad.255603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C, Diaz-Nido J, Avila J. Regulation of a site-specific phosphorylation of the microtubule-associated protein 2 during the development of cultured neurons. Neuroscience. 1998;87:861–870. doi: 10.1016/s0306-4522(98)00195-x. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Diaz-Nido J, Avila J. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol. 2000;61:133–168. doi: 10.1016/s0301-0082(99)00046-5. [DOI] [PubMed] [Google Scholar]
- Schultz C, Link A, Leost M, Zaharevitz DW, Gussio R, Sausville EA, Meijer L, Kunick C. Paullones, a series of cyclin-dependent kinase inhibitors: synthesis, evaluation of CDK1/cyclin B inhibition, and in vitro antitumor activity. J Med Chem. 1999;42:2909–2919. doi: 10.1021/jm9900570. [DOI] [PubMed] [Google Scholar]
- Shi Y, Ethell IM. Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J Neurosci. 2006;26:1813–1822. doi: 10.1523/JNEUROSCI.4091-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider WD. Nerve growth factor enhances dendritic arborization of sympathetic ganglion cells in developing mammals. J Neurosci. 1988;8:2628–2634. doi: 10.1523/JNEUROSCI.08-07-02628.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–538. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Trivedi N, Marsh P, Goold RG, Wood-Kaczmar A, Gordon-Weeks PR. Glycogen synthase kinase-3beta phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J Cell Sci. 2005;118:993–1005. doi: 10.1242/jcs.01697. [DOI] [PubMed] [Google Scholar]
- Troussard AA, Mawji NM, Ong C, Mui A, St.-Arnaud R, Dedhar S. Conditional knock-out of integrin-linked kinase demonstrates an essential role in protein kinase B/Akt activation. J Biol Chem. 2003;278:22374–22378. doi: 10.1074/jbc.M303083200. [DOI] [PubMed] [Google Scholar]
- Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD. Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-Akt pathway to synergistically regulate neuronal survival. J Cell Biol. 1999;146:955–966. doi: 10.1083/jcb.146.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant AR, Zanassi P, Walsh GS, Aumont A, Alonso A, Miller FD. Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron. 2002;34:985–998. doi: 10.1016/s0896-6273(02)00717-1. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, Cline HT. Rho GTPases and activity-dependent dendrite development. Curr Opin Neurobiol. 2004;14:297–304. doi: 10.1016/j.conb.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Vespa A, Darmon AJ, Turner CE, D'souza SJ, Dagnino L. Ca2+-dependent localization of integrin-linked kinase to cell junctions in differentiating keratinocytes. J Biol Chem. 2003;278:11528–11535. doi: 10.1074/jbc.M208337200. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhou Y, Wang X, Evers BM. Glycogen synthase kinase-3 is a negative regulator of extracellular signal-regulated kinase. Oncogene. 2006;25:43–50. doi: 10.1038/sj.onc.1209004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci. 2001;4:151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]
- Yau CY, Wheeler JJ, Sutton KL, Hedley DW. Inhibition of integrin-linked kinase by a selective small molecule inhibitor, QLT0254, inhibits the PI3K/PKB/mTOR, Stat3, and FKHR pathways and tumor growth, and enhances gemcitabine-induced apoptosis in human orthotopic primary pancreatic cancer xenografts. Cancer Res. 2005;65:1497–1504. doi: 10.1158/0008-5472.CAN-04-2940. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci. 2003;6:1169–1177. doi: 10.1038/nn1132. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]