

Abstract
Thyroid transcription factor 1 (TTF1) [also known as Nkx2.1 (related to the NK-2 class of homeobox genes) and T/ebp (thyroid-specific enhancer-binding protein)], a homeodomain gene required for basal forebrain morphogenesis, remains expressed in the hypothalamus after birth, suggesting a role in neuroendocrine function. Here, we show an involvement of TTF1 in the control of mammalian puberty and adult reproductive function. Gene expression profiling of the nonhuman primate hypothalamus revealed that TTF1 expression increases at puberty. Mice in which the Ttf1 gene was ablated from differentiated neurons grew normally and had normal basal ganglia/hypothalamic morphology but exhibited delayed puberty, reduced reproductive capacity, and a short reproductive span. These defects were associated with reduced hypothalamic expression of genes required for sexual development and deregulation of a gene involved in restraining puberty. No extrapyramidal impairments associated with basal ganglia dysfunction were apparent. Thus, although TTF1 appears to fulfill only a morphogenic function in the ventral telencephalon, once this function is satisfied in the hypothalamus, TTF1 remains active as part of the transcriptional machinery controlling female sexual development.
Keywords: TTF1, homeobox genes, conditional gene deletion, hypothalamus, basal ganglia, female puberty
Introduction
Morphogenesis of the basal forebrain is controlled by several homeobox genes showing spatially and temporally overlapping patterns of expression (Alvarez-Bolado et al., 1995; Shimamura et al., 1995). The ventral telencephalon, a forebrain component, gives origin to the basal ganglia and the preoptic area (POA) (Shimamura et al., 1995); the basal ganglia are, in turn, composed of two subdivisions: the striatum, located dorsally, and the pallidum, located ventrally (Gerfen, 1992). The diencephalon, the other forebrain component, also has two subdivisions, the thalamus and the hypothalamus. Development of both the pallidum and the hypothalamus requires the temporally restricted expression of thyroid transcription factor 1 (TTF1) [also known as Nkx2.1 (related to the NK-2 class of homeobox genes) and T/ebp (thyroid-specific enhancer-binding protein)] (Kimura et al., 1996; Sussel et al., 1999), a member of the Nkx family of homeobox genes (Price et al., 1992; Pera and Kessel, 1998). In the absence of TTF1, the pallidum fails to form (Sussel et al., 1999), and development of the hypothalamus is severely impaired (Kimura et al., 1996; Takuma et al., 1998; Marin et al., 2002).
TTF1 expression persists in some postmitotic basal forebrain neurons, suggesting a role for TTF1 in maintaining neuronal differentiated functions (Sussel et al., 1999; Marin et al., 2000, 2002). Such a role might extend to postnatal life, because TTF1 remains expressed after birth in selected striatal/pallidum interneurons (Marin et al., 2000), as well as in defined glial and neuronal subsets of the hypothalamus and the POA (Lee et al., 2001). Specifically, TTF1 is detected in POA neurons that control reproductive function via release of the peptide luteinizing hormone (LH)-releasing hormone (LHRH) (Lee et al., 2001), in preproenkephalinergic neurons of the lateral ventromedial nucleus (Lee et al., 2001; Davis et al., 2004), which restrain the initiation of puberty by transsynaptically inhibiting LHRH neurons (Ojeda and Skinner, 2006), in unidentified neurons of the arcuate nucleus, and in ependymoglial cells of the third ventricle and median eminence (ME). The ME is the final common pathway for LHRH neuronal axons converging to release LHRH into the portal system that connects the hypothalamus to the pituitary gland (Silverman et al., 1994). In female rats, TTF1 mRNA abundance increases in the hypothalamus preceding the natural onset of puberty and after puberty-inducing hypothalamic lesions, suggesting an involvement of TTF1 in the control of female puberty (Lee et al., 2001).
Mutations in the TTF1 gene result in neurological symptoms that include hypotonia, dyskinesia, and choreoathetosis (Krude et al., 2002; Pohlenz et al., 2002), a condition consisting of rapid involuntary and slow writhing movements. These symptoms, which are caused by impairments in basal ganglia function, are also observed in mice carrying only one Ttf1 allele disrupted by gene targeting (Pohlenz et al., 2002). In the absence of both alleles, the pallidum is absent, cholinergic neurons of the basal forebrain fail to develop, and migration of GABAergic neurons from the pallidum to the striatum and cortex is impaired (Sussel et al., 1999; Marin et al., 2002). These abnormalities suggest that loss of TTF1 may result in cognitive deficiencies (because of the absence of cholinergic neurons) (Ginsberg et al., 2006) and behavioral abnormalities (attributable in part to the loss of cortical GABAergic neurons) (Siniscalchi et al., 2003). It is also possible that the functional integrity of these neuronal populations requires continuous TTF1 support. Unfortunately, neither possibility can be addressed using conventional Ttf1 knock-out (KO) mice, because they are born dead (Kimura et al., 1996).
In the present study, we use Cre-loxP technology to demonstrate an involvement of TTF1 in the hypothalamic control of puberty and show that in contrast to the need for TTF1 to sustain this process, TTF1 deficiency instituted after establishment of telencephalic neuronal fates does not diminish differentiated basal ganglia-dependent neurological functions.
Materials and Methods
Animals
Nonhuman primates.
The rhesus monkeys (Macaca mulatta) used in this study were classified into different stages of pubertal development according to the criteria reported by Watanabe and Terasawa (1989). Plasma LH levels obtained from a different set of age-matched, contemporary animals (n = 10 per group) increased from 2.59 ± 0.97 in juvenile animals to 6.48 ± 1.64 ng/ml at midpuberty, reflecting pubertal maturation of the hypothalamic–pituitary axis. Brain tissue was obtained through the Tissue Distribution Program of the Oregon National Primate Research Center. Gene expression profiling and real-time PCR studies were conducted using mediobasal hypothalamus (MBH) and cerebral cortex (CTX) samples from juvenile (1–2 years of age) and midpubertal (3–4 years of age) animals.
Mice.
The mice were housed under a controlled photoperiod (12 h of light and 12 h of darkness; lights on at 07:00 A.M.) and temperature (23–25°C) and were given ad libitum access to rodent chow and tap water. They were used in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The experimental protocols were approved by the Animal Care and Use Committee of the Oregon National Primate Research Center (ONPRC). Mice used for behavioral experiments were transferred from the ONPRC to the Oregon Health and Sciences University (OHSU) main campus and kept under the same environmental conditions described above. All behavioral procedures used were approved by the OHSU Animal Care and Use Committee.
Mice expressing Cre under the control of the Synapsin I promoter were those previously described by Zhu et al. (2001). Because the Synapsin I promoter becomes active only in terminally differentiated neurons (Hoesche et al., 1993), Cre-mediated recombination deletes the Ttf1 gene only from postmitotic neurons. Mice carrying floxed Ttf1 alleles, produced by one of our laboratories, were described recently (Kusakabe et al., 2006). Crossing these two mutant lines generated hemizygote SynCre mice (SynCre+/−) carrying one floxed allele (Ttf1 flox/+). Breeding these animals to Ttf1 flox/flox mice resulted in a progeny consisting of wild-type (WT) animals (SynCre−/−, Ttf1 flox/flox, and SynCre−/− Ttf1 flox/+), heterozygous (HT) animals [SynCre+/− Ttf1 flox/+ (Ttf1SynCre flox/+), and SynCre+/− Ttf1 flox/flox mice (Ttf1SynCre KO)]. A potential problem of using SynCre mice is that the transgene is frequently expressed in the testis and is transmitted to the progeny (Rempe et al., 2006). Although this might represent a problem in some cases, TTF1 is not expressed in the gonads. Its main sites of expression are in the forebrain, circumventricular organs, lungs, and pituitary gland. In our experience, Ttf1SynCre KO mice grow normally, indicating that neither the pituitary–thyroid axis nor the respiratory system is affected. We also observed that male Ttf1SynCre KO mice are reproductively indistinguishable from wild-type animals.
Genotyping
The primers used to detect the WT and floxed alleles (sense 5Neo2, 5′-TGCCGTGTAAACACGAGGAC-3′; and antisense 3Neo2, 5′-GACTCTCAAGCAAGTCCATCC-3′) were those described recently (Kusakabe et al., 2006). The PCR conditions used consisted of an initial activation step of 5 min at 95°C and 36 cycles as follows: 30 s of denaturing at 94°C, 30 s of annealing at 60°C, a 1 min extension at 72°C, and a final extension of 10 min at 72°C. Because tail DNA from Ttf1SynCre flox/+ mice did not always yield a clear 540 bp PCR product that identifies the WT allele, the genotype of these animals was confirmed in a second PCR using primers that amplify a 271 bp fragment from a DNA segment only present in the wild-type allele. The sense primer 5′-CAGCTGGAGGGCGGTGGATTGG-3′ and the antisense primer 5′-TGAACGCCGGTCTCGCTAACTGGA-3′ were located within a Xho–XhoI genomic fragment located downstream from Ttf1 exon 2. This segment was replaced by a loxP-flanked pGK-neo cassette in Ttf1 floxed mice (Kusakabe et al., 2006). The PCR conditions were similar to those described above, except that the annealing temperature was 60°C. The SynCre transgene was identified with a set of primers (sense, 5′-CTTGGTCGCGTCCGTGCA-3′; and antisense, 5′-CAGGCAAATTTTGGTGTA-3′) that amplifies a PCR product of 376 bp. The touch-down PCR program used consisted of an initial activation of 5 min at 95°C, followed by two cycles of denaturing at 95°C for 30 s, annealing at 65°C for 1 min, and extension at 72°C for 2 min. Then, rounds of two cycles were performed, decreasing the annealing temperature by 2°C in each round, until 55°C was reached. The program was completed with 20 more cycles at an annealing temperature of 50°C.
Degree of Cre-mediated recombination
The procedure used to assess degree of Cre-mediated recombination (DCMR) was that reported by Leneuve et al. (2001). DCMR was assessed in genomic DNA extracted from several brain regions, including the MBH, basal ganglia, hippocampus, cerebral cortex, and cerebellum, between postnatal day 6 and 40 in both WT and Ttf1SynCre KO mice. In addition to the 5Neo2 and 3Neo2 primers described above, we used a second sense primer (5′TTF1 Exon2Del; 5′-ATGCTCAAGACTTCAGGGAGCTAAG-3′) corresponding to a sequence upstream of the first loxP site in intron 1 (see Fig. 2a). In the absence of Cre-mediated recombination, the primer pair 5Neo2–3Neo2 amplifies a 220 bp fragment; after recombination, the pair 5′TTF1 Exon2Del–3Neo2 amplifies a 269 bp fragment. The PCR conditions were similar to those described above for genotyping with the primer pair 5Neo2/3Neo2, except that in this instance, we used 30 cycles. To estimate the degree of recombination, the intensity of the 269 bp and 220 bp bands was determined using the Quantity One software, their respective background was subtracted, and the prevalence of the deleted allele was calculated by using the following equation: degree of recombination = VRA/(VRA + VFA), where VRA is the value of the recombined allele, and VFA is the value of unrecombined floxed allele.
Figure 2.

PCR strategies used to genotype and calculate the DCMR in brain tissue. a, Three primers (horizontal arrows) were designed to distinguish among three possible TTF1 alleles. The wild-type allele is identified by a 540 bp PCR product amplified by primers 5Neo2 and 3Neo2. The intact floxed allele is detected by a 220 bp PCR product amplified by the same pair of primers. To estimate DCMR in different brain regions, the 5Neo2 and 3Neo2 primers were used in conjunction with a second forward primer (5′TTF1 Exon2Del). When paired with 3Neo2, this primer amplifies a 269 bp band that identifies a deleted Ttf1 allele. Although three exons have been described in the mouse genome (http://www.ensembl.org/Mus_musculus/transview?db=core&transcript=ENSMUST00000001536), we represent in the diagram only two exons, because the exact location of an exon upstream from the exon containing the main ATG (as shown in the figure) is uncertain (Lonigro et al., 1996; Oguchi and Kimura, 1998). It is also unclear whether just one or more upstream exons might be present. b, DCMR in MBH and basal ganglia. The PCR products shown on the left correspond to the floxed allele (220 bp band) and the recombined allele (269 bp band). The bar graphs on the right represent the DCMR calculated to occur in Ttf1SynCre KO mice at the indicated postnatal ages. Each bar is the mean of 2–3 animals. AU, Arbitrary units; MM, molecular marker.
Tissue collection and RNA isolation
The MBH from rhesus monkeys was dissected with a cut along the posterior border of the optic chiasm, a caudal cut immediately in front of the mammillary bodies, and two lateral cuts along the hypothalamic sulci. The prefrontal cortex was dissected as a shallow slice along the superior frontal gyrus. The thickness of both tissue fragments was ∼4 mm. The collected tissue samples were placed in vials containing RNAlater (Ambion, Austin, TX) and left overnight at 4°C. The following day, tissues were frozen on dry ice and stored at −85°C until RNA extraction. Dissection of mouse brain regions was performed as described previously (Rogers et al., 1991; Ma et al., 1992). The monkey and mouse tissues were homogenized in 1 ml (<100 mg of tissue per milliliter) of TriReagent solution (Molecular Research Center, Cincinnati, OH), and the aqueous and organic phases were separated by the addition of 0.1 vol of bromo-chloropropane (Sigma Chemicals, St. Louis, MO) followed by centrifugation at 4°C. RNA was precipitated from the aqueous phase with 1 vol of isopropanol followed by centrifugation at 13,000 rpm for 15 min at 4°C. The pellets were resuspended in DEPC-treated H2O, and the suspension was incubated with DNA-free DNase I (2 U per reaction; Ambion) for 30 min at 37°C. RNA concentrations were determined spectrophotometrically, and RNA integrity was verified on denaturing agarose gels.
DNA microarrays
Hybridization of monkey hypothalamic total RNA to DNA microarrays containing human cDNAs was performed by the OHSU Gene Microarray Shared Resource. The arrays were prepared from the IMAGE Consortium human cDNA library [Research Genetics, now Invitrogen (Carlsbad, CA)]. The cDNA sequences were amplified by PCR with DyNAzyme EXT (New England Biolabs, Ipswich, MA). Amplified DNA was separated from unincorporated primers and nucleotides using MultiScreen PCR filtration plates (Millipore, Billerica, MA) on a BioRobot 8000 (Qiagen, Valencia, CA). All amplifications were examined for product by agarose gel electrophoresis. PCR products were dried in a CentriVap vacuum evaporator (LabConCo, Kansas City, MO) and then resuspended in 30 μl of 50% DMSO/Tris-EDTA and stored at −80°C. Printing plates were made by transferring 3 μl aliquots of each PCR product to a 384-well microtiter plate (Whatman, Florham Park, NJ). Arrays were printed with a PyxSys 5500XL microarray printer [Cartesian Technologies, now Genomic Solutions (Ann Arbor, MI)] using sixteen CMP3 printing pins (TeleChem, Sunnyvale, CA) and UltraGAPS array slides (Corning, Corning, NY). Control plant PCR products (Stratagene, La Jolla, CA) were printed but not used for these experiments. After printing, slides were baked for 3.5 h at 80°C and then stored in a desiccator cabinet in the dark until use. Total RNA was labeled using the Tyramide Signal Amplification protocol (TSA; PerkinElmer, Boston, MA) with substantial modifications to increase throughput and reproducibility. Briefly, total RNA template was used to make cDNA using either fluorescein–dCTP or biotin–dCTP. Paired samples were hybridized to arrays in TSA hybridization buffer under M-Series LifterSlips (Erie Scientific, Portsmouth, NH) and incubated overnight at 60°C in a water bath using deep-well hybridization chambers (TeleChem). After hybridization, slides were developed as described in the TSA protocol manual, but with modifications for higher throughput and reproducibility. The specific protocol is available for download from www.ohsu.edu/gmsr/smc. Results from this protocol have been validated in other studies (Reddy et al., 2004). Arrays were scanned on a ScanArray 4000 (PerkinElmer). Cyanine 5 (Cy5) and Cy3 results were saved as separate 16-bit tagged image file format images. Paired images were analyzed using ImaGene (BioDiscovery, El Segundo, CA). Low-quality spots were flagged manually for exclusion within ImaGene. Spot values were calculated with 5% trimming of highest and lowest pixel values. Pairwise comparisons of the data were examined in GeneSight (BioDiscovery) using the following protocol: (1) exclude flagged spots; (2) transform data to log2(intensity); (3) five-bin piecewise linear regression of the entire dataset, (4) calculation of log differences for Cy5 versus Cy3, and (5) combination of the replicates. For increased robustness of the analysis, two arrays were printed per slide so that each target RNA pair is interrogated twice. In addition, each RNA pair was hybridized to two slides, providing four measurements per gene analyzed. Because we compared five individuals at midpuberty to juvenile controls, each gene was interrogated a total of 20 times to take into account both assay and biological variability. A basic confidence analysis of the data identified those genes that were significantly changed with 95% confidence. Pairwise comparison did not include scaling across pairs.
Real-time PCR
Monkey TTF1 mRNA and the mRNAs encoding mouse TTF1, preproenkephalin, KiSS1, and LHRH were quantified by real-time PCR, as described previously (Romero et al., 2002; Shahab et al., 2005). After reverse transcription of 200 ng of total RNA, aliquots of each reaction (10 ng of cDNA per microliter) were diluted (1:10 to 1:50) before using 2 μl for PCR amplification. Each sample was run in triplicate along with a relative standard curve. Relative standard curves were generated by diluting one sample 1:10 to 1:10,000; they served to estimate the relative content of 18S ribosomal RNA in each sample. The primers used to detect 18S ribosomal RNA were purchased as a kit (TaqMan Ribosomal RNA Control Reagents Kit; Applied Biosystems, Foster City, CA). All gene-specific primers (Invitrogen) and fluorescent probes (Applied Biosystems) were selected with the assistance of the program Primer Express (Applied Biosystems). The monkey TTF1 primers (XM_001089890) were as follows: forward, 5′-AGCACACGACTCCGTTCTCA-3′; and reverse, 5′-CCCTCCATGCCCACTTTCTT-3′. The internal fluorescent probe was 5′-TGACATCTTGAGTCCCCTGGAGGAAAGC-3′. The mouse TTF1 primers (mmU19755) were as follows: forward, 5′-GCTCGAGCGACGTTTCAAG-3′; and reverse, 5′-GCGTGGGTGTCAGGTGAATC-3′. The internal fluorescent probe sequence was 5′-CGCCGGAGCGCGAGCATC-3′. The LHRH primers (NM_008145) were as follows: forward, 5′-ACTGATGGCCGGCATTCTAC-3′; and reverse, 5′-CCAGGGCGCAACCCATAGG-3′. The internal fluorescent probe was 5′-ACTGTGTGTTTGGAAGGCTGCTCCAGC-3′. The preproenkephalin primers (BC049766) were as follows: forward, 5′-GCCCAGGCGACATCAATTT-3′; and reverse, 5′-ATCCTTGCAGGTCTCCCAGAT-3′. The internal probe was 5′-GAAGGCAGCTGTCCTTCACATTCCAGTG-3′. The KiSS1 primers (NM_178260) used were as follows: forward, 5′-GAGCCGCTGGCAAAAGTG-3′; and reverse, 5′-GCATACCGCGATTCCTTTTC-3′. The internal fluorescence probe was 5′-ATCCACAGGCCAGCAGTCCGGA-3′. All internal fluorescent probes were covalently linked to the fluorescent dye 6-carboxyfluorescein at the 5′ end and to the quencher dye 6-carboxytetramethylrhodamine at the 3′ end.
Real-time PCRs were performed in a total volume of 10 μl, each reaction containing 2 μl of the diluted reverse-transcribed sample or 2 μl of standard, 5 μl of TaqMan Universal PCR Master Mix (Applied Biosystems), 250 nm each gene-specific and ribosomal fluorescent probe, 300 nm each gene-specific primer, and 10 nm each ribosomal primer. The real-time PCR program used consisted of an initial annealing period of 2 min at 50°C, followed by 10 min of denaturing at 95°C and 40 cycles of 15 s at 95°C and 1 min at 60°C.
In situ hybridization
The hybridization procedure was performed as recommended by Simmons et al. (1989) and described by us (Junier et al., 1991; Ma et al., 1994), using a 35S-UTP-mTTF1-labeled cRNA probe transcribed from an mTTF1 cDNA (described below). Control sections were incubated with a sense probe transcribed from the same plasmid but linearized on the 3′ end to transcribe the coding strand of the cDNA template. After an overnight hybridization at 55°C, the slides were washed and processed for cRNA detection (Junier et al., 1991; Ma et al., 1994). After dehydration, the slides were dipped in NTB-2 emulsion and were exposed to the emulsion for 3 weeks at 4°C. At this time, the slides were developed, counterstained with 0.1% thionin, quickly dehydrated in ascending concentrations of alcohol, and coverslipped for microscopic examination.
Immunohistochemistry
To identify the cells and cellular sites containing TTF1 protein in the mouse hypothalamus, we used rabbit polyclonal antibodies previously characterized by Li et al. (1998). Frozen sections (30 μm) derived from the brain of adult WT and Ttf1SynCre KO mice (perfusion fixed with 4% paraformaldehyde-borate, pH 9.5) were incubated overnight at 4°C with TTF1 antibodies diluted 1:2000. The next day, the sections were incubated with a biotinylated donkey anti-rabbit γ globulin (1:250; Vector Laboratories, Burlingame, CA) for 1 h at room temperature, followed by incubation in avidin–biotin complex (Vector Laboratories) for another 1 h. The immunohistochemical reaction was then developed to a brown color with 3,3′-DAB-HCl and 0.005% H2O2.
Combined immunohistochemistry/in situ hybridization
To determine whether TTF1 is expressed in KiSS1 neurons of the mouse hypothalamus, we used a combined immunohistochemistry/in situ hybridization procedure (Jung et al., 1997; Rage et al., 1997). After completing the TTF1 immunohistochemical procedure, the sections were mounted on glass slides and dried overnight under vacuum before hybridization with the 35S-UTP-labeled rKiSS1 (rat KiSS1) cRNA probe described below. All reagents used for the immunohistochemical procedure were prepared in DEPC-treated water.
Gross morphology
To determine whether SynCre-mediated deletion of neuronal TTF1 affects the normal morphology of the forebrain, brains from WT and TTF1 KO mice (n = 3 each) were perfusion-fixed in 4% paraformaldehyde-borate buffer, pH 9.5, sectioned at 30 μm, and stained with 0.1% thionin before microscopic examination.
DNA cloning
To PCR clone a cDNA from the mouse TTF1 mRNA (GenBank accession number mmU19755), we used a sense primer (5′-CGCCGGGTGCTCTTCTCC-3′) corresponding to nucleotides 490–507 and an antisense primer (5′-GCCACCGCTGCCCACTGAG-3′) complementary to nucleotides 915–933. PCR conditions consisted of an initial activation step of 15 min at 95°C, followed by 36 cycles of denaturing at 94°C for 30 s, annealing at 57°C for 30 s, extension at 72°C for 1 min, and a final extension of 10 min at 72°C. The resulting single PCR product of 444 bp was cloned into the plasmid pGEM-T (Promega, Madison, WI) and sequenced from both ends to verify its identity. This cDNA was used as a template to generate either sense or antisense RNA for in situ hybridization assays. To generate a KiSS1 cDNA, we used a sense primer (5′-ATGATCTCGCTGGCTTCTTG-3′) that corresponds to nucleotides 1–20 in rat KiSS1 mRNA (GenBank accession number AY196983) and an antisense primer (5′-CCCGCGCCACCTGCCTCCTG-3′) complementary to nucleotides 359–379. The PCR conditions used were identical to those used for TTF1, except that the annealing step was at 61°C.
Gene reporter constructs
A luciferase (luc) gene reporter construct containing 5 kb of the rat TTF1 gene 5′ flanking region (Oguchi and Kimura, 1998) was used to determine the effect of estradiol on TTF1 gene transcription.
To prepare a reporter vector containing a segment of the human KiSS1 (hKiSS1) gene 5′ flanking region, we PCR amplified a 1298 bp DNA fragment from human genomic DNA (Promega). The 5′ sense primer used (5′-GGACAGGCCAACGTACACATCATC-3′) corresponds to nucleotides 54657264–54657286 in the human chromosome 1 genomic contig NT_004487.18 gi:88943682. The antisense primer (5′-GCTGGGCTCCCGGTCTCAAG-3′) is complementary to nucleotides 54655947–54655966. The PCR was performed using Platinum PFX DNA Polymerase (Invitrogen) following the manufacturer's recommendations. The PCR program used consisted of an initial denaturing step of 2 min of 94°C and 34 cycles as follows: 15 s at 94°C, 30 s at 66°C, 2 min at 68°C, and a final extension for 10 min at 68°C.
The single PCR product obtained was purified (PCR Purification Kit; Qiagen), an A-extension was performed (BD Advantage cDNA Polymerase Mix; Clontech, Mountain View, CA), and the cDNA was cloned into the pCR–TOPOII vector (TOPO TA Cloning Kit; Invitrogen). Before excising the putative promoter region by SacI–XhoI digestion and subcloning it into the BamHI–XhoI sites of the reporter vector pGL2-Basic-luciferase (Promega), the sequence of the hKiSS1 promoter was verified by sequencing.
Promoter assays
Assessment of the transregulatory activity of TTF1 on the hKiss1 promoter was performed as described previously (Ojeda et al., 1999; Lee et al., 2001), using 293T cells. We first verified the transcriptional activity of the promoter alone. Twenty-four hours after seeding 300,000 cells per well in six-well plates, different concentrations of hKiSS1p–pGL2 (100–500 ng per well) were transiently transfected for 5 h using Lipofectamine (Invitrogen) in conjunction with 20 ng/ml reporter plasmid constitutively expressing β-galactosidase (β-gal) (CMV-Sport β-gal; Invitrogen) to normalize for transfection efficiency. The cells were collected 48 h after transfection and assayed for luciferase and β-gal activity, as reported previously (Ojeda et al., 1999). To examine the ability of TTF1 to transregulate the hKiSS1 promoter, 293T cells were cotransfected with 100 ng/ml wild-type hKiSS1p–pGL2 or hKiSS1p mutants lacking one or both putative TTF1 recognition motifs, plus 250 ng/ml an expression vector (pcDNA3.1Zeo; Invitrogen) containing the coding region of rTTF1 (Lee et al., 2001). As before, the cells were collected 48 h later.
To assess the ability of estradiol to transregulate the TTF1 promoter, we used C6 glioma cells stably transfected with plasmids expressing estrogen receptor α (ERα) or ERβ (Mhyre et al., 2006). The cells were grown in DMEM medium supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT), 100 μg/ml penicillin, and 100 μg/ml streptomycin. They were reseeded 24 h before the assay in 12-well plates (at 120,000 cells per well) in 10% charcoal-stripped FBS (Hyclone) DMEM without phenol red or antibiotics and were supplemented with glutamine (20 mm). Twenty-four hours later, the various reporter gene constructs were premixed at a ratio of 1 μg of DNA to 2 μl of Lipofectamine 2000 (Invitrogen) for 30 min before adding the mixture to the cells. Each well was transiently transfected with a control plasmid (pGL2-Basic; Promega), 500 ng of TTF-1 expression vector, or 750 ng of a tkERE–luc plasmid (Mhyre et al., 2006). This plasmid contains estrogen-responsive elements fused to the thymidine kinase promoter and serves as a positive control for the assay. Variability in transfection efficiency was determined by cotransfecting the cells with 250 ng of CMV-Sport-β-gal plasmid (Invitrogen). The total amount of DNA transfected was kept constant at 1 μg per well by adding the appropriate amount of pGL2–Basic to each well. After 6 h of incubation, the cells were washed with PBS, and fresh medium was added. The day after transfection, cell medium was replaced by medium containing the vehicle (ethanol) or 17β-estradiol (Sigma) at 1 or 10 nm. The cells were harvested 48 h after transfection and assayed for luciferase and β-galactosidase, as previously reported (Ojeda et al., 1999; Lee et al., 2001).
Site-directed mutagenesis of TTF1 binding sites
In silico analysis of the predicted 5′ flanking region of the hKiSS1 gene using the Transcription Element Search System tool (http://www.cbil.upenn.edu/cgi-bin/tess/tess) yielded two putative TTF1 binding sites. To test the functional importance of these sites, we prepared two mutants by site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kit; Stratagene). One of them lacked the TTF1 binding motif proximal to the putative transcription start site (nucleotides 54656068–54656076; NT_004487.18); the other lacked both this site and the more distal motif (nucleotides 54656968–54656976; NT_004487.18). The hKiSS1 promoter–TOPOII construct described above was used as the template for the proximal mutant. The forward mutagenesis primer used (5′-CTCCCAAGGTGCCATGCTGAGGGTCTGAGGAGGAGG-3′) corresponds to nucleotides 54656053–54656091 of the Homo sapiens chromosome 1 genomic contig (NT_004487.18). The reverse mutagenesis primer had a sequence complementary to the forward primer. The mutation was verified by sequencing. To prepare a mutant lacking both TTF1 sites, the hKiSS1p–TOPOII construct carrying the proximal deletion was used as the template. The forward mutagenesis primer used (5′-AAGAAACAGTCTCTGCCAGCCCCTTGTCCAAGGG-3′) corresponds to nucleotides 54656951–54656990 of the Homo sapiens chromosome 1 genomic contig (NT_004487.18). The sequence of the antisense mutagenesis primer was complementary to the forward primer. The deletion of the second TTF-1 binding site was also verified by sequencing.
Both KiSS1 promoter mutants were cloned into the KpnI and XhoI sites of the pGL2–Basic–luciferase vector (Promega).
Evaluation of puberty and reproductive performance
Litters consisting of WT, HT, and Ttf1SynCre KO mice were weaned on day 21, the males were discarded, and the females were placed in groups of four per cage. Starting on day 25, the animals were inspected daily for perforation of the vaginal membrane, an event known as vaginal opening. Thereafter, vaginal lavages were performed daily to detect the appearance of cornified cells, which identify the estrous phase of the rodent estrous cycle. Both vaginal opening and cornification of the vaginal epithelium result from the rise in estrogen secretion that accompanies the onset of puberty in rodents (Ojeda and Skinner, 2006). Ovulation normally occurs on the day of estrus, but in mice it is not certain (Stiff et al., 1974; Nelson et al., 1990) unless vaginal cornification is followed by the appearance of a predominance of leukocytes in the vaginal lavage (Prevot et al., 2003a). This abundance of leukocytes occurs in the diestrous phase of the estrous cycle and indicates that a functional corpus luteum was formed after ovulation. Therefore, we recorded the age at both vaginal opening and at first estrus, considering that a true first estrus (and thus the age at first ovulation) occurs only when the cornified cells in the vaginal lavages were replaced by at least 2 d of lavages containing mostly leukocytes (Prevot et al., 2003a). The time of first ovulation was functionally confirmed by mating 30-d-old WT, HT, and Ttf1SynCre KO females (i.e., at the age of vaginal opening) with WT males and determining the length of the mating–delivery interval (MDI) for each group.
To determine whether neuronal deletion of Ttf1 gene affects adult reproductive capacity, young adult (60 d of age) wild-type and mutant females were exposed to a fertile WT male (one male per female), and the MDI, litter size, and weight were recorded. The same parameters were recorded from subsequent litters born during a 1 year period. The percentage of reproducing dams per experimental group was calculated defining a reproductively active female as an animal showing an MDI no longer than the MDI mean of the first litter of the WT group + 2 SD of the mean. Body weights were recorded from postnatal day 6, every 6 d until day 30, and then monthly until the animals were 8 months of age.
Behavioral testing
Fourteen-month-old WT and Ttf1SynCre KO male mice (n = 5 mice per genotype) were tested in the open field and elevated zero maze (week 1), for novel location and novel object recognition (week 2), in the water maze (week 3), and for rotarod performance and passive avoidance (week 4). The person testing the mice was blinded to the genotype of the mice.
Open field.
To evaluate exploratory behavior, mice were placed in a 40.64 × 40.64 cm brightly lit (luminescence, 200 lux) open arena equipped with infrared photocells interfaced with a computer (Hamilton-Kinder, Poway, CA) for a single 10 min session.
Elevated zero maze.
To assess measures of anxiety, the elevated zero maze was used. The custom-built elevated zero maze (Hamilton-Kinder) consisted of two enclosed areas and two open areas. Mice were placed in the closed part of the maze and allowed ad libitum access for 10 min (luminescence, 200 lux). They could spend their time either in a closed safe area or in an open area. A video tracking system (set at six samples per second; Noldus Information Technology, Sterling, VA) was used to calculate the time spent in the open and closed areas.
Novel location and novel object recognition.
For 3 consecutive days, mice were individually habituated (for 5 min) to the open field described above (luminescence, 200 lux). On the fourth day, the mice were first given three 10 min trials with three plastic objects (monkey, horse, and man) placed in different corners of the enclosure and occupying an area of 6 cm2. Objects were chosen based on preliminary data showing that they were of equal interest to naive C57BL/6J wild-type mice (data not shown). There was a 3 min intertrial interval during which mice were placed back in their home cage, and the maze and the objects were cleaned with 5% acetic acid to remove odors. All familiar objects were exchanged with replicas in subsequent trials. For the fourth 10 min trial, the monkey was moved from one corner of the enclosure to another to evaluate novel location recognition. For the fifth 10 min trial, the horse was replaced by a cow to assess novel object recognition. For each trial, the total time the mouse spent exploring each object within 2 cm was recorded by an observer blinded to the genotype and treatment of the mice. The time spent exploring all objects over the first three trials assessed familiarization with the objects. The difference between the percentages of time spent exploring the object in the familiar (trial 3) and novel (trial 4) locations was calculated to assess novel location recognition. The percentage of time spent exploring the novel object during trial 5 was calculated to assess novel object recognition.
Water maze.
The water maze was used to assess spatial learning and memory. A circular pool (diameter, 140 cm) was filled with opaque water (24°C), and mice were trained to locate a submerged platform to escape from the water (luminescence, 40 lux). First, they were trained to locate a clearly marked platform (days 1 and 2). Subsequently, they were trained to locate a platform hidden beneath the surface of water made opaque using white chalk (days 3–5). During the training with the hidden platform, the mice had to navigate to it using the available spatial cues. There were two daily sessions 3.5 h apart, each consisting of three 60 s trials (with 10–15 min intertrial intervals). Mice that failed to find the platform within 60 s were led to the platform by the experimenter and allowed to stay on the platform for 3 s. During the visible platform training, the platform was moved to a different quadrant of the pool for each session. For the hidden platform training, the platform location was kept constant for each group of mice. The mice were assigned to four groups using a randomized block design to avoid any potential quadrant bias. Mice were placed into the water facing the wall at the side of the pool in nine different locations around the pool circumference, and the starting location was changed for each trial. The swimming patterns of the mice were recorded with the Noldus Ethovision video tracking system, set at six samples per second. Time to locate the platform (latency), distance moved, cumulative distance to the platform, and swim speeds were analyzed. Probe trials (no platform present) were used to assess spatial memory retention. Probe trials were conducted 1 h after the last hidden trial on each day of hidden training (total of three probe trials).
Rotarod.
To assess sensorimotor ability, mice were tested using a rotarod apparatus (Hamilton-Kinder). Mice were placed on an elevated rod (7.0 mm in diameter) initially rotating at 4 rpm. Every 15 s, the rod was accelerated by 15 rpm. Fall latency was recorded by timers, which stopped when the mouse broke the photobeams at the bottom of the chamber. Mice received three trials per day for 3 subsequent days.
Passive avoidance.
To assess emotional learning and memory, the passive avoidance test was used. Each mouse was placed in a brightly lighted compartment of a chamber also containing a dark compartment (Hamilton-Kinder). After 5 s of acclimation, the bright house light turned on, and a connecting gate to the dark compartment opened. The mouse, preferring the darkened left side, steps quickly through the gate to enter the dark compartment. After doing so, the mouse received a brief and slight foot shock (0.3 mA for 3 s). Each mouse was trained until it met a learning criterion of three consecutive 120 s trials without entering the dark compartment or up to ten trials, whichever came first. After 24 h, the mouse was again placed in the lighted compartment, and the time to reenter the dark compartment was measured up to 300 s.
Statistical analysis
Differences between two groups were analyzed using the Student's t test. When comparing several groups, the differences were analyzed by one-way ANOVA followed by the Student–Newman–Keuls multiple-comparison test for unequal replications. To analyze the behavioral data, we used ANOVA, followed by Tukey–Kramer post hoc test or by t test as indicated. Data are expressed as mean ± SEM. The null hypothesis was rejected at the 0.05 level for all analyses.
Results
TTF1 gene expression increases in the nonhuman primate hypothalamus at the time of puberty
To identify genes that may participate in the control of primate puberty, we used total RNA and human cDNA arrays containing 8500 probes to interrogate the MBH of female rhesus monkeys at the juvenile and midpubertal stages of development. As in humans, the midpubertal stage of rhesus monkeys precedes the first ovulation by several months (Watanabe and Terasawa, 1989). The CTX was used as a control tissue. The arrays revealed that TTF1 was one of the genes whose expression increased in the MBH during primate puberty (Fig. 1a). This increase was verified using a different set of arrays (Institute for Systems Biology, Seattle, WA) containing 40,000 probes (data not shown) and by real-time PCR (Fig. 1b). TTF1 mRNA was not detected in the CTX (data not shown).
Figure 1.

TTF1 mRNA abundance increases in the female monkey hypothalamus at the time of puberty. a, Puberty-related increase in hypothalamic TTF1 mRNA content detected using human cDNA microarrays. Changes in mRNA content are expressed as fold increase over values present in prepubertal juvenile (JUV) animals. MP, Midpuberty. b, Verification of the array results by real-time PCR. *p < 0.05 versus JUV controls. AU, Arbitrary units. c, Lack of effect of 17β-estradiol-dependent stimulation of ERαs on TTF1 promoter activity measured in transcription activity assays using a luciferase reporter gene driven by the rat TTF1 gene 5′ flanking region. d, Lack of effect of ERβ stimulation by estradiol on TTF1 promoter activity. C6 cells stably expressing either ERα or ERβ were exposed to estradiol (1 or 10 nm), and luciferase activity was measured after 48 h of exposure to the steroid. Tk-ERE, Reporter plasmid in which luciferase expression is driven by estrogen-responsive elements fused to the thymidine kinase promoter. *p < 0.05 versus control wells not exposed to estradiol. In this and the following figures, bars represent group means, error bars represent SEM, and numbers in parentheses indicate the number of animals or independent observations per group.
Estradiol does not transregulate TTF1 gene transcription
Because the puberty-related increases in hypothalamic TTF1 mRNA abundance that we previously detected in female rats (Lee et al., 2001) and have now detected in female monkeys could be attributable to the pubertal rise in estrogen levels, we first analyzed in silico 10 kb of the mouse, rat, and human TTF1 5′ flanking region (http://sdmc.lit.org.sg/ERE-V2/index; home page, http://research.i2r.a-star.edu.sg/promoter/) and found this region to be devoid of detectable putative estrogen-responsive sites. We then performed functional promoter assays using a cell line that stably expresses either ERα or ERβ receptors (Mhyre et al., 2006) and a reporter plasmid in which luciferase expression is driven by 5 kb of the rTTF1 gene 5′ flanking region (Oguchi and Kimura, 1998). Estradiol did not modify TTF1 promoter activity in either ERα- (Fig. 1c) or ERβ-expressing cells (Fig. 1d) when used at a physiological dose (1 nm) or at supraphysiological concentrations (10 nm). In contrast, the tkERE–luc control plasmid responded to estrogen-dependent ERα or ERβ receptor stimulation with distinct increases in luciferase activity. These results indicate that the TTF1 gene is not an estrogen target and suggest that the pubertal increase in hypothalamic TTF1 mRNA is an ovary-independent phenomenon.
Loss of neuronal TTF1 perturbs expression of genes involved in the control of puberty
TTF1 remains expressed after birth not only in hypothalamic neurons but also in ependymoglial cells lining the third ventricle (Lee et al., 2001) and circumventricular organs (Son et al., 2002), precluding the use of conventional gene targeting approaches to identify neuronal versus glial sites of TTF1 action. To circumvent this limitation, we bred mice carrying Ttf1 floxed alleles at the Ttf1 locus (Kusakabe et al., 2006) to transgenic mice in which Cre recombinase is expressed under the control of the synapsin I gene promoter (henceforth referred to as SynCre mice). The SynCre transgene is specifically expressed in postmitotic, terminally differentiated neurons (Zhu et al., 2001). Because synapsin I expression is low during embryonic and perinatal development, reaching a peak by the third postnatal week of life (Hoesche et al., 1993) and because the SynCre transgene is not expressed in glial cells (Zhu et al., 2001), substantial Cre-mediated recombination is expected to occur postnatally and in a neuron-specific manner (Zhu et al., 2001). Figure 2 shows the PCR strategy we used to genotype the mice and to assess the DCMR of the targeted Ttf1 floxed locus in brain tissue. The primers 5Neo2 and 3Neo2, used for genotyping (Kusakabe et al., 2006), amplify PCR products of 540 bp and 220 bp from the WT or Ttf1 floxed allele, respectively (Fig. 2a). To assess the DCMR, we used a multiplex PCR approach (Leneuve et al., 2001) using an additional forward primer (5′TTF1 Exon2Del). The primer pair 5′TTF Exon2Del–5Neo2 amplifies a PCR product of 269 bp if the Ttf1 floxed allele is excised (Fig. 2b). We refer to these animals as Ttf1SynCre KO. We found a DCMR of 50–55% by postnatal day 6 in both brain regions, with peak levels being attained soon afterward (by day 12) in basal ganglia and much later (days 30–40) in the MBH (Fig. 2b). The lack of complete recombination in whole-tissue samples likely reflects the presence of an intact Ttf1 gene in glial cells. In agreement with a previous report using the same SynCre transgene (Zhu et al., 2001), little recombination was observed in the cerebellum (data not shown). In contrast to conventional Ttf1-null mice (Kimura et al., 1996; Sussel et al., 1999), basal forebrain gross morphology was normal in Ttf1SynCre KO mice, a feature illustrated by the similarity that exists between the MBH of WT and Ttf1SynCre KO animals (Fig. 3).
Figure 3.

Gross morphology of the MBH is similar in WT and Ttf1SynCre KO mice. The images depict the morphological aspect of 30 μm tissue sections stained with 0.1% thionin. DMH, Dorsomedial hypothalamic nucleus; VMH, ventromedial hypothalamic nucleus; ARC, arcuate nucleus; V, third ventricle. Scale bar, 400 μm.
Cre-mediated deletion of the Ttf1 floxed alleles resulted in selective loss of TTF1 mRNA in the MBH with no changes in the ME, a region of the hypothalamus that lacks neurons and only contains astroglial and ependymoglial cells. We first quantified these changes by real-time PCR in 60-d-old female mice (Fig. 4a,b) and then verified their cellular distribution by both in situ hybridization (Fig. 4c–f) and immunohistochemistry (Fig. 4g,h). As expected, loss of TTF1 mRNA was evident in both the ventromedial and arcuate nucleus, two neuronal sites of postnatal TTF1 expression in the hypothalamus (Lee et al., 2001; Davis et al., 2004). In contrast, TTF1 mRNA and protein levels appeared to be unchanged in the ME and ependymoglial cells of the ME (Fig. 4c–h). Scattered cells of the pallidum exhibited low levels of immunoreactive TTF1 protein in WT mice (Fig. 4g, inset), and, as observed in the hypothalamus, the staining was very much reduced in Ttf1SynCre KO animals (Fig. 4h, inset)
Figure 4.
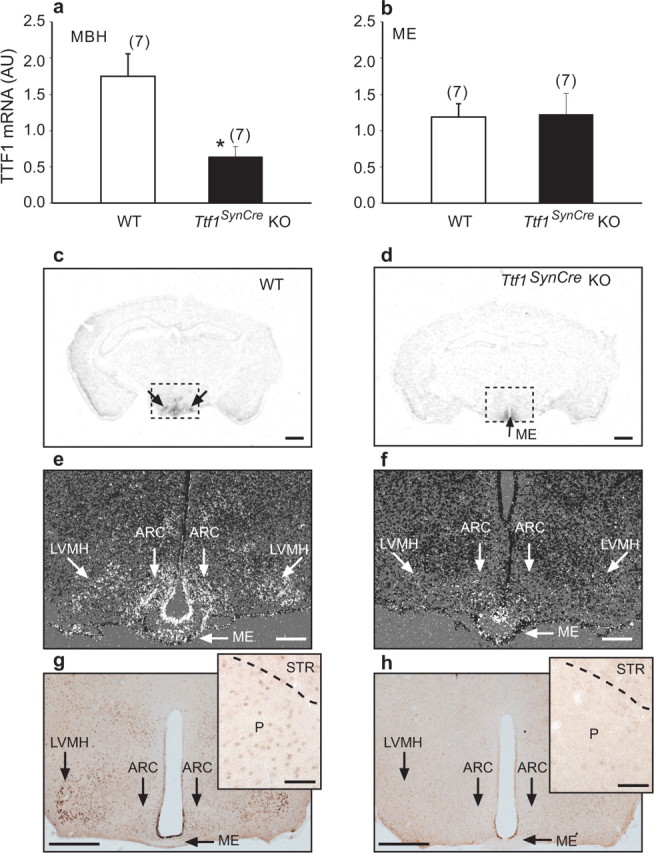
SynCre-mediated deletion of the Ttf1 gene reduces TTF1 expression in hypothalamic neurons but not ependymoglial cells, as assessed by real-time PCR (n = 7 per group), in situ hybridization (n = 3 per group), and immunohistochemistry (n = 3 per group) analysis of the brains from 60-d-old female mice. a, b, TTF1 mRNA content decreases in the MBH (a) but not in the ME (b) of Ttf1SynCre KO mice compared with WT animals, as assessed by real-time PCR. c, d, Coronal sections of the brain at the level of the MBH illustrating the presence of TTF1 mRNA in the MBH of WT mice (c; double arrows) and the loss of expression in the MBH but not the ME (d; single arrow) in Ttf1SynCre KO mice, as determined by in situ hybridization using a 35S-UTP-labeled TTF1 cRNA. e, f, Dark-field, higher-magnification in situ hybridization images showing the presence of TTF1 mRNA in specific MBH subregions including the arcuate (ARC), lateroventral medial nuclei (LVMH), and the ME of WT mice (e) and the selective loss of TTF1 mRNA in the same nuclei of Ttf1SynCre KO mice, without a change of expression in the ME (f). g, Detection of TTF1 protein in the LVMH, ARC, and ME of WT mice. Inset, TTF1 protein is also detected in cells of the pallidum (P), although at lower levels. h, Selective loss of the protein in the LVMH and ARC, with persistent expression in the ME of Ttf1SynCre KO mice, as determined by immunohistochemistry. Inset, Loss of TTF1 in the pallidum of Ttf1SynCre KO mice. STR, Striatum. Scale bars: c, d, 400 μm; e, f, 200 μm; g, h, 400 μm; insets, 50 μm. *p < 0.05 versus WT.
In the hypothalamus, TTF1 displays dual transcriptional activity in regulating the transcription of genes involved in the control of puberty (Lee et al., 2001). It transactivates the LHRH gene, which plays an essential role in controlling sexual development and reproductive function, but it represses the preproenkephalin gene, which restrains the pubertal process (Ojeda and Skinner, 2006). Consistent with these findings, preproenkephalin mRNA content, measured in early adulthood (60 d of age), was increased in the MBH of Ttf1SynCre KO animals, suggesting derepression of transcriptional activity in the absence of TTF1 (Fig. 5a). Although LHRH mRNA content in the POA was not changed at this age (Fig. 5b), the age-related rise in LHRH mRNA abundance observed in 18-month-old WT mice did not occur in Ttf1SynCre KOs (Fig. 5c).
Figure 5.

Hypothalamic expression of TTF1 target genes is altered in 60-d-old Ttf1SynCre KO mice. a, Preproenkephalin mRNA levels are increased in the MBH. Normally, TTF1 represses preproenkephalin gene transcription. b, LHRH mRNA abundance in the POA is similar to WT mice at 60 d. c, However, the age-related increase in LHRH mRNA levels seen in WT mice is abolished in Ttf1SynCre KO animals. Normally, TTF1 transactivates the LHRH promoter. d, KiSS1 mRNA abundance in the MBH is decreased. e, KiSS1 neurons of the arcuate nucleus identified by in situ hybridization (black grains) also express TTF1 protein, identified by immunostaining (brown color). Examples of colocalization are denoted by arrows. Some KiSS1 mRNA-containing cells are TTF1 negative (arrowheads). Scale bar, 20 μm. f, TTF1 transactivates the KiSS1 promoter, as assessed by functional promoter assays using a luciferase reporter gene. g, Deletion of either a single proximal putative TTF1 recognition motif [located at −110 to −100 relative to the presumed transcription start site; mutation 1 (Mut1)] or both this motif and an additional site (located at −1019 to −1010; Mut2) in the 5′ flanking region of the hKiSS1 gene obliterates the transactivating effect of TTF1 on the KiSS1 promoter. *p < 0.05; ***p < 0.001.
Kisspeptin (also known as metastin) is a product of the KiSS1 gene (Kotani et al., 2001; Ohtaki et al., 2001) that serves as a ligand for the receptor G-protein-coupled receptor 54 (GPR54) (Kotani et al., 2001; Muir et al., 2001). Because mutations of the GPR54 gene result in hypothalamic hypogonadism in humans (de Roux et al., 2003; Seminara et al., 2003) and impaired sexual development in mice (Funes et al., 2003; Seminara et al., 2003), it is now clear that activation of GPR54 receptors is a critical transsynaptic input to LHRH neurons required for the initiation of puberty. A major population of KiSS1-expressing neurons is located in the periventricular and arcuate nucleus of the MBH (Gottsch et al., 2004; Shahab et al., 2005; Smith et al., 2005). We found that KiSS1 mRNA levels are significantly reduced in the MBH of 60-d-old Ttf1SynCre KO mice compared with WT controls (Fig. 5d), suggesting that KiSS1 is a TTF1 target gene. To assess this possibility, we first used immunohistochemistry combined with in situ hybridization to determine whether KiSS1 neurons of the arcuate nucleus express TTF1. As shown in Figure 5e, many KiSS1 mRNA-expressing neurons (black grains) also contain the TTF1 protein (brown stained nuclei). Next, we cloned 1.3 kb of the 5′ flanking region of the human KiSS1 gene into a luciferase expression vector. Using this construct in functional promoter assays, we found that TTF1 transactivates the KiSS1 promoter in a dose-related manner (Fig. 5f). A search for transcriptional regulatory motifs revealed the existence of two putative TTF1 binding sites in the KiSS1 promoter (nucleotides −1010 to −1019 and −100 to −109 relative to the putative transcription start site). Both sites conform to the consensus TTF1 binding site sequence GCNCTNNAG (Bohinksi et al., 1994). Deletion of either the proximal site or both sites by site-directed mutagenesis abolished the transactivating effect of TTF1 (Fig. 5g), demonstrating that TTF1 enhances KiSS1 gene expression via specific recognition motifs present in the KiSS1 promoter.
TTF1 is required for the timing of female puberty and maintenance of adult reproductive function
The age at vaginal opening, an indication of increased estrogenic stimulation of the vaginal epithelium, was similar in WT mice, HT animals expressing SynCre but carrying only one floxed Ttf1 allele, and Ttf1SynCre KOs (Fig. 6a). However, the age at puberty, defined by the age at which the first ovulation takes place, was delayed in Ttf1SynCre KOs (Fig. 6b). The length of the first estrous cycle in these animals was almost three times longer than in WT controls, with some lengthening also observed in HT mice (Fig. 6c). After this initial disruption of estrous cyclicity, subsequent cycles assessed during a 30 d period had a slightly longer duration (5–6 d) in Ttf1SynCre KO mice than in WT or HT animals (4–5 d). The delay in puberty was not secondary to differences in body weight gain, because the three experimental groups showed a similar gain in body weight over a 240 d period (Fig. 6d). Representative estrous cycle profiles illustrating the delayed initiation of cyclicity and the slight lengthening of subsequent estrous cycles in Ttf1SynCre KO mice are shown in Figure 6e. The delay in first ovulation was functionally assessed by mating 30-d-old females (i.e., at the age of vaginal opening) of the three different genotypes with WT males and determining the length of the mating–delivery interval for each group. In agreement with the 10 d delay of the first ovulation assessed by the age at first estrus (Fig. 6b), Ttf1SynCre KO mice delivered their pups almost 2 weeks later than WT and HT animals (Fig. 7). This delay does not appear to be related to a defect in mating behavior, because the age at first ovulation calculated from the age at delivery (using 22 d as the length of gestation) is similar (47 vs 42 d) to the age at first ovulation determined by vaginal lavages in mice not exposed to males (compare Figs. 6b, 7). Interestingly, conventional Ttf1+/− mice did not show a delay in the age at first ovulation (data not shown), indicating that the presence of one functional allele is sufficient to maintain normal reproductive development.
Figure 6.

Neuronal deletion of the Ttf1 gene delays the onset of female puberty. a–c, Although the age at vaginal opening occurred at similar ages in WT, HT, and Ttf1SynCre KO mice (a), the age at first ovulation was significantly delayed in Ttf1SynCre KO animals (b), and the first estrous cycle was longer in Ttf1SynCre KO than WT and HT mice (c). d, These changes were not secondary to differences in body weight gain. e, Representative estrous cycle profiles illustrating the delayed onset of normal cyclicity and the slight lengthening of subsequent estrous cycles in Ttf1SynCre KO mice compared with HT and WT mice. P, Proestrus; E, estrus; D1, diestrous day 1; D2, diestrous day 2; VO, vaginal opening. *p < 0.05, **p < 0.01 versus WT controls.
Figure 7.

Mating–delivery interval for the first litter born to WT, HT, and Ttf1SynCre KO female mice. The KO mice delivered their first litter significantly later than both the HT and WT groups, indicating delayed first ovulation. *p < 0.05.
In addition to delaying the time of puberty, neuronal deletion of Ttf1 compromised adult reproductive capacity (Fig. 8). The reproductive life span of Ttf1SynCre KO females was shorter than that of WT and HT mice, with 50% of TTF1-deficient mice failing to become pregnant by 6–9 months of age, a time at which all WT animals and 90% of HT mice were reproductively competent (Fig. 8a). There was also a reduction in the number of litters produced by the Ttf1SynCre KO mice. We determined the total number of litters per dam every 90 d for a period of 1 year and found that by 3 months of age, the number of litters per dam had decreased to almost 50% in the Ttf1SynCre KO group compared with WT and HT controls (Fig. 8b). We also observed that the total number of pups per dam produced in a 1 year period was significantly lower in the TTF1-deficient group than in WT and HT groups (Fig. 8c). Despite these differences, the pups born to TTF1-deficient mice appeared to be normal and showed body weights similar to WT and HT animals (Fig. 8d).
Figure 8.

Reproductive capacity is diminished in Ttf1SynCre KO female mice. a, Fifty percent of Ttf1SynCre KO dams ceased delivering pups by 6–9 months of age, a time during which all WT females and 90% of HT mice were reproducing normally. b, The number of litters produced by each dam every 90 d significantly declined in Ttf1SynCre KO females by 3 months of age. c, Ttf1SynCre KO females produce fewer pups than WT or HT dams in a 1 year period. d, The body weight of the pups at birth was similar in all three experimental groups. **p < 0.01 versus WT controls. No., Number.
Neuronal deletion of the Ttf1 gene does not diminish basal ganglia-dependent functions
To determine whether basal ganglia functions are compromised in TTF1-deficient mice, we used two complementary tests: the open field test to assess general locomotor activity and exploratory behavior in a novel environment (Wilson et al., 1976) and the rotarod test to evaluate sensorimotor function (Jones and Roberts, 1968). In both conventional Ttf1+/− mice (Pohlenz et al., 2002) and patients carrying TTF1 mutations (Krude et al., 2002; Pohlenz et al., 2002), sensorimotor function is impaired. In contrast to these observations, Ttf1SynCre KO mice showed higher, instead of lower, locomotor activity than age-matched WT mice in the open field test (Fig. 9a). Similarly, on the rotarod, KO mice were able to stay longer on the rod than WT mice (Fig. 9b). These data show that basal ganglia function, instead of being diminished, is enhanced in Ttf1SynCre KO mice. Constitutive deletion of the Ttf1 gene results in substantial loss of GABAergic neurons in the cerebral cortex, because these neurons fail to migrate from the pallidum into the cortex in the absence of TTF1 (Sussel et al., 1999). Should this migratory pathway be affected in Ttf1SynCre KO mice, these mice might exhibit changes in anxious behavior (Siniscalchi et al., 2003). To measure anxiety, we used the elevated zero maze test (Shepherd et al., 1994). Surprisingly again, Ttf1SynCre KO mice showed higher, instead of lower, measures of anxiety than WT mice in the elevated zero maze (Fig. 9c). Because most, if not all, tyrosine receptor kinase A (trkA)-positive neurons of the basal forebrain are cholinergic (Sobreviela et al., 1994), and trkA-containing neurons fail to develop in the forebrain of constitutive Ttf1 KO mice (Sussel et al., 1999), it is likely that adult Ttf1SynCre KO mice would suffer from learning and memory deficits, should basal forebrain cholinergic neurons be affected by the loss of TTF1. To evaluate this possibility, we tested Ttf1SynCre KO mice for cognitive function. When object recognition was assessed, WT mice predictably spent more time exploring a familiar object in a novel than a familiar location (Fig. 9d). The response of TTF1-deficient mice was more variable and not statistically different from the response to an old location, indicating reduced novel location recognition (Fig. 9d). However, in a subsequent trial, both WT and KO mice spent more time exploring a novel than a familiar object, indicating intact novel object recognition in both genotypes (Fig. 9e). When spatial learning and memory was assessed in the water maze, there was no genotype difference in acquisition or spatial memory retention (data not shown). Last, emotional learning and memory was assessed in the passive avoidance test. There was no genotype difference in trials to criterion, a measure of passive avoidance learning (WT, 7 ± 1.4; KO, 3.8 ± 1.1) or memory retention 24 h later (WT, 250.6 ± 39.0; KO, 300 ± 0; test cutoff set at 300 s; 0 indicates that none of the KO mice reentered within 300 s).
Figure 9.

Functions directly or indirectly related to basal ganglia activity are not negatively impacted in Ttf1SynCre KO mice. a–e, Locomotor activity in the open field is increased (a), rotarod performance is increased (b), anxiety is increased as assessed by the elevated zero maze (c), and novel location recognition is reduced (d), but novel object recognition is not affected (e). The tests were performed on 14-month-old WT and Ttf1SynCre KO mice. n = 5 mice per genotype. *p < 0.05 versus WT; #p < 0.05 versus old location; ***p < 0.001 versus the two other objects.
Discussion
Our results unveil the existence of a remarkable dichotomy in the manner by which a homeobox gene controls the functional integrity of two basal forebrain regions. Although morphogenesis of both the pallidum and the hypothalamus require the presence of TTF1, only the hypothalamus necessitates the continuous expression of TTF1 to carry out specific differentiated functions. Our study indicates that one such function is the control of the pubertal process and adult reproductive activity. It also suggests that TTF1 exerts this control by regulating the transcriptional activity of genes that, expressed in defined hypothalamic neuronal subsets, are indispensable for the central regulation of reproduction.
Consistent with the fact that SynCre-mediated recombination only occurs in terminally differentiated neurons (Zhu et al., 2001), we observed no overt abnormalities in the gross morphological makeup of either telencephalic or diencephalic derivatives (pallidum and hypothalamus, respectively) in Ttf1SynCre KO mice. It has been known for some time that several cellular subsets of the hypothalamus, including LHRH and preproenkephalin neurons, express TTF1 after birth (Lee et al., 2001). We previously showed that TTF1 controls in an opposite manner the transcription of genes expressed in these cellular subsets. Thus, TTF1 transactivates the LHRH gene, but it represses preproenkephalin gene transcription (Lee et al., 2001). In addition, TTF1 transactivates the erbB2 gene (Lee et al., 2001), which is abundantly expressed in ependymoglial cells (Prevot et al., 2003b). LHRH is essential for reproductive function, and erbB2 is a crucial component of the erbB-dependent signaling complex used by glial cells to facilitate sexual development (Ojeda et al., 2003). In contrast, preproenkephalinergic neurons contribute to restraining the pubertal process (for review, see Ojeda and Skinner, 2006). Because of this dual transcriptional control, it has been hypothesized that TTF1 belongs to an upstream hierarchy of genes that control the pubertal process by differentially regulating the transcription of subordinate genes expressed in different hypothalamic cellular subsets involved in the control of reproductive function (Ojeda et al., 2006). The present results support this view. Not only was puberty delayed in TTF1-deficient mice, but we also demonstrate that the KiSS1 gene [recently shown to play a pivotal role in the hypothalamic control of the pubertal process in humans and mice (de Roux et al., 2003; Seminara et al., 2003)] is subjected to TTF1 transcriptional control. Because kisspeptin, the peptide product of the KiSS1 gene, stimulates LHRH secretion (Gottsch et al., 2004; Navarro et al., 2004), our results add credence to the concept that TTF1 acts as an upstream regulator of the pubertal process by transactivating genes involved in the facilitatory control of puberty and repressing genes that restrain sexual development (Ojeda et al., 2006).
We recently identified a novel gene termed EAP1 (enhanced at puberty) as another upstream regulator of mammalian puberty (S. Heger, C. Mastronardi, G. A. Dissen, A. Lomniczi, R. Cabrera, C. Roth, H. Jung, F. Galimi, and S. R. Ojeda, unpublished results). EAP1 appears to be placed at a higher hierarchical level than TTF1, because it controls TTF1 expression via direct transactivation of the TTF1 promoter and is expressed in a wider spectrum of neuronal subsets involved in the control of puberty than TTF1 (A. Lomniczi, C. Mastronardi, and S. R. Ojeda, unpublished results). In addition to LHRH, preproenkephalinergic, and KiSS1 neurons, EAP1 is expressed in glutamatergic and GABAergic neurons, the two major neuronal networks controlling female sexual development (Ojeda and Skinner, 2006). As predicted by the emerging features of gene networks controlling complex biological systems (Davidson et al., 2002; Basso et al., 2005), TTF1 and EAP1 are not only expressed in separate (but functionally connected) domains of the neuronal network controlling LHRH secretion, but they also share discrete functional domains, because both can transactivate the LHRH promoter and repress the preproenkephalin gene (this study) (Heger, Mastronardi, Dissen, Lomniczi, Cabrera, Roth, Jung, Galimi, and Ojeda, unpublished results).
In addition, the pubertal increase in hypothalamic TTF1 expression (and that of EAP1) (Heger, Mastronardi, Dissen, Lomniczi, Cabrera, Roth, Jung, Galimi, and Ojeda) appears to occur in an estrogen-independent manner, because the transcriptional activity of the TTF1 gene is not affected by the stimulation of ERα or ERβ receptors with either physiological or supraphysiological doses of the steroid. Reinforcing this notion are the following: (1) the absence of recognizable EREs in the TTF1 promoter of rats, mice, and humans, and (2) the finding that in rats, hypothalamic TTF1 mRNA increases during juvenile development (i.e., between days 20–30), when ovarian steroid levels are low, and decreases at the time when plasma estrogen levels increase, preceding the first preovulatory surge of gonadotropins (Lee et al., 2001). We believe that, because of this independence from estrogen, TTF1 can be considered as a component of the centrally driven, gonad-independent machinery that initiates female puberty in mammals.
A surprising finding was that the TTF1 deficiency did not affect LHRH mRNA abundance in young mice but abolished the age-related increase observed in older WT mice. We do not know the reasons for this delayed effect. Perhaps SynCre-mediated recombination is inefficient and/or occurs late in LHRH neurons. Alternatively, LHRH gene transcription might be redundantly controlled by other transcriptional regulators that become less influential in relation to TTF1 as the animal ages. It is noteworthy that the POA (in which LHRH neurons are located) derives from the ventral telencephalon but develops independently of TTF1 control (Sussel et al., 1999). Adding complexity to this issue, LHRH neurons express TTF1, but they originate in either the olfactory placode (Schwanzel-Fukuda and Pfaff, 1989) or elsewhere in the brain (Whitlock, 2005), and thus, they cannot be considered to have a telencephalic origin.
The Ttf1SynCre KO mice did not show impairments in locomotor activity in the open field or sensorimotor function on the rotarod. These data indicate that basal ganglia function is not diminished when TTF1 is deleted from postmitotic differentiated neurons and thus support the concept that continuous expression of TTF1 is not required for postnatal basal ganglia function. A note of caution must be introduced here, because the persistent expression of TTF1 in some neurons of the striatum and pallidum after birth (Marin et al., 2000) suggests that loss of expression in Ttf1SynCre KO mice may have indeed resulted in subtle defects not detected with the methodology we used.
The increased anxiety observed in Ttf1SynCre KO mice cannot be a result of a lack of GABAergic interneuron migration from the pallidum into the cerebral cortex, because such a migratory deficiency, which has been observed in conventional Ttf1 KO mice (Sussel et al., 1999), would not be expected to increase anxiety (Siniscalchi et al., 2003). Instead, the increased anxiety displayed by Ttf1SynCre KO mice in the elevated zero maze might be related to loss of TTF1 from neuronal subsets located in the bed nucleus of the stria terminalis and parts of the septum and the amygdala. TTF1 is expressed in postmitotic neurons of these regions (Sussel et al., 1999), which are implicated in the regulation of anxiety (Gray and Bingaman, 1996; Lee and Davis, 1997).
Dysfunction of cholinergic basal forebrain neurons, a well established defect in Alzheimer's disease, is associated with cognitive impairment (Ginsberg et al., 2006). Although Ttf1SynCre KO mice appeared to have some impairment of novel location recognition, they displayed an intact novel object recognition, as well as normal learning and memory, as assessed by the water maze and passive avoidance tests. Because all of these parameters are affected in aging rats or animals bearing basal forebrain lesions (Paban et al., 2005), our results indicate that basal forebrain cholinergic neuronal function is essentially intact in conditional TTF1-deficient mice. Major behavioral defects measured by the tests we used would have been expected to occur if these neurons were functionally impaired or absent, because it occurs in conventional Ttf1 KOs (Sussel et al., 1999).
In conclusion, our results suggest that beyond its role in regional specification of the ventral forebrain, the continuous presence of TTF1 is required only for the maintenance of postnatal functions inherent to a diencephalic derivative (the hypothalamus) but not those performed by a telencephalic derivative (the basal ganglia). In a broader context, these results also raise the possibility that a similar functional dichotomy may exist in other embryologically distinct brain regions whose developmental fate is controlled by the same homeodomain gene(s).
Footnotes
This work was supported by National Institutes of Health (NIH) Grants HD25123, U54 HD18185 (through cooperative agreement as part of the Specialized Cooperative Center's Program in Reproduction Research, National Institute of Child Health and Human Development/NIH), RR00163 (for the operation of the Oregon National Primate Research Center) (S.R.O.), and AG20904; by Ellison Medical Foundation Grant AG-NS-0201 (J.R.); by German Research Foundation Grant HE 3151/3-1; by the European Commission-PIONEER (S.H.); and by the Intramural Research Program, National Cancer Institute (S.K.). We thank Maria Costa, Leela Goodspeed, and Tomomi Davidson for expert technical assistance. We thank Timothy Pfankuch and Angela Rizk-Jackson for their assistance with the behavioral experiments.
References
- Alvarez-Bolado G, Rosenfeld MG, Swanson LW. Model of forebrain regionalization based on spatiotemporal patterns of POU-III homeobox gene expression, birthdates, and morphological features. J Comp Neurol. 1995;355:237–295. doi: 10.1002/cne.903550207. [DOI] [PubMed] [Google Scholar]
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nat Genet. 2005;37:382–390. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- Bohinksi RJ, Di Lauro R, Whitsett JA. The lung-specific surfactant protein B gene promoter is a target for thyroid transcription factor 1 and hepatocyte nuclear factor 3, indicating common factors for organ-specific gene expression along the foregut axis. Mol Cell Biol. 1994;14:5671–5681. doi: 10.1128/mcb.14.9.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, Otim O, Brown CT, Livi CB, Lee PY, Revilla R, Rust AG, Pan Z, Schilstra MJ, Clarke PJ, Arnone MI, et al. A genomic regulatory network for development. Science. 2002;295:1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- Davis AM, Seney ML, Stallings NR, Zhao L, Parker KL, Tobet SA. Loss of steroidogenic factor 1 alters cellular topography in the mouse ventromedial nucleus of the hypothalamus. J Neurobiol. 2004;60:424–436. doi: 10.1002/neu.20030. [DOI] [PubMed] [Google Scholar]
- de Roux N, Genin E, Carel J-C, Matsuda F, Chaussain J-L, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 2003;100:10972–10976. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funes S, Hedrick JA, Vassileva G, Markowitz L, Abbondanzo S, Golovko A, Yang S, Monsma FJ, Gustafson EL. The KiSS-1 receptor GPR54 is essential for the development of the murine reproductive system. Biochem Biophys Res Commun. 2003;312:1357–1363. doi: 10.1016/j.bbrc.2003.11.066. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Down regulation of trk but not p75 gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer's disease. J Neurochem. 2006;97:475–487. doi: 10.1111/j.1471-4159.2006.03764.x. [DOI] [PubMed] [Google Scholar]
- Gottsch ML, Cunningham MJ, Smith JT, Popa SM, Acohido BV, Crowley WF, Seminara S, Clifton DK, Steiner RA. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology. 2004;145:4073–4077. doi: 10.1210/en.2004-0431. [DOI] [PubMed] [Google Scholar]
- Gray TS, Bingaman EW. The amygdala: corticotropin-releasing factor, steroids, and stress. Crit Rev Neurobiol. 1996;10:155–168. doi: 10.1615/critrevneurobiol.v10.i2.10. [DOI] [PubMed] [Google Scholar]
- Hoesche C, Sauerwald A, Veh RW, Krippl B, Kilimann MW. The 5′-flanking region of the rat synapsin I gene directs neuron-specific and developmentally regulated reporter gene expression in transgenic mice. J Biol Chem. 1993;268:26494–26502. [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ. A rotarod suitable for quantitative measurements of motor incoordination in naive mice. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol. 1968;259:211. doi: 10.1007/BF00537801. [DOI] [PubMed] [Google Scholar]
- Jung H, Shannon EM, Fritschy J-M, Ojeda SR. Several GABAA receptor subunits are expressed in LHRH neurons of juvenile female rats. Brain Res. 1997;780:218–229. doi: 10.1016/s0006-8993(97)01152-9. [DOI] [PubMed] [Google Scholar]
- Junier M-P, Ma YJ, Costa ME, Hoffman G, Hill DF, Ojeda SR. Transforming growth factor alpha contributes to the mechanism by which hypothalamic injury induces precocious puberty. Proc Natl Acad Sci USA. 1991;88:9743–9747. doi: 10.1073/pnas.88.21.9743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, Gonzalez FJ. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 1996;10:60–69. doi: 10.1101/gad.10.1.60. [DOI] [PubMed] [Google Scholar]
- Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E, Brezillon S, Tyldesley R, Suarez-Huerta N, Vandeput F, Blanpain C, Schiffmann SN, Vassart G, Parmentier M. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem. 2001;276:34631–34636. doi: 10.1074/jbc.M104847200. [DOI] [PubMed] [Google Scholar]
- Krude H, Schutz B, Biebermann H, von MA, Schnabel D, Neitzel H, Tonnies H, Weise D, Lafferty A, Schwarz S, DeFelice M, von DA, van LF, DiLauro R, Gruters A. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2–1 haploinsufficiency. J Clin Invest. 2002;109:475–480. doi: 10.1172/JCI14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe T, Kawaguchi A, Hoshi N, Kawaguchi R, Hoshi S, Kimura S. T/EBP/NKX2.1 is required for the maintenance of ordered architecture and function of the differentiated thyroid. Mol Endocrinol. 2006;20:1796–1809. doi: 10.1210/me.2005-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BJ, Cho GJ, Norgren R, Junier M-P, Hill DF, Tapia V, Costa ME, Ojeda SR. TTF-1, a homeodomain gene required for diencephalic morphogenesis, is postnatally expressed in the neuroendocrine brain in a developmentally regulated and cell-specific fashion. Mol Cell Neurosci. 2001;17:107–126. doi: 10.1006/mcne.2000.0933. [DOI] [PubMed] [Google Scholar]
- Lee Y, Davis M. Role of the hippocampus, the bed nucleus of the stria terminalis, and the amygdala in the excitatory effect of corticotropin-releasing hormone on the acoustic startle reflex. J Neurosci. 1997;17:6434–6446. doi: 10.1523/JNEUROSCI.17-16-06434.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leneuve P, Zaoui R, Monget P, LeBouc Y, Holzenberger M. Genotyping of Cre-lox mice and detection of tissue-specific recombination by multiplex PCR. Biotechniques. 2001;31:1156–1162. doi: 10.2144/01315rr05. [DOI] [PubMed] [Google Scholar]
- Li J, Gao E, Mendelson CR. Cyclic AMP-responsive expression of the surfactant protein-A gene is mediated by increased DNA binding and transcriptional activity of thyroid transcription factor-1. J Biol Chem. 1998;273:4592–4600. doi: 10.1074/jbc.273.8.4592. [DOI] [PubMed] [Google Scholar]
- Lonigro R, De Felice M, Biffali E, Macchia PE, Damante G, Asteria C, Di Lauro R. Expression of thyroid transcription factor 1 gene can be regulated at the transcriptional and posttranscriptional levels. Cell Growth Differ. 1996;7:251–261. [PubMed] [Google Scholar]
- Ma YJ, Junier M-P, Costa ME, Ojeda SR. Transforming growth factor alpha (TGFα) gene expression in the hypothalamus is developmentally regulated and linked to sexual maturation. Neuron. 1992;9:657–670. doi: 10.1016/0896-6273(92)90029-d. [DOI] [PubMed] [Google Scholar]
- Ma YJ, Costa ME, Ojeda SR. Developmental expression of the genes encoding transforming growth factor alpha (TGFα) and its receptor in the hypothalamus of female rhesus macaques. Neuroendocrinology. 1994;60:346–359. doi: 10.1159/000126769. [DOI] [PubMed] [Google Scholar]
- Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J Neurosci. 2000;20:6063–6076. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Baker J, Puelles L, Rubenstein JL. Patterning of the basal telencephalon and hypothalamus is essential for guidance of cortical projections. Development. 2002;129:761–773. doi: 10.1242/dev.129.3.761. [DOI] [PubMed] [Google Scholar]
- Mhyre AJ, Shapiro RA, Dorsa DM. Estradiol reduces nonclassical transcription at cyclic adenosine 3′,5′-monophosphate response elements in glioma cells expressing estrogen receptor alpha. Endocrinology. 2006;147:1796–1804. doi: 10.1210/en.2005-1316. [DOI] [PubMed] [Google Scholar]
- Muir AI, Chamberlain L, Elshourbagy NA, Michalovich D, Moore DJ, Calamari A, Szekeres PG, Sarau HM, Chambers JK, Murdock P, Steplewski K, Shabon U, Miller JE, Middleton SE, Darker JG, Larminie CG, Wilson S, Bergsma DJ, Emson P, Faull R, et al. AXOR12, a novel human G protein-coupled receptor, activated by the peptide KiSS-1. J Biol Chem. 2001;276:28969–28975. doi: 10.1074/jbc.M102743200. [DOI] [PubMed] [Google Scholar]
- Navarro VM, Fernandez-Fernandez R, Castellano JM, Roa J, Mayen A, Barreiro ML, Gaytan F, Aguilar E, Pinilla L, Dieguez C, Tena-Sempere M. Advanced vaginal opening and precocious activation of the reproductive axis by KiSS-1 peptide, the endogenous ligand of GPR54. J Physiol (Lond) 2004;561:379–386. doi: 10.1113/jphysiol.2004.072298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JF, Karelus K, Felicio LS, Johnson TE. Genetic influences on the timing of puberty in mice. Biol Reprod. 1990;42:649–655. doi: 10.1095/biolreprod42.4.649. [DOI] [PubMed] [Google Scholar]
- Oguchi H, Kimura S. Multiple transcripts encoded by the thyroid-specific enhancer-binding protein (T/EBP)/thyroid-specific transcription factor-1 (TTF-1) gene: evidence of autoregulation. Endocrinology. 1998;139:1999–2006. doi: 10.1210/endo.139.4.5933. [DOI] [PubMed] [Google Scholar]
- Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, Kanehashi K, Terao Y, Kumano S, Takatsu Y, Masuda Y, Ishibashi Y, Watanabe T, Asada M, Yamada T, Suenaga M, Kitada C, Usuki S, Kurokawa T, Onda H, Nishimura O, et al. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613–617. doi: 10.1038/35079135. [DOI] [PubMed] [Google Scholar]
- Ojeda SR, Skinner MK. Puberty in the rat. In: Neill JD, editor. The physiology of reproduction. Ed 3. San Diego: Academic/Elsevier; 2006. pp. 2061–2126. [Google Scholar]
- Ojeda SR, Hill J, Hill DF, Costa ME, Tapia V, Cornea A, Ma YJ. The Oct-2 POU-domain gene in the neuroendocrine brain: a transcriptional regulator of mammalian puberty. Endocrinology. 1999;140:3774–3789. doi: 10.1210/endo.140.8.6941. [DOI] [PubMed] [Google Scholar]
- Ojeda SR, Prevot V, Heger S, Lomniczi A, Dziedzic B, Mungenast A. Glia-to-neuron signaling and the neuroendocrine control of female puberty. Ann Med. 2003;35:244–255. doi: 10.1080/07853890310005164. [DOI] [PubMed] [Google Scholar]
- Ojeda SR, Lomniczi A, Mastronardi C, Heger S, Roth C, Parent AS, Matagne V, Mungenast AE. Minireview: the neuroendocrine regulation of puberty: is the time ripe for a systems biology approach? Endocrinology. 2006;147:1166–1174. doi: 10.1210/en.2005-1136. [DOI] [PubMed] [Google Scholar]
- Paban V, Chambon C, Jaffard M, Alescio-Lautier B. Behavioral effects of basal forebrain cholinergic lesions in young adult and aging rats. Behav Neurosci. 2005;119:933–945. doi: 10.1037/0735-7044.119.4.933. [DOI] [PubMed] [Google Scholar]
- Pera EM, Kessel M. Demarcation of ventral territories by the homeobox gene NKX2.1 during early chick development. Dev Genes Evol. 1998;208:168–171. doi: 10.1007/s004270050170. [DOI] [PubMed] [Google Scholar]
- Pohlenz J, Dumitrescu A, Zundel D, Martine U, Schonberger W, Koo E, Weiss RE, Cohen RN, Kimura S, Refetoff S. Partial deficiency of thyroid transcription factor 1 produces predominantly neurological defects in humans and mice. J Clin Invest. 2002;109:469–473. doi: 10.1172/JCI14192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevot V, Rio C, Cho GJ, Lomniczi A, Heger S, Neville CM, Rosenthal NA, Ojeda SR, Corfas G. Normal female sexual development requires neuregulin-erbB receptor signaling in hypothalamic astrocytes. J Neurosci. 2003a;23:230–239. doi: 10.1523/JNEUROSCI.23-01-00230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevot V, Cornea A, Mungenast A, Smiley G, Ojeda SR. Activation of erbB-1 signaling in tanycytes of the median eminence stimulates transforming growth factor β1 release via prostaglandin E2 production and induces cell plasticity. J Neurosci. 2003b;23:10622–10632. doi: 10.1523/JNEUROSCI.23-33-10622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M, Lazzaro D, Pohl T, Mattei M-G, Rüther U, Olivo J-C, Duboule D, Di Lauro R. Regional expression of the homeobox gene Nkx-2.2 in the developing mammalian forebrain. Neuron. 1992;8:241–255. doi: 10.1016/0896-6273(92)90291-k. [DOI] [PubMed] [Google Scholar]
- Rage F, Lee BJ, Ma YJ, Ojeda SR. Estradiol enhances prostaglandin E2 receptor gene expression in luteinizing hormone-releasing hormone (LHRH) neurons and facilitates the LHRH response to PGE2 by activating a glia-to-neuron signaling pathway. J Neurosci. 1997;17:9145–9156. doi: 10.1523/JNEUROSCI.17-23-09145.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, McWeeney S, Park BS, Manczak M, Gutala RV, Partovi D, Jung Y, Yau V, Searles R, Mori M, Quinn J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer's disease. Hum Mol Genet. 2004;13:1225–1240. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- Rempe D, Vangeison G, Hamilton J, Li Y, Jepson M, Federoff HJ. Synapsin I Cre transgene expression in male mice produces germline recombination in progeny. Genesis. 2006;44:44–49. doi: 10.1002/gene.20183. [DOI] [PubMed] [Google Scholar]
- Rogers LC, Junier M-P, Farmer SR, Ojeda SR. A sex-related difference in the developmental expression of class II β-tubulin messenger RNA in rat hypothalamus. Mol Cell Neurosci. 1991;2:130–138. doi: 10.1016/1044-7431(91)90005-9. [DOI] [PubMed] [Google Scholar]
- Romero C, Paredes A, Dissen GA, Ojeda SR. Nerve growth factor induces the expression of functional FSH receptors in newly formed follicles of the rat ovary. Endocrinology. 2002;143:1485–1494. doi: 10.1210/endo.143.4.8711. [DOI] [PubMed] [Google Scholar]
- Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338:161–164. doi: 10.1038/338161a0. [DOI] [PubMed] [Google Scholar]
- Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O'Rahilly S, Carlton MB, Crowley WF, Jr, Aparicio SA, Colledge WH. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- Shahab M, Mastronardi C, Seminara SB, Crowley WF, Ojeda SR, Plant TM. Increased hypothalamic GPR54 signaling: a potential mechanism for initiation of puberty in primates. Proc Natl Acad Sci USA. 2005;102:2129–2134. doi: 10.1073/pnas.0409822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JK, Grewal SS, Letcher A, Bill DJ, Dourisch CT. Behavioural and pharmacological characterisation of the elevated “zero-maze” as an animal model of anxiety. Psychopharmacology (Berl) 1994;116:56–64. doi: 10.1007/BF02244871. [DOI] [PubMed] [Google Scholar]
- Shimamura K, Hartigan DJ, Martinez S, Puelles L, Rubenstein JLR. Longitudinal organization of the anterior neural plate and neural tube. Development. 1995;121:3923–3933. doi: 10.1242/dev.121.12.3923. [DOI] [PubMed] [Google Scholar]
- Silverman A-J, Livne I, Witkin JW. The gonadotropin-releasing hormone (GnRH), neuronal systems: immunocytochemistry and in situ hybridization. In: Knobil E, Neill JD, editors. The physiology of reproduction. Ed 2. Vol 1. New York: Raven; 1994. pp. 1683–1709. [Google Scholar]
- Simmons DM, Arriza JL, Swanson LW. A complete protocol for in situ hybridization of messenger RNAs in brain and other tissues with radiolabeled single-stranded RNA probes. J Histotechnol. 1989;12:169–181. [Google Scholar]
- Siniscalchi A, Rodi D, Cavallini S, Marino S, Ferraro L, Beani L, Bianchi C. Effects of cholecystokinin tetrapeptide (CCK(4)) and of anxiolytic drugs on GABA outflow from the cerebral cortex of freely moving rats. Neurochem Int. 2003;42:87–92. doi: 10.1016/s0197-0186(02)00052-9. [DOI] [PubMed] [Google Scholar]
- Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology. 2005;146:3686–3692. doi: 10.1210/en.2005-0488. [DOI] [PubMed] [Google Scholar]
- Sobreviela T, Clary DO, Reichardt LF, Brandabur MM, Kordower JH, Mufson EJ. TrkA-immunoreactive profiles in the central nervous system: colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J Comp Neurol. 1994;350:587–611. doi: 10.1002/cne.903500407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son YJ, Hur MK, Ryu BJ, Park SK, Damante G, D'Elia AV, Costa ME, Ojeda SR, Lee BJ. TTF-1, a homeodomain-containing transcription factor, participates in the control of body fluid homeostasis by regulating angiotensinogen gene transcription in the rat subfornical organ. J Clin Invest. 2002;278:27043–27052. doi: 10.1074/jbc.M303157200. [DOI] [PubMed] [Google Scholar]
- Stiff ME, Bronson FH, Stetson MH. Plasma gonadotropins in prenatal and prepubertal female mice: disorganization of pubertal cycles in the absence of a male. Endocrinology. 1974;94:492–496. doi: 10.1210/endo-94-2-492. [DOI] [PubMed] [Google Scholar]
- Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- Takuma N, Sheng HZ, Furuta Y, Ward JM, Sharma K, Hogan BL, Pfaff SL, Westphal H, Kimura S, Mahon KA. Formation of Rathke's pouch requires dual induction from the diencephalon. Development. 1998;125:4835–4840. doi: 10.1242/dev.125.23.4835. [DOI] [PubMed] [Google Scholar]
- Watanabe G, Terasawa E. In vivo release of luteinizing hormone releasing hormone increases with puberty in the female rhesus monkey. Endocrinology. 1989;125:92–99. doi: 10.1210/endo-125-1-92. [DOI] [PubMed] [Google Scholar]
- Whitlock KE. Origin and development of GnRH neurons. Trends Endocrinol Metab. 2005;16:145–151. doi: 10.1016/j.tem.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Wilson RC, Vacek T, Lanier DL, Dewsbury DA. Open-field behavior in muroid rodents. Behav Biol. 1976;17:495–506. doi: 10.1016/s0091-6773(76)90901-9. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF. Ablation of NF1 function in neuron induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]