

Abstract
KCNQ (KV7) potassium channels underlie subthreshold M-currents that stabilize the neuronal resting potential and prevent repetitive firing of action potentials. Here, antibodies against four different KCNQ2 and KCNQ3 polypeptide epitopes show these subunits concentrated at the axonal initial segment (AIS) and node of Ranvier. AIS concentration of KCNQ2 and KCNQ3, like that of voltage-gated sodium (NaV) channels, is abolished in ankyrin-G knock-out mice. A short motif, common to KCNQ2 and KCNQ3, mediates both in vivo ankyrin-G interaction and retention of the subunits at the AIS. This KCNQ2/KCNQ3 motif is nearly identical to the sequence on NaV α subunits that serves these functions. All identified NaV and KCNQ genes of worms, insects, and molluscs lack the ankyrin-G binding motif. In contrast, vertebrate orthologs of NaV α subunits, KCNQ2, and KCNQ3 (including from bony fish, birds, and mammals) all possess the motif. Thus, concerted ankyrin-G interaction with KCNQ and NaV channels appears to have arisen through convergent molecular evolution, after the division between invertebrate and vertebrate lineages, but before the appearance of the last common jawed vertebrate ancestor. This includes the historical period when myelin also evolved.
Keywords: action potential, axon initial segment, node of Ranvier, neuromodulation, epilepsy, M-current
Introduction
Although classic biophysical studies provide a general model for vertebrate action potential (AP) initiation and propagation (Coombs et al., 1957; Frankenhaeuser and Huxley, 1964), questions remain regarding mechanisms underlying these important processes in mammals. It is agreed that mammalian spikes initiate in the proximal axon (Stuart et al., 1997), but the precise site of initiation and mechanisms regulating threshold there are incompletely defined. The APs of mammalian peripheral myelinated axons lack the prominent “delayed rectifier” current that mediates repolarization and afterhyperpolarization in squid and frog fibers (Chiu et al., 1979). Instead, mammalian axons exhibit a diversity of smaller K+ conductances (Vogel and Schwarz, 1995). Except for juxtaparanodal Kv1.1/Kv1.2 channels, the molecular identities and distinctive functions of these mammalian K+ channels are uncertain (Scherer and Arroyo, 2002).
Biophysical considerations and studies of human disease suggest that these mammalian axonal K+ currents are important. Because K+ conductances shunt excitatory current, K+ channels concentrated near the proximal axonal spike initiation site could powerfully influence the AP threshold (Howard et al., 2005). Similarly, K+ conductances further along the axon are thought to be important for preventing aberrant triggering of APs at more distal nodes (Schwarz et al., 1995). Indeed, mutations in the juxtaparanodal subunit Kv1.1 cause spontaneous epileptic seizures, bouts of uncoordinated movement, and aberrant APs that arise spontaneously in motoneuron axons (myokymia), both in humans and transgenic mice (Adelman et al., 1995; Smart et al., 1998; Zhou et al., 1999; Herson et al., 2003).
Mutations in KCNQ2 and KCNQ3 (Kv7.2 and Kv7.3) also cause seizures and (in the case of KCNQ2) myokymia in humans (Dedek et al., 2001; Singh et al., 2003). Both homomeric KCNQ2 and heteromeric KCNQ2/KCNQ3 channels mediate M-current (IM), a K+ current that activates with slow kinetics at voltages above about −60 mV (Brown and Adams, 1980; Wang et al., 1998; Shapiro et al., 2000; Hadley et al., 2003). KCNQ channels are molecular targets of flupirtine, a marketed nonopioid analgesic, retigabine, an antiepileptic agent in current clinical trials, and of many earlier-stage compounds in development for seizures or pain (Gribkoff, 2003).
Using immunohistochemistry, Devaux et al. (2004) showed that KCNQ2 was precisely colocalized with NaV channels at nodes of Ranvier. Devaux et al. noted similarities between the kinetic and pharmacological properties of KCNQ currents and those of IKs, a slow, low-voltage activated K+ current recorded at mammalian nodes (Röper and Schwarz, 1989). Neuronal axon initial segments (AISs) in spinal cord and brain also stained strongly for KCNQ2 and, in a subset of cells, for KCNQ3. This AIS localization was intriguing, given the subthreshold activation voltage of KCNQ channels, and the above-mentioned role of the proximal axon in AP initiation. Interaction with the large adaptor protein ankyrin-G is the mechanism for selective retention of NaV channels at the AIS plasmalemma (Garrido et al., 2003; Lemaillet et al., 2003). Here, we provide evidence that ankyrin-G interaction also mediates the AIS localization of KCNQ channels. Phylogenetic analysis suggests this shared mechanism for Na+ and K+ channel concentration at the AIS arose by convergent evolution, before the last common ancestor of jawed vertebrate species appeared.
Materials and Methods
Antibodies.
All anti-KCNQ antibodies were raised against synthetic peptides and affinity-purified against the immunogens before use. Rabbit anti-KCNQ2-N (Cooper et al., 2001) and guinea pig anti-KCNQ2-C (Cooper et al., 2000) antibodies were described previously. KCNQ2-N is directed against residues 13-37 from the intracellular N-terminal region of KCNQ2, which appear to be absolutely conserved in mammals (Singh et al., 1998; Pan et al., 2001). The KCNQ2-C immunogen [KSKSGLAFRKDPPPEPSPSKGSPc; termed “KCNQ2C1” in Cooper et al. (2000) and representing residues 397–416 in the splice form described by Singh et al. (1998)] is in a variably spliced region but is highly conserved among mammalian KCNQ2 orthologs. Guinea pig anti-KCNQ3b-N antibodies (Devaux et al., 2004) are directed against residues 36–57 (AGDEERKVGLAPGDVEQVTLALc) from the N-terminal region of splice isoforms (“KCNQ3b”) of KCNQ3 using the longer first exon (Schroeder et al., 1998); this antibody does not recognize splice isoforms (“KCNQ3a”) that include the shorter first exon (Wang et al., 1998). Rabbit anti-KCNQ3-C (Cooper et al., 2000) detects residues 578-604 (STPKHKKSQKGSAFTFPSQQSPRNEPYc) of human KCNQ3 (Schroeder et al., 1998); this peptide is absolutely conserved in mouse (but possibly not in rat; see GenBank AF087454.2). The sodium channel antibody used was PanNaV (clone K58/35; Sigma, St. Louis, MO), a mouse monoclonal antibody against a peptide highly conserved in all voltage-gated sodium channel isoforms (Rasband et al., 1998). Immunodetection of hemagglutinin epitope (HA)-tagged constructs was performed using rat monoclonal (Roche, Basel, Switzerland) or mouse monoclonal (Covance, Princeton, NJ) antibodies. Secondary antibodies, highly purified to minimize cross-reactivity, were from Jackson ImmunoResearch (West Grove, PA).
cDNA constructs.
Generation of cDNAs encoding full-length human KCNQ2 and KCNQ3b (Thomas Jentsch, University of Hamburg, Hamburg, Germany), rat KCNQ3a (David McKinnon, SUNY at Stony Brook, Stony Brook, NY), and human KCNQ5 (Klaus Steinmeyer, Aventis, Frankfurt am Main, Germany) have been described previously (Biervert et al., 1998; Schroeder et al., 1998; Wang et al., 1998; Lerche et al., 2000). Generation of C-terminal green fluorescent protein (GFP)-tagged ankyrin-G (ankyrin-G-GFP) and N-terminal HA-tagged neurofascin (HA-NF) has been described previously (Garver et al., 1997; Zhang et al., 1998). All constructs were subcloned into either pcDNA3 (Invitrogen, San Diego, CA) or pEGFP-N1 (Clontech, Mountain View, CA), and bidirectionally sequenced before experimental use.
The spectrin-binding, serine-rich, and tail domains of full-length ankyrin-G-GFP were deleted through inverted PCR using pfu polymerase (Stratagene) to obtain AnkG-MB-GFP, which contains only the ∼800-aa-long membrane-binding domain (Zhang and Bennett, 1998). Neurofascin fusion constructs were derived from construct HA-NF. Cytoplasmic domain-deleted neurofascin, HA-NF-DelC, was made by inverted PCR with pfu polymerase. To generate fusion proteins, we exploited a SpeI site introduced immediately upstream to the HA-NF-DelC stop codon. For HA-NF-Q2C, a cDNA encoding amino acids 570-844 of KCNQ2 was PCR-amplified to introduce a SpeI cut site at 5′ end and a NotI site at 3′ end. This was subcloned using the SpeI–NotI sites in HA-NF-DelC. A cDNA encoding amino acids 577-872 of rat KCNQ3a was PCR-amplified with introduction of a 5′ ApaI cut site and a 3′ NotI site; this piece was in-frame subcloned into ApaI–NotI sites of HA-NF-Delc to make HA-NF-Q3C. Similarly, a cDNA encoding residues 579-932 of KCNQ5 was PCR-amplified with the introduction of 5′ ApaI and 3′ NotI sites, and subcloned to make HA-NF-Q5C. To test the function of the putative KCNQ2/KCNQ3 ankyrin-G interaction motifs, we used PCR with pfu polymerase to generate mutant full-length or fusion protein constructs in which the residues ESD (810-812 of KCNQ2) or ETD (827–829 of KCNQ3) within the motifs were mutated to AAA. The full-length subunit constructs with ankyrin-G interaction motif mutations are referred to herein as KCNQ2(AAA) and KCNQ3(AAA); fusion protein constructs with these mutations are called HA-NF-Q2C(AAA) and HA-NF-Q3C(AAA).
Immunohistochemistry.
All procedures involving rodents were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Mice were deeply anesthetized with halothane and killed by decapitation. Brains were rapidly dissected, blocked, and embedded using OTC (Sigma). Cryostat sections were cut at 10–20 μm, transferred to precooled SuperfrostPlus slides (Fisher Scientific, Houston, TX), and stored at −20°C overnight or until used. Sciatic nerves were dissected and placed in cold PBS solution. Fibers were teased using fine needles, transferred to slides, and allowed to air dry. Nerves were then stored at −20°C overnight or until used.
Antibody immunoreactions on tissue sections or nerves were performed essentially as described previously (Devaux et al., 2004). Briefly, specimens were permeabilized and extracted, and nonspecific binding sites blocked, by incubation with Tris-buffered saline solution containing 0.5% Triton X-100 and 5% fish skin gelatin for 1 h. Nerves were then incubated with primary antibodies in blocking buffer (with 0.2% Triton X-100) for 15–18 h, washed, incubated with secondary antibodies for 2 h, washed, counterstained with the nucleic acid-binding dye 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Invitrogen), and coverslipped using ProLong antifade reagent (Invitrogen). For all antibody combinations, single-label and secondary-only control samples were processed in parallel with multilabel samples; these revealed no evidence of antibody cross-reactivity.
Cultured transfected hippocampal neurons on coverslips were fixed with 4% paraformaldehyde in PBS and blocked with 4% nonfat milk (in PBS). Surface expression of HA-NF chimeric proteins was then detected with mouse monoclonal anti-HA antibodies (Covance). Cells were then permeabilized (0.2% Triton X-100 in PBS), blocked again, and reacted with antibodies against microtubule associated protein-2 (MAP2) (rabbit polyclonal; Covance), Na channels (PanNaV; Sigma), and endogenous KCNQ2 and KCNQ3 subunits.
Wide-field microscopy: image acquisition and analysis.
Immunolocalization experiments were performed using a Nikon (Melville, NY) TE2000 microscope equipped for epifluorescence and differential interference contrast (DIC), using 4-40× dry and 60×, 1.4 numerical aperture (NA) oil immersion objectives, a z-axis motor with feedback probe (Prior Scientific, Cambridge, UK), and a Spot KE Slider cooled CCD camera (Diagnostic Instruments, Sterling Heights, MI) under the control of Image-Pro Plus 5.0 software (Media Cybernetics, Silver Spring, MD).
In some experiments, as noted below, the subcellular localization or colocalization of proteins was assayed using wide-field deconvolution microscopy (Scriven et al., 2000; Swedlow and Platani, 2002). Images were acquired using the 60× objective and a 1.5× projection lens, giving pixel x–y dimensions of 82 × 82 nm. Coregistration of color channel data in each image axis was established using latex fluorospheres (Invitrogen). For each sample and color channel, 60-90 images were acquired at 0.27 μm z-step intervals. Deconvolution was performed using 10-40 iterations of a blind deconvolution algorithm in AutoDeblur software (AutoQuant, Troy, NY). Where indicated in Results, images showing maximal intensity projections of two to four deconvolved optical sections were generated using the AutoVisualize software plug-in (AutoQuant).
Subcellular redistribution assays.
Methods and theory for this assay have been described previously in detail (Zhang and Bennett, 1998; Zhang et al., 1998). Human embryonic kidney (HEK) 293 cells were cultured in 10% fetal bovine serum in DMEM, and transfected using FuGENE 6 (Roche) following the manufacturer’s protocol. Transfections were performed with pairs of constructs: efficiently membrane-targeted “bait” constructs (including the HA-tagged ectomembrane and transmembrane domains of neurofascin and, where applicable, intracellular portions of KCNQ subunits) and “reporters” consisting of either GFP-tagged full-length ankyrin-G or the ∼800-aa-long, ankyrin-G “membrane-binding” domain (AnkG-MB-GFP) (Zhang et al., 1998). Two days after transfection, cells were washed three times with PBS, and then fixed in freshly diluted 3.5% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for 20 min. Cells were washed three times, blocked (10% normal goat serum, 2% bovine serum albumin in PBS) for 5 min, and then reacted with rat monoclonal anti-HA antibodies (Roche). Cells were washed three times with PBS then incubated with Cy3-conjugated anti-rat IgG antibodies.
Preliminary results in this assay were similar for the full-length ankyrin-G-GFP and AnkG-MB-GFP, but the redistribution of GFP fluorescence to the membrane and clearing of the cytoplasm and nuclei was generally more robust when the smaller AnkG-MB-GFP was used. Because results with full-length ankyrin-G could be confounded by competing interactions between, for example, the native HEK 293 cell cytoskeleton and the spectrin-binding domains, we systematically analyzed and present here results using the AnkG-MB-GFP construct only.
For statistical analysis, we surveyed coverslips for anti-HA-positive cells (i.e., expressing neurofascin or its fusion constructs), and then examined the distribution of green fluorescence. Cells were classified as either possessing or lacking an easily detected cytoplasmic clearing and a surface “edge” in green fluorescence that colocalized with anti-HA staining, representative of membrane-associated AnkG-MB-GFP. Nuclei were labeled with DAPI, allowing identification and exclusion from analysis of smaller cells lacking a clearly defined cytoplasmic compartment. For each condition, we tabulated results for 100 cells/condition/experiment; data shown are average (±SEM) of three separate transfection experiments.
Fluorescence recovery after photobleaching assay for ankyrin-G membrane-binding domain immobilization.
The rationale of the assay is that proteins that are integral to or associated with the membranes of intracellular organelles or the plasmalemma are less mobile than soluble proteins freely diffusing in the cytoplasm (Lippincott-Schwartz et al., 2003). Therefore, interaction between a cytoplasmic, fluorescently tagged reporter protein and the cytoplasm-facing domain of a transfected membrane protein may be detectable as a reduction in mobility of the reporter measurable by a reduced rate of fluorescence recovery after photobleaching (FRAP), and/or an increase in the immobilized fraction. Because the endodomains of proteins destined for delivery to the plasmalemma (such as ion channels) always face the cytoplasmic compartment, the FRAP immobilization assay allows analysis of interaction with intracellular as well as surface-localized proteins. Important assumptions include that competing interactions of the reporter do not dominate, that the binding partners under investigation are truly accessible to each other, and that the concentrations of the binding partners and the interaction kinetics under investigation allow the “bound” fraction signal to be detected.
AnkG-MB-GFP was chosen as the reporter after preliminary experiments in which the mobility of AnkG-MB-GFP and full-length ankyrin-G-GFP constructs were compared, expressed alone without integral membrane protein binding partners. AnkG-MB-GFP exhibited faster and more complete FRAP than full-length ankyrin-G-GFP. This result was as expected based on the size difference between the two constructs and previous results (Zhang et al., 1998; Lippincott-Schwartz et al., 2003). Because the informative signal in our immobilization assay is the difference in FRAP between the more mobile, cytoplasmic ankyrin-G-based reporter and the less mobile, coexpressed integral membrane proteins, AnkG-MB-GFP was used for subsequent experiments.
Cells were transfected with the following constructs: AnkG-MB-GFP alone; AnkG-MB-GFP, and human KCNQ2; AnkG-MB-GFP, human KCNQ2, and rat KCNQ3; or AnkG-MB-GFP, KCNQ2(AAA), and KCNQ3(AAA). Experiments was performed at room temperature on living HEK 293 cells, using an inverted confocal microscope (FV1000 instrument and FV10-ASW software; Olympus Optical, Tokyo, Japan), fitted with a 60×, 1.4 NA oil immersion objective lens, a 25 mW/405 nm violet light emitting diode laser for regional photobleaching, and a 10 mW visible light argon laser for sample illumination/image collection. For each condition, AnkG-MB-GFP and total channel subunit plasmids were transfected at a 1:1 molar ratio. Individual cells exhibiting medium-intensity, homogeneous-appearing green fluorescence were selected for FRAP. Extensive preliminary experiments were used to optimize laser power, duration of photobleaching, and duration of experimental trials. GFP fluorescence was monitored (1 s intervals; 434 ms duration) using the 488 nm laser line (0.4% excitation power). For the experiments shown, baseline data were collected for 4950 ms, bleaching (10% power setting, 405 nm) was performed for 50 ms on the region of interest (ROI) (a 2.5 μm diameter circle), and recovery was monitored over an additional 55 s.
Images collected included the entire cell, and only cells that showed no detectable changes in shape or position during the experiment were further analyzed. To determine FRAP curves (see Fig. 5B), intensity in the UV-laser bleached ROI was first normalized to a second “reference region” of equal size and average initial intensity, to control for bleaching resulting from image collection alone (typically, ∼10%). Recovery was calculated according to the following formula (Tanimura et al., 2003): % Recovery = [rI(t) − rI(post)]/[rI(pre) − rI(post)], where rI = (Ib − Ir), Ib is the average fluorescence intensity of the ROI, Ir is the intensity of the reference region, rI(pre) is the initial (prebleach) relative intensity, rI(post) is the postbleach relative intensity, and rI(t) is the relative intensity at each time point. The final fluorescence intensity of the bleached ROI generally did not reach the normalized final values of the reference region. The difference between 100% and the observed percent recovery in the bleached ROI represents the “immobile fraction” in FRAP analyses (Lippincott-Schwartz et al., 2003). The recovery time constants were calculated by least-squares regression using the following equation: % Recovery = A[1 − exp(−t/τ)], where τ is the time constant and A is the mobile fraction.
Figure 5.

Functional KCNQ2 and KCNQ2/KCNQ3 tetramers interact with ankyrin-G. A, B, FRAP measurement of AnkG-MB-GFP mobility. AnkG-MB-GFP fluorescence in a bleached region recovers quickly when it is expressed alone or coexpressed with KCNQ2/KCNQ3 channels with mutated C3 motifs, but more slowly when coexpressed with either wild-type heteromeric or KCNQ2 homomeric channels. A, Images of GFP fluorescence during the FRAP experiments. The arrows show the bleached region (see Materials and Methods) at indicated times before or after exposure to a high-intensity bleaching light. B, Averaged recovery time courses and bar graph showing immobile fraction for each transection condition (±SEM). C, Western blots show that mutant and wild-type channels are expressed at equal protein levels. Top, Representative blots; bottom, average densitometry results (±SEM). Scale bar, 2.5 μm.
Western blot analysis.
Western blots were performed using HEK 293 cells transfected using conditions identical to those used for FRAP. Two days after transfection, cells were washed with PBS, and then lysed in buffer containing 1% Triton, 1× Complete (Roche) protease inhibitor mixture, and the following (in mm): 40 Tris-HCl, 150 NaCl, and 1 DTT. The lysate was incubated on ice for 30 min and centrifuged at 16,000 × g for 30 min at 4°C. Proteins in the supernatant were denatured in SDS sample buffer, subjected to PAGE and electrotransfer, and blotted using antibodies KCNQ2-N and KCNQ3-C. Westerns were developed using enhanced chemiluminescence reagents and preflashed film (Amersham Biosciences, Arlington Heights, IL). Filters used for channel subunit immunoblots were subsequently reprobed using mouse anti-tubulin antibodies (Sigma). Film images were digitized with an Epson 3400 Perfection scanner. Densitometry was performed using ImageQuant software (Molecular Dynamics, Sunnyvale, CA), and data were exported to Excel for analysis. For each channel immunoblot band, total intensity was normalized to that of the associated tubulin band.
Hippocampal neuronal lipid-mediated transfection and electroporation.
Hippocampal neurons were dissociated from brains of embryonic day 18 (E18) Sprague Dawley rats and cultured in NeuroBasal medium, by methods previously described in detail (Buchhalter and Dichter, 1991). Two methods of introducing cDNAs into hippocampal neurons were used. When the experimental aims required neuronal immunostaining <7–10 d after transfection, we performed lipid-mediated transfection (Lipofectamine 2000; Invitrogen) of neurons that had been cultured on coverslips for 7–10 d before transfection. When immunostaining was planned for >7–10 d after transfection, cDNAs were introduced at the time of dissociation by electroporation, using a rat neuron nucleofector system (Amaxa, Gaithersburg, MD) following the manufacturer’s protocol (Program O-03), and 2 μg plasmid per condition. Neurons were then plated at low density (100,000/dish) under serum-free conditions. The nucleofector system gave much higher transfection efficiencies (50%) than lipid mediated transfection, but cells were too fragile and axons too immature to allow analysis during the first days after nucleofection/plating; therefore, both approaches were used.
To test the detergent resistance of HA-tagged fusion proteins expressed in cultured neurons, a protocol modified after Winckler et al. (1999) was used. Coverslips plated with transfected neurons were washed three times quickly with PBS, incubated with mouse anti-HA antibodies (1:1000) in PBS for 30 min at room temperature, and washed again with PBS. The neurons were then incubated in extraction buffer (30 mm PIPES, 1 mm MgCl2, 5 mm EDTA, 0.5% Triton X-100) for 5 min at 37°C, washed three times quickly with PBS, and fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. After three additional PBS washes, nonspecific sites were blocked by incubation with 4% nonfat dry milk for 5 min in PBS. Then, cells were incubated with chicken anti-MAP2 or rabbit anti-AnkG antibodies in PBS with 4% milk for 1 h at room temperature. Finally, cells were washed, incubated with secondary antibodies, washed, counterstained with DAPI, and mounted on slides in ProLong (Invitrogen).
Immunostaining of fixed neurons was performed as described for HEK 293 cells above, with several modifications. Neurons were fixed (4% formaldehyde in PBS), and then blocked (4% nonfat dry milk in PBS, no detergent). Cells were incubated sequentially with mouse monoclonal anti-HA antibodies (HA1.11; 1:1000; Covance) and Cy3-conjugated anti-mouse IgG to detect surface expressed HA-tagged chimeric proteins. Cells were then permeabilized (0.2% Triton X-100 in PBS) and incubated with anti-MAP2 antibodies (chicken polyclonal; 1:10,000; Covance) and FITC-conjugated anti-chicken IgG. Under FITC illumination, 12-mm-diameter coverslips were systematically surveyed for neurons, identified by morphology of MAP2 staining. In many cases, axons and AISs were readily identifiable by their characteristic thinner profile and low, but detectable MAP2 staining compared with that of dendrites. Identified neurons were then examined for Cy3 (anti-HA) staining. The data are expressed as the number of cells with anti-HA concentration at proximal axons per 100 MAP2-positive neurons.
Electrophysiology.
HEK 293 cells were cotransfected with KCNQ2 and KCNQ3, or KCNQ2(AAA) and KCNQ3(AAA), with or without full-length ankyrin-G-GFP using FuGENE 6. Full-length ankyrin-G-GFP was used for electrophysiological experiments, rather than AnkG-MB-GFP, to more closely mimic in vivo conditions. Whenever cotransfected, ankyrin-G-GFP and total KCNQ (KCNQ2 alone or KCNQ2 and KCNQ3) plasmids were used at equimolar concentrations. When ankyrin-G-GFP was coexpressed, this served as a marker for selection of individual cells for recording. Otherwise, pEGFP-C1 (Clontech), cotransfected at a ratio of 1:20 compared with channel subunits, was used for this purpose. Whole-cell patch-clamp recordings were performed 2–3 d after transfection, on well isolated green fluorescent cells. Patch pipettes (1.5–3.0 MΩ) were filled with the following (in mm): 120 KCl, 5.4 CaCl2, 1.8 MgCl2, 10 HEPES, 10 EGTA, 1.6 Mg-ATP, adjusted to pH of 7.2 with KOH. External solution contained the following (in mm): 4 KCl, 140 NaCl, 2 CaCl2, 1.0 MgCl2, and 10 HEPES, pH 7.4. Only cells with access resistances <10 MΩ were used for recordings. The setup consisted of an Axopatch 200A amplifier, Digidata 1322A (Molecular Devices, Foster City, CA) interface, Nikon TE300 inverted microscope, and Sutter MPC-285 micromanipulator (Sutter Instrument, Novato, CA). Data were acquired (5 kHz) and filtered at 2 kHz using pCLAMP 9.0 software (Molecular Devices) running on a Hewlett-Packard (Palo Alto, CA) personal computer. Capacitance and access resistance were compensated to 90%. All measurements were obtained at room temperature (∼22°C).
To evaluate deactivation kinetics (see Fig. 6Ai,Bi), cells were held at −80 mV for 1 s, depolarized to 0 mV for 1.5 s, and then repolarized to test voltages between −80 mV and 0 mV in 10 mV increments for 1.5 s. To determine activation kinetics (see Fig. 6Aii,Biii), cells were held at −80 mV, and then stepped to test potentials between −80 mV and +50 mV for 1.5 s. Tail current amplitudes were measured at −60 mV from the decaying time course of deactivating current as the difference between the average of a 10 ms segment, taken 10–20 ms into the hyperpolarizing step, and normalized to values obtained at +50 mV. For all protocols, a 17 s interpulse interval at −80 mV was used. Data are represented as mean ± SEM. Voltage dependence was fitted by the Boltzmann equation: I/I(50) = 1/(1 + exp(V0.5 − Vm)/k), where I is the tail current recorded at −60 mV after a step to the membrane potential (Vm), I(50) is the current after a step to +50 mV, V0.5 is the membrane potential at which I is equal to I(50)/2, and k is the slope factor. Clampfit 9.0 (Molecular Devices) was used for fitting steady-state activation and deactivation curves. Monoexponential fitting was performed both for activation and deactivation kinetics.
Figure 6.

Expression of ankyrin-G has very small effects on coexpressed KCNQ2/KCNQ3 channels. A, Representative current traces in response to pulse protocols (shown in insets) used for kinetic analysis: i, deactivation protocol; ii, activation protocol. B, Analysis of voltage gating of wild-type KCNQ2/KCNQ3 heteromeric channels, C3 motif mutants, and wild-type channels coexpressed with ankyrin-G-GFP: i, deactivation time constants; ii, conductance/voltage curves; iii, activation time constants. The kinetics of the three conditions is very similar, with some evidence for slight slowing of activation and deactivation gating in the presence of ankyrin-G. Steady-state voltage dependence of gating of wild-type and mutant are indistinguishable, but ankyrin-G confers a ∼6 mV shift toward depolarized voltages.
Phylogenetic analysis.
We searched National Center for Biotechnology Information (NCBI) protein and genome databases to identify all KCNQ and NaV sequences of invertebrates and vertebrates. First, the full-length KCNQ cDNAs were identified by BLAST searching NCBI databases, using the tBLASTn program and a very highly conserved KCNQ channel peptide sequence (KRKFKESLRPYDVMDVIEQYSAGH, located in the C2 region) (Fig. 2) that includes part of the subunit interaction domain (Schwake et al. 2003). Similarly, NaV clones were identified by tBLASTn searches using the PanNaV immunogen peptide (TEEQKKYYNAM-KKLGSKK), which corresponds to a highly conserved portion of the intracellular linker between homology domains III and IV, responsible for fast inactivation (Stuhmer et al., 1989; Rasband et al., 1998). Sequence alignments of KCNQ and NaV polypeptides were performed using ClustalW, version 1.5, implemented in DS Gene 1.5 (Accelyrs, San Diego, CA). Alignment parameters were as follows: (1) for pairwise alignment: matrix, BLOSUM; open gap penalty, 10; extend gap penalty, 0.1; (2) for multiple alignment: matrix, BLOSUM; gap penalty, 10; extend gap penalty, 0.05; delay divergent (%), 40. After alignment, the phylogenetic tree of NaV and KCNQ member subunits from invertebrates and vertebrates was constructed using DS Gene, using neighbor joining as the tree-building method.
Figure 2.

KCNQ2 and KCNQ3 polypeptides share a homologous domain near their C termini, harboring a candidate ankyrin-G binding motif. A, Topology map of KCNQ2. Transmembrane domains, the subunit interaction domain (SID), and sites for interaction with PIP2, calmodulin (CM), and A-kinase anchoring protein 79/150 (AKAP79/150) are indicated in the proximal portion of the large C-terminal intracellular portion of the polypeptide. The C3 domain (boxed) is at the distal C-terminal region. B, Alignment of functional domains of five human KCNQ subunits, with residue number positions of transmembrane (6 TM), and highly conserved C-terminal regions (C1, C2/SID, and C3) regions indicated. Only KCNQ2 and KCNQ3 have C3 domains. C, The C3 domains contain a putative ankyrin-G binding loop motif. Clustal alignment of KCNQ2, KCNQ3, and KCNQ5 shows high homology of KCNQ2 and KCNQ3 within the C3 domain. Within C3, the arrows identify the beginning and end of the sequence with high homology to the previously identified NaV channel ankyrin-G binding motif. At positions within the alignment where two or more residues are identical, the background is shown as black. At positions where two or more sequences show conservative substitutions, the background is shown as light gray.
Results
KCNQ2 and KCNQ3 are colocalized at neuronal axon initial segments and nodes of Ranvier of many neurons in the mouse
Recently, Devaux et al. (2004) reported that KCNQ2 immunoreactivity was a ubiquitous feature at central and peripheral nodes and AISs. Devaux et al. found KCNQ3 in only a subpopulation of nodes, and at the AISs of only some neuronal types in rat brain and spinal cord. This was surprising for two reasons. First, it is well established that heterologous coexpression of KCNQ2 and KCNQ3 subunits results in much larger currents compared with those resulting from expression of KCNQ2 alone (Schroeder et al., 1998; Yang et al., 1998; Schwake et al., 2000; Selyanko et al., 2001; Surti et al., 2005). In addition, native KCNQ channels in superior cervical ganglion, hippocampal, and striatal medium spiny neurons appear to be predominantly KCNQ2/KCNQ3 heterotetramers (Wang et al., 1998; Selyanko et al., 2002; Shah et al., 2002; Shen et al., 2005). Nonetheless, some recent evidence points to expression of KCNQ2 homotetramers by some neurons (Saganich et al., 2001; Hadley et al., 2003).
To further assess this, we investigated the possibility that our previously reported results reflected differential expression of KCNQ3 splice isoforms. We found cDNA clones in GenBank indicating that two different KCNQ3 isoforms, previously reported but never subjected to comparative study, are expressed in mammalian brain (Schroeder et al., 1998; Wang et al., 1998). The initial portions of the N-terminal intracellular domains of these isoforms are completely distinctive, apparently because of the use of alternative first exons. We called these isoforms, containing shorter and longer N-terminal regions, KCNQ3a and KCNQ3b, respectively. Because the antibody used by Devaux et al. (2004) was specific for KCNQ3b, we reanalyzed the distribution of KCNQ3 immunoreactivity in mouse peripheral nerve, spinal cord, and brain, using both anti-KCNQ3b-N and an antibody [anti-KCNQ3-C (Cooper et al., 2000)] directed against a C-terminal epitope that is conserved in known mouse KCNQ3 variants (Fig. 1). Anti-KCNQ3-C antibodies strongly labeled narrow ∼1 μm patches scattered along teased fibers of mouse sciatic nerve (Fig. 1A). We colabeled fibers with anti-KCNQ3-C and monoclonal antibodies against Kv1.2, which is localized to the juxtaparanodes (Wang et al., 1993). Kv1.2 staining symmetrically flanked the KCNQ3-C-stained patches (Fig. 1A,B), and superimposition of DIC and immunofluorescence images revealed that the KCNQ3-C stain was precisely localized at nodes (Fig. 1B). The intensity of KCNQ3-C staining at sciatic nerve nodes was strikingly variable, unlike the results previously reported for KCNQ2 (Devaux et al., 2004). Thus, some nodes exhibited strong KCNQ3-C staining, whereas at other nodes, KCNQ3-C staining was weak or absent (Fig. 1B, insets). Superimposition of DIC and fluorescence images of nodes colabeled for KCNQ3-C and NaV channels revealed that staining of both antibodies was concentrated at the node (Fig. 1C). The nodal immunostaining for both channel types frequently appeared as parallel lines in high magnification images (Fig. 1C), suggesting that the channels were present at the nodal membrane. Such “near-membrane” nodal staining was previously documented for KCNQ2 by confocal imaging (Devaux et al., 2004). Efforts to use postembedding immunogold methods to localize the KCNQ subunits ultrastructurally proved unsuccessful (see Discussion). To obtain optimal light-level localization of the nodal KCNQ2 and KCNQ3 immunostaining, we processed Z-stacks of high-magnification immunofluorescence images through a deconvolution algorithm. Deconvolved optical sections through the node showed that the two subunits were tightly colocalized, primarily in linear bands likely representing the nodal membrane (KCNQ2-C and KCNQ3-C antibodies) (Fig. 1D).
Figure 1.

KCNQ2, KCNQ3, and NaV channels are concentrated at the node of Ranvier and AIS. A, Teased sciatic nerve fibers show staining for KCNQ3 at nodes of Ranvier. The KCNQ3-C immunostain (red) is located in patches, flanked by staining for Kv1.2 (green), a marker of the juxtaparanodes. B, Superimposed DIC and immunofluorescence images show several teased sciatic nerve fibers, including two nodes of Ranvier (dashed boxed regions in main image). As indicated, the Kv1.2 antibody (green in main image, bottom panels in insets) labels both juxtaparanodes (jpn) strongly. KCNQ3-C antibodies (red in main image, top panels in insets) label the upper node strongly but show only a tiny area of weak staining at the lower node. C, Single teased fiber showing colocalized KCNQ3-C (red) and NaV (green) staining at a node (n). The paranode (p) is partially demyelinated, as seen in the DIC image. D, Longitudinal optical z-section through the center of a single sciatic nerve fiber stained using KCNQ2-C (green) and KCNQ3-C (red) antibodies, obtained by wide-field deconvolution microscopy (see Materials and Methods). Both antibodies show strong colabeling at the membrane of node of Ranvier, but only KCNQ3-C shows light additional staining of myelin. The bottom panels show the nodal region at higher magnification. E, Strong colabeling at AIS of a large motoneuron in the spinal cord ventral horn by KCNQ2-C (red) and KCNQ3-C (green). F, A Purkinje cell AIS is intensely colabeled by NaV (red) and KCNQ3-C (green) antibodies. KCNQ3-C also stains the nucleus of this cell type (pc nuc). Cerebellar cortical layers (mol, molecular; pc, Purkinje cell; gc, granule cell) are indicated. G, Colabeling of cerebellar white matter nodes of Ranvier by NaV (red) and KCNQ3-C (green) antibodies, one with ring appearance. H, The diagram at the left shows the arrangement of somata, dendrites, and axons in hippocampal area CA1. Pyramidal cells, with somata clustered in the stratum pyrimidale (pyr) and apical dendrites (den) in the stratum radiatum (rad), give off axons (green) that enter stratum oriens (ori), acquire myelin, and travel to the subiculum within the alveus (alv). The color micrographs show that CA1 AISs are labeled by KCNQ2-N and KCNQ3b-N antibodies (green), as indicated. Grayscale insets show colabeling of AISs in boxed regions by NaV antibodies. I, Colabeling of CA1 pyramidal cell AISs by KCNQ2-C (green) and NaV antibodies (red). J, Colabeling of AISs of cortical layer 5 neurons by KCNQ3-C (green) and NaV antibodies (red). In these neurons, NaV staining begins more proximally on the axon than does KCNQ3-C (arrows), as previously shown for KCNQ2 (Devaux et al., 2004). Scale bars: A, 10 μm; B, main image, 5 μm; inset, 3 μm; C, 5 μm; D, top image, 3 μm; bottom images, 1 μm; E, 10 μm; F, 5 μm; G, 2.5 μm; H, 20 μm; I, J, 10 μm.
KCNQ3-C antibodies also stained the AISs of all neuronal types systematically examined for the current study, including the AISs of neuronal types [e.g., spinal motoneurons (Fig. 1E), cerebellar Purkinje cells (Fig. 1F)] that lacked detectable AIS staining with KCNQ3b-specific antibodies (Devaux et al., 2004). Many nodes of Ranvier in brain [e.g., cerebellar white matter (Fig. 1G)] also exhibited strong anti-KCNQ3-C stain. AISs of hippocampal CA1 pyramidal cells (Fig. 1H, KCNQ2-N and KCNQ3b-N; I, KCNQ2-C) and cortical layer 5 neurons (Fig. 1J, KCNQ3-C) were also strongly stained.
The detection of KCNQ2 and KCNQ3 immunoreactivity at nodes and AISs required a protocol that, to our knowledge, has been little used in previous studies of ion channel immunolocalization in brain, but which was modified from protocols that have proved useful for revealing the localization of proteins embedded in the myelin sheaths of peripheral nerves (Scherer et al., 1995). Fresh tissue blocks were rapidly frozen, cryosectioned, and extracted with 0.2–0.5% Triton X-100 before reactions with antibodies (see Materials and Methods). Labeling of an apparently intracellular pool of KCNQ channels localized to somata of interneurons, seen previously in aldehyde-fixed sections (Cooper et al., 2001), was abolished in this unfixed, detergent-extracted tissue. Conversely, fixation with aldehydes made detection of KCNQ2 and KCNQ3 staining of AISs and nodes undetectable. Based on labeling with four different antibodies, we conclude that detergent-resistant subpopulations of KCNQ2 and KCNQ3 subunits are specifically concentrated near NaV channels at nodes and AISs, in sciatic nerve, spinal cord, cerebellar cortex, hippocampus, and cerebral cortex.
The markedly variable nodal labeling by KCNQ3 antibodies (Fig. 1B) (Devaux et al., 2004) stood in contrast to the highly restricted and intense staining by KCNQ2 and NaV channel antibodies. Sciatic nerve contains a mixture of motor and sensory myelinated fibers that vary considerably in size, conduction velocity, pharmacology, and physiological function (Schmalbruch, 1986; Gokin et al., 2001; Vleggeert-Lankamp et al., 2004). We could not further assess whether KCNQ3 staining variability was reflecting heterogeneous subunit composition of axonal KCNQ proteins, or mere technical issues, such as selective extraction of KCNQ3 subunits from some nodes by detergent, or the potential inability of our antibodies to detect all KCNQ3 subunit isoforms. Rather than exploring further the possible diversity of nodal KCNQ channels, we elected to search for a mechanism that might mediate the robust targeting and/or stabilization of KCNQ2 and KCNQ3 subunits seen at many AISs and nodes.
KCNQ2 and KCNQ3 share C3, a distal C-terminal domain containing a putative ankyrin-G binding loop
Ankyrin-G and ankyrin-B are large adaptor proteins (190–480 kDa, depending on splicing) that bind multiple integral membrane proteins and mediate their interactions with the detergent-resistant actin-spectrin cortical cytoskeleton (Bennett and Baines, 2001). Given the established importance of ankyrin-G for assembly of AISs (Zhou et al., 1998; Jenkins and Bennett, 2001, 2002) and previous biochemical evidence that some brain KCNQ2/KCNQ3 channels are part of the detergent-resistant protein complex (Cooper et al., 2000), we scrutinized the KCNQ family polypeptides for sequences homologous to known ankyrin interaction motifs (Zhang et al., 1998, 2000; Chang and Low, 2003; Garrido et al., 2003; Lemaillet et al., 2003). The C-terminal regions of all KCNQ subunits include a very proximal conserved segment, C1, containing sites for binding calmodulin and phosphatidylinositol bisphosphate (PIP2), and another conserved region, C2, containing sequences important for tetramerization and subunit interaction (Wen and Levitan, 2002; Yus-Najera et al., 2002; Hoshi et al., 2003; Schwake et al., 2003; Zhang et al., 2003) (Fig. 2). We identified a more distal ∼80 aa section that was highly conserved (51% identity/69% allowing conservative substitutions) in KCNQ2 and KCNQ3, but completely absent in other KCNQ family members (Fig. 2B,C). Within this domain, which we called C3, we found most of the residues of a ∼10 aa ankyrin-G interaction motif (Fig. 2C), recently identified as necessary and sufficient for the concentration of NaV channels at AISs (Garrido et al., 2003; Lemaillet et al., 2003).
Concentration of KCNQ2 and KCNQ3 at AISs is abolished in cerebellar neurons lacking ankyrin-G
To test whether ankyrin-G is necessary for concentration of KCNQ2 and KCNQ3 subunits at AISs, we used mutant mice lacking ankyrin-G expression in most neurons of cerebellar cortex (Zhou et al., 1998). Ankyrin-G-deficient Purkinje cells from these mice fail to concentrate NaV channels and neurofascin at AISs and show reduced action potential firing responses to excitatory stimulation (Zhou et al., 1998). We compared labeling results using antibodies against KCNQ2, KCNQ3, and NaV channels in cerebellar sections cut from mutant mice and littermate controls.
In controls, KCNQ2-N (data not shown), and KCNQ2-C, KCNQ3-C, and PanNaV antibodies (Fig. 3A) strongly labeled the distinctively shaped AISs of cerebellar stellate, basket, Purkinje, and granule cells. In ankyrin-G mutant animals, the staining of these AISs for NaV channels was abolished, as was staining for KCNQ2 and KCNQ3 (Fig. 3B). Staining of AISs and nodes by anti-KCNQ and NaV antibodies was normal outside the cerebellar cortex, consistent with the previously documented regional specificity of the ankyrin-G knock-out (data not shown). In addition, in the granule cell layer only, a few AISs retained strong NaV, KCNQ2, and KCNQ3 staining (Fig. 3B, circles). These AISs were thicker and longer than the very fine, abundant AISs of the granule cells seen in sections from wild-type mice (Fig. 3A, four-headed arrows). Thus, a small population of cerebellar cortical neurons, restricted to the granule cell layer but likely larger in size than the granule cells themselves, may express a distinctive ankyrin-G isoform at the AISs.
Figure 3.
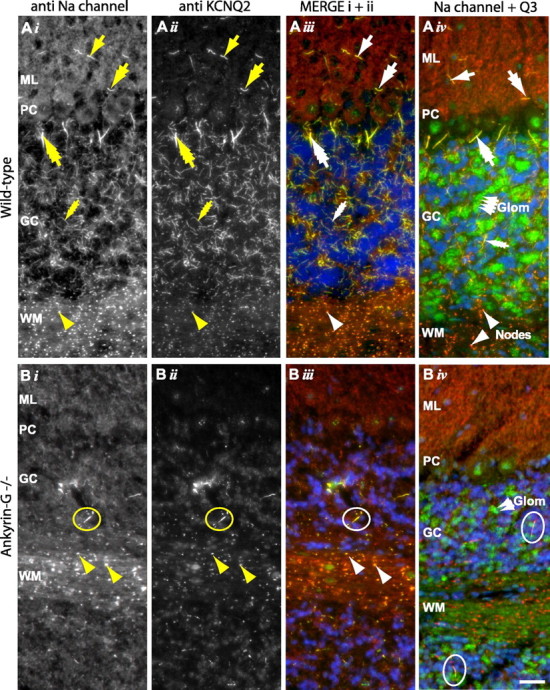
Concentration of KCNQ2, KCNQ3, and NaV channels at AISs of cerebellar cortical neurons requires ankyrin-G. A, Cerebellum of wild-type mice shows intense staining of AISs and nodes using antibodies against NaV channels (i, red in iii and iv), KCNQ2 (ii, green in iii), or KCNQ3 (green in iv). In iii and iv, nuclei are counterstained with the DNA-binding dye DAPI (blue). AISs of stellate cells (single-headed arrow), basket cells (double-headed arrows), Purkinje cells (triple-headed arrows), and granule cells (four-headed arrows) exhibit distinctive morphologies, and are all colabeled by the three antibodies. In white matter, nodes (arrowheads) are also labeled. KCNQ3-C (iv) but not KCNQ2-C (ii) (also, KCNQ3b-N, but not KCNQ2-N, not shown), labels ∼5 μm2 structures in the molecular layer only. The location, appearance, and distribution of these structures suggest they may correspond to cerebellar glomeruli (Glom). B, KCNQ2, KCNQ3, and NaV channel targeting to AISs is absent in ankyrin-G mutant mice. No stellate, basket, Purkinje, or granule cell AISs are seen. Rare AISs are seen in the granule cell layer (circles). These are considerably larger and thicker than granule cell AISs. In white matter, some nodes still stain for all markers; in granule cell layer, putative mossy fiber presynaptic termini also stain for KCNQ3. ML, Molecular layer; PC, Purkinje cell layer; GC, granule cell layer; WM, white matter. Scale bar, 20 μm.
An additional striking finding was that anti-KCNQ3 antibodies, but not anti-KCNQ2 antibodies, very strongly labeled irregular patches, 3–10 μm in diameter, located exclusively in the granule cell layer (Fig. 3Aiv, labeled “Glom”). Colabeling using the blue nucleic acid stain, DAPI, showed that these KCNQ3-immunoreactive patches were not clusters of granule cell somata (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). DAPI labels granule cell somata strongly, but the KCNQ3-stained patches were always located in positions unlabeled by DAPI. Strong KCNQ3 staining of these patches was preserved in the ankyrin-G knock-out mice (Fig. 3Biv and supplemental Fig. S1, available at www.jneurosci.org as supplemental material), despite the fact that KCNQ3-C staining of most granule cell layer AISs, as noted above, was abolished. Although the identity of the structures in these patches labeled by KCNQ3 antibodies was not further elucidated, the shape and distribution of the patches were suggestive of the cerebellar glomeruli, morphologically and functionally complex synaptic structures formed by afferent mossy fiber axon terminal expansions, granule cell dendrites, and Golgi cell axon terminals (Anderson and Flumerfelt, 1984). Thus, channels containing KCNQ3, but not KCNQ2, may be targeted or stabilized at one or more of these glomerular components, independent of expression of the main cerebellar cortical isoform of ankyrin-G.
The C3 domain motif is sufficient for ankyrin-G interaction
To test whether the C3 domain motif of KCNQ2 and/or KCNQ3 was capable of interacting with ankyrin-G, we generated a set of fusion proteins containing the extracellular and transmembrane domains of neurofascin and the distal C-terminal regions of KCNQ2, KCNQ3, and KCNQ5 (see Materials and Methods). We expressed these constructs individually along with the GFP-tagged ankyrin-G membrane binding domain (AnkG-MB-GFP) and studied the subcellular distribution of GFP fluorescence in live and fixed cells (Zhang and Bennett, 1998). In cells expressing AnkG-MB-GFP alone, GFP fluorescence exhibited an inhomogeneous, cytoplasmic (and often intranuclear) pattern (Fig. 4Ai, representative image; ii, intensity histogram). In cells coexpressing AnkG-MB-GFP and full-length neurofascin (HA-NF), GFP fluorescence was strongly redistributed to the cell membrane, resulting in a clearing of the cytoplasmic compartment (and, variably, of the nucleus), and the appearance of a distinct, membrane-associated edge of GFP fluorescence (Fig. 4B). Truncated neurofascin (HA-NF-DelC), lacking the motif that binds AnkG-MB, exhibited a complete loss of ability to redistribute AnkG-MB-GFP fluorescence (Fig. 4C). The neurofascin-KCNQ2C fusion protein (HA-NF-Q2C) strongly redistributed AnkG-MB-GFP fluorescence to the cell surface (Fig. 4D). Mutation of three conserved residues in the KCNQ2 C3 domain putative ankyrin-G binding motif (residues 810–812, ESD→AAA) completely abolished the ability of the HA-NF-Q2C to redistribute AnkG-MB-GFP (Fig. 4E).
Figure 4.

The KCNQ2 and KCNQ3 C3 domain motifs confer ability to bind ankyrin-G. The subcellular distribution of AnkG-MB-GFP fusion protein was detected in cells expressing AnkG-MB-GFP only (A), or coexpressed with HA-NF (B), HA-NF with C-terminal truncation eliminating intracellular ankyrin-G binding site (HA-NF-del) (C), NF-KCNQ2 C3 domain fusion (HA-NF-Q2C) (D), NF-KCNQ2 C3 domain fusion with C3 motif ESD mutated to AAA [HA-NF-Q2C(AAA)], NF-KCNQ3 C3 domain fusion (HA-NF-Q3C), NF-KCNQ3 C3 domain fusion with C3 motif ETD mutated to AAA [HA-NF-Q3C(AAA)]. i, Representative optical sections, obtained by wide-field deconvolution immunofluorescence microscopy, showing distribution of HA-tagged constructs (red) and AnkG-MB-GFP (green). ii, Intensity histograms for images in i. Histograms are shown partially overlapping to facilitate comparison of distribution of the two fluorophores. HA-NF, HA-NF-Q2C3, and HA-NF-Q3C3 strongly redistributed AnkG-MB-GFP to the cell surface, but constructs with truncation of NF or mutation of the C3 domains lack this ability. iii, Counts of cells with and without surface-redistributed GFP fluorescence. In each trial, 100 cells/condition were classified visually as possessing or lacking a detectable cytoplasmic clearing and surface redistribution, or edge. Bar graphs indicate average (±SEM) of three experiments. cyto, Cytoplasm; nuc, nucleus. Scale bar, 10 μm.
A fusion protein of neurofascin and the distal C-terminal region of KCNQ3 (HA-NF-Q3C) also redistributed AnkG-MB-GFP fluorescence to the cell surface and cleared the cytoplasmic compartment (Fig. 4F) but was less effective than either HA-NF or HA-NF-Q2C. Only 33.3 ± 6.8% of HA-NF-Q3C-transfected cells were scored as showing the redistribution, compared with 99 ± 1.0% of cells transfected with HA-NF or HA-NF-Q2C. Nonetheless, fusion proteins mutated at conserved residues in the putative C3 domain motif of KCNQ3 (ETD→827AAA), although efficiently expressed and targeted to the cell surface, exhibited complete loss of ability to redistribute AnkG-MB-GFP from the cytoplasm to the membrane (Fig. 4G). A neurofascin-KCNQ5 C-terminal fusion protein was unable to redistribute AnkG-MB-GFP (data not shown). GFP itself (lacking ankyrin-G-MB) was not redistributed by neurofascin or the neurofascin-KCNQ2/KCNQ3 fusion proteins (data not shown). We conclude that the KCNQ2/KCNQ3 motif, in the context of the C3 domain, is sufficient to bind the MB domain of ankyrin-G in living cells.
The C3 domain motif is necessary for ankyrin-G interaction with intact KCNQ2/KCNQ3 channels
To extend the above findings, we coexpressed full-length KCNQ2 and KCNQ3 and studied the distribution of the proteins by imaging of immunostained cells. We found the proteins to be highly retained intracellularly, without an apparent enrichment (or visible edge) at the cell surface (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Because full-length KCNQ2 and KCNQ3 subunits were thus not suitable for use as bait in the surface redistribution assay, we developed an approach using FRAP to monitor interaction between ankyrin-G and full-length channel subunits.
In our FRAP assay, we expressed the AnkG-MB-GFP reporter either alone in HEK 293 cells or in combination with full-length KCNQ2/KCNQ3 wild-type channels, KCNQ2 homotetrameric channels, or mutant KCNQ2/KCNQ3 heteromeric channels harboring the C3 domain motif mutations that abolished activity in the surface redistribution assay. Membrane proteins are less mobile than freely diffusing soluble proteins; thus, it is expected that interaction between AnkG-MB-GFP and the channel subunits could result in reduced mobility of AnkG-MB-GFP, detectable as slower and/or incomplete FRAP (Fig. 5). Photobleaching of AnkG-MB-GFP, expressed alone, resulted in rapid and nearly complete recovery of fluorescence (τ = 4.49 ± 0.12 s; mobile fraction, 89 ± 2.9%; n = 10) (Fig. 5B). Coexpression of AnkG-MB-GFP with KCNQ2 tetrameric channels, or with KCNQ2/KCNQ3 heteromeric channels, resulted in AnkG-MB-GFP FRAP that was slower and less complete (τ = 7.04 ± 0.84 s; mobile fraction, 71.1 ± 1.6%; n = 7; and τ = 11.16 ± 0.58 s; mobile fraction, 68.3 ± 4.2%; n = 11, respectively). Mutation of conserved residues within the C3 motifs of both KCNQ2 and KCNQ3 (ESD→810AAA in KCNQ2, ETD→827AAA in KCNQ3) resulted in AnkG-MB-GFP kinetics very similar to those of AnkG-MB-GFP expressed alone (τ = 3.13 ± 0.13 s; mobile fraction, 85.3%; n = 10). Western blotting was performed to rule out the possibility that the complete loss of activity in the FRAP assay exhibited by the mutant KCNQ2 and KCNQ3 subunits was attributable to rapid degradation of the mutant mRNAs or polypeptides, and to compare amounts of total steady-state proteins expressed. Western blots (Fig. 5C) showed that mutant KCNQ2 and KCNQ3 subunit cDNAs were expressed at protein levels that were similar to those of wild-type subunits, whereas cells not transfected gave undetectable levels of the subunits.
Ankyrin-G coexpression has slight effects on KCNQ2/KCNQ3 channel gating but is insufficient to account for IKs-like kinetics or voltage dependence
The nodal current, IKs, is similar in many respects to IM, as first noted by Dubois (1983). However, IKs exhibits much faster gating kinetics than IM, and activates in much more hyperpolarized voltage range (IKs has a V1/2 of approximately −65 mV; IM has a V1/2 of approximately −35 to 40 mV) (Röper and Schwarz, 1989; Wang et al., 1998). Because accessory subunit and other protein–protein interactions may contribute to such differences in channel properties (Rettig et al., 1994; Krapivinsky et al., 1995; Nadal et al., 2003), we tested whether coexpression of full-length ankyrin-G-GFP altered the kinetics or voltage dependence of heteromeric KCNQ2/KCNQ3 channels (Fig. 6). We found that KCNQ currents in cells coexpressing full-length ankyrin-G-GFP exhibited slightly depolarized voltage dependence, and slightly slower activation and deactivation kinetics, compared with currents from cells expressing KCNQ2/KCNQ3 with GFP (Fig. 6B). Furthermore, the C3 domain mutations that abolished ankyrin-G interactions in subcellular redistribution and FRAP assays (Figs. 4, 5) also had no major effects on these channel properties (Fig. 6B). Thus, the modest effects of ankyrin-G on KCNQ channel gating kinetics and voltage dependence we observed provided some additional evidence that ankyrin-G and KCNQ2/KCNQ3 channels interact, but the recordings did not reveal any explanation for the much larger biophysical differences between channels formed by KCNQ tetramers and IKs.
The KCNQ2/KCNQ3 C3 domain is sufficient to restrict protein expression to neuronal AISs, and the ankyrin-G binding motif is necessary
The mechanisms involved in establishing and maintaining polarized membrane protein expression on axons and for further restriction to AISs are incompletely understood, but may involve preferential sorting to axonal vesicles, preferential transport along axonal microtubules, or selective endocytotic retrieval from somatodendritic locations (Fache et al., 2004; Winckler, 2004). These issues can be investigated by studies of hippocampal neurons in dissociated culture, which form axons and assemble AISs that exhibit highly restricted localization of native ankyrin-G, neurofascin, and NaV channel proteins (Garrido et al., 2003; Lemaillet et al., 2003). To test whether the KCNQ2/KCNQ3 C3 domain motif could mediate AIS protein targeting and/or retention, we transfected rat hippocampal neurons with HA-neurofascin and fusion cDNA constructs, and studied the subcellular distribution of the proteins at the cell surface by staining for the extracellular HA tag (Fig. 7). Because expression of the transfected proteins was driven by a constitutive viral promoter, transient high protein levels were anticipated. Indeed, for several days after transfection, all constructs were robustly overexpressed at the membrane, in a nonspecific pattern including axons, dendrites, and somata. For example, staining 7 d after lipid-mediated transfection (total, 15 d in vitro) revealed intense HA-NF-KCNQ2C labeling of all axons and dendrites; labeling of native MAP2 proteins was restricted to dendrites (Fig. 7A). At longer intervals after transfection, total protein expression levels declined, and selective AIS retention became apparent for proteins possessing ankyrin-G binding motifs. Thus, even 22 d after transfection, HA-neurofascin (Fig. 7B) showed staining that was highly concentrated at AISs and low on the soma, dendrites, and distal axons.
Figure 7.
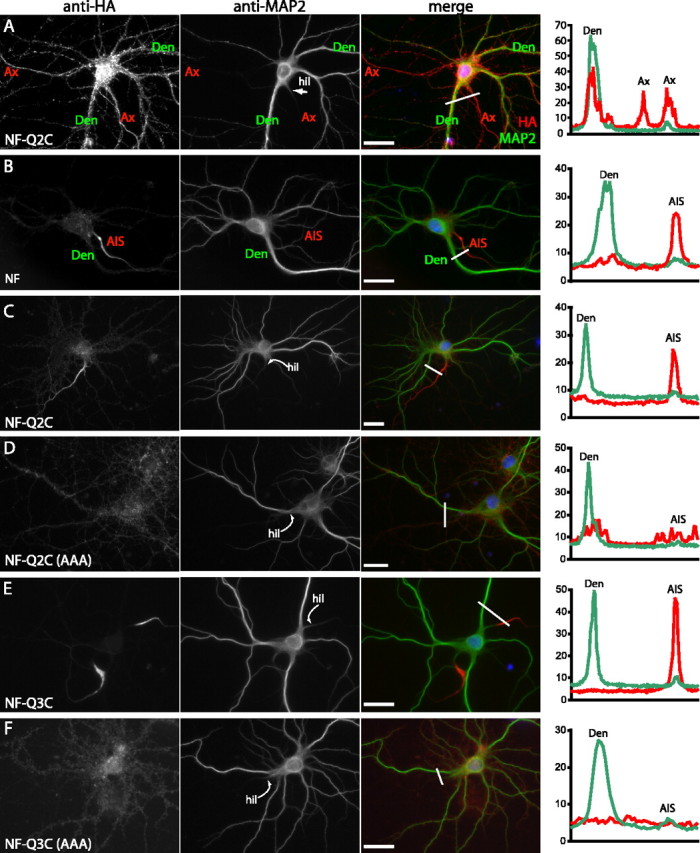
The C3 domain mediates retention at AISs of cultured hippocampal neurons. Hippocampal neurons were dissociated at E18, transfected with the indicated HA-tagged constructs, and immunostained with anti-HA and anti-MAP2 antibodies to determine the subcellular localization of surface HA-tagged proteins. The micrographs show anti-HA (left), anti-MAP2 (center), and merged images (right). Histograms (far right) show the intensity of HA (red) and MAP2 (green) immunofluorescence along the line shown in white in the merged image panels (expressed as a percentage of saturating intensity). A, Neuron transfected with HA-NF-Q2C at 8 d in vitro, stained at 15 d in vitro. HA-NF-Q2C is nonspecifically localized at axonal and somatodendritic membranes. B, Neuron electroporated with HA-NF at E18, and then immunostained after 22 d in vitro. HA-NF is selectively retained at the AIS. C, D, Neurons electroporated with HA-NF-Q2C or HA-NF-Q2C(AAA) at E18, and then immunostained after 17 d in vitro. E, F, Neurons electroporated with HA-NF-Q3C or HA-NF-Q3C(AAA) at E18, and then immunostained after 15 d in vitro. Neurons transfected with constructs including the intact C3 domains from KCNQ2 (C) or KCNQ3 (E) exhibit strong labeling restricted to the proximal axon; constructs with the mutated C3 domain motif show reduced and nonpolarized distribution of fusion protein expression (D, F). Ax, Axon; Den, dendrite; AIS, axon initial segment; hil, axon hillock. Scale bars, 10 μm.
To test whether the KCNQ C3 domain motif could drive such AIS-restricted localization, we performed parallel transfection and immunostaining experiments using the KCNQ2, KCNQ3, and KCNQ5 C-terminal fusion cDNAs. Constructs containing the intact KCNQ2 (Fig. 7C, 17 d after transfection) or KCNQ3 (Fig. 7E, 15 d after transfection) C3 domain motifs exhibited AIS-restricted strong staining. In contrast, electroporations of mutant constructs lacking the putative ankyrin-G interaction motif performed in parallel gave staining that was less intense at these long time points after transfection, and showed no evidence of polarized protein localization (Fig. 7D,F). We quantified these results in two ways. First, we compared the relative intensity of staining for the transfected proteins and endogenous MAP2 at the AISs and proximal dendrites. As shown (Fig. 7B–F, histograms), the staining intensity of MAP2 was high on dendrites (Den), but could be detected above background on axons at the very proximal portion. Staining intensity of HA-neurofascin, or HA-neurofascin-KCNQ2C and -KCNQ3C fusion proteins was both polarized (axons ≫ dendrites) and restricted to the proximal axon, but mutant protein expression less readily detected above background and showed no difference between axons and dendrites. Second, we calculated the percentage of MAP2-positive neurons on coverslips exhibiting such AIS-restricted staining per condition. The wild-type neurofascin-KCNQ2C and -KCNQ3C constructs were present at AISs of 46 ± 4.2 and 47.3% ± 1.0% of neurons, respectively, whereas the C3 motif mutants were at the AISs of only 0.2 ± 0.3% (KCNQ2 mutant) and 3.4 ± 2.6% (KCNQ3 mutant; 200 neurons counted per condition per transfection trial; n = 3). Observation of constructs containing the KCNQ5 C-terminal region revealed only the nonpolarized pattern of staining, but this finding was not analyzed quantitatively (data not shown).
Previous studies have shown that protein bound at the AISs of cultured hippocampal neurons are resistant to brief extraction treatments with detergent (Winckler et al., 1999; Garrido et al., 2003). Indeed, when neurons were treated with 0.5% Triton X-100 before fixation, staining for wild-type HA-NF-Q2C and HA-NF-Q3C proteins (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) localized at AISs were selectively retained, both at early and later intervals after transfection. This suggests that interaction between the KCNQ C3 domain and the endogenous neuronal AIS cytoskeleton was stabilizing the subset of fusion proteins localized to the AISs.
The ankyrin-G binding domains of KCNQ2, KCNQ3, and NaV channels arose early in the vertebrate evolutionary lineage
To better understand the origin of the ankyrin-G interaction mechanism at nodes and AISs, we identified cDNAs and genes for invertebrate and vertebrate KCNQ and NaV channels, aligned the predicted amino acid sequences, and generated a phylogenetic tree (Fig. 8). KCNQ and NaV orthologs were present in numerous invertebrate genomes as shown previously (Plummer and Meisler, 1999; Goldin, 2002; Yu and Catterall, 2004; Wei et al., 2005; Wen et al., 2005). However, we found no examples of invertebrate KCNQ or NaV channels possessing the ankyrin-G binding motif. Invertebrate KCNQ genes possessed the proximal conserved C1 and C2 domains, but lacked distal C3 domains. In contrast, all of the vertebrate NaV and neuronal KCNQ2/KCNQ3 genes identified in bony fish, amphibian, birds, and mammals possessed the ankyrin-G binding motif (Fig. 8A,B). The vertebrate KCNQ ankyrin-G binding motif was in all cases a part of a larger conserved C3-like domain (detected by the presence of highly conserved residues upstream and downstream of the putative ankyrin-G interaction motif, including an absolutely conserved sequence, HEELERS) (Fig. 2 and supplemental Table 1, available at www.jneurosci.org as supplemental material). This larger domain was always absent from NaV genes, which exhibited high sequence diversity in the regions adjoining the ankyrin-G binding motif, as previously noted (Lemaillet et al., 2003) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Thus, the NaV and KCNQ genes with ankyrin-G interaction motifs possess no common ancestor. Therefore, it appears that the ankyrin-G binding motif present in both the vertebrate NaV and KCNQ channels evolved before the appearance of the proximate common ancestor of fish, birds, and mammals, and before the gene duplication events that produced the paralogous subunit genes (NaV1.1–NaV1.9 and KCNQ2–KCNQ3) possessing the motifs. This appears to represent an example of molecular evolutionary convergence at the level of function, mechanism, and sequence (discussed below) (Doolittle, 1994; Zhang and Kumar, 1997; Zakon, 2002).
Figure 8.

The ankyrin-G binding motifs of NaV channel α subunits and KCNQ2/KCNQ3 subunits appear first in lower vertebrates, apparently reflecting a process of convergent molecular evolution. A, Clustal alignment was performed on full-length predicted amino acid sequences of invertebrates and vertebrate NaV α subunits. The positions corresponding to the ankyrin-G binding motifs and the conserved DIII–DIV intracellular linker mediating fast inactivation are shown. Ankyrin-G binding motifs are absent from invertebrates, but are present in teleost fish and mammals. B, Alignments made, as in A, but of KCNQ channel subunits of invertebrates and vertebrates. A portion of the highly conserved KCNQ C2 domain, mediating subunit association, and the ankyrin-G binding motif are shown. The ankyrin-G binding motif is absent from invertebrate genes but is present in KCNQ2 and KCNQ3 of teleost fish, birds, and mammals. For genes with accession numbers AAN63887, CAA11526, CAF90163, and AAV34442, we assigned KCNQ family identity based on high homology with known KCNQ genes identified by BLASTp analysis. For A and B, species name and NCBI protein database accession numbers are indicated in parentheses. C, Phylogenetic tree of Aplysia NaV and representative KCNQ channels. The analysis confirms the very distant evolutionary relationship between NaV and KCNQ channels and illustrates the close paralogous relationship between KCNQ2 and KCNQ3. This suggests that the appearance of the similar ankyrin-G binding motifs in NaV and KCNQ2/KCNQ3 genes took place independently, before the last common ancestor of jawed vertebrates. Scale bar, absolute number of changed residues. D, Diagram showing proposed interactions between NaV channels, KCNQ channels, ankyrin-G, and the actin–βIV spectrin cortical cytoskeleton at AISs and nodes. For simplicity, other known membrane proteins of nodes and AISs (neurofascin and relate cell adhesion molecules; NaV channel β subunits) are not depicted. The stoichiometry of interactions between ankyrin-G and membrane proteins is unknown.
Discussion
In sympathetic neurons, IM, a small current mediated by KCNQ2 homomers and/or KCNQ2/KCNQ3 heteromers (Brown and Adams, 1980; Wang et al., 1998; Hadley et al., 2003) exerts substantial negative control over responsiveness to excitatory inputs. Inhibition of IM by metabotropic neurotransmitter receptors provides a robust, reversible mechanism for increasing this responsiveness (Brown, 1988). KCNQ2 and KCNQ3 mutations cause epilepsy and myokymia, phenotypes that imply roles in brain and in axons of spinal motoneurons (Dedek et al., 2001; Singh et al., 2003). Here, we investigate the basis of these phenotypes and report three main findings. First, KCNQ2 and KCNQ3 are widely colocalized with NaV channels at the AIS and node of Ranvier—in the hippocampus, cerebral and cerebellar cortex, the ventral horn, and the sciatic nerve. Second, interaction between the KCNQ2 and KCNQ3 C3 domain motifs and the ankyrin-G membrane binding domain appears to underlie retention at the AIS. Remarkably, the KCNQ ankyrin-G interaction peptide shows high homology to the NaV channel motif that serves this function. Third, phylogenetic analysis reveals that KCNQ and NaV genes of other gnathostomes (jawed vertebrates) possess the motif, but related invertebrate genes lack it.
Differences in procedure reveal distinct somatic and axonal populations of KCNQ channels
Exceptionally strong immunohistochemical evidence now indicates that channels containing KCNQ2 and KCNQ3 are present at the node and AIS, based on staining by antibodies against four different subunit epitopes and demonstration of a membrane-staining pattern at nodes. Why is the KCNQ2 and KCNQ3 protein distribution reported here (and by Devaux et al., 2004) so different from previous studies using the same antibodies (Cooper et al., 2000, 2001)? In those previous reports, KCNQ2-N, KCNQ2-C, and KCNQ3-C antibodies gave primarily somatic and intracellular staining patterns. Furthermore, certain populations of neurons with large axonal arbors (e.g., aspiny cholinergic striatal neurons, reticular thalamic neurons, parvalbumin-expressing hippocampal interneurons) showed particularly strong, punctate somatic labeling for KCNQ2. AISs and nodes were unlabeled.
Immunohistochemical experiments require striking a balance between tissue destruction sufficient to allow antibodies to access epitopes and structural preservation sufficient for assigning the locations of antibody–antigen complexes within the tissue under investigation. Cooper et al. (2000, 2001) used procedures (aldehyde fixation, paraffin embedding, and microtome sectioning in one study; aldehyde fixation, vibratome sectioning, staining of relatively thick “floating” sections in the other) that preserve tissue structure relatively well. However, fixation, which introduces artifactual cross-links in and between membranes and proteins, can prevent antibody accessibility to some antigenic epitopes. The current study used either isolated, unfixed single nerve fibers, or snap-frozen, unfixed tissue that was cryostat sectioned and thaw-mounted on slides. Subsequent immunostaining procedures (18–24 h) were conducted at room temperature in buffers containing Triton X-100 detergent. We view this method as a subcellular fractionation procedure—indeed, staining for cytoplasmic and readily solubilized proteins that are robustly immunostained in fixed sections (i.e., parvalbumin, synaptophysin) (data not shown) was abolished. Presumably such proteins are removed from tissue sections during the large-volume wash steps.
Brain KCNQ2 and KCNQ3 proteins exhibit unusual detergent resistance, so that brain membranes must be pretreated with agents designed to strip membranes of the cortical cytoskeleton to achieve partial solubilization of the subunits under non-denaturing conditions (Cooper et al., 2000). A distinctive, electron-dense protein undercoat abuts the plasmalemma at AISs and nodes (Palay et al., 1968; Chan-Palay, 1972; Kosaka, 1980). This undercoat likely represents the actin/βIV spectrin/ankyrin-G cytoskeleton, into which the NaV, KCNQ2, KCNQ3, and neurofascin endodomains are inserted (Fig. 8D). Apparently, access to the intracellular epitopes recognized by the four anti-KCNQ2 and anti-KCNQ3 antibodies used in our studies is prevented by fixation and enhanced by freeze–thaw and detergent extraction steps. KCNQ subunit biochemical detergent resistance, and the requirement for “antigen retrieval” for immunohistochemistry, parallels the behavior of the classical neurotransmitter receptors that are localized to postsynaptic densities by similar interactions with scaffold proteins (Müller et al., 1996; Fritschy et al., 1998).
The current studies support the titular conclusion of Devaux et al. (2004), that “KCNQ2 is a nodal channel,” but add that, sometimes, so is KCNQ3. Here, an additional antibody, KCNQ3-C, against an epitope common to both KCNQ3a and KCNQ3b isoforms, stains many rat sciatic nerve nodes, motoneuron AISs, and many CNS nodes. Nevertheless, some nodes lack staining by KCNQ3-C (Fig. 1B). Channels containing KCNQ2 but lacking KCNQ3 subunits may be expressed at some nodes, as seen previously at somata of immature sympathetic neurons (Hadley et al., 2003). KCNQ subunits differ in their sensitivity to extracellular tetraethylammonium (Wang et al., 1998; Hadley et al., 2000). Voltage-clamp studies of peripheral nerves using this blocker may shed light on the subunit composition of axonal KCNQ channels.
A common ankyrin-G mechanism retains axonal KCNQ and NaV channels
We identified a motif near the C termini of KCNQ2 and KCNQ3 that was very similar in sequence to the peptide recently shown to mediate ankyrin-G binding and axonal retention of NaV channels (Garrido et al., 2003; Lemaillet et al., 2003; Fache et al., 2004). Considerable evidence suggests that this motif plays analogous roles on KCNQ2- and KCNQ3-containing channels as described for NaV channels. First, targeting of both the K+ and Na+ channels to AISs is abolished in cerebellar neurons of transgenic mice lacking ankyrin-G. Second, surface redistribution assays show that the C-terminal regions of KCNQ2 and KCNQ3 (but not KCNQ5) interact with the ankyrin-G-MB domain, in a manner dependent on the motif. Third, intact KCNQ2/KCNQ3 channels interact with ankyrin-G, as demonstrated by FRAP, and by slight changes in voltage dependence and kinetics of KCNQ2/KCNQ3 channels resulting from ankyrin-G coexpression. Fourth, the C3 domain motif confers both resistance to detergent extraction and selective retention at AISs upon fusion proteins transfected into cultured hippocampal neurons.
Evolution of the ankyrin-G mechanism retaining KCNQ2/KCNQ3 and NaV channels
Given their related structure and functions, we were intrigued how a common mechanism for Na+ and K+ channel axonal retention might have evolved. Although voltage-gated Na+ and K+ channel genes are thought to share distant common ancestry, they are clearly present in invertebrates as separate gene families (Lock et al., 1994; Plummer and Meisler, 1999; Goldin, 2002). We found no examples of invertebrate KCNQ or NaV genes containing the ankyrin-G interaction motif (Fig. 8A,B). These phylogenetic results are fascinating in two respects. First, they suggest that ancestral NaV and KCNQ genes independently evolved ankyrin-G binding sequences and functionality. Such “convergent molecular evolution” at the functional, mechanistic, and sequence levels is extraordinary (Doolittle, 1994; Zakon, 2002). Second, because the NaV and KCNQ2/KCNQ3 polypeptides of jawed fish possess and those of invertebrates appear to lack the ankyrin-G interaction motif, this mechanism appears to have emerged at about the same period of evolutionary history as other key components required for saltatory conduction (Yasargil et al., 1982; Bullock et al., 1984). Axonal myelination affords markedly improved energy efficiency and conduction speed; its introduction is regarded as among the more fundamental enabling innovations of early vertebrate evolution (Zalc and Colman, 2000). Therefore the emergence of myelination and a mechanism for coordinate nodal and AIS retention of KCNQ and NaV channels at a similar period in evolutionary history is a remarkable coincidence. The phenotypes linked to hypofunctional KCNQ2 and KCNQ3 mutations, seizures and myokymia, suggest the advantages afforded by colocalization of balanced numbers of KCNQ and NaV channels at sites of initiation and propagation of saltatory action potentials, which may have provided a survival advantage favoring the shared ankyrin-G binding mechanism during the evolution of the myelinated fiber. Our analysis can be refined by sequencing the KCNQ and NaV genes of jawless vertebrates (e.g., lamprey and hagfish), which lack myelinated axons, and primitive, jawed fish (e.g., sharks), which possess them.
A hypothesis regarding KCNQ channel function at axon initial segments and nodes
Understanding axonal KCNQ channel function will require quantitative analysis of channel currents and associated neuronal activity. Because the present study uses only immunohistochemistry, molecular methods in preparations of reduced complexity, and phylogenetic analysis, inferences about function are preliminary. Nonetheless, our findings—anatomical, cell biological, and evolutionary—illuminate from new angles the hypothesis, implicit in Devaux et al. (2004), that KCNQ channels are key partners with NaV channels at AISs and nodes of Ranvier. As discussed above, a current that helps regulate the excitability of peripheral nerve nodes, IKs, exhibits KCNQ-like features. The functional contribution of KCNQ subunits to IKs can be tested by voltage-clamp and in vivo threshold tracking studies of peripheral nerve (Röper and Schwarz, 1989; Burke et al., 2001). Our findings imply that small IKs-like currents are also functional at the initial segments and nodes of central axons. Because such channels activate at more hyperpolarized voltages than transient NaV channels do, the “subthreshold” activity of a small number localized on proximal axons could potentially exert inhibitory influence on AP initiation. Classical and peptide transmitters, acting both via synapses and at a distance, can modulate neuronal excitability through effects on KCNQ channels (Jan and Jan, 1982; Brown, 1988; Hille, 2001). Modulation of KCNQ channels strategically localized to the proximal axon could help control the “gain” relationship between synaptic excitation and AP firing. New tools, including KCNQ-selective drugs (Gribkoff, 2003) and animal models permitting temporal control over KCNQ channel activity (Peters et al., 2005), may allow definitive experimental testing of this hypothesis.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants R01 NS043174 (S.S.S.), R01 NS49119 and P30 HD26979 (E.C.C.), and T32-GM07517 and R01 NS41811 (S.D.C.); the Whitaker Foundation (S.D.C.); the University of Pennsylvania McCabe Fund (E.C.C.); and a Miles Family Fund Clinician–Scholar Award (E.C.C.). V.B. is an investigator of the Howard Hughes Medical Institute. FRAP experiments were performed at the University of Pennsylvania Dynamic Imaging Center (NIH Grant P30 NS047321). We thank Thomas Jentsch, David McKinnon, and Klaus Steinmeyer for KCNQ cDNAs; Margie Maronski and Marc Dichter for cultured hippocampal neurons and assistance with electroporation; Peter Mohler for genotyping ankyrin-G transgenic mice; Matthew Rasband for advice and protocols for neuronal transfection and immunostaining; Philip Haydon, Steve Moss, Lily Y. Jan, Kleopas Kleopa, Leif Finkel, and Jerome Devaux for helpful discussion; Brian Litt for support (S.D.C.); and our anonymous reviewers for useful critical feedback.
References
- Adelman JP, Bond CT, Pessia M, Maylie J (1995). Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron 15:1449–1454. [DOI] [PubMed] [Google Scholar]
- Anderson WA, Flumerfelt BA (1984). Time course of mossy fiber degeneration following pontine ablation in the rat. J Comp Neurol 227:401–413. [DOI] [PubMed] [Google Scholar]
- Bennett V, Baines AJ (2001). Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev 81:1353–1392. [DOI] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK (1998). A potassium channel mutation in neonatal human epilepsy. Science 279:403–406. [DOI] [PubMed] [Google Scholar]
- Brown D (1988). M-currents: an update. Trends Neurosci 11:294–299. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR (1980). Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neuron. Nature 283:673–676. [DOI] [PubMed] [Google Scholar]
- Buchhalter JR, Dichter MA (1991). Electrophysiological comparison of pyramidal and stellate nonpyramidal neurons in dissociated cell culture of rat hippocampus. Brain Res Bull 26:333–338. [DOI] [PubMed] [Google Scholar]
- Bullock TH, Moore JK, Fields RD (1984). Evolution of myelin sheaths: both lamprey and hagfish lack myelin. Neurosci Lett 48:145–148. [DOI] [PubMed] [Google Scholar]
- Burke D, Kiernan MC, Bostock H (2001). Excitability of human axons. Clin Neurophysiol 112:1575–1585. [DOI] [PubMed] [Google Scholar]
- Chang SH, Low PS (2003). Identification of a critical ankyrin-binding loop on the cytoplasmic domain of erythrocyte membrane band 3 by crystal structure analysis and site-directed mutagenesis. J Biol Chem 278:6879–6884. [DOI] [PubMed] [Google Scholar]
- Chan-Palay V (1972). The tripartite structure of the undercoat in initial segments of Purkinje cell axons. Z Anat Entwicklungsgesch 139:1–10. [DOI] [PubMed] [Google Scholar]
- Chiu SY, Ritchie JM, Rogart RB, Stagg D (1979). A quantitative description of membrane currents in rabbit myelinated nerve. J Physiol (Lond) 292:149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs JS, Curtis DR, Eccles JC (1957). The generation of impulses in motoneurones. J Physiol (Lond) 139:232–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EC, Aldape KD, Abosch A, Barbaro NM, Berger MS, Peacock WS, Jan YN, Jan LY (2000). Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci USA 97:4914–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EC, Harrington E, Jan YN, Jan LY (2001). M-channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci 21:9529–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedek K, Kunath B, Kananura C, Reuner U, Jentsch TJ, Steinlein OK (2001). Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc Natl Acad Sci USA 98:12272–12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC, Scherer SS (2004). KCNQ2 is a nodal K+ channel. J Neurosci 24:1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle RF (1994). Convergent evolution: the need to be explicit. Trends Biochem Sci 19:15–18. [DOI] [PubMed] [Google Scholar]
- Dubois JM (1983). Potassium currents in the frog node of Ranvier. Prog Biophys Mol Biol 42:1–20. [DOI] [PubMed] [Google Scholar]
- Fache MP, Moussif A, Fernandes F, Giraud P, Garrido JJ, Dargent B (2004). Endocytotic elimination and domain-selective tethering constitute a potential mechanism of protein segregation at the axonal initial segment. J Cell Biol 166:571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankenhaeuser B, Huxley AF (1964). The action potential in the myelinated nerve fiber of Xenopus laevis as computed on the basis of voltage clamp data. J Physiol (Lond) 171:302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM, Weinmann O, Wenzel A, Benke D (1998). Synapse-specific localization of NMDA and GABA(A) receptor subunits revealed by antigen-retrieval immunohistochemistry. J Comp Neurol 390:194–210. [PubMed] [Google Scholar]
- Garrido JJ, Giraud P, Carlier E, Fernandes F, Moussif A, Fache MP, Debanne D, Dargent B (2003). A targeting motif involved in sodium channel clustering at the axonal initial segment. Science 300:2091–2094. [DOI] [PubMed] [Google Scholar]
- Garver TD, Ren Q, Tuvia S, Bennett V (1997). Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol 137:703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokin AP, Philip B, Strichartz GR (2001). Preferential block of small myelinated sensory and motor fibers by lidocaine: in vivo electrophysiology in the rat sciatic nerve. Anesthesiology 95:1441–1454. [DOI] [PubMed] [Google Scholar]
- Goldin AL (2002). Evolution of voltage-gated Na+ channels. J Exp Biol 205:575–584. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK (2003). The therapeutic potential of neuronal KCNQ channel modulators. Expert Opin Ther Targets 7:737–748. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA (2000). Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. Br J Pharmacol 129:413–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, Brown DA (2003). Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci 23:5012–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herson PS, Virk M, Rustay NR, Bond CT, Crabbe JC, Adelman JP, Maylie J (2003). A mouse model of episodic ataxia type-1. Nat Neurosci 6:378–383. [DOI] [PubMed] [Google Scholar]
- Hille B (2001). In: Ionic channels of excitable membranes Ed 3 Sunderland, MA: Sinauer.
- Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H (2003). AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci 6:564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard A, Tamas G, Soltesz I (2005). Lighting the chandelier: new vistas for axo-axonic cells. Trends Neurosci 28:310–316. [DOI] [PubMed] [Google Scholar]
- Jan LY, Jan YN (1982). Peptidergic transmission in sympathetic ganglia of the frog. J Physiol (Lond) 327:219–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V (2001). Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol 155:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V (2002). Developing nodes of Ranvier are defined by ankyrin-G clustering and are independent of paranodal axoglial adhesion. Proc Natl Acad Sci USA 99:2303–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T (1980). The axon initial segment as a synaptic site: ultrastructure and synaptology of the initial segment of the pyramidal cell in the rat hippocampus (CA3 region). J Neurocytol 9:861–882. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE (1995). The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature 374:135–141. [DOI] [PubMed] [Google Scholar]
- Lemaillet G, Walker B, Lambert S (2003). Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J Biol Chem 278:27333–27339. [DOI] [PubMed] [Google Scholar]
- Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K (2000). Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem 275:22395–22400. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Altan-Bonnet N, Patterson GH (2003). Photobleaching and photoactivation: following protein dynamics in living cells. Nat Cell Biol Suppl, S7–S14. [PubMed]
- Lock LF, Gilbert DJ, Street VA, Migeon MB, Jenkins NA, Copeland NG, Tempel BL (1994). Voltage-gated potassium channel genes are clustered in paralogous regions of the mouse genome. Genomics 20:354–362. [DOI] [PubMed] [Google Scholar]
- Müller BM, Kistner U, Kindler S, Chung WJ, Kuhlendahl S, Fenster SD, Lau LF, Veh RW, Huganir RL, Gundelfinger ED, Garner CC (1996). SAP102, a novel postsynaptic protein that interacts with NMDA receptor complexes in vivo. Neuron 17:255–265. [DOI] [PubMed] [Google Scholar]
- Nadal MS, Ozaita A, Amarillo Y, Vega-Saenz de Miera E, Ma Y, Mo W, Goldberg EM, Misumi Y, Ikehara Y, Neubert TA, Rudy B (2003). The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron 37:449–461. [DOI] [PubMed] [Google Scholar]
- Palay SL, Sotelo C, Peters A, Orkand PM (1968). The axon hillock and the initial segment. J Cell Biol 38:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Selyanko AA, Hadley JK, Brown DA, Dixon JE, McKinnon D (2001). Alternative splicing of KCNQ2 potassium channel transcripts contributes to the functional diversity of M-currents. J Physiol (Lond) 531:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D (2005). Conditional transgenic suppression of M channels in mouse brains reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci 8:51–60. [DOI] [PubMed] [Google Scholar]
- Plummer NW, Meisler MH (1999). Evolution and diversity of mammalian sodium channel genes. Genomics 57:323–331. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Trimmer JS, Schwarz TL, Levinson SR, Ellisman MH, Schachner M, Shrager P (1998). Potassium channel distribution, clustering, and function in remyelinating rat axons. J Neurosci 18:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O (1994). Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature 369:289–294. [DOI] [PubMed] [Google Scholar]
- Röper J, Schwarz JR (1989). Heterogeneous distribution of fast and slow potassium channels in myelinated rat nerve fibres. J Physiol (Lond) 416:93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saganich MJ, Machado E, Rudy B (2001). Differential expression of genes encoding subthreshold-operating voltage-gated K+ channels in brain. J Neurosci 21:4609–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Arroyo EJ (2002). Recent progress on the molecular organization of myelinated axons. J Peripher Nerv Syst 7:1–12. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Deschenes SM, Xu YT, Grinspan JB, Fischbeck KH, Paul DL (1995). Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci 15:8281–8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalbruch H (1986). Fiber composition of the rat sciatic nerve. Anat Rec 215:71–81. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Kubisch C, Stein V, Jentsch TJ (1998). Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K channels causes epilepsy. Nature 396:687–690. [DOI] [PubMed] [Google Scholar]
- Schwake M, Pusch M, Kharkovets T, Jentsch TJ (2000). Surface expression and single channel properties of KCNQ2/KCNQ3, M-type K+ channels involved in epilepsy. J Biol Chem 275:13343–13348. [DOI] [PubMed] [Google Scholar]
- Schwake M, Jentsch TJ, Friedrich T (2003). A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep 4:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JR, Reid G, Bostock H (1995). Action potentials and membrane currents in the human node of Ranvier. Pflugers Arch 430:283–292. [DOI] [PubMed] [Google Scholar]
- Scriven DR, Dan P, Moore ED (2000). Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J 79:2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Brown DA (2001). Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J Physiol (Lond) 534:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Delmas P, Hadley JK, Tatulian L, Wood IC, Mistry M, London B, Brown DA (2002). Dominant-negative subunits reveal potassium channel families that contribute to M-like potassium currents. J Neurosci 22: RC212(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P (2002). Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol (Lond) 544:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B (2000). Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J Neurosci 20:1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Hamilton SE, Nathanson NM, Surmeier DJ (2005). Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J Neurosci 25:7449–7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales TO, Peiffer A, Anderson VE, Leppert M (1998). A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet 18:25–29. [DOI] [PubMed] [Google Scholar]
- Singh NA, Westenskow P, Charlier C, Pappas C, Leslie J, Dillon J, Anderson VE, Sanguinetti MC, Leppert MF (2003). KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain 126:2726–2737. [DOI] [PubMed] [Google Scholar]
- Smart S, Lopantsev V, Zhang CL, Robbins C, Wand H, Chiu SY, Schwartzkroin PA, Messing A, Tempel B (1998). Deletion of the Kv1.1 potassium channel causes epilepsy in mice. Neuron 20:809–820. [DOI] [PubMed] [Google Scholar]
- Stuart G, Spruston N, Sakmann B, Hausser M (1997). Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends Neurosci 20:125–131. [DOI] [PubMed] [Google Scholar]
- Stuhmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, Kubo H, Numa S (1989). Structural parts involved in activation and inactivation of the sodium channel. Nature 339:597–603. [DOI] [PubMed] [Google Scholar]
- Surti TS, Huang L, Jan YN, Jan LY, Cooper EC (2005). Identification by mass spectrometry and functional characterization of two phosphorylation sites of KCNQ2/KCNQ3 channels. Proc Natl Acad Sci USA 102:17828–17833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedlow JR, Platani M (2002). Live cell imaging using wide-field microscopy and deconvolution. Cell Struct Funct 27:335–341. [DOI] [PubMed] [Google Scholar]
- Tanimura N, Nagafuku M, Liddicoat DR, Hamaoka T, Kosugi A (2003). Analysis of the mobility of signaling molecules in lymphocytes using fluorescence photobleaching techniques. Sci STKE 2003:110. [DOI] [PubMed] [Google Scholar]
- Vleggeert-Lankamp CL, van den Berg RJ, Feirabend HK, Lakke EA, Malessy MJ, Thomeer RT (2004). Electrophysiology and morphometry of the Aalpha- and Abeta-fiber populations in the normal and regenerating rat sciatic nerve. Exp Neurol 187:337–349. [DOI] [PubMed] [Google Scholar]
- Vogel W, Schwarz JR (1995). Voltage-clamp studies in axons: macroscopic and single-channel currents. In: The axon (Waxman SG, Kocsis JD, Stys PK, eds) , pp. 257–280. New York: Oxford UP.
- Wang H, Kunkel DD, Martin TM, Schwartzkroin PA, Tempel BL (1993). Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature 365:75–79. [DOI] [PubMed] [Google Scholar]
- Wang H-S, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282:1890–1893. [DOI] [PubMed] [Google Scholar]
- Wei AD, Butler A, Salkoff L (2005). KCNQ-like potassium channels in Caenorhabditis elegans. Conserved properties and modulation. J Biol Chem 280:21337–21345. [DOI] [PubMed] [Google Scholar]
- Wen H, Levitan IB (2002). Calmodulin is an auxiliary subunit of KCNQ2/KCNQ3 potassium channels. J Neurosci 22:7991–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Weiger TM, Ferguson TS, Shahidullah M, Scott SS, Levitan IB (2005). A Drosophilia KCNQ channel essential for early embryonic development. J Neurosci 25:10147–10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winckler B (2004). Scientiae forum/models and speculations pathways for axonal targeting of membrane proteins. Biol Cell 96:669–674. [DOI] [PubMed] [Google Scholar]
- Winckler B, Forscher P, Mellman I (1999). A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature 397:698–701. [DOI] [PubMed] [Google Scholar]
- Yang WP, Levesque PC, Little WA, Conder ML, Ramakrishnan P, Neubauer MG, Blanar MA (1998). Functional expression of two KvLQT1-related potassium channels responsible for an inherited idiopathic epilepsy. J Biol Chem 273:19419–19423. [DOI] [PubMed] [Google Scholar]
- Yasargil GM, Greeff NG, Luescher HR, Akert K, Sandri C (1982). The structural correlate of saltatory conduction along the Mauthner axon in the tench (Tinca tinca L.): identification of nodal equivalents at the axon collaterals. J Comp Neurol 212:417–424. [DOI] [PubMed] [Google Scholar]
- Yu FH, Catterall WA (2004). The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE 2004:re15. [DOI] [PubMed] [Google Scholar]
- Yus-Najera E, Santana-Castro I, Villarroel A (2002). The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J Biol Chem 277:28545–28553. [DOI] [PubMed] [Google Scholar]
- Zakon HH (2002). Convergent evolution on the molecular level. Brain Behav Evol 59:250–261. [DOI] [PubMed] [Google Scholar]
- Zalc B, Colman DR (2000). Origins of vertebrate success. Science 288:271–272. [DOI] [PubMed] [Google Scholar]
- Zhang D, Kiyatkin A, Bolin JT, Low PS (2000). Crystallographic structure and functional interpretation of the cytoplasmic domain of erythrocyte membrane band 3. Blood 96:2925–2933. [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, Logothetis DE (2003). PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37:963–975. [DOI] [PubMed] [Google Scholar]
- Zhang J, Kumar S (1997). Detection of convergent and parallel evolution at the amino acid sequence level. Mol Biol Evol 14:527–536. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bennett V (1998). Restriction of 480/270-kD ankyrin G to axon proximal segments requires multiple ankyrin G-specific domains. J Cell Biol 142:1571–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Davis JQ, Carpenter S, Bennett V (1998). Structural requirements for association of neurofascin with ankyrin. J Biol Chem 273:30785–30794. [DOI] [PubMed] [Google Scholar]
- Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V (1998). AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol 143:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Messing A, Chiu SY (1999). Determinants of excitability at transition zones in Kv1.1-deficient myelinated nerves. J Neurosci 19:5768–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]