

Abstract
Psychiatric illnesses, particularly schizophrenia, are associated with disrupted markers for interneuronal function and interneuron-mediated brain rhythms such as gamma frequency oscillations. Here we investigate a possible link between these two observations in the entorhinal cortex and hippocampus by using a genetic and an acute model of psychiatric illness. Lysophosphatidic acid 1 receptor-deficient (LPA1-deficient) mice show psychomotor-gating deficits and neurochemical changes resembling those seen in postmortem schizophrenia studies. Similar deficits are seen acutely with antagonism of the NMDA subtype of glutamate receptor. Neither model induced any change in power or frequency of gamma rhythms generated by kainate in hippocampal slices. In contrast, a dramatic decrease in the power of gamma oscillations was seen in superficial, but not deep, medial entorhinal cortex layers in both models. Immunolabeling for GABA, parvalbumin, and calretinin in medial entorhinal cortex from LPA1-deficient mice showed an ∼40% reduction in total GABA- and parvalbumin-containing neurons, but no change in the number of calretinin-positive neurons. This deficit was specific for layer II (LII). No change in the number of neurons positive for these markers was seen in the hippocampus. Acute NMDA receptor blockade, which selectively reduces synaptic drive to LII entorhinal interneurons, also disrupted gamma rhythms in a similar manner in superficial entorhinal cortex, but not in hippocampus. These data demonstrate an area-specific deficit in gamma rhythmogenesis in animal models of psychiatric illness and suggest that loss, or reduction in function, of interneurons having a large NMDA receptor expression may underlie the network dysfunction that is seen.
Keywords: schizophrenia, LPA1 receptor, inhibition, entorhinal cortex, gamma oscillation, ketamine
Introduction
Schizophrenia (and related psychiatric illnesses) is associated with a number of quantifiable changes in cognitive performance, neurochemistry, psychophysiology, and neuroanatomy. Deficits in working memory (Park and Holzman, 1992) and sensorimotor gating (Geyer and Braff, 1987; Braff et al., 1992) are seen, with the magnitude of reduction correlating with the severity of psychosis (Meincke et al., 2004). Neurochemical changes involve monoaminergic, glutamatergic, and GABAergic systems (Weinberger, 1997). Psychophysiological changes include deficits in mismatch negativity, a component of (usually) auditory evoked potentials related to gamma frequency EEG activity (Marshall et al., 1996). Gamma rhythms are disrupted both in their generation and the cortical synchrony they facilitate during sensory stimulation (Spencer et al., 2003, 2004). Neuroanatomical changes include a disorganized arrangement of stellate cells in entorhinal cortex (Falkai et al., 2000) and changes in markers for interneuron function, particularly interneurons containing parvalbumin (for review, see Arnold, 2000; Lewis et al., 2005).
No animal models are available that capture all of these aspects of schizophrenia and related psychoses. However, some models are available that display some or many of these deficits. Sensorimotor-gating deficits are seen in mGluR5 (Brody et al., 2004) and lysophosphatidic acid 1 receptor (LPA1) knock-out mice (Harrison et al., 2003) and mice with mutations in proline dehydrogenase or reelin (Gogos et al., 1999; Tueting et al., 1999). Psychosis induced by the NMDA receptor antagonist ketamine is associated with sensorimotor-gating deficit in humans and rats (de Bruin et al., 1999; Oranje et al., 2002). In addition, LPA1 receptor knock-out mice show many neurochemical changes associated with postmortem analysis of brains from schizophrenics (Roberts et al., 2005), and ketamine models both the hyperdopamine and hypoglutamate putative mechanisms of schizophrenia (Seeman et al., 2005).
A number of lines of evidence implicate the entorhinal cortex as a site for dysfunction in schizophrenia and related psychiatric illnesses (Arnold, 2000). Sensorimotor gating involves entorhinal cortical processing (Goto et al., 2002), and disruption in gross anatomy is associated with positive symptoms of schizophrenia (Prasad et al., 2004). At the cellular level changes in synaptic-related marker mRNAs are found in entorhinal layers II/III (LII/LIII) in postmortem brains from schizophrenics (Hemby et al., 2002), peptide levels associated with interneurons are reduced (Bachus et al., 1997), and developmental changes in LII neurons are seen (Falkai et al., 2000). The medial entorhinal cortex generates gamma frequency oscillations in vivo, in the whole brain preparation, and in vitro (Chrobak and Buzsáki, 1998; Dickson et al., 2000; Cunningham et al., 2003). These oscillations are associated with cognitive function via facilitation of entorhinal/hippocampal interactions (Fell et al., 2001).
Gamma oscillations generated by metabotropic receptor activation (Whittington et al., 1995; Fisahn et al., 1998) involve synchronous outputs from predominantly parvalbumin-positive, fast-spiking interneurons (Gloveli et al., 2005). We therefore wished to test whether a relationship between gamma power and interneuron function exists in the entorhinal cortex and hippocampus in two animal models of symptoms associated with psychiatric illness, a developmental model involving the ablation of the gene encoding the LPA1 receptor and an acute model (ketamine) in entorhinal cortical/hippocampal slices.
Materials and Methods
Slices (450 μm) containing hippocampus and entorhinal cortex were prepared from young adult male LPA1 receptor knock-out mice (LPA1−/−) and wild-type (WT) litter mates, as well as adult male Wistar rats and C57BL/6J mice. All procedures were performed according to the requirements of the United Kingdom Animals Scientific Procedures Act (1986). Animals were anesthetized with inhaled isoflurane, immediately followed by an intramuscular injection of ketamine (≥ 100 mg/kg) and xylazine (≥ 10 mg/kg). Animals were perfused intracardially with ∼50 ml of modified artificial CSF (ACSF), which was composed of the following (in mm): 252 sucrose, 3 KCl, 1.25 NaH2PO4, 24 NaHCO3, 2 MgSO4, 2 CaCl2, and 10 glucose. All salts were obtained from BDH Chemicals (Poole, UK). The brain was removed and submerged in cold (4–5°C) ACSF during dissection. Horizontal slices were cut and transferred to a recording chamber maintained at 34°C at the interface between ACSF [containing the following (in mm): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 24 NaHCO3, 2 MgSO4, 2 CaCl2, and 10 glucose] and warm, moist carbogen gas (95% O2/5% CO2). Slices were permitted to equilibrate for 45 min before any recordings commenced. All slices (hippocampal and entorhinal) were taken from an ∼2-mm-deep region containing medial entorhinal cortex and temporal hippocampus (interaural, 2.56–4.36 mm; bregma, –7.44 to –5.64 mm).
Recording electrodes were pulled from borosilicate glass (Harvard Apparatus, Edenbridge, UK), filled with ACSF, and had resistances in the range of 2–5 MΩ (extracellular) and 50–100 MΩ (intracellular). Neuronal subtypes were characterized electrophysiologically (interneurons having fast, nonaccommodating spiking patterns and stellate cells having a pronounced sag on injection of hyperpolarizing current) and anatomically post hoc after biocytin injection. For stimulation experiments bipolar platinum electrodes (25-μm-diameter wire; tip separation, ∼200 μm) were used to activate synaptic pathways electrically (60 μs duration; 1–10 V). The stimulation electrode was positioned in the superficial layers of the medial entorhinal cortex. The following drugs were added to the perfusion medium for various experimental conditions: bicuculline methochloride (5 μm), d,l-2-amino-5-phosphonovalerate (d-AP5; 50 μm), 6-nitro-7-sulfamoylbenz-[S]-quinoxalinedione (NBQX; 10 μm), (2S)-3[[(1S)-1-(3,4-dichlorophenyl)-ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid hydrochloride (CGP 55845A; 2 μm), all from Tocris Bioscience (Avonmouth, UK), plus kainic acid (200–400 nm) and ketamine (10–50 μm) from Sigma–Aldrich (Poole, UK).
Peak frequency and power values were obtained from power spectra generated with Fourier analysis in the Axograph software package (Molecular Devices, Sunnyvale, CA). Power for a given frequency band was determined as the area under the peak in the power spectra between 20 and 80 Hz for gamma frequency oscillations. All values are given as the mean ± SE where distributed normally; otherwise, data are expressed as the median (interquartile range). Power spectra were constructed off-line from digitized data (digitization frequency, 10 kHz), using a 60 s epoch of recorded activity. We measured the kinetics of EPSPs and IPSPs with Mini Analysis (Jaejin Software, Leonia, NJ); we obtained >200 events per slice and pooled them for additional analysis.
Previously electrophysiologically characterized entorhinal cortex and hippocampal slices from LPA1−/− and WT were sandwiched between two Millipore filters (Bedford, MA) to avoid deformations and were fixed for 1–7 d in a solution containing 4% paraformaldehyde in PBS (0.1 m, pH 7.4). Then these slices were gelatin-embedded and resectioned at 60 μm thickness. Vibratome sections next were immersed for 20 min in 50 mm ammonium chloride in PBS and rinsed in PBS. Incubation in 10 mm sodium citrate in distilled water (dH2O), pH 8.4 (1 h, at 80°C), was used to improve the labeling. This was followed by rinses in PBS, the quenching of endogenous peroxidases with 0.3% H2O2 in 10% methanol in PBS, and a blocking in 0.1% bovine gelatin/0.1% Triton X-100 in PBS. The primary antibodies were prepared in the blocking solution (rabbit anti-calretinin, 1:2000, Swant, Bellinzona, Switzerland; rabbit anti-parvalbumin, 1:10,000, Calbiochem, La Jolla, CA; rabbit anti-GABA, 1:1000, Sigma–Aldrich). The sections were incubated for 48 h at 4°C. After rinses the sections were incubated with biotinylated goat anti-rabbit IgG (H+L) (1:200; 2 h at room temperature; Vector Laboratories, Peterborough, UK) in PBS, followed by ABC Elite Kit (1:200; 2 h at room temperature; Vector Laboratories). The peroxidase reaction was revealed by incubating the sections in 0.05% 3,3′-diaminobenzidine and 0.01% H2O2. Sections were mounted on glass slides, dehydrated, and coverslipped with DPX (VWR Scientific, Poole, UK) before being examined with a standard light microscope. Control experiments, in which the primary antibody or each single step in the protocol was omitted, gave no signal (supplemental material, available at www.jneurosci.org).
Quantifications were performed by various individuals, independently, and blind to experimental conditions and to the origin of the sections from LPA1−/− or wild-type mice. Cells were considered immunopositive when they showed a heavy brown labeling corresponding to the peroxidase reaction product. Only positive cells were counted in microscopic view fields at 10× magnification within the 60 μm sections of the entorhinal cortex and hippocampus.
Results
Medial entorhinal cortex gamma rhythms
Bath application of 200–400 nm kainate generated persistent field gamma rhythms in all layers of the medial entorhinal cortex (mEC), with peak power in LII/LIII, as reported previously (Cunningham et al., 2003). However, there was a marked reduction in the kainate-induced gamma rhythms in superficial layers in slices from LPA1−/− mice (Fig. 1). In field recordings from LII the gamma rhythm was almost absent in LPA1-deficient tissue (WT area power, 303.9 ± 73.0 μV2; LPA1−/− area power, 85.5 ± 33.3 μV2; p < 0.001; n = 10), but what gamma remained was not significantly different in frequency from that seen in slices from WT mice (WT, 41.5 ± 2.8 Hz; LPA1−/−, 40.5 ± 4.1 Hz; p > 0.05; n = 10). In contrast, gamma power in the deep mEC was not significantly different in slices from WT and LPA1−/− mice (WT area power, 182.9 ± 33.1 μV2; LPA1−/− area power, 138.1 ± 31.4 μV2; p > 0.05; n = 10). No significant difference in frequency of population gamma was seen in the deep layers (WT, 39.8 ± 3.0 Hz; LPA1−/−, 28.5 ± 7.0 Hz; p > 0.05; n = 10).
Figure 1.
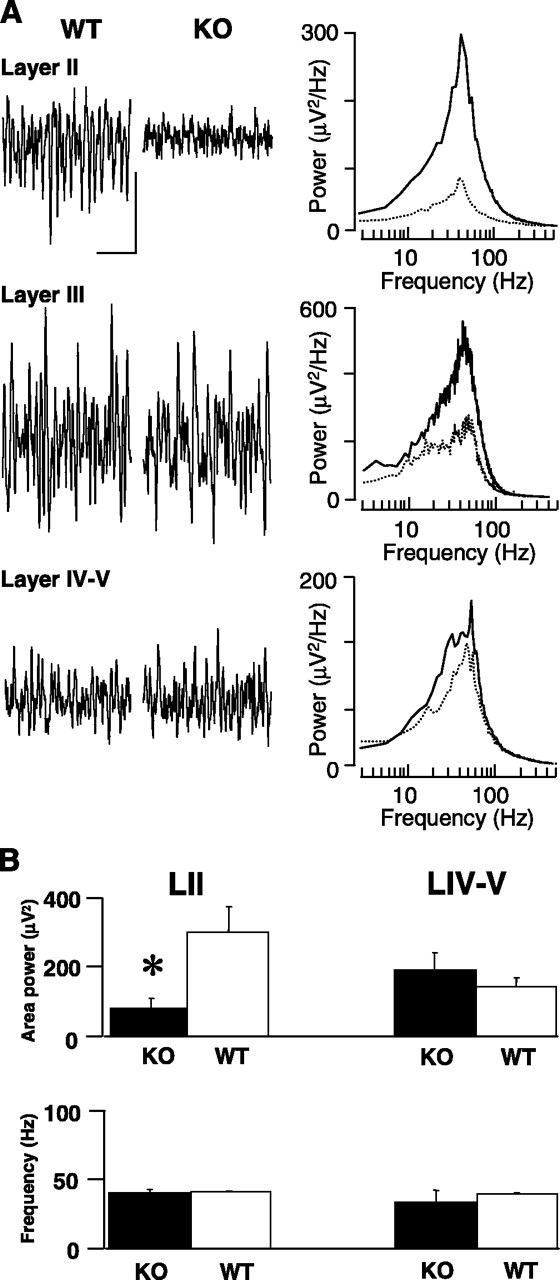
Kainate-induced field gamma rhythms were disrupted in superficial mEC layers but not in deep layers. A, Example field potential traces (0.5 s) showing oscillatory activity in superficial layers II and III and deep layer IV/V in slices from control (WT) and LPA1−/− (KO) mice. The right panel shows pooled power spectra (n = 5) of 60 s epochs of field activity from WT (solid line) and KO (dashed line) slices. Calibration: 0.05 mV, 100 ms. B, Bar graphs showing pooled data (mean ± SEM) from superficial (left panel) and deep (right panel) entorhinal cortex. Note that only the area power (not the frequency) is changed significantly (*) when slices from WT and KO mice are compared.
Because gamma rhythms in the mEC are dependent on the output from fast-spiking interneurons (Cunningham et al., 2003), we examined the profile of IPSPs in principal cells in the superficial layers to determine whether the reduction in the field was accompanied by a reduction in phasic inhibitory input. During population gamma rhythms in WT slices the principal cell intracellular recordings typically took the form of rhythmic trains of IPSPs. IPSPs were phase-locked to the local field potential rhythm and occurred at the same frequency (Fig. 2). In cells from LPA1−/− mice the trains of IPSPs were far more erratic, with individual IPSPs occurring far less often than in cells from WT mice and being of a smaller amplitude, on average. IPSPs seen during gamma oscillations in WT slices had a mean amplitude of 9.1 ± 0.1 mV (at −30 mV resting membrane potential). In slices from LPA1−/− mice mean amplitude was slightly smaller at 8.1 ± 0.1 mV. Cumulative probability analysis of IPSP amplitude revealed a highly significant difference (p < 0.05; n = 4), although the kinetics of these IPSPs was not significantly different in tissue from the two types of mice (WT decay time, 37.3 ± 4.7 ms; LPA1−/− decay time, 24.7 ± 12.7 ms; p > 0.05; n = 4). Cumulative probability analysis of inter-IPSP intervals revealed a highly significant increase (p < 0.05; n = 4) (Fig. 2B). To establish whether these findings were attributable to a failure of the kainate model to activate interneurons, we also recorded stimulus-evoked IPSPs in principal cells under nonoscillating conditions in which GABAB receptor-mediated and ionotropic glutamate receptor-mediated events were blocked pharmacologically. At three stimulus intensities (2.5, 5, 10 V) delivered to the superficial mEC proximal to the recorded principal cells, evoked IPSPs in slices from LPA1−/− mice were significantly smaller than those in WT slices (Fig. 2C). Mean reduction in IPSP amplitude was 45.0 ± 7.5% (p < 0.05; n = 10). No alteration was seen in the kinetics of evoked IPSPs in either condition.
Figure 2.
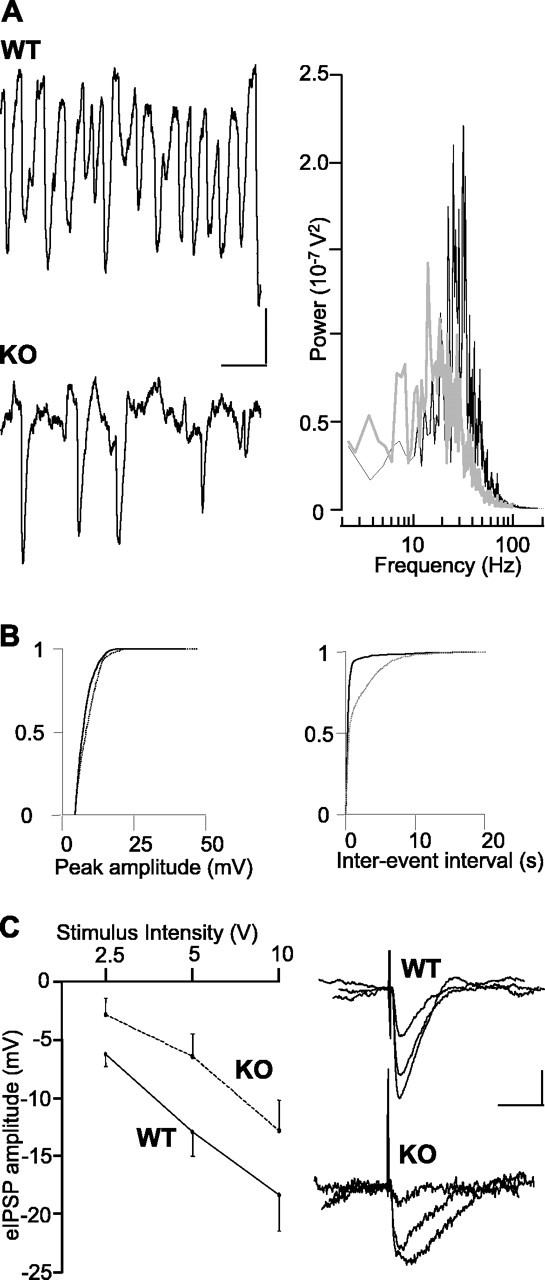
Disrupted field gamma rhythms are associated with disrupted phasic inhibition in superficial principal cells. A, The pattern of IPSP trains in LII stellate cells was disrupted in LPA1−/− mice (KO) compared with controls (WT) at −30 mV. IPSPs were seen less frequently and with more variable inter-IPSP intervals. Power spectra of IPSP trains showed a much reduced inhibitory gamma frequency input to these cells. WT data are plotted as a solid line; KO data are plotted as a gray line. Calibration: 2 mV, 50 ms. B, Cumulative probability plots for IPSP recordings showing a small but significant decrease in amplitude (left graph) and large increase in interevent interval (right graph) in stellate cells during population gamma rhythms. KO data are plotted as dashed lines. C, Stimulus–response relationship for monosynaptically evoked IPSPs (eIPSPs) onto stellate cells in nonoscillating slices. At each stimulus intensity, the applied IPSP amplitudes in slices from LPA1−/− mice (KO) were ∼30–50% smaller than those evoked in control (WT) slices. Data are illustrated as mean ± SEM. Calibration: 5 mV, 10 ms.
Such a large reduction in both stimulus-evoked and oscillatory GABAA receptor-mediated inhibition would be expected to increase the excitability of principal cells in the superficial mEC. In LII stellate cells (where the decrease in kainate-induced gamma field power was largest) we saw an increase in ongoing action potential generation during the gamma rhythm (Fig. 3A). WT stellate cells generated action potentials with a frequency of 1.3 ± 0.5 Hz, whereas stellate cells in slices from LPA1−/− mice generated action potentials at 5.5 ± 0.5 Hz (p < 0.05; n = 4). This increase in output was not accompanied by a significant change in mean membrane potential during the gamma oscillation (WT membrane potential, −59 ± 3 mV; LPA1−/− membrane potential, −56 ± 3 mV; p > 0.05; n = 5). However, a significant increase in input resistance was seen (LPA1−/−, 31 ± 3 MΩ; WT, 24 ± 2 MΩ; p < 0.05; n = 7) along with a large increase in the frequency of EPSPs in slices from LPA1−/− mice during the gamma rhythm (Fig. 3A). No change in kinetics (WT decay time, 69.8 ± 10.6 ms; LPA1−/− decay time, 69.6 ± 19.3 ms; p > 0.05; n = 6) or mean amplitude (WT peak amplitude, 2.5 ± 0.3 ms; LPA1−/− peak amplitude, 2.4 ± 0.4 ms; p > 0.05; n = 6) of these EPSPs was seen, but a highly significant increase in frequency from 1.7 ± 0.1 to 2.5 ± 0.1 Hz was apparent when WT was compared with LPA1−/− tissue (p < 0.05; n = 6) (Fig. 3B).
Figure 3.
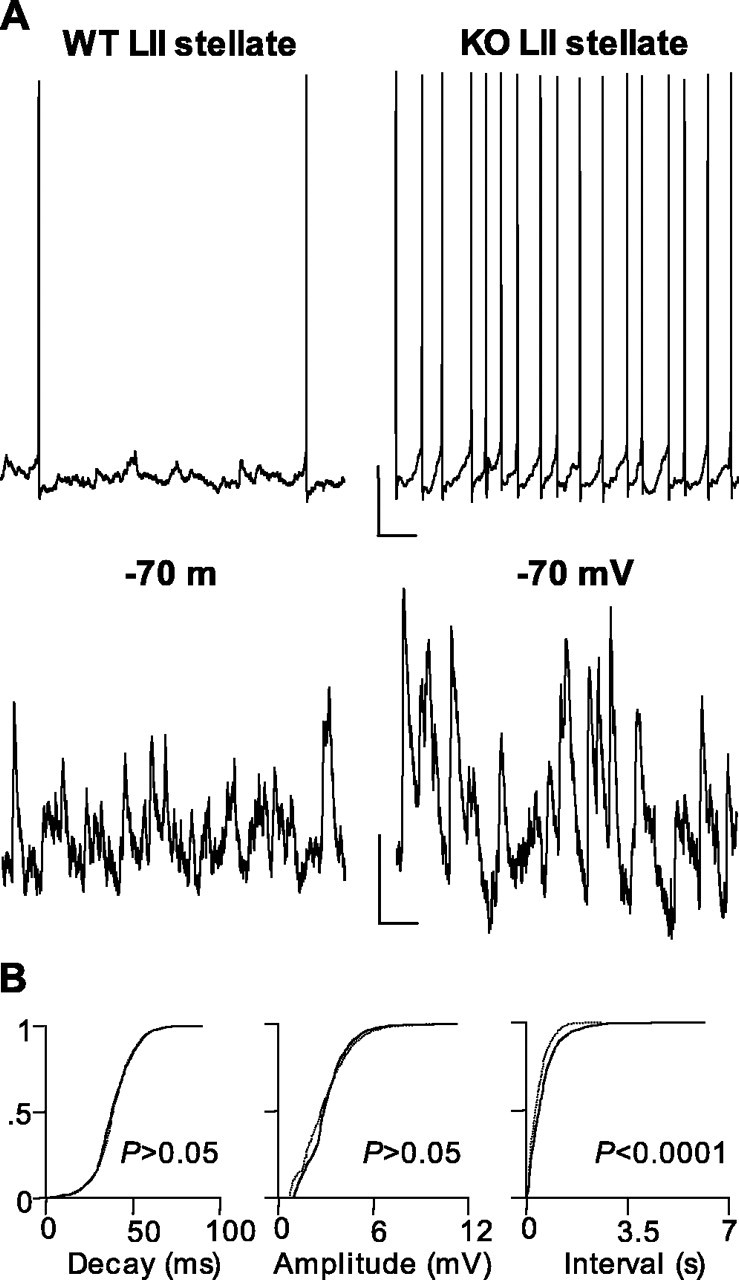
Elevated excitatory activity in superficial mEC layers in slices from LPA1−/− mice. A, Example traces showing altered spike generation and EPSP inputs to stellate cells during population gamma oscillations in control (WT, left panel) and LPA1−/− (KO, right panel) slices. Spike trains were taken at resting membrane potential (−58 mV) in each case. Cells were held at −70 mV for EPSP recordings. Note the large increase in both excitatory input and spike output in these cells. Calibration: 10 mV, 200 ms (action potentials, top panels); 2 mV, 50 ms (EPSPs, bottom panels). B, Cumulative probability plots for EPSP data showing no significant change in decay time or peak amplitude but a large increase in the rate of EPSP invasion of stellate cells from LPA1−/− mice (KO) compared with controls (WT).
Other models of psychosis in rodents have reported specific reductions in parvalbumin-positive interneurons in prelimbic areas (Cochran et al., 2002, 2003), so we examined whether the decrease in functional inhibition seen in mEC tissue from LPA1−/− mice also could be attributable to a reduction in the number of these interneurons. mEC slices from LPA1−/− mice showed that the total number of GABA-reactive cells was only 53% of WT number (WT, 84 ± 12; LPA1−/−, 45 ± 6; p < 0.05; n = 8) (Fig. 4). This reduction in GABAergic interneuron number appeared to be predominantly attributable to a loss of parvalbumin-positive neurons. In slices from LPA1−/− mice the parvalbumin-immunoreactive cells were only 63% of the number found in WT slices (WT, 120 ± 15; LPA1−/−, 77 ± 8; p < 0.05; n = 8), whereas calretinin-immunoreactive cell numbers were not significantly different (WT, 84 ± 10; LPA1−/−, 89 ± 9; p > 0.05; n = 5).
Figure 4.
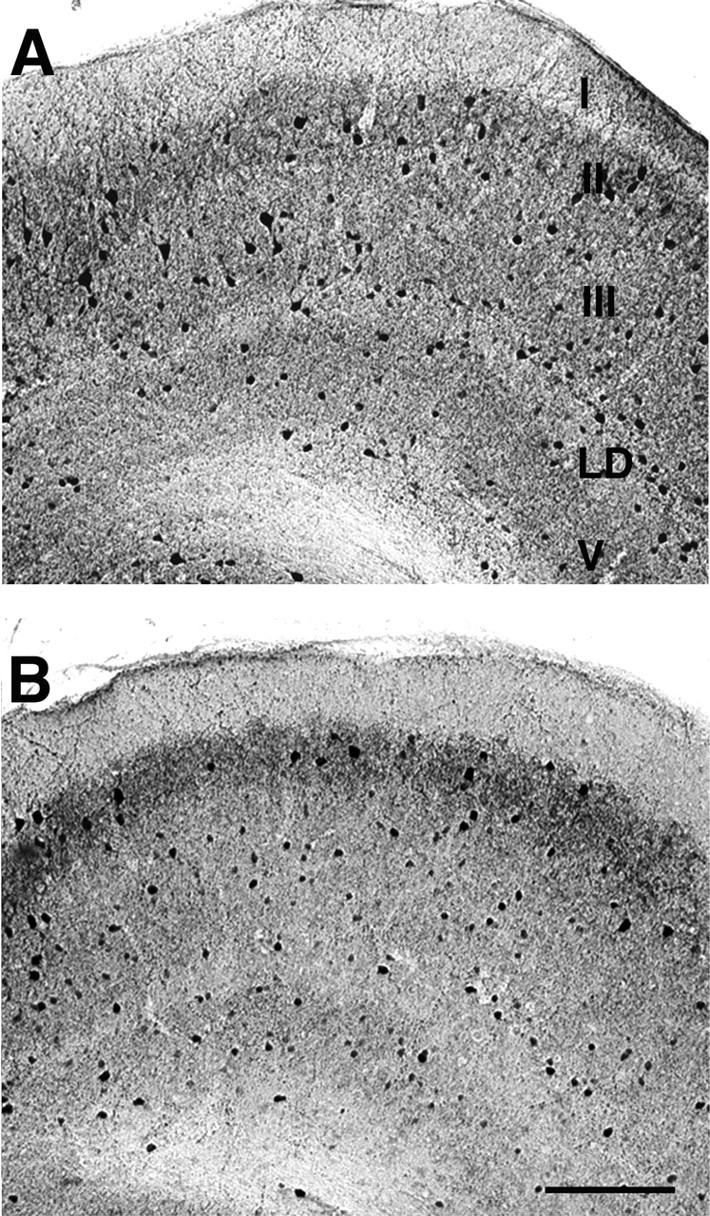
LPA1−/− mice show reduced parvalbumin-immunoreactive interneuron numbers in mEC. Shown is the distribution of parvalbumin-immunoreactive neurons in the entorhinal cortex of WT (A) and KO (B) mice. Parvalbumin-immunoreactive cells were counted in microscopic view fields, as shown in example panels. KO entorhinal cortex (B) showed a decrease in the number of parvalbumin-immunoreactive neurons in layer II compared with WT (A) (see Results). I–III, V, Layers I–III and V; LD, lamina dissecans. Scale bar: (in B) A, B, 65 μm.
Because the decrease in inhibition-based gamma rhythms was seen only in superficial mEC in tissue from LPA1−/− mice, we further analyzed the incidence of the above interneuron markers in a lamina-specific manner. Cell bodies immunoreactive for GABA, parvalbumin, and calretinin were quantified in LII/LIII and deep layers below lamina dissecans. In LPA1−/− tissue a decrease in the number of GABA-reactive cells per unit area was seen in LII when compared with WT slices (LPA1−/−, 3020 ± 285 mm−2; WT, 4656 ± 391 mm−2; p < 0.05). The majority of this decrease in GABA-reactive neurons was correlated selectively with a decrease in parvalbumin-immunoreactive cell bodies (LPA1−/−, 3687 ± 206 mm−2; WT, 5198 ± 336 mm−2; p < 0.05), but not calretinin (LPA1−/−, 3097 ± 365 mm−2; WT, 3255 ± 407 mm−2; p > 0.05). This decrease in parvalbumin-positive GABAergic neurons was selective for LII. No significant differences were seen for parvalbumin-reactive neurons (LPA1−/−, 5187 ± 512 mm−2; WT, 5366 ± 570 mm−2; p > 0.05) or calretinin-reactive neurons (LPA1−/−, 3089 ± 340 mm−2; WT, 2958 ± 261 mm−2; p > 0.05) in LIII. In deep layers, where no change in gamma rhythms was seen, no significant change in parvalbumin-reactive cell body numbers was seen either (LPA1−/−, 2960 ± 226 mm−2; WT, 2811 ± 197 mm−2; p > 0.05). Interestingly, a decrease in the mean number of calretinin-immunopositive cell bodies was seen; however, this change did not reach significance in the present study (LPA1−/−, 2006 ± 195 mm−2; WT, 2409 ± 174 mm−2; p > 0.05).
Hippocampal gamma rhythms
In contrast to the superficial layers of the mEC, bath application of kainate (50–300 nm) generated similar robust field oscillations in the gamma band in slices from both WT and LPA1−/− mice (Fig. 5A). Peak frequency was 25.8 ± 2.1 Hz for slices from WT mice and 26.0 ± 1.9 Hz for LPA1−/− tissue (p > 0.05; n = 12). A small decrease in mean peak power and area of the kainate-induced gamma rhythm was seen at a concentration of 200 nm, but this did not reach significance (WT area, 4856.1 ± 1130.3 μV2; LPA1−/− area, 3507.4 ± 1021.3 μV2; p > 0.05; n = 12) (Fig. 5B).
Figure 5.
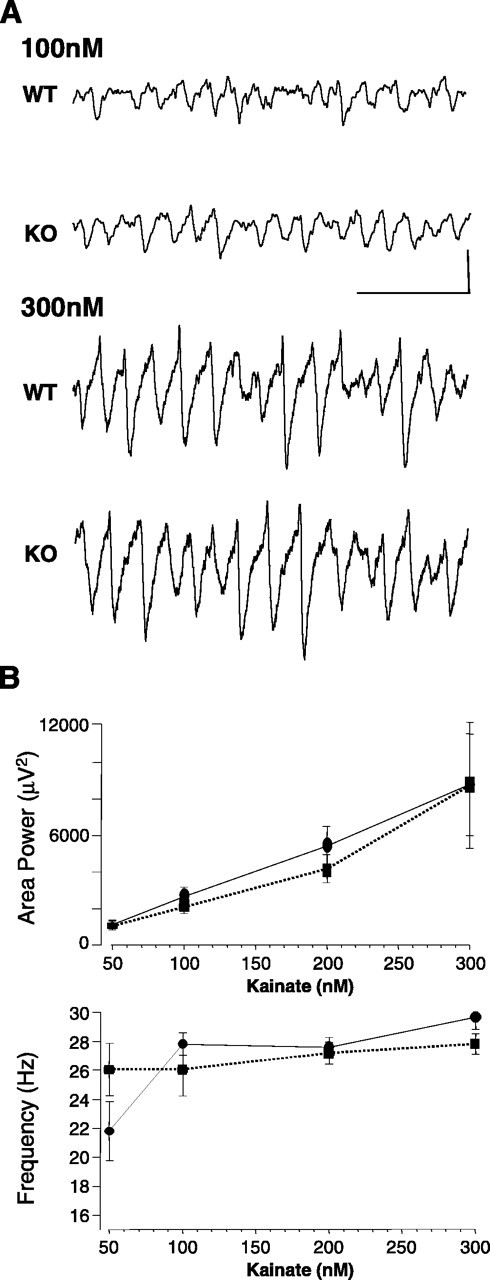
Power of hippocampal gamma rhythms is unchanged in slices from LPA1−/− mice. A, Example extracellular recordings showing lack of difference in gamma rhythms generated by 100 and 300 nm kainate in hippocampal area CA3 stratum radiatum. Calibration: 0.1 mV, 100 ms. B, Pooled data plots (mean ± SEM; n = 5) for the area power and peak frequency of field potential oscillations as a function of kainate concentration. No effects are seen when slices from control mice (solid lines) and LPA1−/− mice are compared (dashed lines).
Generation of gamma rhythms in the hippocampus has been shown to be related closely to the output from fast-spiking interneurons (Fisahn et al., 1998; Gloveli et al., 2005). We therefore also quantified the number of GABA-, calretinin-, and parvalbumin-positive interneurons in cornu ammonis in slices after the above electrophysiological measurements and compared these values with those for the total number of GABA-positive cells and calretinin-positive cells (Fig. 6). No significant difference was found in the total number of GABA-positive neurons that were labeled (WT GABA neurons, 70 ± 7; LPA1−/− GABA neurons, 66 ± 10; p > 0.5; n = 7). No change in the number of parvalbumin-positive interneurons was seen (WT, 39 ± 8; LPA1−/−, 36 ± 12; p > 0.5; n = 8) or the number of calretinin-positive cells (WT, 64 ± 5; LPA1−/−, 61 ± 11; p > 0.5; n = 5).
Figure 6.
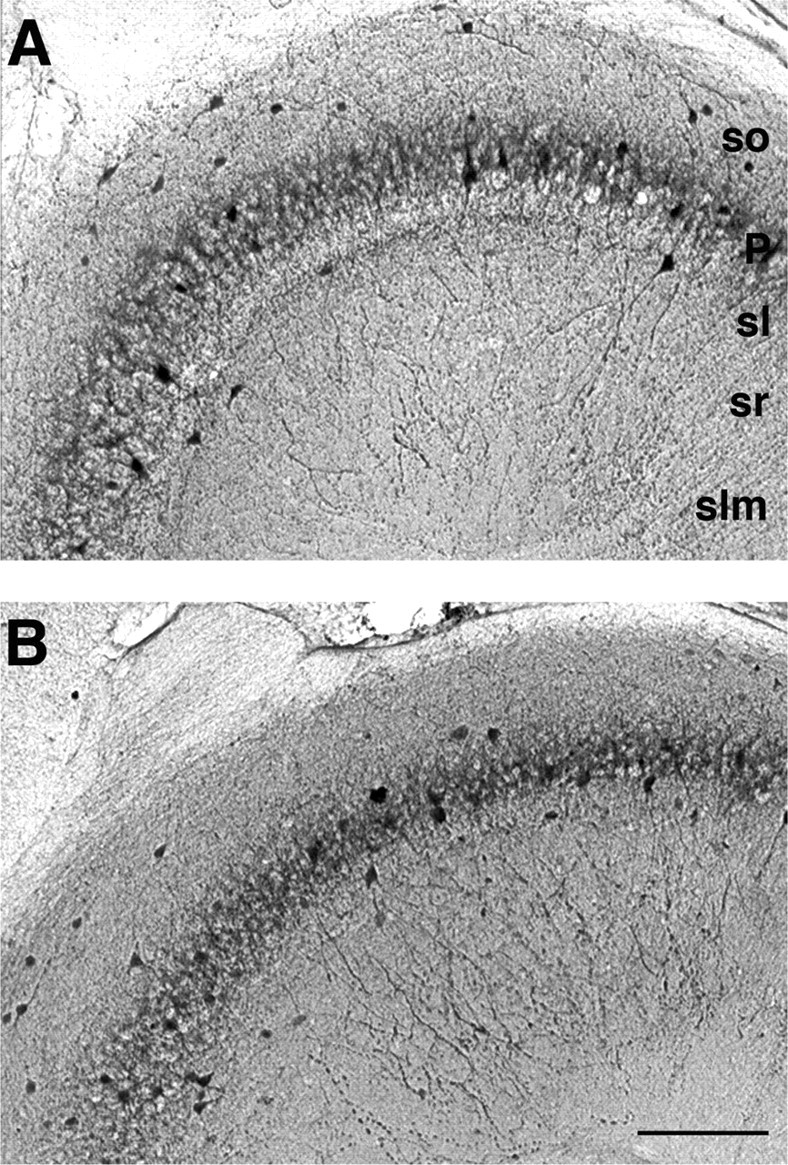
Numbers of parvalbumin-immunopositive interneurons are unchanged in area CA3 from LPA1−/− mice. The distribution of parvalbumin-immunoreactive neurons in area CA3 of WT (A) and KO (B) mice is shown. Parvalbumin-immunopositive cells were counted in microscopic view fields, as shown in example panels of CA3 region. No significant changes in the number of parvalbumin-immunopositive neurons were observed between WT (A) and KO (B) hippocampus (see Results). so, Stratum oriens; P, stratum pyramidale; sl, stratum lucidum; sr, stratum radiatum; slm, stratum lacunosum-moleculare. Scale bar: (in B) A, B, 140 μm.
Comparison with acute ketamine application
Acute administration of noncompetitive NMDA receptors antagonists is accepted as a good acute model of psychosis and both positive and negative symptoms of schizophrenia in both humans (Adler et al., 1999) and animals (Adams and Moghaddam, 1998). Given the similarities between the behavioral and neurochemical profile of LPA1−/− mice and clinical observations from schizophrenic patients (Harrison et al., 2003) and the existence of specific interneuron subtypes in LII of the entorhinal cortex with strong NMDA receptor-mediated drive (Jones and Buhl, 1993), we compared the main findings above from slices from LPA1−/− mice with effects of acute exposure of control slices (from C57BL/6J mice and Wistar rats) to ketamine (10–50 μm). In the superficial layers of the mEC the concentrations of ketamine at or above 20 μm produced a significant reduction in the power of field gamma oscillations generated by 400 nm kainate (Fig. 7A). Bath application of 20 μm ketamine reduced the area power in superficial mEC from 352.2 ± 64.3 to 199.7 ± 55.7 μV2 (p < 0.01; n = 7) and caused a small but nonsignificant change in mean frequency from 45 ± 3 to 39 ± 6 Hz (p > 0.05; n = 7). In contrast, no significant change in the power of field gamma oscillations in the deep mEC was seen at any ketamine concentration that was tested (p > 0.05; n = 7). Similarly, ketamine bath application produced no significant change in the power (Fig. 7B) and area power of kainate-induced persistent field gamma oscillations in area CA1 or CA3 in the hippocampus (control area, 11352 ± 583.0 μV2; 20 μm ketamine area, 10414.3 ± 511.5 μV2; p > 0.05; n = 5). These data showed that the anatomical profile of deficiencies in gamma rhythmogenesis in LPA1−/− mice was similar to that generated by acute, noncompetitive blockade of NMDA receptors. In the LPA1−/− model the decrease in gamma activity in superficial layers was associated with a decrease in IPSPs in principal cells and a concurrent increase in neuronal output. This also was seen with bath application of ketamine (Fig. 8) and again was accompanied by an increase in stellate cell membrane resistance (control, 27 ± 3; ketamine, 38 ± 2 MΩ; p < 0.05; n = 5). The same pattern of effects of ketamine application was seen in slices from both rats and mice.
Figure 7.
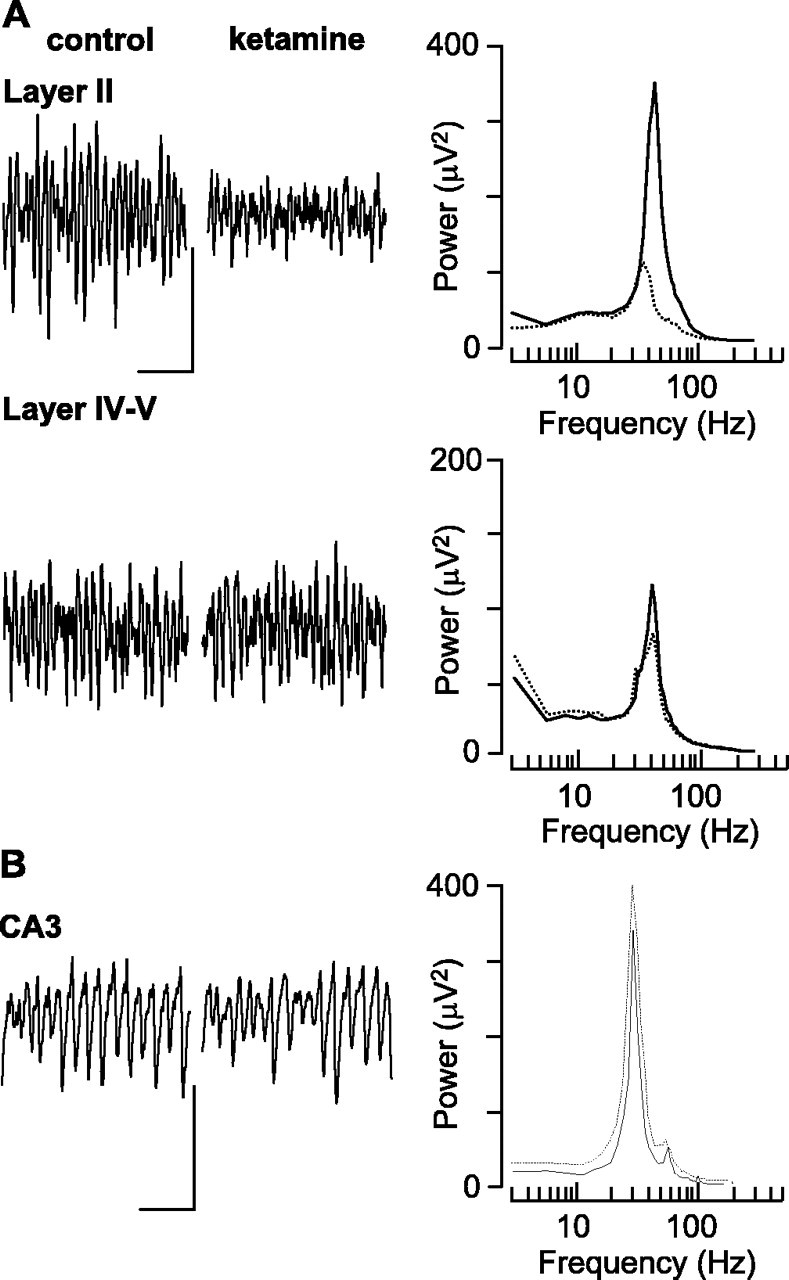
Ketamine application to control slices mimics the anatomical profile of disrupted gamma rhythms seen in LPA1−/− slices. A, Example traces show 0.5 s of field potential activity in superficial (layer II) and deep (layer IV/V) laminas in the presence and absence of ketamine (20 μm). Calibration: 50 μV, 100 ms. Pooled power spectra (n = 7) illustrate the selective reduction in gamma frequency oscillations in superficial entorhinal cortex. B, Example traces show 0.5 s of field potential activity in hippocampal area CA3 in the presence and absence of ketamine (20 μm). Calibration: 50 μV, 100 ms. Pooled power spectra (n = 7) show lack of effect of ketamine on hippocampal gamma rhythms. Control data are plotted as solid lines; data gathered in the presence of ketamine are plotted as dashed lines.
Figure 8.
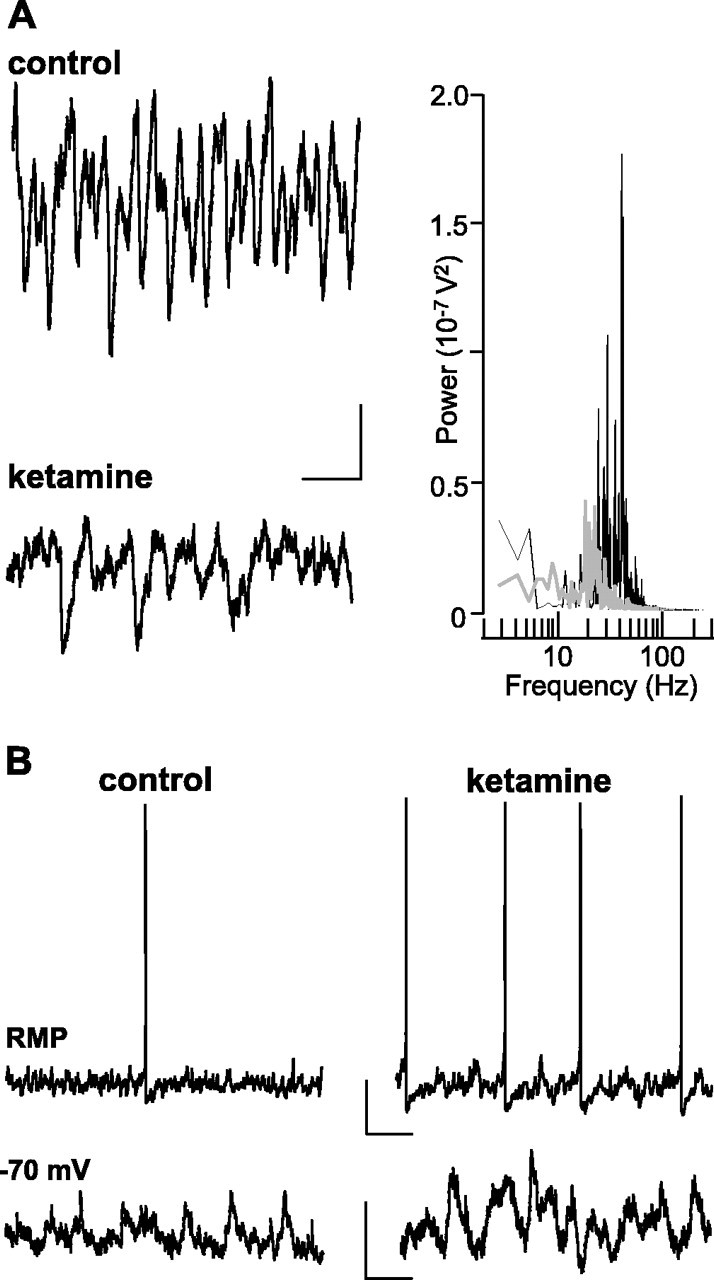
Ketamine-induced reduction in gamma rhythmogenesis is accompanied by disinhibition. A, IPSP trains associated with the field gamma rhythm are disrupted in the presence of ketamine. Example traces show stellate cell IPSPs recorded from −30 mV membrane potential in the presence and absence of ketamine (20 μm). Calibration: 2 mV, 50 ms. The graph shows power spectra from 60 s epochs of stellate cell IPSP trains in control (solid line) and ketamine (gray line) conditions. Note the large reduction in activity within the gamma band. B, Ketamine-induced disruption in gamma oscillations is associated with increased stellate cell output. Example traces show stellate cell spiking at resting membrane potential (−57 and −59 mV) in the presence and absence of ketamine (20 μm; top traces). Bottom traces show EPSPs at −70 mV membrane potential in the two conditions. Note the different time axis in the spike and EPSP traces. Calibration: 10 mV, 200 ms (spike traces); 2 mV, 50 ms (EPSP traces).
Discussion
Using two animal models of symptoms of schizophrenia, we have demonstrated a spatially selective disruption in the generation of gamma frequency network oscillations in the entorhinal cortex. In the case of both LPA1−/− mice and a bath application of ketamine the gamma oscillations were reduced in power in the superficial layers, whereas deep layer oscillations were relatively spared. In LPA1−/− mice this disruption in gamma rhythms was accompanied by a selective reduction in parvalbumin-immunopositive GABAergic cell bodies in LII, and in the presence of ketamine the superficial interneuron output was reduced. In neither model was a change in gamma activity or parvalbumin immunoreactivity seen in isolated hippocampal slices.
Gamma oscillations have been associated with sensory processing for a number of years. Original studies by Singer and Gray (1995) demonstrated that a synchronous pattern of neuronal output from neurons in anatomically disparate areas of the cortex occurs at gamma frequencies in response to object perception in the sensorium (Engel et al., 1991; Rodriguez et al., 1999). Motor performance also is associated with changes in gamma activity (Aoki et al., 1999), and the interaction between sensory and motor systems probed by prepulse inhibition of startle tests (which reveal sensorimotor-gating deficits in schizophrenia) involves the entorhinal cortex (Goto et al., 2002). More recent work has indicated that this pattern of synchronous network activity at gamma frequencies is linked inherently to processes involved in aspects of short-term memory (working memory, declarative memory) (Miltner et al., 1999), context representation and attention (Tiitinen et al., 1993; Borgers et al., 2005), and the mismatch negativity response (Marshall et al., 1996). Each of these processes also is disrupted in patients with psychiatric illness (Weinberger et al., 1986; Elvevag and Goldberg, 2000). In terms of deficits in gamma rhythmogenesis specifically related to schizophrenia, work from McCarley and coworkers has shown a deficit in gamma rhythm generation and associated synchrony in the neocortex during visual and auditory processing tasks (Spencer et al., 2003, 2004).
The generation of persistent gamma rhythms in the entorhinal cortex has mechanistic similarities to those seen in hippocampus (Whittington et al., 1995; Fisahn et al., 1998; Cunningham et al., 2003). Basically, ongoing axonal activity in principal neuronal axons provides a drive to GABAergic interneurons. The resulting output from a subset of these interneurons targeting principal cell somata and axonal initial segments serves to modulate principal cell firing patterns phasically so that the highest probability of spike generation occurs approximately once every 25 ms (Whittington and Traub, 2003). The interneuron subtypes implicated in this mechanism all target the principal cell perisomatic region, and most are immunopositive for parvalbumin (Gloveli et al., 2005; Mann et al., 2005). Markers for these interneuron subtypes are reduced significantly in prefrontal cortical areas in schizophrenics, and some similar changes have been reported in entorhinal cortex (Arnold, 2000). Glutamic acid decarboxylase isoform 67 (GAD67), a general marker for GABAergic interneurons, is reduced selectively in gamma-generating layers of cortex (Akbarian et al., 1995). Also, GABA transporter 1 (GAT1) is decreased in the same areas (Volk et al., 2001), suggesting that both synthesis and reuptake of GABA are disrupted in schizophrenics. The origins of this disruption appear to coincide with a reduction in parvalbumin signal (Hashimoto et al., 2003). Although the number of parvalbumin-positive neurons is unchanged, the function of these neurons appears to be disrupted significantly. In particular, chandelier cells, which provide very strong feedback inhibition to axons via chains of inhibitory synapses (cartridges), do so to a much lesser extent in schizophrenia (Woo et al., 1998). It is not clear as yet why subtypes of interneuron are susceptible in schizophrenia and the LPA1 knock-out mice. Evidence for differences in excitatory amino acid receptors on interneurons has been suggested in hippocampus (Grunze et al., 1996). The similarities between the functional deficits in LPA1−/− mice and acute application of ketamine suggest a role for NMDA receptors, but which subtypes are responsible remains to be elucidated.
The present study provides evidence for a similar mechanism at work in the entorhinal cortex. The loss of parvalbumin signal in LII medial entorhinal cortex of LPA1−/− mice, coupled with the enhanced excitability of superficial principal neurons and the reduction in inhibitory control over principal neurons (both during gamma rhythms and evoked by electrical stimulation), suggests that a selective reduction in perisomatic inhibition may underlie the deficits in gamma rhythmogenesis. LPA1 receptor expression is regulated developmentally (Weiner et al., 1998). LPA1 expression is high before birth, disappears at birth, then reappears during the first postnatal week and peaks at postnatal day 18. The expression profile of LPA1 after birth parallels the expression profile of other markers associated with interneuron function. For instance, parvalbumin expression and the Kv3.1b potassium channel expression in interneurons start toward the end of the first postnatal week and tend to peak in the second–third week, similar to LPA1 (Bergmann et al., 1991; Du et al., 1996). LPA1 expression also has been associated with the generation of neurons from progenitor cells and their subsequent migration and survival (Kingsbury et al., 2003). Although not specific for parvalbumin-positive neurons, the absence of LPA1 receptors may have a predominant effect on network function associated with these cells because they are highly active and have such a large effect on population responses and there are, relatively, very few of them. The correlation among the degree of loss of parvalbumin signal, principal cell inhibition, and the power of gamma rhythms (∼40% in each case) supports this notion.
The similarity between the deficits in network function seen in the LPA1−/− mice and with acute application of ketamine suggests that the function of LII parvalbumin-immunopositive interneurons also was disrupted in this model. Noncompetitive NMDA receptor antagonists generate psychomotor-gating deficits and other behavioral responses closely associated with schizophrenia (Ellison, 1995) and selectively enhance c-fos expression in entorhinal and prefrontal areas (Vaisanen et al., 2004). Parvalbumin-immunopositive basket cells in LII entorhinal cortex are activated strongly by NMDA receptor agonists (Jones and Buhl, 1993), and these cells possess a marked tonic excitatory component to their drive during gamma oscillations (Cunningham et al., 2003). Blockade of NMDA receptors with ketamine reduces the output from superficial basket cells concurrently with a selective drop in gamma power in superficial layers. Disinhibition of the entorhinal cortex also has been reported in slices from rats pretreated with NMDA receptor antagonists (Dugladze et al., 2004). It is not clear why ketamine produces a similar pattern of selective reduction of gamma power in superficial layers. Too little is known at present about interneuron subtypes and distribution to suggest different inhibitory circuits in deep and superficial layers. However, it is interesting to note that spontaneous inhibitory events are different in the two layers (Woodhall et al., 2005) and that synaptic plasticity also is expressed differentially (Solger et al., 2004).
The lack of effect of either model on hippocampal gamma rhythmogenesis is in stark contrast to the observations in entorhinal cortex. The hippocampus has many parvalbumin-positive interneurons (Gulyas et al., 1991), but their number appears to be inert to the removal of the LPA1 receptor. The reasons for this remain to be resolved, but it is interesting to consider that, in contrast to parvalbumin-positive interneurons in the superficial entorhinal cortex, neurons with this signal in adult hippocampus receive relatively little drive via NMDA receptors (Nyiri et al., 2003) (A. Rozov and H. Monyer, personal communication). This also may be related to the lack of effect of NMDA receptor antagonists on hippocampal gamma (Fisahn et al., 1998). Postmortem studies of hippocampi from schizophrenic patients show selective reduction in parvalbumin-positive neurons in all hippocampal subfields, whereas calretinin-positive cells remain unchanged (Zhang and Reynolds, 2002; Knable et al., 2004). No such changes in parvalbumin signal were seen in either of the two models that were used, suggesting that the LPA1 receptor transduction pathway is not responsible for the above changes in humans. Ketamine has been reported to reduce hippocampal parvalbumin signal after repeated exposure (Keilhoff et al., 2004) in rodents, suggesting that the duration of exposure in the present study (4–8 h) was insufficient to precipitate such changes. However, in vivo the change in superficial layer network activity would be expected to influence hippocampal rhythmicity. Ketamine administration or lesion of entorhinal–hippocampal connectivity results in a change in the profile of oscillations in cornu ammonis (Charpak et al., 1995; Ylinen et al., 1995).
In summary, two models of psychiatric illness associated with sensorimotor-gating deficit generated a pattern of disruption in gamma rhythms specific to the superficial entorhinal cortex. In each case the disruption was accompanied by an apparent reduction in parvalbumin-immunopositive interneurons or a decrease in output of fast-spiking superficial entorhinal interneurons. Given that these interneurons have been shown to have a relatively powerful input via NMDA receptors, we speculate that specific functional deficits in this subset of fast-spiking inhibitory cells may underlie some of the behavioral correlates of cortical dysfunction seen in psychosis.
Footnotes
This work was supported by the Medical Research Council (United Kingdom) and The Wellcome Trust.
References
- Adams B, Moghaddam B (1998). Corticolimbic dopamine neurotransmission is temporally dissociated from the cognitive and locomotor effects of phencyclidine. J Neurosci 18:5545–5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler CM, Malhotra AK, Elman I, Goldberg T, Egan M, Pickar D, Breier A (1999). Comparison of ketamine-induced thought disorder in healthy volunteers and thought disorder in schizophrenia. Am J Psychiatry 156:1646–1649. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE Jr, Jones EG (1995). Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry 52:258–266. [DOI] [PubMed] [Google Scholar]
- Aoki F, Fetz EE, Shupe L, Lettich E, Ojemann GA (1999). Increased gamma-range activity in human sensorimotor cortex during performance of visuomotor tasks. Clin Neurophysiol 110:524–537. [DOI] [PubMed] [Google Scholar]
- Arnold SE (2000). Cellular and molecular neuropathology of the parahippocampal region in schizophrenia. Ann NY Acad Sci 911:275–292. [DOI] [PubMed] [Google Scholar]
- Bachus SE, Hyde TM, Herman MM, Egan MF, Klienman JE (1997). Abnormal cholecystokinin mRNA levels in entorhinal cortex of schizophrenics. J Psychiatr Res 31:233–256. [DOI] [PubMed] [Google Scholar]
- Bergmann I, Nitsch R, Frotscher M (1991). Area-specific morphological and neurochemical maturation of non-pyramidal neurons in the rat hippocampus as revealed by parvalbumin immunocytochemistry. Anat Embryol (Berl) 184:403–409. [DOI] [PubMed] [Google Scholar]
- Borgers C, Epstein S, Kopell NJ (2005). Background gamma rhythmicity and attention in cortical local circuits: a computational study. Proc Natl Acad Sci USA 102:7002–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff DL, Grillon C, Geyer MA (1992). Sensorimotor gating and schizophrenia. Arch Gen Psychiatry 49:206–215. [DOI] [PubMed] [Google Scholar]
- Brody SA, Dulawa SC, Conquet F, Geyer MA (2004). Assessment of a prepulse inhibition deficit in a mutant mouse lacking mGluR5 receptors. Mol Psychiatry 9:35–41. [DOI] [PubMed] [Google Scholar]
- Charpak S, Pare D, Llinás R (1995). The entorhinal cortex entrains fast CA1 hippocampal oscillations in the anaesthetized guinea-pig: role of the monosynaptic component of the perforant path. Eur J Neurosci 7:1548–1557. [DOI] [PubMed] [Google Scholar]
- Chrobak JJ, Buzsáki G (1998). Gamma oscillations in the entorhinal cortex of the freely behaving rat. J Neurosci 18:388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran SM, Fujimura M, Morris BJ, Pratt JA (2002). Acute and delayed effects of phencyclidine upon mRNA levels of markers of glutamatergic and GABAergic neurotransmitter function in the rat brain. Synapse 46:206–214. [DOI] [PubMed] [Google Scholar]
- Cochran SM, Kennedy M, McKerchar CE, Steward LJ, Pratt JA, Morris BJ (2003). Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology 28:265–275. [DOI] [PubMed] [Google Scholar]
- Cunningham MO, Davies CH, Buhl EH, Kopell N, Whittington MA (2003). Gamma oscillations induced by kainate receptor activation in the entorhinal cortex in vitro J Neurosci 23:9761–9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin NM, Ellenbroek BA, Cools AR, Coenen AM, van Luijtelaar EL (1999). Differential effects on gating of auditory evoked potentials and prepulse inhibition in rats. Psychopharmacology (Berl) 142:9–17. [DOI] [PubMed] [Google Scholar]
- Dickson CT, Biella G, de Curtis M (2000). Evidence for spatial modules mediated by temporal synchronization of carbachol-induced gamma rhythm in medial entorhinal cortex. J Neurosci 20:7846–7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhang L, Weiser M, Rudy B, McBain CJ (1996). Developmental expression and functional characterization of the potassium channel subunit Kv3.1b in parvalbumin-containing interneurons of the rat hippocampus. J Neurosci 16:506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugladze T, Lepsveridze E, Breustedt J, Kehrer C, Heinemann U, Gloveli T (2004). Effects of phencyclidines on signal transfer from the entorhinal cortex to the hippocampus in rats. Neurosci Lett 354:185–188. [DOI] [PubMed] [Google Scholar]
- Ellison G (1995). The N-methyl-d-aspartate antagonists phencyclidine, ketamine and dizocilpine as both behavioral and anatomical models of the dementias. Brain Res Brain Res Rev 20:250–267. [DOI] [PubMed] [Google Scholar]
- Elvevag B, Goldberg TE (2000). Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol 14:1–21. [PubMed] [Google Scholar]
- Engel AK, Konig P, Kreiter AK, Singer W (1991). Interhemispheric synchronization of oscillatory neuronal responses in cat visual cortex. Science 252:1177–1179. [DOI] [PubMed] [Google Scholar]
- Falkai P, Schneider-Axmann T, Honer WG (2000). Entorhinal cortex pre-alpha clusters in schizophrenia: quantitative evidence of a developmental abnormality. Biol Psychiatry 47:937–943. [DOI] [PubMed] [Google Scholar]
- Fell J, Klaver P, Lehnertz K, Grunwald T, Schaller C, Elger CE, Fernandez G (2001). Human memory formation is accompanied by rhinal–hippocampal coupling and decoupling. Nat Neurosci 4:1259–1264. [DOI] [PubMed] [Google Scholar]
- Fisahn A, Pike FG, Buhl EH, Paulsen O (1998). Cholinergic induction of network oscillations at 40 Hz in the hippocampus in vitro Nature 394:186–189. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Braff DL (1987). Startle habituation and sensorimotor gating in schizophrenia and related animal models. Schizophr Bull 13:643–668. [DOI] [PubMed] [Google Scholar]
- Gloveli T, Dugladze T, Saha S, Monyer H, Heinemann U, Traub RD, Whittington MA, Buhl EH (2005). Differential involvement of oriens/pyramidale interneurons in hippocampal network oscillations in vitro J Physiol (Lond) 562:131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogos JA, Santha M, Takacs Z, Beck KD, Luine V, Lucas LR, Nadler JV, Karayiorgou M (1999). The gene encoding praline dehydrogenase modulates sensorimotor gating in mice. Nat Genet 21:434–439. [DOI] [PubMed] [Google Scholar]
- Goto K, Ueki A, Iso H, Morita Y (2002). Reduced prepulse inhibition in rats with entorhinal cortex lesions. Behav Brain Res 134:201–207. [DOI] [PubMed] [Google Scholar]
- Grunze HC, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW, Greene RW (1996). NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci 16:2034–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas AI, Toth K, Danos P, Freund TF (1991). Subpopulations of GABAergic neurons containing parvalbumin, calbindin D28k, and cholecystokinin in the rat hippocampus. J Comp Neurol 312:371–378. [DOI] [PubMed] [Google Scholar]
- Harrison SM, Reavill C, Brown G, Brown JT, Cluderay JE, Crook B, Davies CH, Dawson LA, Grau E, Heidbreder C, Hemmati P, Hervieu G, Howarth A, Hughes ZA, Hunter AJ, Latcham J, Pickering S, Pugh P, Rogers DC, Shillam CS, Maycox PR (2003). LPA1 receptor-deficient mice have phenotypic changes observed in psychiatric disease. Mol Cell Neurosci 24:1170–1179. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA (2003). Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 23:6315–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemby SE, Gisberg SD, Brunk B, Arnold SE, Tojanowski JQ, Eberwine JH (2002). Gene expression profile for schizophrenia: discrete neuron transcription patterns in the entorhinal cortex. Arch Gen Psychiatry 59:631–640. [DOI] [PubMed] [Google Scholar]
- Jones RS, Buhl EH (1993). Basket-like interneurons in layer II of the entorhinal cortex exhibit a powerful NMDA-mediated synaptic excitation. Neurosci Lett 149:35–39. [DOI] [PubMed] [Google Scholar]
- Keilhoff G, Becker A, Grecksch G, Wolf G, Bernstein HG (2004). Repeated application of ketamine to rats induces changes in the hippocampal expression of parvalbumin, neuronal nitric oxide synthase and cFOS similar to those seen in human schizophrenia. Neuroscience 126:591–598. [DOI] [PubMed] [Google Scholar]
- Kingsbury MA, Rehen SK, Contos JJ, Higgins CM, Chun J (2003). Non-proliferative effects of lysophosphatidic acid enhance cortical growth and folding. Nat Neurosci 6:1292–1299. [DOI] [PubMed] [Google Scholar]
- Knable MB, Barci BM, Webster MJ, Meador-Woodruff J, Torrey EF (2004). Molecular abnormalities of the hippocampus in severe psychiatric illness: postmortem findings from the Stanley Neuropathology Consortium. Mol Psychiatry 9:609–620. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW (2005). Cortical inhibitory interneurons and schizophrenia. Nat Rev Neurosci 6:312–324. [DOI] [PubMed] [Google Scholar]
- Mann EO, Suckling JM, Hajos N, Greenfield SA, Paulsen O (2005). Perisomatic feedback inhibition underlies cholinergically induced fast network oscillations in the rat hippocampus in vitro Neuron 45:105–117. [DOI] [PubMed] [Google Scholar]
- Marshall L, Molle M, Bartsch P (1996). Event-related gamma band activity during passive and active oddball tasks. NeuroReport 7:1517–1520. [DOI] [PubMed] [Google Scholar]
- Meincke U, Light GA, Geyer MA, Braff DL, Gouzoulis-Mayfrank E (2004). Sensitization and habituation of acoustic startle reflex in patients with schizophrenia. Psychiatry Res 126:51–61. [DOI] [PubMed] [Google Scholar]
- Miltner WH, Braun C, Arnold M, Witte H, Taub E (1999). Coherence of gamma-band EEG activity as a basis for associative learning. Nature 397:391–393. [DOI] [PubMed] [Google Scholar]
- Nyiri G, Stephenson FA, Freund TF, Somogyi P (2003). Large variability in synaptic N-methyl-d-aspartate receptor density on interneurons and a comparison with pyramidal cell spines in the rat hippocampus. Neuroscience 119:347–363. [DOI] [PubMed] [Google Scholar]
- Oranje B, Gispen-de Wied CC, Verbaten MN, Kahn RS (2002). Modulating sensory gating in healthy volunteers: the effects of ketamine and haloperidol. Biol Psychiatry 52:887–895. [DOI] [PubMed] [Google Scholar]
- Park S, Holzman PS (1992). Schizophrenics show spatial working memory deficits. Arch Gen Psychiatry 49:975–982. [DOI] [PubMed] [Google Scholar]
- Prasad KM, Patel AR, Muddasani S, Sweeney J, Keshavan MS (2004). The entorhinal cortex in first-episode psychotic disorders: a structural magnetic resonance imaging study. Am J Psychiatry 161:1612–1619. [DOI] [PubMed] [Google Scholar]
- Roberts C, Winter P, Hughes ZA, Langmead C, Maycox PR, Dawson LA (2005). Neurochemical changes in LPA1 receptor-deficient mice—a putative model of schizophrenia. Neurochem Res 30:371–377. [DOI] [PubMed] [Google Scholar]
- Rodriguez E, George N, Lachaux JP, Martinerie J, Renault B, Varela FJ (1999). Perception's shadow: long-distance synchronisation of human brain activity. Nature 397:430–433. [DOI] [PubMed] [Google Scholar]
- Seeman P, Ko F, Tallerico T (2005). Dopamine receptor contribution to the action of PCP, LSD, and ketamine psychotomimetics. Mol Psychiatry 10:877–883. [DOI] [PubMed] [Google Scholar]
- Singer W, Gray CM (1995). Visual feature integration and the temporal correlation hypothesis. Annu Rev Neurosci 18:555–586. [DOI] [PubMed] [Google Scholar]
- Solger J, Wozny C, Manahan-Vaughan D, Behr J (2004). Distinct mechanisms of bidirectional activity-dependent synaptic plasticity in superficial and deep layers of rat entorhinal cortex. Eur J Neurosci 19:2003–2007. [DOI] [PubMed] [Google Scholar]
- Spencer KM, Nestor PG, Niznikiewicz MA, Salisbury DF, Shenton ME, McCarley RW (2003). Abnormal neural synchrony in schizophrenia. J Neurosci 23:7407–7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer KM, Nestor PG, Perlmutter R, Niznikiewicz MA, Klump MC, Frumin M, Shenton ME, McCarley RW (2004). Neural synchrony indexes disordered perception and cognition in schizophrenia. Proc Natl Acad Sci USA 101:17288–17293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiitinen H, Sinkkonen J, Reinikainen K, Alho K, Lavikainen J, Naatanen R (1993). Selective attention enhances the auditory 40 Hz transient response in humans. Nature 364:59–60. [DOI] [PubMed] [Google Scholar]
- Tueting P, Costa E, Dwivedi Y, Guidotti A, Impagnatiello F, Manev R, Pesold C (1999). The phenotypic characteristics of heterozygous reeler mouse. NeuroReport 10:1329–1334. [DOI] [PubMed] [Google Scholar]
- Vaisanen J, Ihalainen J, Tanila H, Castren E (2004). Effects of NMDA receptor antagonist treatment on c-fos expression in rat brain areas implicated in schizophrenia. Cell Mol Neurobiol 24:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DR (1997). The biological basis of schizophrenia: new directions. J Clin Psychiatry 58:22–27. [PubMed] [Google Scholar]
- Weinberger DR, Berman KF, Zec RF (1986). Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Arch Gen Psychiatry 43:114–124. [DOI] [PubMed] [Google Scholar]
- Weiner JA, Hecht JH, Chun J (1998). Lysophosphatidic acid receptor gene vzg-1/lpA1/edg-2 is expressed by mature oligodendrocytes during myelination in the postnatal murine brain. J Comp Neurol 398:587–598. [PubMed] [Google Scholar]
- Whittington MA, Traub RD (2003). Interneuron diversity series: inhibitory interneurons and network oscillations in vitro Trends Neurosci 26:676–682. [DOI] [PubMed] [Google Scholar]
- Whittington MA, Traub RD, Jefferys JG (1995). Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature 373:612–615. [DOI] [PubMed] [Google Scholar]
- Woo T-U, Whitehead RE, Melchitzky DS, Lewis DA (1998). A subclass of gamma aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci USA 95:5341–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhall GL, Bailey SJ, Thompson SE, Evans DI, Jones RS (2005). Fundamental differences in spontaneous synaptic inhibition between deep and superficial layers of the rat entorhinal cortex. Hippocampus 15:232–245. [DOI] [PubMed] [Google Scholar]
- Ylinen A, Bragin A, Nadasdy Z, Jando G, Szabo I, Sik A, Buzsáki G (1995). Sharp wave-associated high frequency oscillation (200 Hz) in the intact hippocampus: network and intracellular mechanisms. J Neurosci 15:30–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk D, Austin M, Pierri J, Sampson A, Lewis D (2001). GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am J Psychiatry 158:256–265. [DOI] [PubMed] [Google Scholar]
- Zhang ZJ, Reynolds GP (2002). A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res 55:1–10. [DOI] [PubMed] [Google Scholar]