

Abstract
The receptor tyrosine kinase (RTK) Ret is activated by the formation of a complex consisting of ligands such as glial cell line-derived neurotrophic factor (GDNF) and glycerophosphatidylinositol-anchored coreceptors termed GFRαs. During activation, Ret translocates into lipid rafts, which is critical for functional responses to GDNF. We found that Ret was rapidly ubiquitinated and degraded in sympathetic neurons when activated with GDNF, but, unlike other RTKs that are trafficked to lysosomes for degradation, Ret was degraded predominantly by the proteasome. After GDNF stimulation, the majority of ubiquitinated Ret was located outside of lipid rafts and Ret was lost predominantly from nonraft membrane domains. Consistent with the predominance of Ret degradation outside of rafts, disruption of lipid rafts in neurons did not alter either the GDNF-dependent ubiquitination or degradation of Ret. GDNF-mediated survival of sympathetic neurons was inhibited by lipid raft depletion, and this inhibitory effect of raft disruption on GDNF-mediated survival was reversed if Ret degradation was blocked via proteasome inhibition. Therefore, lipid rafts sequester Ret away from the degradation machinery located in nonraft membrane domains, such as Cbl family E3 ligases, thereby sustaining Ret signaling.
Keywords: growth factor, signal transduction, lipid microdomains, sympathetic, neurotrophic, lysosome
Introduction
The activity of receptor tyrosine kinases (RTKs) is tightly regulated, given that several cancers are linked to the impaired downregulation of RTKs (Peschard and Park, 2003). A prevalent mechanism for the downregulation of RTKs is targeted degradation of the receptor itself. Ligand-dependent degradation of RTKs, such as epidermal growth factor receptor (EGFR), is accomplished via ubiquitination of the receptor by E3 ligases such as Cbl. Monoubiquitination acts as an internalization signal that leads, ultimately, to the delivery of activated receptors to lysosomes to be degraded (Bonifacino and Weissman, 1998; Dikic and Giordano, 2003; Hicke, 2001). A mechanism that is responsible for the degradation of cytoplasmic proteins, the proteasomal pathway, is initiated by the addition of chains of ubiquitin of four molecules or more, a process called polyubiquitination (Aguilar and Wendland, 2003). These polyubiquitin chains target these proteins to the proteasome, a large multiprotein complex that degrades proteins by the sequential removal of amino acids.
Ret is the receptor for the glial cell line-derived neurotrophic factor (GDNF) family ligands (GFLs), consisting of the four homologous growth factors GDNF, neurturin, artemin, and persephin (Baloh et al., 2000; Airaksinen and Saarma, 2002). GFLs do not bind directly to Ret to activate it but instead bind with high affinity to coreceptor molecules called GFRαs, which, as a GFL–GFRα complex, bind to and activate Ret. The functions of GDNF are remarkably pleotropic. GDNF is a potent survival- and growth-promoting factor for neurons of both the CNS and peripheral nervous system (Henderson et al., 1994; Arenas et al., 1995; Buj-Bello et al., 1995; Li et al., 1995; Mount et al., 1995; Oppenheim et al., 1995; Trupp et al., 1995). GDNF is also required for the proliferation and the migration of cranial parasympathetic and enteric neuronal precursors (Moore et al., 1996; Pichel et al., 1996; Sanchez et al., 1996; Enomoto et al., 2000). Furthermore, GDNF has functions outside the nervous system, such as in the ureteric bud branching that is critical for kidney genesis (Moore et al., 1996; Pichel et al., 1996; Sanchez et al., 1996).
Lipid rafts are membrane microdomains that are rich in cholesterol and sphingolipids, creating a more ordered membrane environment (Simons and Ikonen, 1997; Brown and London, 2000), which concentrates proteins that are modified with saturated lipids, such as glycerophospholipid (GPI)-anchored proteins such as the GFRαs. Lipid rafts are critical for signal transduction processes and are enriched in signaling proteins (Brown and London, 1998; Simons and Toomre, 2000; Tsui-Pierchala et al., 2002b). In the absence of ligand, Ret is located outside of lipid rafts in nonordered membrane regions (Tansey et al., 2000; Paratcha et al., 2001). Binding of ligand forms the active signaling complex including the GFRα coreceptors and induces Ret translocation into lipid rafts, and this translocation is required for maximal signal transduction and function of Ret (Tansey et al., 2000). However, whether lipid rafts influence the downregulation of Ret is unknown. Furthermore, whether ubiquitination and degradation have physiologic functions in Ret signaling, which were the topics of this investigation, is unknown.
Materials and Methods
Sympathetic neuron cultures and treatments.
Sympathetic neurons from the SCG of postnatal day 1 Sprague Dawley rats (Harlan Bioproducts for Science, Indianapolis, IN) were enzymatically dissociated and seeded onto collagen-coated 35 mm plates or two-well chamber slides (Nunc, Naperville, IL). The neurons were maintained in medium containing NGF (50 ng/ml; Harlan Bioproducts for Science) for 10–14 d before experimental treatments, as described previously (Easton et al., 1997). Treatments were performed as described in detail in the figure legends, and, in general, neurons were deprived of NGF for 24–48 h before stimulation with GDNF. For pharmacologic inhibition of the proteasome, clasto-lactacystin β-lactone (10 μm) and epoxomicin (5 μm; Biomol Research Laboratories, Plymouth Meeting, PA) were added to the medium 30 min before GDNF treatment. To inhibit lysosomal proteases, ammonium chloride (2 mm; Sigma, St. Louis, MO) and concanamycin (200 nm; Biomol) were added to the medium 1 h before GDNF treatment.
Immunoprecipitations.
Ret51, Ret9, and ubiquitin immunoprecipitations were performed as reported previously (Tsui-Pierchala et al., 2002a). Briefly, after the described treatments, sympathetic neurons were washed twice with ice-cold PBS, pH 7.4, and extracted with lysis buffer (Tris-buffered saline, pH 7.4, 1% Nonidet P-40, 10% glycerol, 500 μm sodium orthovanadate, and protease inhibitors) by gentle agitation for 20 min at 4°C. The lysates were then cleared and subjected to immunoprecipitation by addition of 10 μl of goat anti-Ret51 (C20; Santa Cruz Biotechnology, Santa Cruz, CA), goat anti-Ret9 (C19-G; Santa Cruz Biotechnology), or 5 μl of anti-multiubiquitin (Stressgen, Victoria, British Columbia, Canada). Next, 25 μl of protein A slurry and 50 μl of protein G slurry (Invitrogen, Carlsbad, CA) were added, and the samples were rotated at 4°C for 3 h. The immunoprecipitates were separated from the supernatant via centrifugation (2000 × g) for 3 min in a Microfuge, and these pellets were then washed three times with lysis buffer. The pellets were next prepared for SDS-PAGE by adding 25 μl of 2× sample buffer (Tris, pH 6.8, 20% glycerol, 4% SDS, 0.2% bromophenol blue, and 10% β-mercaptoethanol) and boiled for 5–10 min. Cbl and Cbl-b immunoprecipitations were performed as just described (C-15 and C-20, respectively; Santa Cruz Biotechnology).
Preparation of detergent-resistant membranes.
For the purposes of this study, we define a lipid raft as a membrane preparation that is resistant to extraction with 1% Triton X-100 at 4°C, which will also be called a detergent-resistant membrane (DRM). After the described treatments, the dishes were placed on ice and washed twice with ice-cold PBS. The neurons were then detergent extracted by adding DRM lysis buffer (TBS containing 1% Triton X-100, 500 μm sodium orthovanadate, and protease inhibitors) and rocking the plates gently at 4°C for 20 min. The crude lysates were then scraped into Eppendorf (Eppendorf Scientific, Westbury, NY) tubes and spun at 16110 × g in a 4°C Microfuge for 10 min. The supernatants were rapidly removed from the pellets and placed into new Eppendorf tubes. The pellets, which contain the DRMs, were washed quickly with ice-cold DRM lysis buffer, centrifuged again, and this wash buffer was rapidly removed. Both the insoluble pellets and the supernatants were then treated with 2× sample buffer and boiled for 5 min. In some cases, proteins were immunoprecipitated from both the soluble and insoluble fractions and to do this, the lipid rafts were disrupted with glucoside and diluted in DRM lysis buffer to the same volume as the supernatant fraction. An equivalent amount of glucoside was added to the supernatant as well, such that the detergent concentrations were the same between the soluble and insoluble protein fractions. The immunoprecipitations were then performed as described for Nonidet P-40 lysis of neurons described in the previous section.
Flotation gradients.
A second method used to analyze lipid rafts biochemically was density centrifugation with the Optiprep reagent (Sigma). For these experiments, sympathetic neurons were maintained in vitro on precoated collagen 35 mm culture dishes (BD Biosciences, San Diego, CA). After 7–10 d in vitro (DIV), these neurons were deprived of NGF for 24 h and treated with GDNF as described in the figure legends. They were then washed twice with ice-cold PBS and extracted by agitating the dishes gently at 4°C in TXNE (TBS containing 0.2% Triton X-100, protease inhibitors, and 100 mm sodium orthovanadate). The dishes were scraped with a cooled rubber policeman, and this mixture was transferred to Eppendorf tubes and homogenized by using a hand-held dounce homogenizer. These crude homogenates were mixed with 60% Optiprep containing 0.2% Triton X-100 to reach 35% Optiprep. Two hundred fifty microliters of this were placed into the bottom of an ultracentrifuge tube and 3.2 ml of 30% Optiprep (containing 0.2% Triton X-100, sodium orthovanadate, and protease inhibitors) was then layered on top, followed by a layer of 200 μl of TXNE. These gradients were centrifuged at 200,000 × g for 4 h, and six fractions of 600 μl were carefully removed from the top of the gradient. The fractions next were precipitated with TCA, washed twice with a 1:1 mixture of ethanol and ether, and the pellets allowed to dry overnight. Sample buffer was added to the protein precipitates and boiled for 10 min, and these extracts were analyzed by SDS-PAGE and immunoblotting.
Immunoblotting.
Protein samples were subjected to SDS-PAGE by using 4–12% Tris–glycine gradient gels (Invitrogen) for the immunoblotting for all proteins except for caveolin, which required separation on either 16% Tris–glycine gels or 4–12% NuPAGE gels (Invitrogen). Separated proteins were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) and then washed briefly in TBST (10 mm Tris, pH 7.4, 100 mm sodium chloride, 0.1% Tween 20). The blots were next incubated with TBST containing 2% BSA for 45 min followed by a 2–3 h incubation with the primary antibody in this same blocking solution at room temperature. The immunoblots were washed three times with TBST containing 0.5% BSA and then incubated with a secondary antibody in the blocking solution for 45 min. After washing three times in TBST, the blots were developed using a chemiluminescent substrate (Supersignal; Pierce, Rockford, IL). In some cases, these blots were stripped and reprobed with a different primary antibody. This was accomplished by incubating these blots with a glycine stripping buffer (100 mm glycine, pH 2.5) twice for 15 min. The blots were then washed three times with copious amounts of TBST and the procedure for immunoblotting repeated as just described. When blots were probed with GFRα1 and GFRα2 antibodies, 5% milk was used instead of BSA for blocking. The antibodies were acquired from the following companies: anti-Ret9 (C19; rabbit; Santa Cruz Biotechnology), anti-Ret51 (C20; goat; Santa Cruz Biotechnology), anti-Cbl (C-15; Santa Cruz Biotechnology), anti-Cbl-b (H-121; Santa Cruz Biotechnology), anti-phosphotyrosine (clone 4G10; Upstate Biotechnology, Lake Placid, NY), anti-actin (I19; Santa Cruz Biotechnology), anti-caveolin, anti-flotillin-1, and anti-transferrin receptor (Zymed, San Francisco, CA), anti-GFRα1 (R & D Systems, Minneapolis, MN), and anti-GFRα2 (Proteus, San Diego, CA). Anti-phospho-1062Ret and anti-Ret51 (rabbit polyclonals) were generated previously in our laboratory (Tsui-Pierchala et al., 2002c). All antibodies were used at a 1:1000 dilution except for anti-phosphotyrosine, anti-transferrin receptor, and anti-flotillin-1, which were used at a 1:3000 dilution. For the quantification of immunoblots, Un-Scan-It densitometry software was used (Silk Scientific, Orem, UT). Two representative experiments were quantified and described in the Results section as the mean ± range.
Disruption of lipid rafts.
Lipid rafts were acutely disrupted by either the depletion of membrane cholesterol or the depletion of membrane sphingomyelin using β-methylcyclodextrin (MCD) or neutral sphingomyelinase (Smase), respectively. To deplete cholesterol, 10 mm MCD (Sigma) was added to the neurons in serum-free medium for 15 min at 37°C. The neurons were then washed once and the experimental treatments conducted. For the depletion of plasma membrane sphingomyelin, neurons were treated with 1–4 mU/ml of Smase (Sigma) in serum-free culture medium for 90 min before treatment with GDNF. For long-term depletion of cholesterol, lovastatin (5 μm; Sigma) was added to the neuronal culture medium for 12 h before treatment with GDNF. During lovastatin treatment, the neurons were switched to a serum-free medium and the survival experiments also conducted in the continued presence of lovastatin in serum-free medium.
MTT reduction assays.
Sympathetic neurons maintained in vitro for 5 DIV were deprived of NGF for 24 h in the presence of boc-aspartyl(OMe)-fluoromethylketone (Enzyme Systems Products, Livermore, CA) to inhibit apoptosis. The neurons were then washed twice and treated with NGF, GDNF, or epoxomicin as described in the figure legends. After 4 h, the neurons were washed once with warmed L15 medium containing 0.1% BSA and then incubated in this medium containing the MTT reagent (0.4 mg/ml;Sigma). This medium was removed, DMSO added to the wells and agitated gently, and these extracts were transferred to a translucent microtiter plate. The absorbance at 550 nm was measured by using a microplate reader and subtracted from the background absorbance measured at 650 nm. Each condition was performed on triplicate wells of sympathetic neurons from two independent cultures. Each data point was graphed as the mean ± SEM.
Survival assays.
Survival assays were performed on 5 DIV neurons, which were plated into chamber slides. After the treatments, the neurons were washed once with ice-cold PBS and incubated in PBS containing 4% paraformaldehyde for 48 h at 4°C. The neurons were then Nissl stained with Toluidine Blue O (Sigma), dehydrated, and the slides coverslipped. The number of Nissl-positive neurons in each condition were counted by a naive observer and graphed as a percentage of the number of neurons in NGF-containing medium. Only neurons that had round, smooth cell bodies and Nissl staining that was similar to neurons maintained in NGF were counted. Multiple independent wells were counted from two independent cultures and the data graphed as the mean value with error bars representing the SEM. Significant differences were tested using a Student’s t test.
Results
Activation of Ret induces its proteasome-dependent degradation
We observed previously that after activation with GDNF, Ret autophosphorylation declines over time in sympathetic neurons (Tsui-Pierchala et al., 2002c). Ret is alternatively spliced to yield two major isoforms, Ret9 and Ret51, which differ only in their C termini. Ret9 has nine unique amino acids after residue 1063, whereas Ret51 has 51 alternative amino acids, which include two additional tyrosine residues (Tahira et al., 1990), one of which is autophosphorylated (Coulpier et al., 2002; Tsui-Pierchala et al., 2002c). Both Ret9 and Ret51 phosphorylation decline after GDNF stimulation in sympathetic neurons (Tsui-Pierchala et al., 2002c). To determine to what extent this downregulation of Ret phosphorylation is caused by the degradation of Ret itself, total cellular extracts were produced from sympathetic neurons treated with GDNF for various lengths of time. Immunoblotting revealed that both Ret9 and Ret51 levels declined rapidly after GDNF stimulation, with the majority of both species lost within hours (Fig. 1A). The loss of Ret9 was slower than the decline of Ret51, which was more apparent when the data were quantified (Fig. 1B). Interestingly, the degradation of Ret51 obeyed first-order kinetics (t1/2 = 0.52 h) (Fig. 1C), in contrast to Ret9 degradation, which demonstrated zero-order kinetics, suggesting that Ret9 and Ret51 are degraded by different mechanisms. Recently, the activity-dependent degradation of Ret9 and Ret51 has been observed in cell lines as well (Scott et al., 2005). Neurturin, a second member of the GFLs, which activates Ret predominantly via GFRα2, also led to the degradation of Ret9 and Ret51 with kinetics that were similar to GDNF (data not shown). Therefore, GFLs led to the rapid degradation of Ret in sympathetic neurons with kinetics that differed depending on the Ret isoform.
Figure 1.
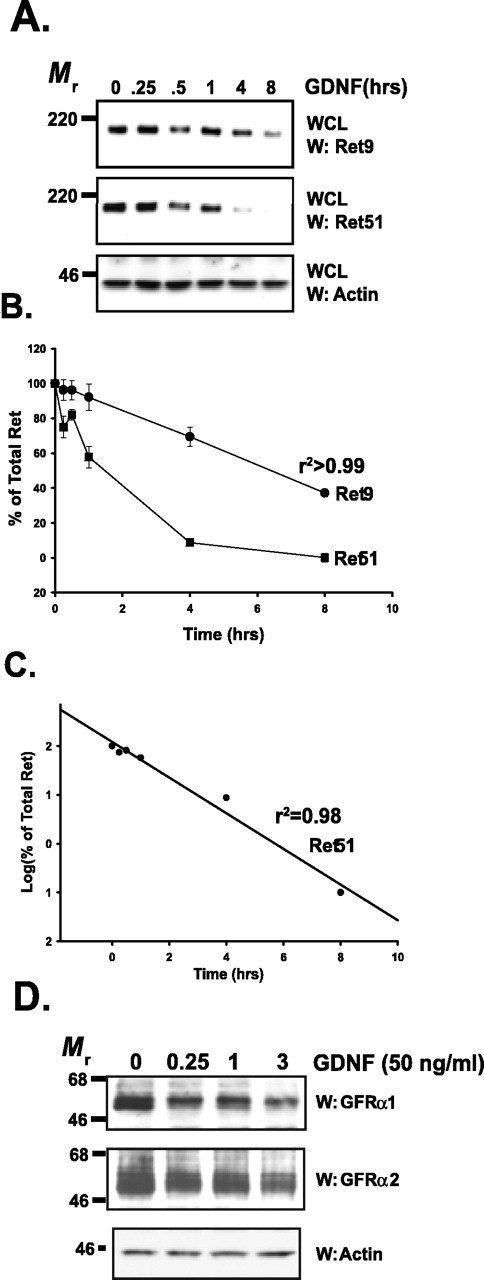
Ret and GFRαs are degraded rapidly after activation. A, Sympathetic neurons were treated with GDNF (50 ng/ml) for various lengths of time, whole-cell extracts were produced from them, and the extracts were subjected to immunoblotting. Ret9 (top) and Ret51 (middle) immunoblotting revealed that both of these Ret isoforms are lost rapidly after GDNF stimulation. Actin immunoblotting (bottom) was used to confirm equal loading of cell extracts. WCL, Whole-cell lysate. B, Quantification of the experiment shown in A. Three separate experiments were conducted, quantified, and graphed as the mean ± SEM. C, Semilog plot of the data for Ret51 degradation kinetics shown in B. D, Sympathetic neurons were treated with GDNF (50 ng/ml) for various lengths of time (labeled on top), and whole-cell extracts were prepared. These extracts were immunoblotted with antibodies to GFRα1 (top) and GFRα2 (middle). Equal loading of proteins was confirmed with actin immunoblotting (bottom). This experiment was conducted twice with identical results. W, Western blot.
To determine whether the GFRα coreceptors required for Ret activation are also degraded after the ligand-dependent activation of Ret, whole-cell extracts were prepared from sympathetic neurons after various lengths of GDNF treatment and analyzed. The levels of both GFRα1 and GFRα2 rapidly declined after GDNF stimulation, and the majority of both GFRα1 and GFRα2 were lost within 3 h (Fig. 1D). Quantification of the immunoblots revealed that 78 and 61% of GFRα1 and GFRα2, respectively, were lost within 3 h of GDNF treatment. Although GDNF acts predominantly through GFRα1 in vivo, GDNF can also efficiently activate Ret via GFRα2 in vitro, which explains why the levels of GFRα2 decline after GDNF treatment as well. The GFRαs are GPI-anchored proteins and do not traverse the plasma membrane and, thus, most likely are not ubiquitinated. These results suggest that either Ret is internalized as a GDNF–GFRα–Ret complex in order for ligand activation of Ret to promote the concomitant degradation of GFRα1 and GFRα2, or GFRα1 and GFRα2 are liberated from the cell surface after formation of the receptor complex via cleavage of their GPI anchor.
The two major organelles responsible for protein degradation are the lysosome and the proteasome. RTKs such as Kit and EGFR are degraded predominantly through the lysosome, which is accomplished by monoubiquitination, often on multiple lysines, of the receptor after its activation (Waterman and Yarden, 2001; Dikic and Giordano, 2003). Monoubiquitination acts as a signal that prompts the internalization of the target protein into endosomes and leads, ultimately, to its delivery to lysosomes (Hicke, 2001). Proteins targeted for destruction by the proteasome are polyubiquitinated, in chains of four or more ubiquitin molecules; this modification leads to the trafficking of proteins to the proteasome. Consistent with Ret being degraded by one of these pathways, Ret51 was rapidly ubiquitinated after its activation in sympathetic neurons, as determined by the identification of Ret51 in ubiquitin immunoprecipitates (Fig. 2A). The incorporation of ubiquitin shifted the molecular weight of Ret51 dramatically (Fig. 2A). As expected, concurrent with the dramatic increase in ubiquitinated Ret51 after exposure to ligand, the level of nonubiquitinated Ret51 decreased (Fig. 2A, middle). The ubiquitination of Ret51 after its activation by GDNF was also observed when Ret immunoprecipitates were analyzed by immunoblotting with antibodies to ubiquitin (data not shown).
Figure 2.

Ret is ubiquitinated and degraded predominantly by the proteasome. A, Sympathetic neurons were treated with GDNF (50 ng/ml) or medium alone for 15 min, lysed, and ubiquitinated proteins were immunoprecipitated. The immunoprecipitates were analyzed by Ret51 immunoblotting (top). The supernatants from the immunoprecipitation were subjected to Ret51 and actin immunoblotting to determine the amount of nonubiquitinated Ret51 and to confirm that equal amounts of protein were analyzed, respectively. B, Neurons were pretreated with epoxomicin (5 μm), concanamycin (10 μm), or both for 30–60 min before stimulation with GDNF for 3 h. As a control, some neurons were pretreated with medium containing the vehicle alone (DMSO) for 1 h. After the treatments, the neurons were detergent extracted, and Ret51 was immunoprecipitated. These immunoprecipitates were subjected to Ret51 immunoblotting (top). The blots were stripped and reprobed with phosphotyrosine antibodies (P-Tyr; middle) and the supernatants of the immunoprecipitates analyzed with actin immunoblotting (bottom) to confirm that similar amounts of protein were analyzed. C, Sympathetic neurons were treated with GDNF, epoxomicin, or concanamycin for 8 h, similar to the analysis in B. Ret51 analysis was performed as described in B. D, Neurons were either not infected (No Virus) or infected with lentivirus to express either wild-type (WT) ubiquitin or ubiquitin containing a K48R mutation for 4 d. The medium was then replaced, and 24 h later, these neurons were treated with GDNF (50 ng/ml) for 2 h. The levels of Ret51 were then examined as described in B. E, Neurons that were treated with GDNF (50 ng/ml) for 15 min were subjected to ubiquitin immunoprecipitation, as in A. These precipitates were immunoblotted for Ret9 (top), and the supernatants were subjected to Ret9 (middle) and actin (bottom) immunoblotting. F, Sympathetic neurons were subjected to an identical analysis as in B. Whole-cell lysates (WCLs) were made from the treated neurons, and these were analyzed by Ret9 (top) and actin (bottom) immunoblotting. All of the experiments described in this figure were performed two to four times with identical results. IP, Immunoprecipitate; Ub, ubiquitin; W, Western blot.
To identify the pathway that accounts for Ret51 degradation, a pharmacologic approach was taken. Sympathetic neurons were treated with epoxomicin, a potent and highly selective proteasome inhibitor, or concanamycin, a selective inhibitor of lysosomal proteases, before stimulation with GDNF. Ret51 levels were monitored at various times after GDNF treatment and compared with neurons that were not treated with these inhibitors. Inhibition of the proteasome blocked a significant portion of GDNF-dependent Ret51 degradation (47 ± 6%), in contrast to inhibition of lysosomal proteases, which consistently had no effect on their own (16 ± 8% of Ret remaining with concanamycin and GDNF compared with 21 ± 7% of Ret remaining with GDNF treatment alone) (Fig. 2B). Inhibition of both the proteasome and the lysosome simultaneously led to nearly complete inhibition of Ret51 degradation by GDNF (77 ± 5%) (Fig. 2B), indicating that both pathways can contribute to Ret degradation, with the proteasome being the predominant pathway for Ret degradation in sympathetic neurons. Identical results were obtained when either lactacystin or ammonium chloride was used to inhibit the proteasome and lysosomal proteases, respectively (data not shown). To further characterize this observation, sympathetic neurons were stimulated with GDNF for a longer period of time, and the effects of proteasome and lysosome inhibition were determined. Treatment of sympathetic neurons with GDNF for 8 h induced the degradation of the vast majority of Ret51 (Fig. 2C). Inhibition of the proteasome during Ret activation again inhibited Ret51 degradation, whereas inhibition of the lysosome had no effect (Fig. 2C). The simultaneous blockade of both pathways inhibited the majority of Ret51 degradation initiated by GDNF treatment (Fig. 2C). Therefore, although neither the proteasomal nor the lysosomal degradation pathways completely account for ligand-mediated Ret downregulation, the proteasome has a more predominate function than does the lysosome. The observation that lysosomal inhibitors only have a discernable effect on Ret degradation when the proteasome is also inhibited suggests a possible “shunting” of Ret to the lysosome when the proteasome is inhibited. Interestingly, inhibition of Ret degradation resulted in the accumulation of Ret that was autophosphorylated (Fig. 2B), indicating that the degradation of Ret is the major mechanism for downregulating Ret signaling in sympathetic neurons.
To further investigate the role of the proteasome in the ligand-dependent degradation of Ret51, neurons were infected with a lentiviral vector encoding a mutated form of ubiquitin, ubiquitin K48R, which lacks the lysine required for the formation of ubiquitin chains that target proteins specifically to the proteasome. This mutation does not inhibit its incorporation into proteins as a monomer and, thus, can still function as an internalization signal that leads to lysosomal degradation of membrane proteins. Ectopic overexpression of K48R ubiquitin significantly slowed the degradation of Ret51 after GDNF stimulation compared with either overexpression of nonmutated ubiquitin or noninfected neurons (Fig. 2D). This is consistent with the results using pharmacologic inhibitors and indicates that the proteasome accounts for the majority of Ret51 degradation after GDNF-dependent activation in sympathetic neurons.
Because of the significant differences in the degradation kinetics of Ret9 and Ret51, we examined the extent of Ret9 ubiquitination after activation by GDNF. Ret9 was ubiquitinated rapidly after GDNF stimulation in sympathetic neurons, albeit to a lesser extent than Ret51 (Fig. 2E). The prominent shift in molecular weight that occurs when Ret51 was ubiquitinated was not observed with Ret9 (Fig. 2E), suggesting that Ret9 is polyubiquitinated to a lesser extent than is Ret51. To determine whether Ret9 degradation was proteasome dependent or lysosome dependent, neurons were treated with epoxomicin or concanamycin, respectively, before GDNF stimulation. Both proteasome inhibition and lysosome inhibition reduced Ret9 degradation after its activation (Fig. 2F), suggesting that both of these mechanisms regulate Ret9 levels after activation in neurons. In summary, Ret9 is ubiquitinated rapidly after GDNF-dependent activation in sympathetic neurons and is subsequently degraded in a proteasome- and lysosome-dependent manner, in contrast to Ret51 degradation, which does not require the lysosome.
The rapidity of Ret degradation after ligand stimulation suggests that this destruction of activated receptors may limit the functions of Ret that require sustained signaling, such as survival or trophism. To determine whether the GDNF-dependent degradation of Ret limits the trophic properties of GDNF, sympathetic neurons were investigated using an MTT assay, which monitors changes in cellular metabolism. Sympathetic neurons were deprived of NGF to lower their metabolic rate to a basal level (defined as 100%) and were then subjected to brief, 4 h treatments of GDNF or NGF to monitor their metabolism-enhancing activities. Treatment of NGF-deprived sympathetic neurons with GDNF increased their metabolic activity by ∼20% within 4 h (Fig. 3). However, treatment of sympathetic neurons with GDNF in the presence of proteasome inhibition caused a significantly greater increase in metabolic activity (42%; p < 0.005), similar to the level of MTT reduction observed with NGF treatment (Fig. 3). The treatment of deprived neurons with epoxomicin alone had no effect on their metabolic status (Fig. 3). Together, the trophic effects of GDNF are limited by the proteasome-dependent downregulation of Ret that occurs after its activation in sympathetic neurons.
Figure 3.

Ligand-dependent Ret activation limits the trophic activity of GDNF on sympathetic neurons. The metabolic activity of neurons was determined using an MTT reduction assay as described in Materials and Methods. Neurons were treated with NGF (50 ng/ml), GDNF (50 ng/ml), epoxomicin (Epox; 5 μm), or GDNF in the presence of epoxomicin for 4 h. MTT reduction was then measured and graphed as a percentage of neurons treated with medium alone (No Treat). Error bars represent SEM.
Ret translocation into lipid rafts is sustained in sympathetic neurons
After activation in various cell lines, Ret translocates from detergent-soluble, “nonraft” membrane domains into DRMs, or lipid rafts, via the GFRα coreceptors that are localized constitutively in the lipid rafts (Tansey et al., 2000). This translocation enhances tremendously the signal transduction of Ret, and this requirement of lipid rafts is, at least in part, because of the necessity for Src in Ret signaling, which is concentrated in lipid rafts (Encinas et al., 2001). Whether Ret translocation into lipid rafts occurs and is sustained in neurons that normally express and require GFL receptors for development and maintenance is unclear. Furthermore, to what extent lipid rafts participate in the ligand-mediated degradation of Ret has not been investigated. For the purposes of this study, we define a lipid raft as either a membrane preparation that is resistant to extraction with 1% Triton X-100 or a membrane fraction that is buoyant on an Optiprep density gradient. Primary sympathetic neurons were treated with GDNF for various lengths of time, and lipid rafts were analyzed by density gradient centrifugation. When neurons were not treated with GDNF, the vast majority of Ret was located in dense, or nonordered, membrane domains and very little Ret was observed in buoyant, lipid raft fractions (Fig. 4). The opposite was true for GFRαs: the majority of GFRα2 was located in buoyant membrane domains. When neurons were treated with GDNF, Ret translocated into lipid rafts and the level of Ret in buoyant fractions was sustained for hours (Fig. 4). The level of Ret in nonraft fractions declined within 3 h of GDNF treatment, and the amount of Ret in fractions 5 and 6 was 43% less than the amount of Ret in these fractions from neurons not treated with GDNF (medium alone), when normalized to the levels of transferrin receptor, a nonraft resident protein.
Figure 4.

Ret translocates into lipid rafts after activation with GDNF. Sympathetic neurons were treated with medium alone or with GDNF (50 ng/ml) for either 1 or 3 h. The neurons were then lysed and subjected to Optiprep density centrifugation as described in Materials and Methods. Six equal fractions were taken from the top of the tubes and labeled from 1 to 6, fraction 6 being the densest (raft and nonraft fractions are labeled above the fraction numbers). These fractions were analyzed by immunoblotting for Ret51 (top) and GFRα2 (middle). GFRα2 confirmed the location of the buoyant, lipid raft fractions, and immunoblotting with antibodies to transferrin receptor (TrfR; bottom) indicated the nonraft membrane fractions. This experiment was performed twice with similar results. W, Western blot.
To confirm these results with another biochemical method, sympathetic neurons were extracted with ice-cold Triton X-100 various times after GDNF treatment. Similar to the observations using density centrifugation, Ret was located outside of DRMs before GDNF treatment and rapidly moved into DRMs after GDNF treatment (Fig. 5A). The translocation of Ret into lipid rafts peaked within 1 h and was sustained for hours (Fig. 5A). Both Ret9 and Ret51 were lost predominantly from the nonraft fractions, similar to what was observed by density centrifugation. Quantification of the immunoblots revealed that Ret9 and Ret51 in the Triton X-100-insoluble fraction remained at levels of 188 ± 12 and 116 ± 8%, respectively, 3 h after GDNF treatment, compared with the amount of Ret that moved into the lipid raft fraction within 15 min. In contrast, after 3 h of GDNF treatment only 29 ± 8 and 22 ± 10% of Ret9 and Ret51, respectively, remained in the nonraft fractions. Ret that translocated into lipid rafts was highly autophosphorylated, as was Ret located outside of lipid rafts, as determined by PY1062Ret immunoblotting (Fig. 5A). Note that the level of phosphorylated Ret declined in lipid rafts where Ret levels were more stable, indicating that both degradation and dephosphorylation limit Ret signaling under these conditions. In summary, GDNF treatment leads to the rapid and sustained movement of Ret into lipid raft microdomains with the concomitant loss of Ret predominantly from nonraft domains.
Figure 5.

Ubiquitinated Ret resides outside of lipid rafts in GDNF-stimulated sympathetic neurons. A, Sympathetic neurons were stimulated with GDNF (50 ng/ml) for various lengths of time, and the neurons were then extracted with Triton X-100. The extracts were centrifuged, and the insoluble pellets containing lipid rafts (left 4 lanes) and the soluble fraction containing nonraft membrane fractions (right 4 lanes) were then separated by SDS-PAGE and electroblotted. The blots were probed with antibodies to Ret9 (top 2 panels), Ret51 (next 2 panels), and PY1062Ret (5th panel). Shorter exposures of the Ret9 and Ret51 immunoblots are shown (labeled as “Light,” panels 2 and 4) to better observe the loss of Ret protein in these fractions. To confirm the relative purity of the lipid-raft fractionation, blots were probed with anti-transferrin receptor (6th panel) and anti-caveolin-1 (bottom panel), which are markers of nonraft fractions and lipid-raft fractions, respectively. B, Sympathetic neurons were treated with GDNF (50 ng/ml) for 15 min and then Triton X-100 was extracted as described in A. The fractions were then treated with glucoside to solubilize the rafts and subjected to ubiquitin immunoprecipitation. The supernatants of this immunoprecipitation were used for a second Ret51 immunoprecipitation, and all of the immunoprecipitates were subjected to Ret51 immunoblotting (top). The activation state of the complexes was determined with phosphotyrosine immunoblotting (second), and the purity of the raft and nonraft fractions was confirmed by probing the supernatants with antibodies to transferrin receptor and caveolin-1 (bottom 2 panels). These immunoblots also confirmed that equal amounts of protein were analyzed. The experiments in this figure were conducted two or three times with similar results. INSOL, Insoluble pellets containing lipid rafts; SOL, soluble fraction containing nonraft membrane fractions; IP, immunoprecipitate; TrfR, transferrin receptor; Cav, caveolin; Ub, ubiquitin; P-Tyr, phosphotyrosine; W, Western blot.
Lipid rafts and caveolas are membrane locations involved in the internalization of proteins, a process that, for some molecules, requires monoubiquitination. To determine whether the ubiquitination of Ret occurs within lipid rafts, sympathetic neurons were treated with GDNF, then Triton X-100 extracted to produce DRMs and non-DRMs, and both of these fractions were then subjected to ubiquitin immunoprecipitation followed by immunoblotting. The vast majority of ubiquitinated Ret was located in nonraft fractions and was highly autophosphorylated (Fig. 5B, left). Immunoprecipitation of Ret remaining in the extracts after the ubiquitin immunoprecipitations revealed that Ret indeed translocated into lipid rafts but that this fraction of Ret was apparently not ubiquitinated because it was not immunoprecipitated with ubiquitin antibodies (Fig. 5B, right). These data suggest that either the majority of Ret is not ubiquitinated in lipid rafts or that, after ubiquitination, Ret rapidly moves out of lipid rafts into nonordered membrane regions.
Lipid rafts are not required for the ligand-dependent degradation of Ret
The observation that Ret was not ubiquitinated in lipid rafts and that the degradation of Ret appeared to occur outside of lipid rafts suggested that rafts may be dispensable for the ligand-dependent degradation of Ret. To test this hypothesis, sympathetic neurons were exposed to two structurally distinct agents that deplete plasma membrane lipid rafts. Lipid rafts are rich in cholesterol and sphingolipids, which are critical for the formation of lipid rafts. Depletion of membrane cholesterol with brief treatments of β-methyl-cyclodextrin (MCD) leads to the loss of lipid rafts, as observed by the inability of Ret to move into these membrane fractions (Fig. 6A). The treatment of sympathetic neurons with MCD for several hours did not alter their morphology, suggesting that there were no readily apparent toxic effects of cholesterol depletion with MCD (data not shown). The disruption of lipid rafts from sympathetic neurons by using MCD did not alter the GDNF-dependent degradation of either Ret9 or Ret51 (Fig. 6B). An alternative method for the disruption of lipid rafts located in the plasma membrane is the depletion of sphingomyelin from these domains (Scheek et al., 1997). Treatment of cells with neutral sphingomyelinase (Smase) by adding this enzyme directly into the culture medium led to the loss of lipid rafts within hours, and, as expected, the translocation of Ret into DRMs was disrupted as well (Fig. 6C). Similar to the depletion of rafts with MCD, Smase treatment, even at levels much higher than necessary to disrupt lipid rafts, did not alter the degradation kinetics of Ret9 or Ret51 after GDNF stimulation for 3 h (Fig. 6D). In addition, the depletion of lipid rafts with either MCD or Smase did not alter the ubiquitination of Ret after activation with GDNF, indicating that lipid rafts are not required for the ubiquitination of Ret (Fig. 6E). Together, these data indicate that lipid rafts were not required for the ubiquitination or degradation of Ret after ligand stimulation, implying that rafts do not serve as the membrane domain where the initial steps of Ret downregulation occur. This does not, however, rule out the possibility that lipid rafts may have some protective effect on the fraction of Ret that translocates into rafts, which would not be easily observed from whole cell extracts at the short time points examined here.
Figure 6.
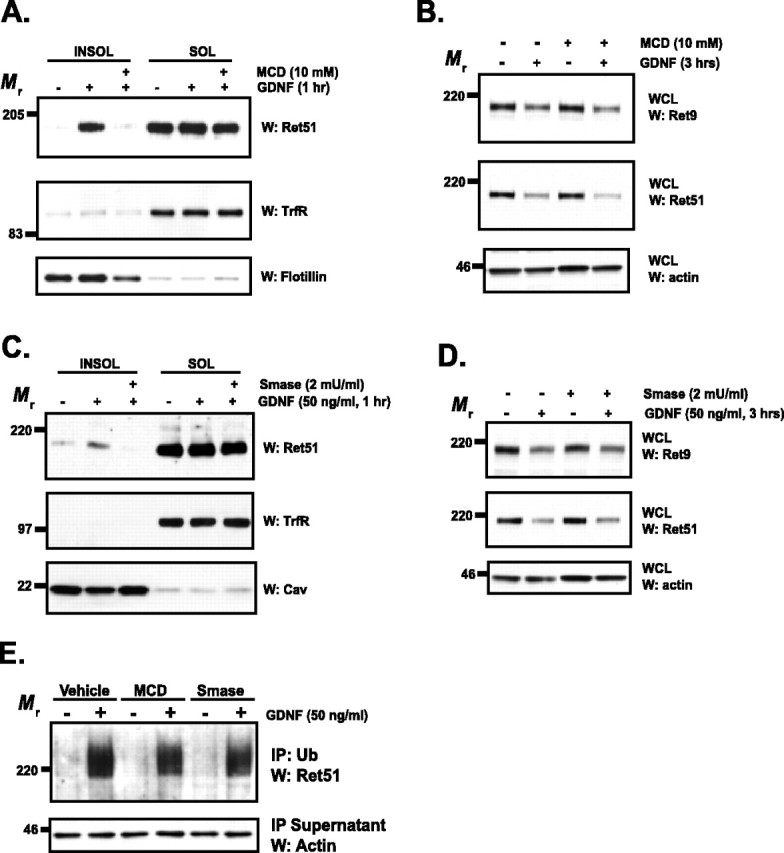
Lipid rafts are not required for the GDNF-induced ubiquitination or degradation of Ret. A, Sympathetic neurons were treated with MCD (10 mm), or medium alone, for 15 min, washed, and then stimulated with GDNF (50 ng/ml) or medium alone for 1 h. The neurons were extracted with 1% Triton X-100, and the insoluble pellets (left) containing lipid rafts were collected via centrifugation from the soluble, nonraft fractions (right). These fractions were separated by SDS-PAGE and immunoblotted with antibodies to Ret51. To confirm the relative purity of the insoluble and soluble fractions, flotillin and transferrin receptor immunoblotting, respectively, was conducted. B, Neurons were treated with MCD as in A, washed, and then stimulated with GDNF (50 ng/ml), or medium alone, for 3 h. Whole-cell extracts were then produced from the neurons, and the extracts were subjected to Ret9 (top) and Ret51 (2nd panel) immunoblotting. Actin immunoblotting confirmed that equal amounts of protein were analyzed. C, Sympathetic neurons were treated with medium alone or with neutral Smase (2 mU/ml) for 90 min. The neurons were then stimulated with GDNF (50 ng/ml) or vehicle alone for 1 h in the continued presence of Smase. These neurons were then subjected to Triton X-100 extraction and immunoblotting as described in A. D, Similar to the treatments described in C, sympathetic neurons were treated with Smase before treatment with GDNF or medium alone for 3 h. Total cellular protein was harvested, and analyzed as was done as in B. E, Cells were treated with MCD, Smase, or vehicle only, as done in the previous panels. After a 15 min treatment with GDNF (50 ng/ml), the neurons were detergent extracted, and ubiquitinated proteins were immunoprecipitated. These immune complexes were analyzed with Ret51 immunoblotting, and actin immunoblotting of the supernatants confirmed that equal amounts of protein were analyzed. The experiments in this figure were performed two to four times with similar results. INSOL, Insoluble pellets containing lipid rafts; SOL, soluble fraction containing nonraft membrane fractions; IP, immunoprecipitate; TrfR, transferrin receptor; Cav, caveolin; W, Western blot; WCL, whole-cell lysate.
Lipid rafts enhance GDNF signaling by rescuing activated Ret from degradation
An important function of lipid rafts is to act as signaling platforms in which signaling molecules and receptors are brought within close proximity, thus enhancing interaction and activation. In the case of Ret, this function is important because lipid rafts are enriched in Src, which is critical for Ret signal transduction (Encinas et al., 2001). The data described above led to the hypothesis that an additional function of lipid rafts in GFL signaling may be to sequester Ret away from the degradation machinery in nonraft membrane domains, thus prolonging the half-life of activated Ret and, subsequently, the signals emanating from it. If this hypothesis is correct, the inhibitory effects of lipid raft disruption on Ret function should be reversed, at least partially, by the inhibition of Ret degradation. This possibility was tested by examining the survival-promoting effects of GDNF on sympathetic neurons. Sympathetic neurons absolutely require NGF for survival, and removal of this neurotrophic factor from the medium led to the apoptotic death of the neurons within 24–36 h (Fig. 7). Addition of GDNF to NGF-deprived neurons maintained the survival of ∼35% of the neurons (Fig. 7). When Ret degradation was blocked by inhibition of the proteasome, GDNF maintained the survival of >95% of the neurons (Fig. 7). These data, along with the data on GDNF-dependent enhancement of metabolism (Fig. 3), indicate that one of the limiting determinants of Ret function is its targeted destruction after activation in sympathetic neurons. Treatment of NGF-deprived neurons with proteasome inhibitors alone rescued only 20% of sympathetic neurons from apoptosis, indicating that the dramatic effect of epoxomicin on GDNF-mediated survival was not a nonspecific effect of blocking proteasomal protein degradation (Fig. 7).
Figure 7.

Lipid rafts enhance the survival-promoting activity of GDNF by protecting Ret from degradation. Sympathetic neurons were maintained in NGF, deprived of NGF for 24 h, or deprived of NGF in the presence of GDNF (50 ng/ml). Some neurons that were treated with GDNF were concurrently treated with the proteasome inhibitor epoxomicin (Epox; 5 μm) or were depleted of cholesterol by treatment with lovastatin (Lova; 5 μm) for 12 h before GDNF treatment in the continued presence of the inhibitor. As a control, NGF-deprived neurons were saved with potassium depolarization alone (KCl; 25 mm) or in the presence of lovastatin. The neurons were then fixed, Nissl stained, and the number of surviving neurons in each condition counted. These experiments were plotted as the mean ± SEM. The asterisks indicate statistically significant differences (p < 0.05) from the two indicated conditions and GDNF or GDNF and lovastatin treatments, respectively.
When lipid rafts were disrupted by cholesterol depletion by using lovastatin, the survival-promoting ability of GDNF was lost completely, indicating that lipid rafts are required for Ret activation to promote survival in sympathetic neurons (Fig. 7). To determine whether the inhibitory effects of lipid raft disruption on GDNF-mediated survival is because of an accelerated loss of activated Ret, raft-depleted, GDNF-maintained neurons were subjected to proteasome inhibition to blunt the loss of activated Ret. The blockade of Ret degradation by proteasome inhibition reversed significantly the effects of lipid raft depletion on GDNF-dependent survival (Fig. 7) but did not rescue all of the neurons. These data suggest that one of the major functions of lipid rafts is to sequester some portion of activated Ret away from its targeted degradation in nonraft membrane regions. Furthermore, the observation that proteasome inhibition could not completely block the effects of raft depletion suggests that lipid rafts provide additional functions for Ret signal transduction, such as enhancing the exposure of Ret to signaling molecules located in rafts. The depletion of cholesterol from neurons may have deleterious side effects unrelated to their effects on Ret degradation. To test whether other aspects of sympathetic neuron survival are also affected by lovastatin treatment, we maintained the survival of NGF-deprived neurons with potassium depolarization, which has potent survival-promoting effects on sympathetic neurons. Cholesterol depletion of potassium-depolarized neurons had no effect on their survival (Fig. 7), indicating that the effects of cholesterol depletion on survival are selective to GDNF and are not a nonspecific effect of the inhibition of 3-hydroxy-3-methylglutaryl CoA reductase. Together, these results suggest that the ligand-dependent degradation of Ret limits the survival-promoting ability of GDNF in sympathetic neurons.
Cbl proteins are excluded from lipid rafts
To understand the mechanism by which lipid rafts protect Ret from activity-dependent degradation, the expression of Cbl proteins were examined. Cbl-b, which is highly expressed in sympathetic neurons, was not detected in Triton X-100-insoluble fractions before or after GDNF stimulation of sympathetic neurons (Fig. 8). In contrast, Cbl-b was readily detected in nonraft membrane domains (Fig. 8). Importantly, GDNF stimulation led to the tyrosine phosphorylation of Cbl-b, and the association of Ret with Cbl-b was readily detected in nonraft domains, suggesting that Cbl-b likely regulates Ret ubiquitination (Fig. 8). Cbl-1, a second E3 ligase that is expressed in sympathetic neurons, was excluded from lipid rafts but did not appear to associate with Ret either before or after Ret activation (data not shown). Together, these data suggest that lipid rafts protect Ret from degradation after its activation by sequestering Ret away from the Cbl-1 and Cbl-b E3 ligases that are located predominantly outside of rafts. Recently, it was observed that the extent of Cbl association with Ret51, which is recruited to Ret as a complex with Grb2, coincided with more rapid kinetics of degradation (Scott et al., 2005). Thus, one of the predominant mechanisms regulating the scope of Ret signaling and function may be through modulation of the access of E3 ligases to Ret.
Figure 8.
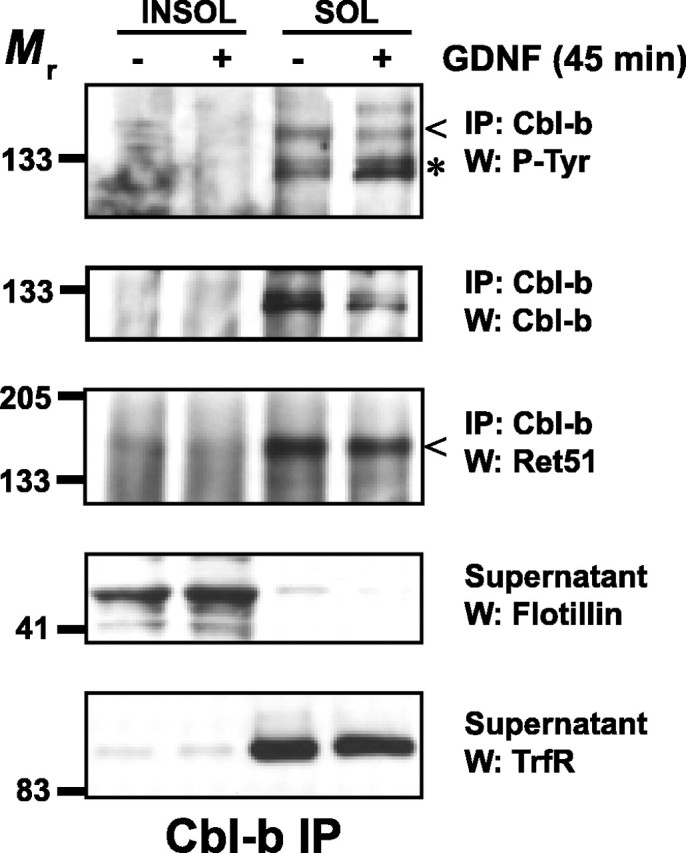
Lipid rafts protect Ret from degradation by sequestering Ret away from Cbl proteins. Sympathetic neurons were treated with medium alone or with medium containing GDNF (50 ng/ml) for 45 min. Detergent-resistant membranes were purified as described in Materials and Methods. Cbl-b was immunoprecipitated from both Triton X-100-insoluble and -soluble fractions, and these immunoprecipitates were analyzed by immunoblotting using the antibodies listed to the right of each blot. The relative purity of the lipid raft fractions were confirmed with flotillin-1 and transferrin receptor immunoblotting of the supernatants from the immunoprecipitations. This experiment was performed twice with similar results. INSOL, Insoluble pellets containing lipid rafts; SOL, soluble fraction containing nonraft membrane fractions; IP, immunoprecipitate; TrfR, transferrin receptor; P-Tyr, phosphotyrosine; W, Western blot.
Discussion
In this study, we investigated the mechanisms responsible for the downregulation of the GFL receptor Ret after its activation. Ret is rapidly ubiquitinated and degraded after its activation in sympathetic neurons, and this degradation is executed predominantly by the proteasome (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Ret moves rapidly into lipid rafts after GDNF treatment, and the level of Ret in lipid rafts is stable for several hours, in contrast to Ret located in nonraft domains, where Ret appears to be degraded. Indeed, the vast majority of ubiquitinated Ret is located in nonraft fractions and lipid rafts do not function as the membrane location where Ret ubiquitination and degradation is initiated after its activation. These results uncovered a novel function of lipid rafts, namely that lipid rafts sequester activated Ret away from the degradation machinery, such as Cbl proteins, active in nonraft domains. Thus, activated Ret located in lipid rafts has a longer half-life, which augments its signal output and enhances GDNF functions such as neuronal survival.
Proteasomal versus lysosomal degradation of RTKs
Most studies on the mechanism of RTK downregulation after their activation indicate that the proteasome does not function directly in the degradation of RTKs. Proteasome inhibitors, for example, do not block the degradation of c-Kit or T-cell receptor and have only a minor effect on platelet-derived growth factor receptor degradation (Mori et al., 1995a,b; Bonifacino and Weissman, 1998). Proteasome inhibitors block Met degradation significantly, but evidence suggests that the effects of these inhibitors are caused by blocking the internalization and trafficking of Met rather than destruction of the receptor itself (Jeffers et al., 1997; Hammond et al., 2001). Proteasome inhibition blocks a significant portion of, but not all, Ret degradation initiated by GDNF. In contrast to the function of the proteasome, the function of lysosomes in RTK degradation has been demonstrated repeatedly. Inhibitors of lysosomes, which typically act by deacidification of the lysosome, prevent, or at least delay, the degradation of all of the RTKs mentioned above, including Met (Bonifacino and Weissman, 1998; Hammond et al., 2001; Waterman and Yarden, 2001; Bonifacino and Traub, 2003). Ret appears to be a unique contradiction to these previous observations. Inhibition of the lysosome had no effect on Ret degradation, even with multiple inhibitors used at levels much higher than is necessary to block lysosomal proteases, indicating that lysosomes were not the sole endpoint for Ret degradation. A lysosomal contribution to Ret degradation could only be observed subsequent to proteasome inhibition, indicating that lysosomes can participate in Ret degradation when the proteasome is inhibited. Thus, lysosomes may act as a shunting pathway for the degradation of Ret under certain circumstances. Together, these data suggest that the proteasome is the predominant mechanism required for the degradation of Ret, although how a membrane-spanning protein could be degraded by the proteasome is unclear.
One possible explanation for the difference between the degradation of Ret and the degradation of other RTKs is that Ret is predominantly localized to the axons of neurons. Degradation characteristics of other RTKs have been analyzed, for the most part, in cells that lack the specialized molecular environment of neuronal processes. Lysosomes appear to be essentially absent from the distal axons of sympathetic neurons, suggesting that to degrade Ret, Ret would either have to be transported retrogradely to the cell bodies in which lysosomes are much more abundant (Overly and Hollenbeck, 1996), or Ret is degraded locally by other mechanisms such as the proteasome. Proteasomes have been identified in axons (Watts et al., 2003; Zhai et al., 2003) and participate in local signaling functions such as axon pruning (Watts et al., 2003). Under the conditions of these experiments, it is unlikely that Ret must be retrogradely transported back to cell bodies to be degraded in lysosomes, given how long axons are in sympathetic neurons in vitro and given how rapidly Ret, after GFL stimulation, is degraded. It will be of interest to investigate whether proteasome-dependent degradation is the predominant pathway for the downregulation of signaling receptors that are located in neuronal processes.
Lipid rafts and protein degradation
The nature and even the existence of lipid rafts in living cells has been debated (Kenworthy, 2002; Munro, 2003). Although this is not the proper forum to review the evidence for and against the existence of rafts, it would be remiss not to mention this controversy here. Techniques involving the visualization of rafts in living cells have yielded varied results, and debate will continue as to whether this evidence supports or denies the existence of rafts (Subczynski and Kusumi, 2003; Glebov and Nichols, 2004; Sharma et al., 2004). Analysis of lipid rafts in Ret signaling in neurons necessitated the use of biochemical methods, given the difficulty of the techniques required to observe rafts in living primary sympathetic neurons. Extraction with 1% Triton X-100 or density centrifugation, whether or not detergents are used, consistently yield low buoyancy membranes, which are highly enriched in cholesterol, sphingolipids, proteins that are modified with saturated lipids, and signaling molecules (Simons and Ikonen, 1997; Brown and London, 2000). Interestingly, the existence of caveolas is not debated; caveolas are well characterized, flask-shaped membrane invaginations found in several cell types that have the biochemical characteristics of lipid rafts (Anderson, 1998; Dobrowsky, 2000; Fielding and Fielding, 2000). Another compelling observation that supports a model of membrane subdomains in cells is that under resting conditions, some molecules are excluded from rafts and under stimulated conditions, they move into rafts; the Ret receptor is an example of this (Tansey et al., 2000; Paratcha et al., 2001). Unactivated Ret is primarily excluded from DRMs, whereas after formation of the mature signaling complex, Ret translocates into these membrane fractions. Furthermore, the behavior of Ret differs between these two membrane compartments, and these differences in Ret between raft and nonraft domains can be directly related to the signaling capacity of the receptor. Depletion of cholesterol abrogates GDNF-dependent survival selectively, which can be reversed by increasing the half-life of the Ret (Fig. 7). Ultimately, the size, constitution, and the dynamics of rafts in living neurons may not be understood for some time, but undoubtedly, biochemical membrane microdomains exist that are extremely useful for explaining the behavior of receptor function.
Lipid rafts are important for the ubiquitination and degradation of several proteins, and several receptors that reside in rafts are ubiquitinated there, such as FcϵR1 (Lafont and Simons, 2001; Legler et al., 2003). Furthermore, after IgE engagement, the E3 ligases Cbl and Nedd4, which participate in the ubiquitination of several cell surface receptors, translocate into lipid rafts, suggesting that they ubiquitinate FcϵR1 in rafts (Lafont and Simons, 2001). This is in contrast to Ret: ubiquitinated Ret was not detectable in rafts, and disruption of lipid rafts did not inhibit the GDNF-dependent ubiquitination of Ret, indicating that rafts are not required for the initiation of Ret degradation. Furthermore, Cbl and Cbl-b were not located in rafts and did not translocate into rafts after GDNF stimulation of neurons. Thus, whether lipid rafts are involved in ubiquitination of their constituent proteins may depend on the stimulus, cell type, and on which E3 ligase is required.
Lipid rafts appear to sequester and protect Ret from degradation, and this lengthening of the half-life of activated Ret in turn enhances survival promotion by GDNF. This suggests that, in addition to their function as signaling platforms, a function of lipid rafts may be to sequester active receptors from downregulation. Whether other activities of GDNF such as migration and proliferation require lipid rafts to extend the signaling duration of activated Ret is unclear. In fact, the functions of GFLs in vivo that require lipid rafts have not been determined and remain an important question for the future.
Footnotes
This work was supported by National Institutes of Health Grants R37AG-12947 and AG-13729 (E.M.J.), AG-13730 (J.M.), and K01 NS-045221 (B.A.P.). We thank Dr. Toshiyuki Araki for expert assistance. We also thank Drs. Cynthia Tsui and Patricia Osborne for their thoughtful scientific discussions and critical reading of this manuscript, and Mary Bloomgren for secretarial assistance.
References
- Aguilar RC, Wendland B (2003). Ubiquitin: not just for proteasomes anymore. Curr Opin Cell Biol 15:184–190. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Saarma M (2002). The GDNF family: signalling, biological functions and therapeutic value. Nat Rev 3:383–394. [DOI] [PubMed] [Google Scholar]
- Anderson RG (1998). The caveolae membrane system. Annu Rev Biochem 67:199–225. [DOI] [PubMed] [Google Scholar]
- Arenas E, Trupp M, Akerud P, Ibanez CF (1995). GDNF prevents degeneration and promotes the phenotype of brain noradrenergic neurons in vivo. Neuron 15:1465–1473. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Enomoto H, Johnson EM Jr, Milbrandt J (2000). The GDNF family ligands and receptors-implications for neural development. Curr Opin Neurobiol 10:103–110. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM (2003). Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 72:395–447. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Weissman AM (1998). Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol 14:19–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, London E (1998). Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol 14:111–136. [DOI] [PubMed] [Google Scholar]
- Brown DA, London E (2000). Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem 275:17221–17224. [DOI] [PubMed] [Google Scholar]
- Buj-Bello A, Buchman VL, Horton A, Rosenthal A, Davies AM (1995). GDNF is an age-specific survival factor for sensory and autonomic neurons. Neuron 15:821–828. [DOI] [PubMed] [Google Scholar]
- Coulpier M, Anders J, Ibanez CF (2002). Coordinated activation of autophosphorylation sites in the RET receptor tyrosine kinase. J Biol Chem 277:1991–1999. [DOI] [PubMed] [Google Scholar]
- Dikic I, Giordano S (2003). Negative receptor signalling. Curr Opin Cell Biol 15:128–135. [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT (2000). Sphingolipid signalling domains: floating on rafts or buried in caves? Cell Signal 12:81–90. [DOI] [PubMed] [Google Scholar]
- Easton RM, Deckwerth TL, Parsadanian AS, Johnson EM (1997). Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from Bax deletion. J Neurosci 17:9656–9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas M, Tansey MG, Tsui-Pierchala BA, Comella JX, Milbrandt J, Johnson EM Jr (2001). c-Src is required for glial cell line-derived neurotrophic factor (GDNF) family ligand-mediated neuronal survival via a phosphatidylinositol-3 kinase (PI-3K)-dependent pathway. J Neurosci 21:1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Heuckeroth RO, Golden JP, Johnson EM Jr, Milbrandt J (2000). Development of cranial parasympathetic ganglia requires sequential actions of GDNF and neurturin. Development 127:4877–4889. [DOI] [PubMed] [Google Scholar]
- Fielding CJ, Fielding PE (2000). Cholesterol and caveolae: structural and functional relationships. Biochim Biophys Acta 1529:210–222. [DOI] [PubMed] [Google Scholar]
- Glebov OO, Nichols BJ (2004). Lipid raft proteins have a random distribution during localized activation of the T-cell receptor. Nat Cell Biol 6:238–243. [DOI] [PubMed] [Google Scholar]
- Hammond DE, Urbe S, Vande Woude GF, Clague MJ (2001). Down-regulation of MET, the receptor for hepatocyte growth factor. Oncogene 20:2761–2770. [DOI] [PubMed] [Google Scholar]
- Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simmons L, Moffet B, Vandlen RA, Simpson LC, Moffet B, Vandlen RA, Koliatsos VE, Rosenthal A (1994). GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science 266:1062–1064. [DOI] [PubMed] [Google Scholar]
- Hicke L (2001). Protein regulation by monoubiquitin. Nat Rev 2:195–201. [DOI] [PubMed] [Google Scholar]
- Jeffers M, Taylor GA, Weidner KM, Omura S, Vande Woude GF (1997). Degradation of the Met tyrosine kinase receptor by the ubiquitin-proteasome pathway. Mol Cell Biol 17:799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy A (2002). Peering inside lipid rafts and caveolae. Trends Biochem Sci 27:435–437. [DOI] [PubMed] [Google Scholar]
- Lafont F, Simons K (2001). Raft-partitioning of the ubiquitin ligases Cbl and Nedd4 upon IgE-triggered cell signaling. Proc Natl Acad Sci USA 98:3180–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legler DF, Micheau O, Doucey M-A, Tschopp J, Bron C (2003). Recruitment of TNF Receptor 1 to lipid rafts is essential for TNFa-mediated NF-kB activation. Immunity 18:655–664. [DOI] [PubMed] [Google Scholar]
- Li LX, Wu WT, Lin L-FH, Lei M, Oppenheim RW, Houenou LJ (1995). Rescue of adult mouse motoneurons from injury-induced cell death by glial cell line-derived neurotrophic factor. Proc Natl Acad Sci USA 92:9771–9775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MW, Klein RD, Farinas I, Sauer H, Armanini M, Phillips H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A (1996). Renal and neuronal abnormalities in mice lacking GDNF. Nature 382:76–79. [DOI] [PubMed] [Google Scholar]
- Mori S, Kanaki H, Tanaka K, Morisaki N, Saito Y (1995a). Ligand-activated platelet-derived growth factor β-receptor is degraded through proteasome-dependent proteolytic pathway. Biochem Biophys Res Commun 217:224–229. [DOI] [PubMed] [Google Scholar]
- Mori S, Tanaka K, Omura S, Saito Y (1995b). Degradation process of ligand-stimulated platelet-derived growth factor β-receptor involves ubiquitin-proteasome proteolytic pathway. J Biol Chem 270:29447–29452. [DOI] [PubMed] [Google Scholar]
- Mount HTJ, Dean DO, Alberch J, Dreyfus CF, Black IB (1995). Glial cell line-derived neurotrophic factor promotes the survival and morphologic differentiation of Purkinje cells. Proc Natl Acad Sci USA 92:9092–9096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S (2003). Lipid rafts: elusive or illusive? Cell 115:377–388. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Houenou LJ, Johnson JE, Lin LF, Li L, Lo AC, Newsome AL, Prevette DM, Wang S (1995). Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature 373:344–346. [DOI] [PubMed] [Google Scholar]
- Overly CC, Hollenbeck PJ (1996). Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J Neurosci 16:6056–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paratcha G, Ledda F, Baars L, Coulpier Baars, Besset V, Anders J, Scott R, Ibanez F (2001). Released GFRα1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron 29:171–184. [DOI] [PubMed] [Google Scholar]
- Peschard P, Park M (2003). Escape from Cbl-mediated downregulation: a recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell 3:519–523. [DOI] [PubMed] [Google Scholar]
- Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, Sariola H, Westphal H (1996). Defects in enteric innervation and kidney development in mice lacking GDNF. Nature 382:73–76. [DOI] [PubMed] [Google Scholar]
- Sanchez MP, Silos-Santiago I, Frisen J, He B, Lira SA, Barbacid M (1996). Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature 382:70–73. [DOI] [PubMed] [Google Scholar]
- Scheek S, Brown MS, Goldstein JL (1997). Sphingomyelin depletion in cultured cells blocks proteolysis of sterol regulatory element binding proteins at site 1. Proc Natl Acad Sci USA 94:11179–11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RP, Eketjall S, Aineskog H, Ibanez CF (2005). Distinct turnover of alternatively spliced isoforms of the RET kinase receptor mediated by differential recruitment of the Cbl ubiquitin ligase. J Biol Chem 280:13442–13449. [DOI] [PubMed] [Google Scholar]
- Sharma P, Varma R, Sarasij RC, Ira RC, Gousset K, Krishnamoorthy G, Rao M, Maor S (2004). Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell 116:577–589. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E (1997). Functional rafts in cell membranes. Nature 387:569–572. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D (2000). Lipid rafts and signal transduction. Nat Rev 1:31–41. [DOI] [PubMed] [Google Scholar]
- Subczynski WK, Kusumi A (2003). Dynamics of raft molecules in the cell and artificial membranes: approaches by pulse EPR spin labeling and single molecule optical microscopy. Biochem Biophys Acta 1610:231–243. [DOI] [PubMed] [Google Scholar]
- Tahira T, Ishizaka Y, Itoh F, Sugimura T, Nagao M (1990). Characterization of ret proto-oncogene mRNAs encoding two isoforms of the protein product in a human neuroblastoma cell line. Oncogene 5:97–102. [PubMed] [Google Scholar]
- Tansey MG, Baloh RH, Milbrandt J, Johnson EM Jr (2000). GFRα-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron 25:611–623. [DOI] [PubMed] [Google Scholar]
- Trupp M, Ryden M, Jornvall H, Funakoshi H, Timmusk T, Arenas E, Ibanez CF (1995). Peripheral expression and biological activities of GDNF, a new neurotrophic factor for avian and mammalian peripheral neurons. J Cell Biol 130:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Milbrandt J, Johnson EM Jr (2002a). NGF utilizes c-Ret via a novel GFL-independent, inter-RTK signaling mechanism to maintain the trophic status of mature sympathetic neurons. Neuron 33:261–273. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM Jr (2002b). Lipid rafts in neuronal signaling and function. Trends Neurosci 25:412–417. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Ahrens RC, Crowder RJ, Milbrandt J, Johnson EM Jr (2002c). The long and short isoforms of Ret function as independent signaling complexes. J Biol Chem 277:34618–34625. [DOI] [PubMed] [Google Scholar]
- Waterman H, Yarden Y (2001). Molecular mechanisms underlying endocytosis and sorting of ErbB receptor tyrosine kinases. FEBS Lett 490:142–152. [DOI] [PubMed] [Google Scholar]
- Watts RJ, Hoopfer ED, Luo L (2003). Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron 38:871–885. [DOI] [PubMed] [Google Scholar]
- Zhai Q, Wang J, Kim A, Liu Q, Watts RJ, Hoopfer ED, Mitchison T, Luo L, He Z (2003). Involvement of the ubiquitin-proteasome system in early stages of Wallerian degeneration. Neuron 39:217–225. [DOI] [PubMed] [Google Scholar]