

Abstract
In neurons, L-type calcium channels (CaV1.2 and CaV1.3) regulate an extensive range of functions. However, the roles of CaV1.3-containing L channels, which are physiologically and pharmacologically distinct from the better understood CaV1.2 channels, are only beginning to be determined. We find that CaV1.3 channels are modulated by the insulin-like growth factor-1/receptor tyrosine kinase (IGF-1/RTK) through a signaling pathway that involves phospholipase C, calcium release from IP3-sensitive internal stores, and calcium/calmodulin kinase II. In addition, we find that the IGF-1-induced modulation requires phosphorylation of a specific serine residue, S1486, in the EF hand motif of the CaV1.3 subunit. This modulation alters CaV1.3 activity, causing a left shift in the current–voltage relationship and strongly potentiating peak currents at hyperpolarized membrane potentials. We also find that CaV1.3 channels and their RTK-dependent potentiation contribute to the regulation of the survival-promoting transcription factor cAMP response element-binding protein (CREB): in both cortical and hippocampal neurons, depolarization and IGF-1 rapidly increase phospho-CREB levels in a manner that requires CaV1.3 activity and the S1486 phosphorylation site to achieve a full effect. Although the full effects of CaV1.3 channels remain to be determined, their preferential localization to dendritic shafts and spine heads coupled with their ability to activate at relatively hyperpolarized and even subthreshold potentials suggests that CaV1.3 activity may subserve different cellular functions from CaV1.2 and, in particular, may be important in transducing signals initiated by excitatory neurotransmission.
Keywords: CaV1.3, CaMKII, IP3, IGF-1, Shank, CREB
Introduction
Calcium influx through the related L-type calcium channels CaV1.2 and CaV1.3 performs an array of functions that include promoting neuronal survival (Galli et al., 1995; Blair et al., 1999; West et al., 2001; Marshall et al., 2003), controlling calcium signaling to the nucleus (Deisseroth et al., 1998; Dolmetsch et al., 2001; Chen et al., 2003), influencing the rate of neurite outgrowth (Redmond et al., 2002), stabilizing dendritic arbors (Lohmann et al., 2002), and regulating spike activity (Hernandez-Lopez et al., 2000; Olson et al., 2005; Yamamoto et al., 2005). We showed previously that insulin-like growth factor-1 (IGF-1) rapidly potentiates neuronal CaV1.2 channel activity via an IGF-1 receptor tyrosine kinase (RTK)-, phosphatidylinositol-3 kinase (PI-3 kinase)-, and Akt-dependent pathway that culminates in Src-dependent phosphorylation of a specific tyrosine residue on CaV1.2 (Blair and Marshall, 1997; Blair et al., 1999; Bence-Hanulec et al., 2000). In the long term, IGF-1 and CaV1.2 have been shown to promote neuronal survival via regulation of specific transcription factors, including cAMP response element-binding protein (CREB) (Finkbeiner and Greenberg, 1998; Brunet et al., 2001; Marshall et al., 2003). Although recent studies have shown that CaV1.3 can also regulate CREB (Zhang et al., 2005), little is known about how CaV1.3 activity is controlled.
CaV1.3 L channels differ from those of CaV1.2 in important respects. In particular, CaV1.3 channels activate at more hyperpolarized membrane potentials (Koschak et al., 2001; Xu and Lipscombe, 2001; Lipscombe et al., 2004) and therefore are expected to respond to physiological stimuli, such as modest synaptic stimulation, that could not open CaV1.2 channels. This property is a key determinant of evoked spike activity in striatal neurons (Hernandez-Lopez et al., 2000; Olson et al., 2005). CaV1.3 channels also display fast activation kinetics and a different subcellular distribution, with CaV1.3 being primarily dendritic and frequently colocalizing with synaptic markers (Westenbroek et al., 1998; Simon et al., 2003; Olson et al., 2005). Conversely, CaV1.2 is expressed at high levels in somata. Pharmacologically, CaV1.3 channels are less sensitive to dihydropyridine antagonists (Lipscombe et al., 2004; Helton et al., 2005). In addition, CaV1.3 exists in two isoforms that differ at their C termini and, potentially, could be differentially regulated: a short isoform (CaV1.3b) that terminates immediately after the EF hand and calmodulin-binding IQ motifs, and a long isoform (CaV1.3a) that extends an additional 512 amino acids and contains Src homology 3 (SH3) and postsynaptic density-95/Discs large/zona occludens-1 (PDZ) binding motifs. Importantly, the C-terminal extension of the long isoform permits Shank binding and synaptic localization of CaV1.3a (Olson et al., 2005).
Understanding the signaling pathways regulating CaV1.3 is essential to providing insight into the regulation of neuronal survival, excitability, and arborization. In at least some neurons, CaV1.3 modulation by D2 dopaminergic and metabotropic glutamate (mGlu) G-protein-coupled receptors can regulate action potential activity (Hernandez-Lopez et al., 2000; Yamamoto et al., 2005); interestingly, both of these pathways use IP3 signaling. L channels also contribute to the formation of dendritic arbors, causing in a calcium/calmodulin kinase IIβ (CaMKIIβ)-dependent manner increased outgrowth in immature neurons and the rapid stabilization of dendritic arbors via an IP3-sensitive pathway in more mature neurons (Lohmann et al., 2002; Konur and Ghosh, 2005; Lohmann and Wong, 2005); importantly, however, it remains to be determined if CaV1.2, CaV1.3, or both contribute to these processes. We demonstrate here that CaV1.3 channel activity is rapidly potentiated by IGF-1 and that, unlike the IGF-1 regulation of CaV1.2, the CaV1.3 signaling pathway involves IP3-sensitive stores, CaMKII, and phosphorylation of S1486 in the CaV1.3 EF hand motif. Mechanistically, we find that the potentiation arises from increased activation of CaV1.3 at hyperpolarized membrane potentials and, within minutes, produces increased levels of phospho-CREB (pCREB), a transcription factor strongly associated with promoting dendritic development and neuronal survival.
Materials and Methods
Constructs.
cDNAs encoding CaV1.3 [CaV1.3a and CaV1.3b; gifts from Dr. D. Lipscombe (Brown University, Providence, RI)] were in pcDNA6 expression vectors. To remove the PDZ binding site, the 24 C-terminal amino acids were deleted (CaV1.3a-del). α2δ and β1b constructs [gifts from Dr. T. Snutch (University of British Columbia, Vancouver, British Columbia, Canada)] were in pcDNA3. The wild-type myc-tagged and green fluorescent protein (GFP)-tagged Shank-1a constructs [gifts from Drs. R. Huganir (Johns Hopkins School of Medicine, Baltimore, MD) and T. Boeckers (Ulm University, Ulm, Germany)] were in pGW1. To disrupt the SH3 domain, tryptophan-520 was mutated to alanine (W520A-Shank-1a). To disrupt the ability of Shank to bind to Homer, proline-1495 within the Homer ligand site was mutated to leucine (P1495L-Shank-1a).
Cell culture and transfection.
SH-SY5Y human neuroblastoma cells were maintained in 10% FCS in DMEM. Hippocampal and cortical neurons were cultured from embryonic day 18 rats (Goslin and Banker, 1989) and maintained in Neurobasal medium supplemented with B27 and Glutamax. For SH-SY5Y cells, cDNAs encoding CaV1.3a or CaV1.3b were cotransfected with 2δ and β1b at a 2:1:1 ratio with GFP using Lipofectamine Plus (Invitrogen, San Diego, CA). For neurons, cDNA encoding hemagglutinin (HA)-tagged CaV1.3a were cotransfected with Q-GFP using Lipofectamine 2000 (Invitrogen). All cells were tested 1 d after transfection. Because serum and serum supplements contain a large number of growth factors, cells were switched to serum-free media before testing to eliminate potential pre-exposure to IGF-1.
Electrophysiology.
Calcium channel currents were monitored as described previously (Blair and Marshall, 1997; Blair et al., 1999; Bence-Hanulec et al., 2000), using the permeabilized patch variation of standard whole-cell patch recording techniques (Hamill et al., 1981; Rae et al., 1991) and barium as the charge carrier. An EPC-9 patch-clamp amplifier (List Biological Laboratories, Campbell, CA) was used in conjunction with HEKA Elektronik (Lambrecht/Pfalz, Germany) data acquisition and analysis software [low-pass filter, 2–5 kHz (−3 dB, digital Gaussian filter); sample interval, 50 μsec; P/n leak subtraction, n = 5; Instrutech, Mineola, NY]. Barium currents were evoked by depolarizing voltage pulses (−70 to +40 mV, in 10 mV increments) from a holding potential of −80 mV; to minimize rundown, pulse duration was minimized (30–50 ms). For each cell, the pulse protocol was run a minimum of three times before IGF-1 addition to ensure that currents were stable. IGF-1 diluted in extracellular recording saline was then superfused over the cells, and currents were recorded for an additional 0.5–5 min. The estimation of rate constants was as follows. The rising phase of each evoked current was fit with a single exponential. Similarly, inactivation during the test pulse was assessed by fitting the time-dependent reduction in current by a single exponential. At the end of each test pulse, the membrane potential was returned to −80 mV, revealing tail currents that were fit with the sum of two exponentials. All curve fitting was done using the HEKA software, allowed to run to 1000 iterations.
Patch electrodes were filled with (in mm) 150 CsCl, 5 Na-HEPES, pH 7.4, and amphotericin B (final concentration, 0.25 mg/ml) to permeabilize the patch and allow low-resistance electrical access without breaking the patch membrane (electrode resistance, ∼3 MΩ). For SH-SY5Y cells, the extracellular recording saline included barium as a charge carrier plus a mixture of inhibitors to block other voltage-gated channels: 100 mm NaCl, 20 mm BaCl2, 20 mm tetraethylammonium chloride, 5 mm 4-aminopyridine, 1 μm tetrodotoxin, 1 μm nimodipine, 400 nm ω-conotoxin-GVIA, and 10 mm Na-HEPES, pH 7.4. For cortical neurons, 300 nm ω-agatoxin-IVA was also included to inhibit P/Q calcium channels. Nimodipine stock solutions were made at near saturating concentrations in DMSO; when 100 μm nimodipine was used to inhibit CaV1.3 activity, the final DMSO concentration in test salines was 0.002%. The low (1 μm) concentration of nimodipine used to inhibit CaV1.2 activity (Reeve et al., 1994; Bence-Hanulec et al., 2000) (supplemental Fig. 2E, time point 3, available at www.jneurosci.org as supplemental material) was not expected to affect CaV1.3 activity (Koschak et al., 2001; Xu and Lipscombe, 2001) and, in our hands, had no detectable effects on CaV1.3 currents. Under these conditions, CaV1.3 currents could be recorded for 5 min without displaying significant “rundown.”
Figure 2.
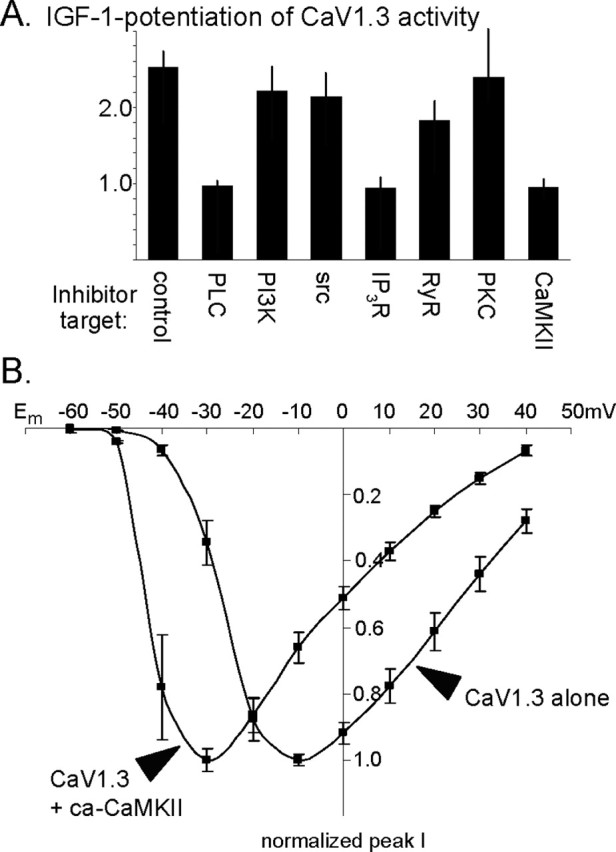
IGF-1/RTK potentiation of CaV1.3 requires CaMKII, phospholipase C, and calcium release from IP3-sensitive stores. A, SH-SY5Y cells expressing wild-type CaV1.3b were pretreated for 30 min with specific inhibitors or vehicle and tested for their ability to respond to IGF-1. To determine the fold change in CaV1.3 current levels for each cell, the peak current 1 min after IGF-1 was compared with that before IGF-1 (test potential, −30 mV). The value 1.0 indicates a complete block of potentiation. Currents in vehicle-treated control cells exhibited a more than twofold potentiation, whereas inhibiting phospholipase C with 10 μm U73122, release from IP3-sensitive intracellular stores with 100 μm 2-APB, or CaMKII with 10 μm KN93 fully blocked potentiation. Inhibiting release from RyR-sensitive stores with 100 μm ruthenium red slightly but not significantly decreased potentiation (p > 0.90). Inhibiting PI-3 kinase or Src (intermediates required for potentiating CaV1.2) with 100 nm wortmannin or 10 μm PP2 had no effect, nor did inhibiting PKC with 10 μm bisindolylmaleimide I. PLC, phospholipase C; IP3R, IP3 receptor. B, Evoked CaV1.3 current–membrane voltage relationship indicates that when coexpressed with a ca-CaMKII, CaV1.3 channel activity was indistinguishable from IGF-1-potentiated activity. Compared with unpotentiated CaV1.3 activity, CaV1.3b channels in the presence of ca-CaMKII produce larger currents at hyperpolarized membrane potentials and display a left shift, with current amplitudes peaking at −30.8 ± 1.8 mV (n = 16 cells). No differences with observed with CaV1.3a plus ca-CaMKII (−32.3 ± 2.6 mV; n = 11 cells).
Immunocytochemistry.
To determine total levels of CaV1.3 or pCREB, cultured cortical or hippocampal neurons were fixed with 2% paraformaldehyde plus 3% sucrose, washed, and permeabilized with 0.01% Triton X-100, and nonspecific binding sites were blocked with 4% goat serum. Cells were then incubated with an anti-GFP monoclonal antibody (mAb) (1:100, QBiogene AFP; QBiogene, Irvine, CA) and either an anti-CaV1.3 rabbit polyclonal antibody (pAb), which recognizes both a and b isoforms and the antigenic site of which is intracellular (1:400; Alamone Laboratories, Jerusalem, Israel), or an anti-pCREB rabbit pAb (anti-phospho-S133-CREB, 1:500; Upstate Biotechnology, Lake Placid, NY). After washing off unbound primary Ab, cells were incubated with tetramethylrhodamine isothiocyanate (TRITC)-conjugated goat anti-rabbit Ab and FITC-conjugated goat anti-mouse Ab (Jackson ImmunoResearch, West Grove, PA). Labeling was visualized under fluorescence optics using a Zeiss (Thornwood, NY) Axioskop or a Leica (Nussloch, Germany) TCS SP2 AOBS laser-scanning confocal microscope.
To identify surface expression of CaV1.3, we inserted two extracellular HA tags into the wild-type and S1486A-CaV1.3a constructs; the epitope-tagged constructs expressed well in neurons and in SH-SY5Y cells where they produced currents and IGF-1 responses (or nonresponses) indistinguishable from untagged constructs. To immunolabel surface CaV1.3, neurons were transfected with HA-tagged CaV1.3 for 1 d and processed as above, omitting the permeabilization step. The extracellular epitope was detected using an anti-HA mAb (1:1000; Covance, Denver, PA), followed by incubation in TRITC-conjugated anti-mouse secondary Ab (Jackson ImmunoResearch).
Results
CaV1.3 activity is rapidly modulated by IGF-1
Within 30 s of bath application, low (10–100 ng/ml) concentrations of IGF-1 strongly potentiated the activity of both CaV1.3a- and CaV1.3b-containing L channels (Fig. 1A–D) (n = 20 of 20 cells for CaV1.3a; n = 50 of 51 cells for CaV1.3b). Specifically, CaV1.3a or CaV1.3b was coexpressed with the auxiliary subunits α2δ and β1b and, to identify transfectants, with GFP in SH-SY5Y human neuroblastoma cells. In the presence of inhibitors to block other voltage-gated channels, CaV1.3 channel currents were evoked by depolarizing voltage pulses. No differences were observed when a different β subunit was used (n = 6 of 6 β2a-expressing cells were potentiated). Calcium imaging confirmed these results (Fig. 1E). When CaV1.3-expressing SH-SY5Y cells were depolarized by elevating extracellular potassium, those that had been treated immediately prior with IGF-1 showed larger responses. Note also that the addition of IGF-1 caused a small, transient increase in free intracellular calcium; because this effect was observed in saline that would be predicted to create a transmembrane potential of approximately −85 mV, no CaV1.3 channels should be active, suggesting that in addition to potentiating the activity of voltage-sensitive CaV1.3 channels, IGF-1 also induces calcium release from internal stores.
Figure 1.

IGF-1 rapidly potentiates CaV1.3 activity. A, Wild-type CaV1.3a-containing channels respond within 30 s to IGF-1 (arrowheads). At hyperpolarized membrane potentials, channels are potentiated (top; −30 mV), but little potentiation is observed at moderately depolarized potentials (middle; 0 mV) and none when the cell is strongly depolarized (bottom; +30 mV). Recordings are from an SH-SY5Y neuroblastoma cell transfected with GFP, CaV1.3a, and the auxiliary subunits α2δ and β1b. Channel currents were evoked by depolarizing voltage steps from a holding potential of −80 mV before and 30 s after the addition of 20 ng/ml IGF-1. B, Current–voltage relationships for CaV1.3a channels. Data are plotted as normalized current versus membrane potential (Em). Note that although CaV1.3 currents are small at strongly hyperpolarized Em, the fold increase in response to IGF-1 is much greater at −40 and −30 mV than at more depolarized potentials. ▪, Currents evoked before IGF-1; ♦, 30 s after 20 ng/ml IGF-1 (arrowhead; n = 20 cells; mean ± SEM). Because individual transfectants express different CaV1.3 levels, data for each cell were normalized to the peak current of that cell obtained before IGF-1 addition. For each cell, the peak current value before IGF-1 was determined; in most cells, the pre-IGF-1 peak value was obtained at −10 mV. CaV1.3a currents in that cell were then measured at each potential before and after IGF-1 and divided by its pre-IGF-1 peak value. Peak current densities at −30 mV were −14.4 ± 2.9 pA/pF before IGF-1 and −50.3 ± 7.1 pA/pF after 30 s in IGF-1. C, D, Identical results are obtained with the short C-terminal isoform CaV1.3b channels. Experiments were performed and analyzed as in A and B (n = 51 cells; peak current densities at −30 mV were −2.4 ± 0.7 pA/pF before IGF-1 and −12.6 ± 4.3 pA/pF after 30 s in IGF-1). E, IGF-1 potentiation measured by increased intracellular calcium levels. Calcium imaging of fluo-4-loaded SH-SY5Y cells expressing CaV1.3a is shown. Depolarization (depol; filled arrowheads) in 90 mm KCl (calculated Em= −5 mV) induces a larger increase in free intracellular calcium levels in cells treated for 1 min with 20 ng/ml IGF-1 (E2) than in untreated cells from sister cultures (E1). Note that the cells responded to the IGF-1 (E2, open arrowhead) with a slight rise in intracellular calcium.
Significantly, the IGF-1-modulation shifts the voltage dependence of CaV1.3 activation (Fig. 1B,D). In the absence of IGF-1, CaV1.3 channel currents were undetectable at −60 mV, and peak current densities were typically obtained at approximately −10 mV (−8 ± 2 mV for CaV1.3a; −12 ± 2 mV for CaV1.3b). However, when the same cells were analyzed after brief (30 s) exposure to IGF-1, whole-cell currents could be detected at potentials as hyperpolarized as −60 mV, and current levels peaked at approximately −20 mV (−19 ± 2 mV for CaV1.3a; −24 ± 3 mV for CaV1.3b). In particular, the IGF-1/RTK effect was most prominent at strongly hyperpolarized membrane potentials, disappearing with increasing depolarization and becoming undetectable at positive membrane potentials.
Mechanistically, the potentiation appears to result from more rapid activation of the channels: for CaV1.3a before IGF-1, τrise was 5.7 ± 0.7 ms, whereas 30 s after IGF-1, τrise was significantly faster at 2.3 ± 0.3 ms; for CaV1.3b before IGF-1, τrise was 6.7 ± 0.8 ms, whereas 30 s after IGF-1, τrise was 2.8 ± 0.4 ms (Em= −30 mV) (Fig. 1A,C, top). No differences in the rates of inactivation or deactivation were demonstrable when barium was used as a charge carrier, suggesting that the primary kinetic mechanism involves increasing the speed of activation.
The CaV1.3 potentiation signaling pathway is distinct from the CaV1.2 pathway
IGF-1 is known to rapidly potentiate the activity of neuronal CaV1.2 channels via a signaling pathway leading from the IGF-1/RTK, to stimulating the lipid kinase PI-3 kinase, to stimulating the serine/threoninc kinase, Akt, and an Src family tyrosine kinase, and resulting in Src-dependent phosphorylation of a specific tyrosine residue in the C terminus of the CaV1.2 subunit (Blair and Marshall, 1997; Blair et al., 1999; Bence-Hanulec et al., 2000). Because of the high homology between L-channel subtypes, we first tested whether any of these intermediates regulated the modulation of CaV1.3 channels. However, neither specific inhibition of PI-3 kinase with either wortmannin or LY294002 [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one] nor inhibition of Src family kinases with PP2 blocked IGF-1-induced potentiation (Fig. 2A), indicating that the modulation of CaV1.3 uses a unique pathway.
The signaling pathway regulating CaV1.3: phospholipase C, IP3-sensitive stores, and CaMKII
The CaV1.3 modulation appears to require phospholipase C-γ, calcium release from IP3-sensitive stores, and CaMKII. Specifically, we found that inhibiting the activation of phospholipase C with U73122 (10 μm) prevented the IGF-1-induced potentiation (Fig. 2A). CaV1.3-expressing SH-SY5Y cells were pretreated with inhibitor or vehicle (0.02% ethanol in extracellular saline) for 30 min, and the ability of IGF-1 to induce potentiation was tested. All U73122-treated cells failed to respond (n = 0 of 7 cells), whereas all of the sister control cells (n = 4 of 4 cells) displayed a prominent increase in CaV1.3 currents at hyperpolarized membrane potentials. U73122 inhibits several phospholipase C isoforms (γ, β, and ε). However, because the isoforms are activated by different mechanisms [phospholipase C-γ by RTKs, phospholipase C-β by G-protein-coupled receptors, phospholipase C-ε by ras (Kelley et al., 2001)], it is likely that phospholipase C-γ mediates the IGF-1/RTK effect.
Activating phospholipase C leads to increased PKC activity and increased calcium release from IP3-sensitive intracellular stores (Rhee, 2001), raising the possibility that downstream signaling components could include either PKC or intracellular calcium. We therefore tested the effects of specifically inhibiting PKC with bisindolylmaleimide I (10 μm). No block of IGF-1 potentiation was detectable, with all treated cells responding (Fig. 2A) (n = 6 of 6 cells). In contrast, inhibiting store release completely blocked the IGF-1 potentiation (Fig. 2A), indicating that elevated internal calcium is required for CaV1.3 potentiation. These data are consistent with our observation that, in conditions in which there would be no voltage-sensitive calcium channel activity, IGF-1 causes a transient increase in intracellular free calcium (Fig. 1E; see Fig. 4D).
Figure 4.
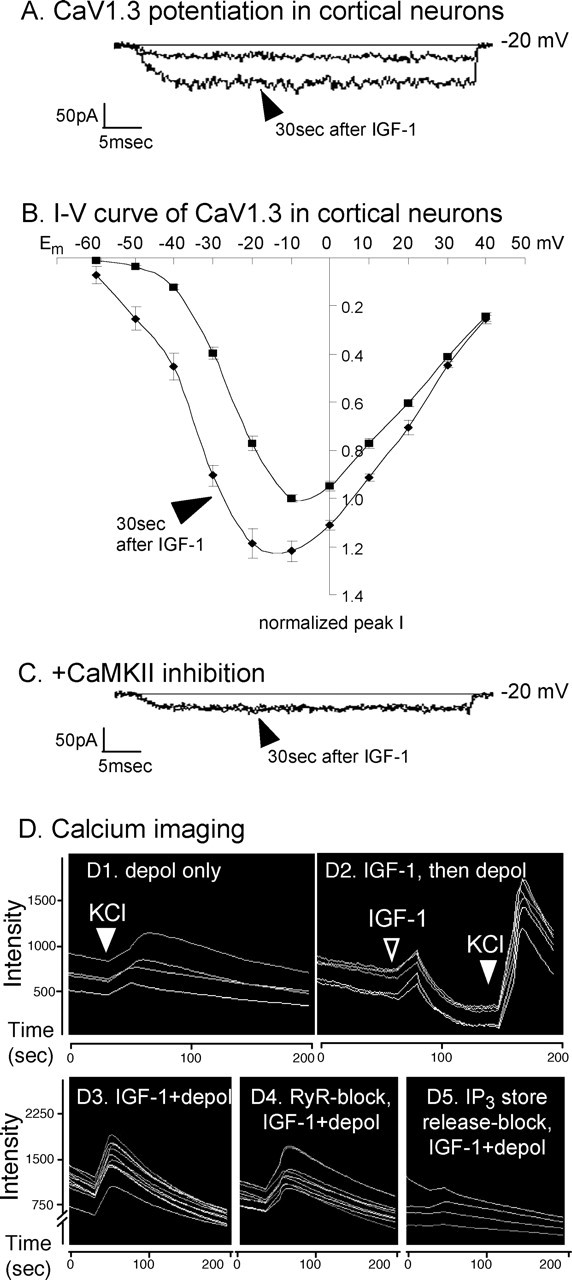
IGF-1 rapidly potentiates CaV1.3 activity in cortical neurons. A, At hyperpolarized membrane potentials, CaV1.3 channels respond within 30 s to IGF-1 (arrowhead). Recordings are from the soma of a cortical pyramidal neuron in which other voltage-sensitive channel activity has been blocked. CaV1.3 currents were evoked by depolarizing voltage steps from a holding potential of −80 mV before and 30 s after the addition of 20 ng/ml IGF-1. B, Current–voltage relationships for native CaV1.3 channels in cortical neurons. Data are plotted as normalized current versus membrane potential (Em). Similar to the neuroblastoma cells, the effect of IGF-1 on CaV1.3 in cortical neurons is much greater between −50 and −30 mV than at more depolarized potentials. ▪, Currents evoked before IGF-1; ♦, 30 s after 20 ng/ml IGF-1 (arrowhead; n = 7 neurons; mean ± SEM). Because of the variability of CaV1.3 current levels in the cortical neurons, data for each neuron were normalized to the peak current of that neuron obtained before IGF-1 addition. For each neuron, the peak current value before IGF-1 was determined; in most neurons, the pre-IGF-1 peak value was obtained at −10 mV. CaV1.3 currents in that neuron were then measured at each potential before and after IGF-1 and divided by its pre-IGF-1 peak value. C, Inhibiting CaMKII with 10 μm KN93 fully blocks the IGF-1-induced potentiation. D, IGF-1 potentiation measured by increased intracellular calcium levels. Dependence on release from IP3-sensitive stores is shown. Calcium imaging of fluo-4-loaded cortical neurons in which CaV1.3 channels were pharmacologically isolated is shown. D1, D2, Depolarization (depol; filled arrowheads) in 90 mm KCl (calculated Em= −5 mV) induces a larger increase in free intracellular calcium levels in cells treated for 1 min with 20 ng/ml IGF-1 (D2) than in untreated cells from sister cultures (D1). Similar to the neuroblastoma cells expressing CaV1.3a in Fig. 1E, the neurons responded to the IGF-1 (D2, open arrowhead) with a slight rise in intracellular calcium. D3–D5, Requirement for calcium release from IP3- but not RyR-sensitive stores in cortical neurons. Depolarization in the presence of IGF-1 causes a large, transient increase in free intracellular calcium (D3) even when RyR-sensitive store release is inhibited with 100 μm ruthenium red (D4). In contrast, pretreatment with 100 μm 2-APB to block IP3-sensitive store release primarily blocks the IGF-1+ depolarization-induced increase (D5). Results with sister cultures are shown. All neurons in each test field were imaged and calcium-fluophore intensities calculated using MetaMorph software (n = 4 independent experiments each performed in triplicate). Quantification was as follows. Because of culture–culture variabilities and because fluo-4 is an indicator of relative changes in intracellular calcium levels, data for each set of sister cultures were normalized to the mean maximum response (i.e., that obtained in depolarizing saline in the presence of IGF-1). Compared with the relative increase in free intracellular calcium obtained in IGF-1+ depolarization (100%), depolarization in the absence of IGF-1 yielded only a 17.2 ± 1.8% increase, whereas IGF-1 alone (in saline with a calculated Em= −85 mV) produced a 24.7 ± 0.7% increase; values are means ± SEMs. Inhibiting RyR-sensitive store release failed to block the IGF-1+ depolarization-induced increase (97.1 ± 5.4%), but inhibiting IP3-sensitive store release eliminated the IGF-1+ depolarization effect (2.7.1 ± 0.1%).
The IGF-1 effect appears to be mediated via release from IP3-sensitive intracellular stores. Pretreatment with the IP3 store release inhibitor 2-APB (100 μm) blocked the ability of CaV1.3b channels to respond to IGF-1 (Fig. 2A) (n = 0 of 12 cells). Conversely, pretreatment with the ryanodine receptor (RyR) inhibitor ruthenium red (100 μm) had little or no effect, with all cells responding to IGF-1 (Fig. 2A) (n = 9 of 9 cells). No differences were observed when CaV1.3a-containing channels were tested (n = 0 of 5 cells were potentiated during IP3 store release inhibition; n = 9 of 10 cells during RyR inhibition). However, a minority of RyR-inhibited cells (n = 1 of 10 cells) showed diminished responses, raising the possibility that RyR-mediated store release might make a marginal contribution; other possibilities include that RyRs were not fully inhibited or that, for other reasons, IGF-1 induced less potentiation in those particular cells. Together, the results clearly suggest that IP3-sensitive intracellular stores are the primary source for calcium release leading to IGF-1 potentiation.
The requirement for calcium store release coupled with the known association of calcium-sensitive CaM and CaMKII with neuronal CaV1.2 channels (Peterson et al., 1999; Hudmon et al., 2005) led us to test whether CaMKII was a signaling intermediate. We found that inhibiting CaMKII with 10 μm KN62 or 10 μm KN93 fully inhibited the potentiation of CaV1.3b channel activity (Fig. 2A) (n = 0 of 10 cells); the inactive analog, KN92 (10 μm), failed to block potentiation in sister cultures (n = 7 of 7 cells potentiated). Again, identical results were observed with CaV1.3a channels when CaMKII activity was blocked (n = 1 of 11 cells potentiated).
Elevating CaMKII activity exerted effects opposite to CaMKII inhibition: coexpressing a constitutively active CaMKII (ca-CaMKII) appears to mimic the IGF-1 effect, causing, in the absence of IGF-1, an increase in CaV1.3 currents at hyperpolarized membrane potentials (Fig. 2B). Although moderate/strong depolarization evoked currents that were indistinguishable (at Em= −10 mV; peak current density without ca-CaMKII, −26.2 ± 3.9 pA/pF vs −27.8 ± 2.1 pA/pF when coexpressing ca-CaMKII), huge differences were observed at relatively hyperpolarized test potentials (at Em= −40 mV; peak current density without ca-CaMKII, −6.8 ± 3.7 pA/pF; peak current density when coexpressing ca-CaMKII, −52.0 ± 10.9 pA/pF). Moreover, subsequent superfusion of IGF-1 was incapable of inducing further potentiation, suggesting that the ca-CaMKII bypasses the requirement for IGF-1. Together, these data indicate that CaMKII is required for IGF-1 potentiation of CaV1.3, acting downstream of the IGF-1/RTK.
Phosphorylation of serine-1486 of CaV1.3 is required
To identify potential modulatory sites on CaV1.3, we first considered those within CaMKII consensus sequences, RXX(S/T) (Soderling, 1996). Because we found that both CaV1.3a and CaV1.3b channels are potentiated, we focused on sites present in both isoforms. Two sites were conserved: S860 in the II–III linker, which by alignment is near a CaMKII phosphorylation site in T-type calcium channels (Welsby et al., 2003), and S1486 in the C terminus. S860 was tested and found not to contribute to the potentiation. Point mutation of this site failed to block the rapid IGF-1 effect (n = 5 of 5 cells potentiated for CaV1.3a; n = 9 of 9 cells potentiated for CaV1.3b). Note also that the PKA site in the C terminus of CaV1.2, which increases its currents in a voltage-independent manner (Naguro et al., 2001), is not conserved in CaV1.3.
S1486, found in both CaV1.3a and CaV1.3b, was a particularly attractive site because it lies within the EF-hand region that interacts with the CaM-binding IQ motif to regulate calcium-dependent inactivation (Kim et al., 2004). Although it should be noted that we do not yet know whether CaMKII phosphorylates this site in vivo, we do find that substitution of alanine for serine at this position (S1486A-CaV1.3) prevents the IGF-1 increase in the amplitude of CaV1.3 currents and the left shift in the current–voltage relationship (Fig. 3A) (n = 0 of 14 cells responded to IGF-1). Interestingly, CaV1.2 lacks a phosphorylatable residue at this position (Fig. 3C).
Figure 3.

Serine-1486, a potential CaMKII phosphorylation site on CaV1.3, is required for IGF-1-induced potentiation. A, S1486A-CaV1.3a channels are unable to respond to IGF-1. After 30 s in IGF-1 (arrowhead), currents fail to increase and a slight rundown is observed. The cumulative results from 10 SH-SY5Y cells transfected with S1486A-CaV1.3a (+ α2δ + β1b + GFP) are shown. Experiments were performed and analyzed as in Figure 1A–D. B, Cells transfected with a phospho-mimic at position 1486 (S1486D-CaV1.3a) have larger currents at hyperpolarized membrane potentials than those expressing wild-type CaV1.3. Cumulative data show that in the absence of IGF-1, S1486D-CaV1.3a channel currents peak at a relatively hyperpolarized potentials (−27.0 ± 3.7 mV; n = 9 cells transfected with S1486D-CaV1.3a). Experiments were performed and analyzed as in Figure 1A–D. C, CaV1.3 channels show almost 100% amino acid identity with CaV1.2 in this region, with the exception of S1486: CaV1.2 has an alanine in the corresponding position.
We also found that a phospho-mimic (S1486D-CaV1.3) behaves similarly to coexpressing ca-CaMKII with wild-type CaV1.3 (Fig. 3B). In the absence of IGF-1, large S1486D-CaV1.3 currents were observed at hyperpolarized membrane potentials, and the current–voltage relationship was shifted to the left (currents peaked at −27.0 ± 3.7 mV; n = 17 of 17 cells). Importantly, the phospho-mimic bypasses IGF-1: exposure to IGF-1 caused no additional potentiation (n = 0 of 9 cells for CaV1.3a; n = 0 of 8 cells for CaV1.3b), suggesting that in the wild-type CaV1.3, S1486 is required for IGF-1-induced potentiation.
IGF-1 rapidly potentiates CaV1.3 in cortical neurons
The ability of IGF-1 to modulate native CaV1.3 channels in cultured cortical neurons was determined. After pharmacological inhibition of all other voltage-gated ion channels and, in particular, in the presence of 1 μm nimodipine to block native CaV1.2 channel activity, CaV1.3 activity in cortical neurons was evoked and IGF-1 responsiveness was tested as for the neuroblastoma cells. We found that IGF-1 (20 ng/ml) strongly potentiates the activity of CaV1.3 channels within 30 s (Fig. 4A) (n = 7 of 7 cells). We also found that the voltage dependence of CaV1.3 activation is shifted, with potentiation being very prominent at hyperpolarized membrane potentials and disappearing with increased depolarization (Fig. 4B). Such a shift in CaV1.3 activity raises the possibility that stimulating neuronal IGF-1/RTKs might significantly increase the ability to respond to synaptic stimulation.
Modulation of native neuronal CaV1.3 channels also requires CaMKII. As with heterologously expressed channels in neuroblastoma cells, pretreatment with 10 μm KN93 fully inhibited the potentiation of cortical neuronal CaV1.3 channel activity (Fig. 4C) (n = 0 of 8 neurons potentiated), indicating that CaMKII is a requisite signaling intermediate. Moreover, when endogenous CaV1.3 levels or when surface expression of epitope-tagged CaV1.3a were assessed immunocytochemically, we found that CaV1.3 was easily detectable in dendritic shafts and spines of cortical and hippocampal neurons and that surface CaV1.3a frequently colocalized with CaMKII in the tips of dendritic spines (Fig. 5). Specifically, when compared with all spine heads in which immunofluorescently labeled surface HA-CaV1.3a was detected, GFP-tagged CaMKII was also detected in 71.2 ± 5.2%.
Figure 5.

Surface CaV1.3 colocalizes with CaMKII in the heads of dendritic spines of pyramidal neurons. A, Surface CaV1.3a, epitope tagged extracellularly with HA, was detected immunocytochemically (TRITC) and found to primarily colocalize with the spontaneous fluorescence of the GFP-tagged CaMKII. The overlay of CaMKII and CaV1.3a shows numerous orange and yellow spine tips, indicating the overlap of green and red markers. B, To reveal surface localization of CaV1.3a in relation to spine structure, neurons in sister cultures were transfected with GFP- and HA-tagged CaV1.3a and surface HA was detected immunocytochemically (TRITC). The overlay shows that CaV1.3a is prominently localized to dendritic shafts and spine tips.
Calcium imaging confirmed the IGF-1 potentiation and indicated a dependence on IP3-sensitive intracellular store release in cortical neurons. When CaV1.3 channels were depolarized by elevating extracellular potassium, those that had been treated immediately prior with IGF-1 showed larger responses (Fig. 4D). We additionally observed that at a predicted membrane potential of approximately −85mV (in which no CaV1.3 channels should be active), exposure to IGF-1 caused a small, transient increase in free intracellular calcium (Fig. 4D2). This suggests that in addition to potentiating the CaV1.3 channels, IGF-1 also causes calcium release from internal stores.
Release from IP3-sensitive intracellular stores appears to be crucial for IGF-1 modulation in neurons (Fig. 4D3–D5). Pretreatment with thapsigargin to deplete stores or with the IP3 store release inhibitor 2-APB (100 μm) blocked the ability of pharmacolocially isolated CaV1.3 channels to respond to depolarization in the presence of IGF-1 (Fig. 4D5) (n = 0 of 37 2-APB-treated neurons; n = 0 of 53 thapsigargin-treated neurons). Conversely, pretreatment with the RyR inhibitor ruthenium red (100 μm) had little or no effect, with all cells responding to IGF-1 (Fig. 4D4) (n = 44 of 44 neurons). However, a minority of the RyR-inhibited neurons (N = 3 of 44 neurons) showed weak responses, raising the possibility that RyR-mediated store release might make a minor contribution. Together, the results clearly imply that IP3-sensitive intracellular stores are the primary source for calcium release in the pathway leading to IGF-1 potentiation.
IGF-1 potentiation of CaV1.3a increases pCREB
The transcription factor CREB is required for IGF-1- and neurotrophin-mediated neuronal survival (Riccio et al., 1999; Brunet et al., 2001; West et al., 2001). Particularly relevant here is that stimulating CaV1.2 L-channel activity is known to promote the formation of activated phospho-S133 CREB (Deisseroth et al., 1998; Dolmetsch et al., 2001; West et al., 2001; Chen et al., 2003). We found that, in cortical and hippocampal neurons, IGF-1 potentiation of CaV1.3 increases pCREB, whereas S1486A-CaV1.3a or small interfering RNA to endogenous CaV1.3a decrease resting pCREB levels and essentially block the ability of IGF-1 to stimulate pCREB (Fig. 6A–G).
Figure 6.

CaV1.3a regulates pCREB in pyramidal neurons: requirements for IGF-1 potentiation of S1486, IP3 store release, and CaV1.3 interaction with Shank. A–L, Levels of activated pCREB were assayed by indirect immunofluorescence. To assess any regulation specifically attributable to CaV1.3 activity, all other calcium channel activity was inhibited pharmacologically (see Materials and Methods). To stimulate the activity of voltage-sensitive CaV1.3 channels, neurons were depolarized for 20 min in iso-osmotic 65 mm KCl, fixed immediately, and processed for immunocytochemistry (n = 8 independent experiments with each test condition assayed in triplicate). Changes in pCREB levels were analyzed using the Intensity module of the MetaMorph image analysis software; the fold change in pCREB was calculated using the mean level observed in the absence of any treatment (A) as the reference value of 1.0; values are means ± SEMs. A, In the absence of any treatment, pCREB levels were moderate/low. B, IGF-1 (20 ng/ml) alone has little effect. Fold change in pCREB levels were 1.04 ± 0.07. C, D, Depolarization in the absence of IGF-1 increases pCREB levels 1.6 ± 0.1-fold (C), but, in the presence of IGF-1, pCREB levels are very high (D), increasing by 3.1 ± 0.1-fold. E, F, Reducing CaV1.3 activity with 100 μm nimodipine (E) or CaV1.3a expression with RNAi specific for CaV1.3a (F; see Fig. 6M,N and supplemental Fig. 2 for controls) strongly reduces the elevation of pCREB induced by depolarization plus IGF-1. F, Left, The RNAi transfectant was identified by GFP coexpression. Right, pCREB levels are low in the transfectant (arrowhead) but high in two neighboring nontransfected neurons. Both treatments resulted in pCREB levels below that of the reference condition: high nimodipine, 0.8 ± 0.1; RNAi-CaV1.3a, 0.7 ± 0.1. G, Overexpressing S1486A-CaV1.3a prevents the IGF-1+ depolarization-induced increase in pCREB, producing a fold increase of only 1.3 ± 0.1. Left, Transfected neuron indicated by GFP fluorescence. Right, pCREB levels in all neurons in the same field with the GFP+ neuron (arrowhead). H, Requirement for Shank: overexpressing Shank-1a with a W520A point mutation to disrupt its SH3 domain blocks the ability of IGF-1+ depolarization to increase pCREB; the fold change in pCREB levels was 1.1 ± 0.1. Left, Transfected neuron (GFP). Right, pCREB levels in all neurons in the field with the W520A-Shank-1a+ neuron (open arrowhead). Inset, Unlike W520A-Shank-1a+ neurons, overexpressing wild-type (wt) Shank-1a in a sister culture results in high pCREB levels (filled arrowhead); the fold increase was 3.3 ± 0.1. I, Requirement for Shank–Homer interaction: overexpressing Shank-1a with a P1495L point mutation to disrupt its binding to Homer blocks the ability of IGF-1+ depolarization to increase pCREB; the fold change in pCREB was 0.9 ± 0.1. Left, Transfected neuron (GFP). Right, pCREB levels in all neurons in the field with the P1495L-Shank-1a+ neuron (open arrowhead). J–L, Ca2+ store dependence. Depleting stores with 1 μm thapsigargin (I) or blocking IP3-sensitive store release with 100 μm 2-APB (L) prevents the IGF-1+ depolarization-induced increase in pCREB, whereas inhibiting ryanodine (RyR)-sensitive store release (K) with 100 μm ruthenium red has little effect, producing a fold increase of 2.9 ± 0.1. For both store depletion and blocking IP3-sensitive store release, the fold change in pCREB levels was 0.9 ± 0.1. M, N, In cortical pyramidal neurons, RNAi to CaV1.3 strongly reduced CaV1.3 levels (M) and evoked CaV1.3 activity (N). M, Left, GFP cotransfection reveals a neuron expressing RNAi to CaV1.3a. Right, Total CaV1.3 (surface plus intracellular) in that neuron (open arrowhead indicates the soma). N, Somal CaV1.3 currents are evident in the absence of RNAi (N1) but undetectable in an RNAi-transfected neuron (N2). Recordings were performed in the presence of 1 μm nimodipine to inhibit CaV1.2 activity. See supplemental Fig. 2 (available at www.jneurosci.org as supplemental material) for additional information.
Phospho-S133 CREB (pCREB) levels in cultured hippocampal and cortical pyramidal neurons were assessed by indirect immunofluorescence after a 20 min exposure to IGF-1, to depolarizing extracellular saline (calculated Em= −20 mV) or to depolarization in the presence of IGF-1. To ensure that any regulation was specifically attributable to CaV1.3, all other calcium channels were inhibited pharmacologically; importantly, 1 μm nimodipine was used to inhibit native CaV1.2 activity. We found that in the absence of depolarization, pCREB levels were moderate/low (Fig. 6A) and that IGF-1 treatment without depolarization produced only a slight increase (Fig. 6B). In contrast, depolarization resulted in a considerable increase (Fig. 6C), and depolarization in the presence of IGF-1 caused a massive rise (Fig. 6D).
To establish CaV1.3 dependence, CaV1.3 activity was inhibited pharmacologically (Fig. 6E) and CaV1.3a expression was reduced with a short hairpin interfering RNA (RNAi) (Fig. 6F,M,N; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Specifically, neurons were transfected with RNAi-CaV1.3a, or untransfected neurons were treated with 100 μm nimodipine, a concentration that should inhibit CaV1.3 activity (Lipscombe et al., 2004), and were depolarized in the presence of IGF-1. Reducing CaV1.3 activity and CaV1.3a levels both reduced the elevation of pCREB (Fig. 6E,F). We also found that the IGF-1+ depolarization-induced increase requires S1486: overexpressing an epitope-tagged S1486A-CaV1.3a strongly reduced the ability of IGF-1+ depolarization to elevate pCREB (Fig. 6G; supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
We also examined the potential roles of Shank and Homer scaffolding proteins. Shank has recently been demonstrated to interact with the C terminus of CaV1.3a (Olson et al., 2005; Zhang et al., 2005) and is known to be important in recruiting Homers to mGlu receptors (mGluRs) (Tu et al., 1999). Homers, in turn, act to tether group I mGluRs to IP3-sensitive stores as well as increase IP3 receptors in dendritic spines (Tu et al., 1998; Sala et al., 2001). Based on our observation that CaV1.3 modulation requires calcium release from IP3-sensitive stores, we hypothesized that the CaV1.3-induced regulation of pCREB might arise through Shank interactions with CaV1.3, which could, via Homer, link the channels to IP3-sensitive calcium stores.
We found that overexpressing Shank-1a with a disrupted SH3 domain (W520A-Shank-1a) both reduced the ability of depolarization and IGF-1 to elevate pCREB in cortical neurons (Fig. 6H) and reduced Shank colocalization with plasmalemmal CaV1.3a (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Similarly, depolarization in the presence of IGF-1 was unable to stimulate formation of pCREB in neurons overexpressing a Shank-1a mutant (P1495L) unable to bind Homer (Fig. 6I). In contrast, when wild-type Shank-1a was overexpressed, IGF-1in the depolarizing saline strongly enhanced pCREB (Fig. 6H, inset). We also found that calcium release from intracellular stores was required. Depleting stores with thapsigargin (Fig. 6J) or blocking IP3-sensitive store release (Fig. 6L) prevented the IGF-1- and depolarization-induced increase in pCREB, whereas inhibiting ryanodine-sensitive store release (Fig. 6K) had little effect. Together, these results indicate that IGF-1-induced potentiation of cortical CaV1.3 channels via phosphorylation of a specific serine residue, S1486, may be a mechanism for regulating CREB (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Discussion
Together, the data indicate that CaV1.3 activity is rapidly potentiated by IGF-1 in a pathway that involves stimulating phospholipase C-γ to produce IP3, calcium release from IP3-sensitive internal stores, activating CaMKII, and the phosphorylation of a CaMKII consensus site in the C termini of CaV1.3a and CaV1.3b (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Surprisingly, this modulatory pathway is distinctly different from that of CaV1.2, in which IGF-1 stimulation of its RTK activates PI-3 kinase, Akt, and Src and leads to phosphorylation of a C-terminal tyrosine residue on CaV1.2 (Blair and Marshall, 1997; Blair et al., 1999; Bence-Hanulec et al., 2000). Moreover, the required serine, S1486, is not conserved in CaV1.2, and the tyrosine required for CaV1.2 potentiation is not present in either CaV1.3a or CaV1.3b. Nonetheless, for CaV1.2 and both CaV1.3 isoforms, IGF-1 stimulation results in increased evoked currents at hyperpolarized membrane potentials, increased rates of activation at hyperpolarized potentials, and a left shift in peak evoked current–membrane potential relationships.
CaMKII-dependent phosphorylation of S1486 could potentially increase CaV1.3 currents by either inducing a facilitation-like state or by reducing inactivation. Intriguingly, S1486, which is found in both CaV1.3a and CaV1.3b, is located within the EF hand, a region that interacts with the CaM-binding IQ motif to regulate calcium-dependent inactivation in the highly homologous CaV1.2 (Kim et al., 2004). Inactivation is an important mechanism for controlling calcium entry, and, although not yet completely understood, recent studies have revealed many essential features of calcium- and voltage-dependent inactivation. The EF hand appears to act as a signal transducer to mediate calcium-dependent inactivation, and deletion of the EF hand motif strongly affects both calcium-dependent and voltage-dependent inactivation in CaV1.2 (Peterson et al., 2000; Kim et al., 2004; Cens et al., 2005), raising the possibility that phosphorylation of S1486 might influence inactivation.
Conversely, relatively little is known of the molecular mechanisms governing facilitation. In cardiac myocytes, L-channel activity is facilitated by a CaMKII-dependent mechanism (Dzhura et al., 2000), but in hippocampal neurons, L-channel activity in dendritic spines is facilitated by β2 adrenergic receptors via a PKA pathway (Davare et al., 2001; Hoogland and Saggau, 2004). Adrenochromaffin cells appear to use another means, whereby facilitation occurs via recruitment of quiescent, but otherwise identical, L channels (Artalejo et al., 1994). However, this mechanism is unlikely to explain our results. We observed large increases in peak currents only at hyperpolarized membrane potentials and no potentiation at strongly depolarized potentials. The multiplicity of L-channel subtypes in neurons has also made it difficult to determine whether facilitation occurs via CaV1.2, CaV1.3, or both. However, in striatal medium spiny neurons, D2 dopaminergic receptor-induced reduction of L currents via a phospholipase C-β–IP3–calcineurin signaling pathway (Hernandez-Lopez et al., 2000) appears to affect CaV1.3 specifically (Olson et al., 2005), raising the possibility that phospho-S1486 could be the target of calcineurin.
Downstream effects of activating CaV1.3 channels include increased IP3-sensitive internal store release via a CaV1.3–Shank–Homer interaction, potentially creating a signaling microdomain (Jacob et al., 2005) that leads to increased levels of the transcription factor pCREB (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). If the pathway we propose for the IGF-1 modulation of CaV1.3 activity is correct, a positive feedback loop would be created. Calcium release from IP3-sensitive stores appears to be important for activating CaMKII to phosphorylate the channel, with the resultant increased channel activity leading to increased calcium release from IP3 stores. However, CaMKII may also provide a potential braking mechanism via phosphorylation and reduced activity of the IP3 receptors (Matifat et al., 2001; Bare et al., 2005).
In the long term, IGF-1 modulation of CaV1.3 may regulate processes such as dendritic growth and neuronal survival by regulating transcription. CaV1.2-mediated calcium influx is known to lead, via the MAPK (mitogen-activated protein kinase) pathway, to elevation of survival promoting phospho-S133 pCREB levels (Dolmetsch et al., 2001; Weick et al., 2003) and, via a CaMKIV and CRM1–exportin-1 pathway, to decreased nuclear levels of the pro-apoptotic transcription factor C/EBPβ (Marshall et al., 2003). For the regulation of pCREB we observe here, CaV1.3a, the extended C-terminal isoform, appears to be crucial. The truncated isoform, CaV1.3b, lacks both the SH3 and PDZ binding sites thought to mediate Shank binding to CaV1.3a (Olson et al., 2005; Zhang et al., 2005). We found in neurons and heterologous cells that CaV1.3a and Shank-1a colocalize and enhance pCREB levels. Furthermore, expressing a CaV1.3a with a C-terminal truncation to remove the PDZ binding site or expressing wild-type CaV1.3a with a Shank-1a, the SH3 domain of which is nonfunctional, eliminates colocalization and the ability of CaV1.3a activity to stimulate pCREB. This dependence on Shank, taken with the absence of Shank interactions with CaV1.3b (Zhang et al., 2005), indicates that CaV1.3a is responsible for the downstream regulation of CREB.
How CaV1.3 and CaV1.2 channel activities both contribute to regulating CREB is as yet unknown. Like CaV1.3, IGF-1 also potentiates the CaV1.2 channels (Blair and Marshall, 1997; Blair et al., 1999; Bence-Hanulec et al., 2000) (supplemental Fig. 2E, available at www.jneurosci.org as supplemental material). At the simplest level, these could be back-up pathways (i.e., system redundancy); in fact, knockdown of CaV1.2 enhances CaV1.3 expression in cardiac myocytes (Xu et al., 2003), and we found that RNAi knockdown of CaV1.3 enhances CaV1.2 expression in cortical neurons. In addition, the differences in the biophysical properties and distribution of CaV1.2 and CaV1.3 channels raise the possibility that channels in different subcellular compartments are regulated by different stimuli. The relatively higher levels of surface CaV1.3a in dendrites and spines suggest that glutaminergic synaptic activity may be their major source of depolarization-dependent activation, whereas CaV1.2 channel activity may primarily be significant during action potential spiking. Moreover, the finding that CaV1.3a in dendritic spine heads frequently colocalizes with synaptic markers and with CaMKII suggests that IGF-1 modulation of CaV1.3 may modulate the efficacy of synaptic transmission.
The different L channels may also differentially regulate the targets of transcription. CREB, as a result of calcium influx, is phosphorylated at multiple sites. CaV1.2 activity is well established to promote phosphorylation of S133 (Dolmetsch et al., 2001; Kornhauser et al., 2002; Weick et al., 2003). Our results as well as a recent study by Zhang et al. (2005) show that CaV1.3 similarly stimulates formation of phospho-S133 CREB. However, Kornhauser et al. (2002) also demonstrated that low concentrations of nimodipine fully blocked depolarization-induced phosphorylation of S142. Because low nimodipine concentrations were sufficient, the data suggest that S142 phosphorylation may be independent of CaV1.3 activity. Their data additionally indicate that phosphorylation of S142 disrupts CREB interactions with the transcriptional coactivator CBP (Kornhauser et al., 2002). Together, these results raise the possibility that CaV1.2 activity may favor CREB transcription not requiring CBP as a cofactor, whereas CaV1.3 activity may principally influence CREB/CBP-mediated transcription. Likewise, another possibility is that CaV1.3 and CaV1.2 channel activities might differentially recruit additional transcription factors, such as CREST (Aizawa et al., 2004; Konur and Ghosh, 2005).
Interestingly, IGF-1 and calcium influx regulation of CREB, CBP, and CREST are essential for normal dendritic outgrowth in the brain (Niblock et al., 2000; Cheng et al., 2003; Aizawa et al., 2004; Konur and Ghosh, 2005). In the shorter term, mGluR-induced release from IP3-sensitive stores has been shown recently to regulate the stability of dendritic arborization and to enhance in a Homer-dependent manner L-channel-mediated calcium influx (Lohmann et al., 2002; Yamamoto et al., 2005). Although these studies did not attempt to separate CaV1.3 from CaV1.2 activities, our observations of IP3 store dependence in the IGF-1/RTK-induced modulation of CaV1.3 raise the tantalizing possibility of cross talk between receptor systems.
Footnotes
*L.G. and L.A.C.B. contributed equally to this work.
This work was supported by National Institutes of Health Grants RO1 NS39063 and NS39309 (J.M.) and RO1 NS37676 (L.A.C.B.) and by Centers of Biomedical Research Excellence Grant P20 RR 15578 (L.A.C.B.). We thank Dr. Diane Lipscombe (Brown University, Providence, RI) for generously providing us with the wild-type CaV1.3a and CaV1.3b constructs, Drs. Richard Huganir, Gareth Thomas (Johns Hopkins School of Medicine, Baltimore, MD), and Tobias Boeckers (Ulm University, Ulm, Germany) for Shank-1a constructs, and Dr. Terry Snutch (University of British Columbia, Vancouver, British Columbia, Canada) for the rat brain α2δ and β1b clones.
References
- Aizawa H, Hu S-C, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A (2004). Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 303:197–202. [DOI] [PubMed] [Google Scholar]
- Artalejo CR, Adams ME, Fox AP (1994). Three types of Ca2+ channel trigger secretion with different efficacies in chromaffin cells. Nature 367:72–76. [DOI] [PubMed] [Google Scholar]
- Bare DJ, Kettlun CS, Liang M, Bers DM, Mignery GA (2005). Cardiac type 2 inositol 1,4,5-trisphosphate receptor: interaction and modulation by calcium/calmodulin-dependent protein kinase II. J Biol Chem 28:15912–15920. [DOI] [PubMed] [Google Scholar]
- Bence-Hanulec KK, Marshall J, Blair LAC (2000). Potentiation of neuronal L calcium channels by IGF-1 requires phosphorylation of the α1 subunit on a specific tyrosine residue. Neuron 27:121–131. [DOI] [PubMed] [Google Scholar]
- Blair LAC, Marshall J (1997). IGF-1 modulates N and L calcium channels in a PI 3-kinase-dependent manner. Neuron 19:421–429. [DOI] [PubMed] [Google Scholar]
- Blair LAC, Bence-Hanulec KK, Mehta S, Franke TF, Kaplan D, Marshall J (1999). Akt-dependent potentiation of L channels by insulin-like growth factor-1 is required for neuronal survival. J Neurosci 19:1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME (2001). Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11:297–305. [DOI] [PubMed] [Google Scholar]
- Cens T, Rousset M, Leyris JP, Fesquet P, Charnet P (2006). Voltage- and calcium-dependent inactivation in high voltage-gated Ca2+ channels. Prog Biophys Mol Biol 90:104–117. [DOI] [PubMed] [Google Scholar]
- Chen WG, West AE, Tao X, Corfas G, Szentirmay MN, Sawadogo M, Vinson C, Greenberg ME (2003). Upstream stimulatory factors are mediators of Ca2+-responsive transcription in neurons. J Neurosci 23:2572–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CM, Mervis RF, Niu S-L, Salem N, Witters LA, Tseng V, Reinhardt R, Bondy CA (2003). Insulin-like growth factor 1 is essential for normal dendritic growth. J Neurosci Res 73:1–9. [DOI] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW (2001). A β2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science 293:98–101. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW (1998). Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 392:198–202. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME (2001). Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294:333–339. [DOI] [PubMed] [Google Scholar]
- Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME (2000). Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol 2:173–177. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Greenberg ME (1998). Ca2+ channel-regulated neuronal gene expression. J Neurobiol 37:171–189. [PubMed] [Google Scholar]
- Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G (1995). Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci 15:1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin K, Banker G (1989). Experimental observations on the development of polarity by hippocampal neurons in culture. J Cell Biol 108:1507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981). Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch 391:85–100. [DOI] [PubMed] [Google Scholar]
- Helton TD, Xu W, Lipscombe D (2005). Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci 25:10247–10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ (2000). D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLCβ1–IP3–calcineurin-signaling cascade. J Neurosci 20:8987–8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland TM, Saggau P (2004). Facilitation of L-type Ca2+ channels in dendritic spines by activation of β2 adrenergic receptors. J Neurosci 24:8416–8427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS (2005). CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol 171:537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob SN, Choe CU, Uhlen P, DeGray B, Yeckel MF, Ehrlich BE (2005). Signaling microdomains regulate inositol 1,4,5-trisphosphate-mediated intracellular calcium transients in cultured neurons. J Neurosci 25:2853–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley GG, Reks SE, Ondrako JM, Smrcka AV (2001). Phospholipase Ce: a novel Ras effector. EMBO J 20:743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ghosh S, Nunziato DA, Pitt GS (2004). Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron 41:745–754. [DOI] [PubMed] [Google Scholar]
- Konur S, Ghosh A (2005). Calcium signaling and the control of dendritic development. Neuron 46:401–405. [DOI] [PubMed] [Google Scholar]
- Kornhauser JM, Cowan CW, Shaywitz AJ, Dolmetsch RE, Griffith EC, Hu LS, Haddad C, Xia Z, Greenberg ME (2002). CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron 34:221–233. [DOI] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J (2001). α1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J Biol Chem 276:22100–22106. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Helton TD, Xu W (2004). L-type calcium channels: the low down. J Neurophysiol 92:2633–2641. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Wong ROL (2005). Regulation of dendritic growth and plasticity by local and global calcium transients. Cell Calcium 37:403–409. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Myhr KL, Wong ROL (2002). Transmitter-evoked local calcium release stabilizes developing dendrites. Nature 418:177–181. [DOI] [PubMed] [Google Scholar]
- Marshall J, Dolan BM, Garcia EP, Sathe S, Tang X, Mao Z, Blair LAC (2003). Calcium channel and NMDA receptor activities differentially regulate nuclear C/EBPβ levels to control neuronal survival. Neuron 39:625–639. [DOI] [PubMed] [Google Scholar]
- Matifat F, Hague F, Brûulé G, Collin T (2001). Regulation of InsP3-mediated Ca2+ release by CaMKII in Xenopus oocytes. Eur J Physiol 441:796–801. [DOI] [PubMed] [Google Scholar]
- Naguro I, Nagao T, Adachi-Akahane S (2001). Ser1901 of α1C subunit is required for the PKA-mediated enhancement of L-type Ca2+ channel currents but not for the negative shift of activation. FEBS Lett 489:87–91. [DOI] [PubMed] [Google Scholar]
- Niblock MM, Brunso-Bechtold JK, Riddle DR (2000). Insulin-like growth factor 1 stimulates dendritic growth in the primary somatosensory cortex. J Neurosci 20:4165–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ (2005). G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci 25:1050–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP, Yue DT (1999). Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 22:549–558. [DOI] [PubMed] [Google Scholar]
- Peterson BZ, Lee JS, Mulle JG, Wang Y, de Leon M, Yue DT (2000). Critical determinants of Ca2+-dependent inactivation within an EF-hand motif of L-type Ca2+ channels. Biophys J 78:1906–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M (1991). Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods 37:15–26. [DOI] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, Ghosh A (2002). Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34:999–1010. [DOI] [PubMed] [Google Scholar]
- Reeve HL, Vaughan PF, Peers C (1994). Calcium channel currents in undifferentiated human neuroblastoma (SH- SY5Y) cells: actions and possible interactions of dihydropyridines and w-conotoxin. Eur J Neurosci 6:943–952. [DOI] [PubMed] [Google Scholar]
- Rhee SG (2001). Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem 70:281–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD (1999). Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286:2358–2361. [DOI] [PubMed] [Google Scholar]
- Simon M, Perrier JF, Hounsgaard J (2003). Subcellular distribution of L-type Ca2+ channels responsible for plateau potentials in motoneurons from the lumbar spinal cord of the turtle. Eur J Neurosci 18:258–266. [DOI] [PubMed] [Google Scholar]
- Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31:115–130. [DOI] [PubMed] [Google Scholar]
- Soderling TR (1996). Structure and regulation of calcium/calmodulin-dependent protein kinases II and IV. Biochim Biophys Acta 1297:131–138. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF (1998). Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron 21:717–726. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23:583–592. [DOI] [PubMed] [Google Scholar]
- Weick JP, Groth RD, Isaksen AL, Mermelstein PG (2003). Interactions with PDZ proteins are required for L-type calcium channels to activate cAMP response element-binding protein-dependent gene expression. J Neurosci 23:3446–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML, Barrett PQ (2003). A mechanism for the direct regulation of T-type calcium channels by Ca2+/calmodulin-dependent kinase II. J Neurosci 23:10116–10121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001). Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 98:11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Hoskins L, Catterall WA (1998). Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J Neurosci 18:6319–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Welling A, Paparisto S, Hofmann F, Klugbauer N (2003). Enhanced expression of L-type Cav1.3 calcium channels in murine embryonic hearts from Cav1.2-deficient mice. J Biol Chem 278:40837–40841. [DOI] [PubMed] [Google Scholar]
- Xu W, Lipscombe D (2001). Neuronal CaV1.3α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21:5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Sakagami Y, Sugiura S, Inokuchi K, Shimohama S, Kato N (2005). Homer 1a enhances spike-induced calcium influx via L-type calcium channels in neocortex pyramidal cells. Eur J Neurosci 2:1338–1348. [DOI] [PubMed] [Google Scholar]
- Zhang H, Maximov A, Fu Y, Xu F, Tang TS, Tkatch T, Surmeier DJ, Bezprozvanny I (2005). Association of CaV1.3 L-type calcium channels with Shank. J Neurosci 25:1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]