

Abstract
Regulation of synaptic efficacy by nerve terminal excitability has not been extensively studied. We performed genetic and pharmacological dissections for presynaptic actions of K+ channels in Drosophila neuromuscular transmission by using electrophysiological and optical imaging techniques. Current understanding of the roles of the Shab IK channel and its mammalian Kv2 counterparts is relatively poor, as compared with that for Shaker IA channels and their Kv1 homologues. Our results revealed the striking effect of Shab mutations during high-frequency synaptic activity, as well as a functional division in synaptic regulation between the Shaker and Shab channels. Shaker channels control the basal level of release, indicated by a response to single nerve stimulation, whereas Shab channels regulate repetitive synaptic activities. These observations highlight the crucial control of nerve terminal excitability by Shaker and Shab channels to confer temporal patterns of synaptic transmission and suggest the potential participation of these channels, along with the transmitter release machinery, in activity-dependent synaptic plasticity.
Keywords: cumulative inactivation, terminal action potential backfiring, presynaptic Ca2+ dynamics, frequency-dependent enhancement, basal transmitter release, motor pattern generation
Introduction
Modulation of synaptic efficacy during patterned activities of presynaptic neurons is important for motor control and for other neuronal network functions (Paulsen and Sejnowski, 2000). In principle, synaptic efficacy can be modulated by regulating membrane excitability and transmitter release in the presynaptic terminals (Kandel et al., 1987; Zhou and Poo, 2004), although modulation of the transmitter release mechanisms has been more extensively studied (Zucker and Regehr, 2002). Potassium currents play crucial roles in regulating membrane excitability and could thus contribute to the control of synaptic efficacy. Both rapidly inactivating A-type (IA) and delayed rectifier (IK) K+ currents have been documented in presynaptic terminals of different species. In some preparations, including the vertebrate calyx of Held (Forsythe, 1994), and the squid giant synapse (Augustine, 1990), delayed rectifier is the predominant K+ current, whereas in the hippocampal mossy fiber (Geiger and Jonas, 2000) and pituitary hypophysis nerve terminals (Thorn et al., 1991), IA is prominent. However, the biological regulation and functional significance of such differential K+-channel expression in the presynaptic terminal still await elucidation.
The Drosophila Shaker (Sh) gene and its vertebrate homologs (Kv1 members) are the best studied among the diverse K+-channel subunit genes thus far identified (Coetzee et al., 1999). Drosophila Sh channels mediate inactivating IA in both in vivo (Salkoff and Wyman, 1981; Wu and Haugland, 1985; Baker and Salkoff, 1990) and heterologous expression preparations (Iverson et al., 1988; Timpe et al., 1988a,b). Several vertebrate Kv1 channels also mediate inactivating IA (Stuhmer et al., 1989; Heinemann et al., 1996; Kalman et al., 1998). Loss of Kv1.1 function in gene-knockout mice leads to epileptiform bursting discharge and seizure (Smart et al., 1998). Drosophila Sh mutants also display seizure-like leg shaking under ether anesthesia (Kaplan and Trout, 1969), abnormal spike bursting in motor circuits (Engel and Wu, 1992), and greatly enhanced neurotransmission at neuromuscular junctions (Jan et al., 1977; Ganetzky and Wu, 1982, 1983).
Although the Shab gene is homologous to Sh (Butler et al., 1989), the in vivo functions of Shab and its vertebrate homologs, the Kv2 members, are much less known. Heterologous expression of Shab produces sustained delayed rectifier K+ currents (Wei et al., 1990), consistent with the phenotypes of Shab mutants (i.e., reduced IK in neurons) (I.-F. Peng and C.-F. Wu, unpublished observation) and muscle cells (Singh and Singh, 1999; Chopra et al., 2000). Additionally, dominant-negative Kv2 expression reduces IK in vertebrate cultured neurons (Blaine and Ribera, 2001; Malin and Nerbonne, 2002). However, the role of Shab and Kv2 channels in the control of presynaptic terminal excitability and neurotransmission has not been established.
Here, we report strikingly different phenotypes of Shab and Sh mutations in neurotransmission, coupled with altered nerve terminal excitability and central pattern generation in Drosophila larvae. Sh IA is responsible for regulating basal-release levels, whereas Shab IK plays a more important role in transmission-level control during high-frequency activity. Our results demonstrate a functional division between Sh IA and Shab IK in regulation of synaptic transmission within different temporal domains and frequency ranges.
Materials and Methods
Drosophila stocks.
The Drosophila melanogaster stocks used included Shab9g, Shab1, Shab2, Sh133, and ShM mutants, and a wild-type (WT) strain Canton-Special. In these mutants, sustained K+ current IK is known to be either decreased in amplitude (Shab1, Shab2) or not detectable (Shab9g) in muscle cells (Singh and Singh, 1999) and cultured neurons (I.-F. Peng and C.-F. Wu, unpublished observations). Inactivating K+ current IA is not detectable in muscle cells (Wu and Haugland, 1985) and decreased in amplitude in the soma of cultured neurons (Zhao et al., 1995) (I.-F. Peng and C.-F. Wu, unpublished observation) in both Sh alleles. Shab stocks were provided by Dr. S. Singh (State University of New York, Buffalo, NY) (Chopra et al., 2000) and the Sh strains were from the original collection of Dr. S. Benzer (California Institute of Technology, Pasadena, CA) (Wu and Haugland, 1985). The multiple mutant alleles of independent isolates used provided control for possible unidentified second-site effects. Results from different alleles of the mutants were consistent unless otherwise noted. Therefore, data from different alleles were combined for the simplicity of display. A fly strain carrying UAS-GCaMP-9, a Ca2+-responsive indicator for Ca2+ imaging, was a generous gift from Drs. Yalin Wang and Yi Zhong (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY) (Wang et al., 2004). All stocks were raised at room temperature on standard medium.
Larval locomotion.
Recordings of larval locomotor behavior on agar plates were performed as described previously (Wang et al., 1997; 2002). Plates made of 0.7% agarose in distilled water were used as the locomotion substrate. Agarose was degassed by boiling and subsequently poured into 14 cm Petri dishes. Wandering stage larvae were removed from the wall of a food vial, washed briefly with distilled water, and transferred by brush to the agar plate. Larvae were allowed to crawl over the agar plate for 2 min or until reaching the edge of the plate. The resultant larval locomotor track was photographed by placing the agar plate on a photographic paper and subjecting it to even illumination from above. The photograph was later enlarged with a dissection microscope equipped with a digital camera to examine the morphology of mouthhook prints.
Electrophysiology.
Postfeeding third instar larvae crawling on the wall of food vials were chosen for recording. Dissections were performed in HL3.1 saline containing (in mm) 70 NaCl, 5 KCl, 4 MgCl2, 10 NaHCO3, 5 trehalose, 115 Sucrose, and 5 HEPES, at pH 7.2, with or without 1 mm CaCl2 (Feng et al., 2004). [Ca2+]o in recording saline varied in different experiments as specified. To evoke nerve action potentials and excitatory junctional potentials (ejps), the segmental nerve was severed from the ventral ganglion and stimulated through the cut end with a suction electrode (10 μm inner diameter). The stimulation voltage was adjusted to 2–2.5 times the threshold voltage to ensure action potential initiation. For ejp recordings, intracellular glass microelectrodes were filled with 3 m KCl and had a resistance of ∼60 MΩ. Ejps were recorded mostly from muscles 1, 6, 7, and 9 with a direct current preamplifier (model M701 microprobe system; WPI, Sarasota, FL).
Action potentials were recorded extracellularly from the segmental nerve en passant in HL3.1 saline with a suction pipette (Wu et al., 1978). For single motor unit recording, the recording site was near the entry to muscle fields or distal to the branching point that sends a collateral to muscle 3. In this manner, all the unit activities recorded are from motor axons. We obtained a consistent pattern of activity from both recording sites. Signals were picked up by a differential alternating-current amplifier (DAM-5A; WPI).
Compound action potential recording was performed to monitor patterns of spike-bursting activities that corresponded to muscle contraction waves during locomotor activity of the larval preparation. The CNS and segmental nerves were maintained intact during larval dissection. Propagating waves of muscle contractions from posterior to anterior segments or from anterior to posterior segments were observed when the preparation was perfused with an HL3 saline containing 1.5 mm Ca2+ (Stewart et al., 1994) at a rate of 0.5∼1 ml/min (Fox et al., 2006). Spontaneous, ongoing nerve activity was recorded en passant (Fox et al., 2006) using a suction electrode and a differential amplifier (DAM-5A; WPI) and was then displayed on a chart recorder (Gould 220; Gould, Cleveland, OH).
Both ejps and action potentials were stored on VCR tapes with a pulse code modulator (model Neuro-Corder DR-384; Neuro Data, New York, NY). Data were digitized with pClamp 5 (Molecular Devices, Burlingame, CA) and analyzed in an IBM (White Plains, New York)-compatible computer.
Ca2+ imaging.
Calcium imaging was performed on an upright microscope (Eclipse E600FN; Nikon, Tokyo, Japan) equipped with a xenon short arc light source (UXL-75XE; Ushio, Tokyo, Japan). Fluorescence from the larval preparation was collected with a water-immersion objective lens (40×, Fluoro; numerical aperture, 0.80), captured with a CCD camera (SM256; SciMeasure Analytical Systems, Decatur, GA), and stored and analyzed using the computer software NEUROCCD-SM256 system (Red Shirt Imaging, New Haven, CT). To monitor Ca2+ dynamics in motor terminals, the Ca2+-sensitive fluorescent protein G-CaMP (Nakai et al., 2001) has been used to monitor Ca2+ dynamics in motorneurons (Reiff et al., 2005). A motorneuron-specific Gal4 enhancer trap C164 (Torroja et al., 1999) was used to drive expression of the transgene UAS-GCaMP-9 (Wang et al., 2004) in motor terminals. (Ga14 is a transcription factor that drives expression of genes placed downstream of the activation signal sequence UAS.) GCaMP has high-affinity for Ca2+ (Kd = 235 nm) and a large Hill coefficient (3.4), yielding a high signal-to-noise ratio at physiological Ca2+ concentrations in vivo. ΔF/F ranges to a maximum of 4.0 in human embryonic kidney cells (Nakai et al., 2001), and 1.0 in the Drosophila brain (Wang et al., 2003, 2004). Background fluorescence of the muscle fiber surrounding presynaptic boutons was subtracted from fluorescence intensities of the presynaptic boutons. The baseline fluorescence of boutons measured before stimulation (F) and the deflection of fluorescent intensity from the baseline (ΔF) were determined. Thus, ΔF/F indicates intracellular Ca2+ level changes caused by nerve activity and was determined by averaging the readings of individual pixels (usually 4–9) that covered single boutons.
Results
Abnormal motor control of Shab and Sh mutants
Drosophila has been a favored model animal for genetic analysis of nervous system development and function. The availability of mutations affecting identified channel subunits that mediate transient Sh IA and sustained Shab IK in Drosophila provides an opportunity for genetic dissection of the roles of different K+ channels in neuromuscular physiology underlying motor behavior. When ether anesthetized, Sh flies exhibited continuous high-frequency leg shaking, which was often followed by wing buzz before regaining a standing posture (Kaplan and Trout, 1969). We found that ether also induced abnormal motor behaviors in Shab mutant flies, but with a characteristic pattern distinct from that of Sh. The leg shaking of Shab flies was milder and occurred during recovery after a prolonged period (>10 min.) of immobilization. During an extended recovering phase (up to 10 min), Shab flies did not display wing buzz, but assumed an unusual standing posture. Their legs extended laterally and they moved by dragging their body on the floor.
Abnormal motor behavior could be observed in Sh and Shab larvae without ether treatment. Previous computer-assisted morphometric analysis has shown that Sh and several ion channel mutations affect different aspects of larval locomotor behavior (Wang et al., 1997, 2002). We found a new phenotype of Shab and Sh larvae motor control that was indicated by the abnormal patterns of mouth-hook impression left on the agar surface in their crawling trails (cf. Wu et al., 1978). Figure 1A shows that the mouth-hook prints of the WT larvae were more regular during forward linear locomotion, whereas the prints of Shab and Sh larvae were irregular. Notably, the interval between mouth-hook prints was more variable for Sh and the force of the mouth-hook impression was more irregular for Shab.
Figure 1.

Abnormal larval locomotion and central motor patterns in Shab and Sh. A, Mouthhook prints left on agar plates by locomotor activity of third instar larvae. WT larvae marked regular patterns during forward locomotion but Shab and Sh left irregular patterns with deep and shallow mouthhook prints. Scale bar, 1 mm. Numbers of larvae were 9–15 for each genotype. B, Burst firing of segmental nerves associated with propagating waves of muscle contraction in the semi-intact larval preparation, in which the CNS and segmental nerves were kept intact. WT displayed periodic bursts of spike activities coupled with posterior-anterior (b) or anterior-posterior (f) muscular contraction waves, resembling the peristaltic movement of intact larvae. In Shab and Sh, the nerve firing was often unassociated with the muscular contraction waves (brackets) and also lost rhythmicity. Scale bar, 1 min. The number of larvae was 6–7 for each genotype.
We further used a semi-intact larval neuromuscular preparation to monitor locomotor patterns of spike activities in segmental nerves that innervate the body wall muscles while observing corresponding muscle contraction waves along the body axis (Fox et al., 2006). In WT larvae, the motor pattern generated by the CNS drives a propagating wave of muscle contractions from posterior to anterior segments, corresponding to the forward crawling (Fig. 1B, b). The rhythmicity of contraction waves and nerve spike bursting was stable and could be maintained for >20 min, with occasional reversals in the direction of propagation (anterior to posterior, correlated with a different bursting pattern) (Fig. 1B, f). In contrast, Shab often displayed uncoordinated local twitching of individual muscles in different segments, interposed with long periods of pause, and rarely showed rhythmic contraction waves. Correspondingly, the nerve spike activity also consisted of prolonged inactive periods along with long bursts that were uncorrelated with contraction waves (Fig. 1B, brackets). The same was true for Sh larvae, in which we rarely observed rhythmic contraction waves and regular bursting activity in the segmental nerve (Fig. 1B).
Enhanced synaptic transmission at the Shab and Sh neuromuscular junctions
The same larval neuromuscular preparation was subjected to electrophysiological examination for abnormality in nerve and muscle excitability as well as in neuromuscular transmission. This preparation has been extensively studied for synaptic function (Jan et al., 1977; Wu et al., 1978), development (Broadie and Bate, 1993; Kidokoro and Nishikawa, 1994; Yoshihara et al., 1997, 2005; Keshishian and Kim, 2004), plasticity (Budnik et al., 1990; Zhong and Wu, 1991; Zhong et al., 1992; Davis et al., 1996; Sigrist et al., 2003), and transmitter release mechanisms (Ramaswami et al., 1994; Kuromi and Kidokoro, 2000; Song et al., 2002; Kidokoro et al., 2004), and is therefore ideal for characterization of new mutant phenotypes. We found striking abnormalities in Shab as well as Sh larvae, although their defects were most pronounced under different stimulus conditions (Fig. 2). Previous reports demonstrate that excitatory junctional potentials (ejps) in Sh are increased in amplitude, a phenomenon most striking at low Ca2+ levels (Jan et al., 1977; Ganetzky and Wu, 1982, 1983). As Figure 2 shows, single nerve stimuli lead to greatly increased ejps in Sh mutants recorded intracellularly in a bath solution containing 0.1 mm Ca2+ (a few tens of millivolts in contrast to less than a few millivolts in WT). In response to single nerve stimuli or low-frequency stimulation (0.5 Hz), Shab, like WT, produced only small ejps of the quantal size, mixed with intermittent release failure. When 10 Hz nerve stimulation was applied, both WT and Shab showed augmentation in ejp amplitude. Nevertheless, Shab ejps displayed a sudden jump in amplitude within tens of seconds of 10 Hz stimulation, unlike WT ejps, which gradually increased in amplitude and reached a low plateau level. This “big bang” of ejps in Shab even surpassed the amplitude of Sh ejps. These giant Shab ejps were characterized by a prolonged duration and a slower rising phase with one or more notches, contrary to the smoother rising phase of the Sh ejps (Fig. 2).
Figure 2.

Enhanced augmentation of synaptic transmission in Shab and increased basal release in Sh. Augmentation of synaptic transmission was examined by evoking ejps via high-frequency (10 Hz) nerve stimulation (underline) after test stimuli at a frequency of 0.5 Hz or lower. WT displayed a gradual increase in ejp amplitudes during 10 Hz stimulation (from b to c), whereas Shab showed a sudden jump in ejp amplitudes (big bang; c) after an initial phase of gradual increase (b). Ejp amplitudes in Sh were larger than those in WT after single stimuli and remained relatively stable, even at 10 Hz stimulation (b, c). [Ca2+] = 0.1 mm. The number of muscles was 13–30 for each genotype.
Because postsynaptic muscle cells also express Shab IK (Singh and Singh, 1999), electrical activity in muscle cells could be affected by Shab mutations. Under normal conditions, larval muscles do not produce all-or-none action potentials (Wu and Haugland, 1985). When Sr2+ was added to the saline, muscle action potentials were readily triggered by depolarizing current injection (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), because Sr2+ is a more effective charge carrier through the Ca2+ channels and suppresses Ca2+ channel inactivation (Hille, 2001). The amplitudes and threshold of such muscle action potentials were not affected by Sh and Shab mutations, but the action potential was significantly prolonged and fired repetitively in Shab larvae, demonstrating an important role of Shab K+ channels in regulating repetitive spike activity.
The abnormal giant ejps in Shab evoked by repetitive stimulation (Fig. 2) were distinguishable from the postsynaptic regenerative Ca2+ spike in muscles, because these prolonged ejps were not overshooting and were not terminated by hyperpolarizing the muscle cell via current injection (data not shown). These results indicate that the abrupt change in ejps induced by high-frequency stimulation reflects a qualitative change in presynaptic terminal properties. However, this phenomenon was not related to activity-dependent long-term modulation of the release mechanisms, such as post-tetanic potentiation (Zhong and Wu, 1991), because the amplitude of these giant ejps in Shab larvae were not maintained after cessation of repetitive stimulation.
Distinct effects on basal transmitter release and paired-pulse facilitation by Sh and Shab
Spontaneous miniature ejps in the absence of nerve stimulation appeared similar in terms of frequency and amplitudes among WT, Sh, and Shab larvae (data not shown). We next compared how Shab and Sh affect the basal-transmitter release at different Ca2+ levels, as defined by the response to a single stimulus. At all Ca2+ concentrations examined, Shab ejps were not significantly distinguished from those in WT (stimulated at 0.5 Hz or lower frequencies) (Figs. 3A,B). However, ejps in Sh larvae were considerably greater in amplitude than those in WT. The differences were most striking at low Ca2+ concentrations and became indistinguishable from WT at concentrations >0.5 mm, at which ejp amplitudes reached saturation. These results indicate that basal release triggered by individual nerve stimuli is predominantly regulated by Sh but not by Shab currents.
Figure 3.

Basal transmitter release and pair-pulse facilitation. A, B, Basal transmitter release was examined by monitoring ejp responses to individual nerve stimuli (≤0.5 Hz). A, Superimposed sample traces. B, Box plot of ejp amplitudes. Note that ejps in Sh were significantly larger than WT at 0.1 and 0.2 mm external Ca2+, whereas those in Shab did not differ from WT. C, D, Pair-pulse facilitation of ejps. Ejp responses to pair-pulse stimulation were examined at different interpulse intervals (IPIs) (20, 50, and 100 ms). Shab displayed significantly greater facilitation than WT at shorter IPIs, whereas Sh showed depression. [Ca2+] = 0.2 mm. The number of muscles examined is indicated. Dots indicate outliers.
Therefore, a clear Shab ejp phenotype depended on previous activity during repetitive stimulation and the underlying process was first examined by a paired-pulse stimulus protocol. Twin-pulse stimulation with varying interpulse intervals (20, 50, and 100 ms IPIs) was applied to examine short-term facilitation of transmitter release in Shab. At 0.2 mm Ca2+, Shab exhibited greater facilitation than WT, especially at shorter IPIs (Fig. 3C,D). We interpret the more extreme phenotype at shorter IPIs to reflect a lesser degree of Sh channel recovery from inactivation in the absence of Shab IK, and thus allowing even more Ca2+ influx in the presynaptic terminals. In contrast, the Sh ejps of much greater amplitudes showed less facilitation than WT and even tended to display synaptic depression at shorter IPIs (Fig. 3C,D). These results demonstrated that Shab channels play a progressively more important role in transmitter release as Sh channels inactivate.
Frequency-dependent regulation of transmitter release by Sh and Shab
An important difference in the operation of Sh and Shab channels results from their distinct kinetic properties of inactivation. Sh channels, but not Shab channels, are sensitive to the activity cycles during which inactivation and recovery from inactivation set the stage for the effectiveness of channel function for the next round of activity. In addition to short IPIs in a twin-pulse stimulation protocol, suppression of Sh channel recovery from inactivation could be achieved by the process of cumulative inactivation after repetitive stimulation (Baukrowitz and Yellen, 1995; Roeper et al., 1997; Kalman et al., 1998). If the big-bang phenomenon of Shab ejps during high-frequency stimulation is caused by cumulative inactivation of Sh channels, it should occur with progressively shorter onset times with increasing stimulus frequencies. When we extended the frequency range of repetitive stimulation from 10 to 20 to 30 Hz, the onset time of giant ejps at 0.1 mm Ca2+ in Shab became correspondingly shorter, consistent with our hypothesis (Fig. 4). In contrast, the increase of ejp amplitudes in WT was gradual, reaching a plateau of greater amplitude with increasing stimulus frequencies. As expected, there was no progressive increase in Sh ejps at all stimulation frequencies from the initial large amplitude.
Figure 4.
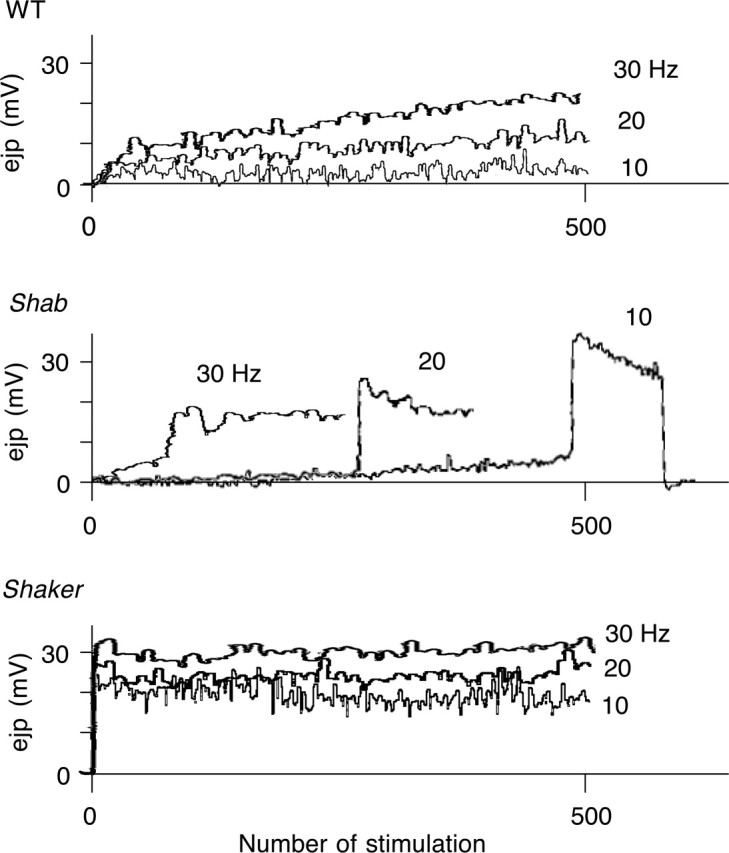
Frequency-dependent regulation of transmitter release. Augmentation of synaptic transmission was examined by evoking ejps at different frequencies of nerve stimulation. WT displayed a smooth increase in ejp amplitudes during stimulation with a larger plateau amplitude at higher stimulus frequencies. Shab exhibited a jump in ejp amplitudes earlier, as frequency increased. Sh exhibited large ejps at the onset of repetitive stimulation, regardless of stimulus frequency. [Ca2+] = 0.1 mm. The number of muscles was 3–10 for each genotype.
Pharmacological dissection of the roles of Sh IA and Shab IK
To further confirm that the synaptic transmission phenotypes observed in Shab and Sh larvae result from alterations in channel function rather than secondary developmental regulations, we applied well studied drugs to examine the acute effects of blocking Sh and Shab channels (Fig. 5A). We found that giant ejps similar to those observed in Shab could be induced by repetitive nerve stimulation in WT larvae treated with quinidine, which is known to block specifically Shab currents at low concentrations in larval muscles (Singh and Singh, 1999) and in cultured neurons (I.-F. Peng and C.-F. Wu, unpublished observation). In contrast, WT larvae treated with 4-AP (200 μm to 1 mm) displayed a significantly increased ejp (tens of millivolts) immediately after a single nerve stimulus (Fig. 5A) (cf. Jan et al., 1977; Ganetzky and Wu, 1982, 1983). Such ejps did not further increase in amplitude during 10 Hz repetitive stimulation, as in the case of ejps in Sh larvae (compare Figs. 2, 5A). Note that quinidine (20–100 μm) induction of giant ejps resembled the big-bang phenomenon in Shab larvae. In particular, these ejps displayed a slower rising with several notches, resembling the giant ejps in Shab, but distinct from the fast rising Sh ejps (compare Figs. 2, 5A).
Figure 5.

Pharmacological dissection of the roles of Sh and Shab K+ channel subunits. A, In the bath containing the Shab channel blocker quinidine (100 μm), WT displayed a sudden jump in ejp amplitudes (b) during high-frequency nerve stimulation (underline, 5 Hz), but the basal release revealed by individual test stimuli (dots, 0.2 Hz) remained small (a), as in Shab mutants (Fig. 2). WT larvae treated with the Sh channel blocker 4-AP (1 mm) displayed large ejps (a) after single stimulation (dots, 0.2 Hz). Furthermore, the ejps remained similar in amplitude (b) during high-frequency stimulation (underline, 10 Hz), reminiscent of Sh ejps. B, Synergistic effects of Sh and Shab channel blockade by mutations and drugs. Bath application of 4-AP (1 mm) on Shab, quinidine (20 μm) on Sh, and 4-AP (200 μm) + quinidine (20 μm) on WT, produced large ejps with a prolonged duration in response to single nerve stimuli (dots). [Ca2+] = 0.1 mm. The number of muscles was 12–21 for each condition except for three in WT + 4-AP.
Strikingly, simultaneous reduction of both inactivating IA and sustained IK, by adding quinidine to Sh, 4-AP to Shab, or both quinidine and 4-AP to WT, led to giant ejps, characterized by a slower rising phase and more prolonged duration, immediately after single nerve stimuli, resembling the big-bang induction in Shab larvae (Fig. 5B). This observation supports the hypothesis that cumulative inactivation of Sh channels during high-frequency spike activities in Shab larvae progressively remove IA and, thus, create a condition in which both IA and IK are substantially reduced in the motor axon terminals (Figs. 2, 4).
The consequences of enhanced presynaptic nerve terminal excitability on transmitter release are most likely mediated by increased intracellular Ca2+ activity. The effects of such pharmacological manipulations on intracellular Ca2+ accumulation could be directly demonstrated at motor terminals using the Ca2+ imaging technique (Fig. 6). We monitored intracellular Ca2+ increases in individual synaptic boutons using the C164 GAL4-UAS GCaMP line, in which expression of the Ca2+-sensitive fluorescent probe (GCaMP) is driven by a motor neuron-specific driver (C164 GAL4). The results support the hypothesis that with removal of inactivating IA or sustained IK by drug treatment, the enhanced synaptic transmission is caused by increased Ca2+ influx in nerve terminals. Consistent with ejp recordings, WT terminals showed Ca2+ accumulation only at high-frequency (20 Hz) stimulation. After quinidine treatment, a substantial Ca2+ signal accumulated during10-Hz stimulation, and the rise in Ca2+ signals became more rapid at 20 Hz stimulation. In contrast, 4-AP treatment gave rise to small but detectable Ca2+ signals, even at 0.2 Hz stimulation, and at either 10 or 20 Hz stimulation, the rise in Ca2+ concentrations was very rapid and reached a plateau level higher than untreated control terminals. Significantly, with the combined effect of 4-AP and quinidine, a large Ca2+ response could be observed reliably after single nerve stimuli. These observations highlight the immediate effect of removing IA by 4-AP on synaptic transmission and the delayed responses after quinidine treatment that were unmasked only after cumulative inactivation of IA during repetitive stimulation. Thus, the synergism becomes conspicuous when both IA and IK are blocked by 4-AP and quinidine, leading to large responses to individual stimuli.
Figure 6.
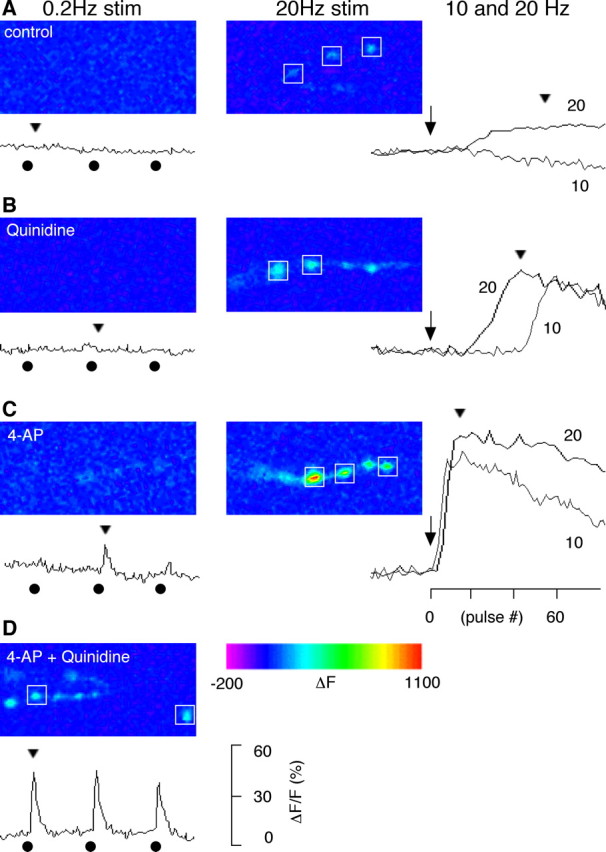
Altered Ca2+ dynamics in the presynaptic boutons treated with blockers of Sh and Shab channels. Presynaptic Ca2+ dynamics were monitored with a Ca2+-sensitive green fluorescent protein expressed in motorneurons (GCaMP; see Materials and Methods). A, In the absence of K+ channel blockers, fluorescent intensity in WT boutons did not change with single nerve stimulation (delivered at 0.2 Hz), but increased during high-frequency stimulation (detectable at 20 Hz but not at 10 Hz). This indicates that high-frequency nerve stimulation causes accumulation of cytosolic Ca2+. B, In the presence of the Shab channel blocker quinidine (30 μm), a sudden increase of fluorescence was observed during high-frequency stimulation (10 and 20 Hz). Note that the onset of the increase was faster for 20 Hz compared with 10 Hz. Single stimulation was not sufficient to trigger Ca2+ increase. C, In the presence of the Sh channel blocker 4-AP (200 μm), single nerve stimulation could sometimes trigger a small Ca2+ transient. However, the presynaptic Ca2+ accumulated very rapidly with high-frequency stimulation. D, When blockers for both Sh and Shab channels were applied, single nerve stimuli consistently triggered a fluorescent transient greater than those enhanced by 4-AP alone. Both the kinetics and amplitudes of fluorescent signals from individual boutons were similar within a nerve terminal branch, therefore, the sample traces shown are the average of signals from boutons indicated by a box (□). The onset of 10 and 20 Hz repetitive stimulation (arrows), the delivery of the 0.2 Hz test stimuli (•), and the time point for the displayed image frames (arrow heads) are indicated along the ΔF/F traces. The number of nerve terminal branches was 3–6 for each condition.
Supernumerary motor axon spikes concomitant with giant ejps
The above hypothesis prompted a direct examination of motor-axon activity by recording action potentials en passant in the segmental nerves that innervate the body-wall muscles (Fig. 7). This approach provided evidence for heightened presynaptic excitability in Shab mutants responsible for the generation of giant ejps during repetitive stimulation. In WT control larvae, simultaneous recording showed that a single nerve stimulus triggered an ejp of small amplitude, correlated with a compound action potential, which contains the spike activity of the motor axons (Fig. 7A). In the case of Sh larvae, additional spikes (mostly one, rarely two) occurred coincidentally with an ejp of increased amplitudes. High-frequency nerve stimulation did not increase the ejp amplitude or the number of supernumerary spikes in Sh larvae (Fig. 7A). In Shab larvae, single nerve stimuli evoked only a compound action potential and a small ejp as in WT larvae. However, 10 Hz nerve stimulation recruited supernumerary spikes (>3) in the motor axon concurrent with the induction of the giant ejp. The supernumerary spikes in the motor axon during the big bang correlated temporally with the notches in the rising phase of the giant ejps (Fig. 7A), demonstrating a drastic consequence of cumulative inactivation of Sh IA in Shab larvae where IK is weakened. Similarly, supernumerary motor axon spikes concurrent with giant ejps could be induced by single nerve stimuli when simultaneous reduction of IA and IK was achieved by adding quinidine to Sh, 4-AP to Shab, or both quinidine and 4-AP to WT larvae (Figs. 7A, 8).
Figure 7.

Increased nerve terminal excitability in Shab and Sh. A, Supernumerary spike activities accompanying giant ejps. Simultaneous recordings of motor axon action potentials (top traces) and ejps (bottom traces of each panel) were performed. At low-frequency stimulation (≤0.5 Hz) (without K+-channel blockers), WT and Shab larvae displayed only a compound action potential (▴) and a quantal level ejp, whereas Sh displayed an additional spike (•) and larger ejp. After 10 Hz stimulation, Shab displayed supernumerary spikes coupled with a step-wise increase in ejp amplitudes, whereas the nerve spike activities in WT and Sh did not change. Significantly, when both Shab and Sh channels were blocked (WT + 200 μm 4-AP and 30 μm Quinidine, Shab + 200 μm 4-AP, or Sh + 20 μm Quinidine), supernumerary spikes coupled with prolonged ejps were triggered even at low-frequency stimulation (≤0.5 Hz). [Ca2+] = 0.1 mm. See Discussion for details. The number of experiments was 4–6 for each condition. B, Back propagation of action potentials. At low-frequency nerve stimulation, an orthodromic compound action potential (▴) was observed in Shab mutant segmental nerves (first panel). When stimulation frequency was increased to 10 Hz (second panel) or when 4-AP (1 mm) was applied to the bath (third panel), back propagating action potentials were observed. The backfiring action potentials were suppressed by the Ca2+ channel blocker Co2+ (1 mm; bottom panel). The number of nerves examined was 4 for control, 3 for 4-AP, and 2 for 4-AP + Co2+ treatment. “p” and “d” are the proximal and distal portions of the nerve, respectively.
Figure 8.
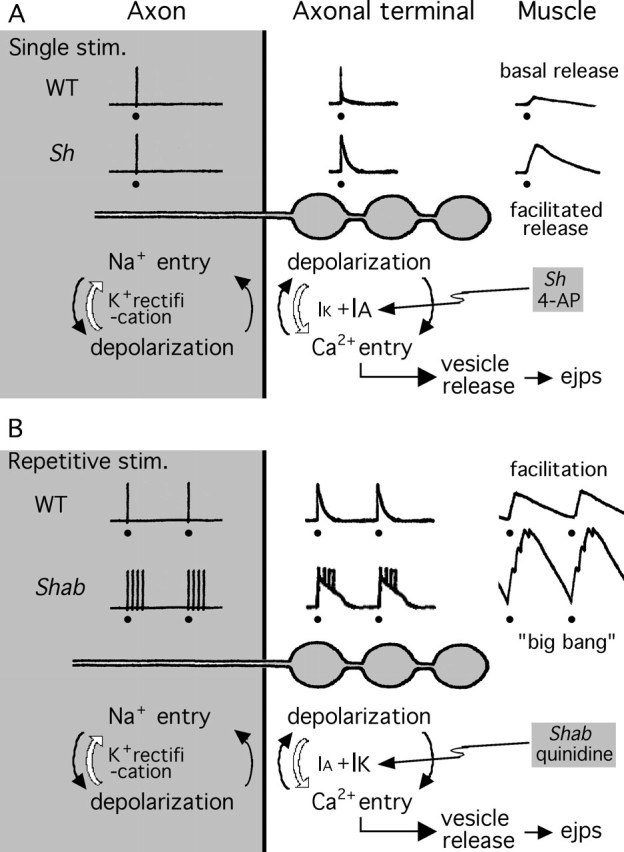
Proposed functional division between Sh IA and Shab IK in the regulation of axonal terminal excitability and synaptic transmission in different frequency ranges. Traces above the schematic drawing of the axon and axonal terminal boutons depict Na+ action potentials in the axon, regenerative Na+ and Ca2+ potentials in the axonal terminal, and muscle ejps. A, Sh IA effectively controls the basal-level release triggered by a single nerve stimulus. Removal of Sh IA results in facilitated release, leading to larger ejps. When the axonal action potential invades the WT terminal, depolarization elicits Ca2+ influx, which further depolarizes the axonal terminal boutons. Subsequent recruitment of voltage-activated IA and IK terminates the depolarization of the axonal terminals. Apparently, Sh IA plays a more important role than Shab IK in this process, because removal of Shab IK does not substantially increase the basal release (see Fig. 3). Weakening of IA by Sh mutations or 4-AP treatment prolongs axonal terminal depolarization and Ca2+ influx and, thus, facilitates transmitter release. B, Shab IK is important in the regulation of high-frequency repetitive synaptic activity. Removal of Shab IK enables the big-bang phenomenon during repetitive nerve stimulation. In WT axonal terminals, cumulative inactivation of Sh IA during high-frequency depolarization cycles weakens the repolarizing force and hence facilitates Ca2+ influx and transmitter release (frequency-dependent facilitation) (Fig. 4). However, the remaining IK effectively counterbalances the depolarization to limit additional increase in Ca2+ influx and transmitter release. When IK is weakened by Shab mutations or quinidine treatment, arrival of an Na+ spike is sufficient to elicit a regenerative Ca2+ spike in the nerve terminal. The slower kinetics of the Ca2+ spike outlasts the refractory period of the Na+ spike and can thus trigger additional Na+ spikes in the adjacent axonal patch. These supernumerary spikes further lengthen axonal terminal depolarization and backfire, propagating antidromically along the axon (Fig. 7B). Such feedback cycles between Na+ and Ca2+ elongate the terminal depolarization and triggers a big bang. Nerve stimuli are indicated by filled circles (•). This figure is modified from Ganetzky and Wu (1982).
Although Sh and Shab mutations may affect the various regions of the motor neuron, the source for supernumerary spike generation appears to reside in the presynaptic nerve terminal. Simultaneous recordings along the segmental nerve revealed the surprising fact that these supernumerary spikes propagated antidromically from the neuromuscular junction, suggesting that generation of such backfiring may involve special excitability properties of nerve terminals. Figure 7B compares the temporal sequence of events recorded at the two recording sites. Note that spikes at the proximal recording site lagged behind those at the distal site. It is known that presynaptic terminals are enriched with Ca2+ channels in addition to Na+ channels (Hille, 2001), both contributing to the initiation of regenerative potentials (see Discussion) (Fig. 8). Apparently, interplay between these channels could sustain prolonged depolarization of the synaptic terminal. This hypothesis is supported by the observation that supernumerary spike activity was inhibited by the Ca2+ channel blocker Co2+, even though the Na+ spike generation (i.e., the compound action potential) was unimpaired.
Discussion
Differential contributions of Sh and Shab in different frequency domains
Both Sh and Shab subunits (Schwarz et al., 1990; Tejedor et al., 1997), as well as their vertebrate homologues Kv1 and Kv2 (Trimmer, 1991; Sheng et al., 1992; Coetzee et al., 1999), are widely expressed in both the central and peripheral nervous systems. However, the role of Shab and Kv2 channels in synaptic function is less well understood than that of Sh and Kv1 channels. Our study provides a first demonstration of the involvement of the Shab channel, and its interaction with the Sh channel, in the regulation of synaptic transmission. Severe deficiencies in motor-pattern generation (Fig. 1) indicate that these channels regulate membrane excitability and synaptic transmission in many central neurons, in addition to the neuromuscular junction. The results from mutational and pharmacological analyses illustrate the salient temporal characteristics of synaptic transmission that are regulated by Sh IA and Shab IK channels. Their complementary properties in voltage dependence and gating kinetics enable a functional division between these two channels. Sh IA undergoes rapid inactivation with a slower recovery time course and could be progressively removed during rapid repetitive depolarizing pulses (Baukrowitz and Yellen 1995). In contrast, inactivation of Shab IK is limited and slow, with more rapid recovery kinetics (Wu and Haugland, 1985; Tsunoda and Salkoff, 1995; Singh and Singh, 1999). As summarized in Figure 8, our data indicate that Sh IA undergoes cumulative inactivation during repetitive depolarization, leading to frequency-dependent facilitation of synaptic transmission. Under this condition, further removal of Shab IK results in misregulation of nerve terminal excitability and uncontrolled, explosive transmission, such as the big-bang phenomenon observed at Shab neuromuscular junctions (Fig. 8). In conclusion, Sh channels effectively control the basal level release triggered by single nerve stimuli, whereas Shab channels are more important in the regulation of repetitive synaptic activities. These distinctions highlight the crucial cooperation between Sh and Shab channels in nerve terminal excitability regulation to confer the patterns of synaptic transmission.
Results from pharmacological experiments demonstrate an effective phenocopy of the Sh and Shab phenotypes by the IA and IK blockers 4-AP and quinidine, respectively (Fig. 5). This suggests that the elevated basal transmission levels in Sh and the big-bang phenomena in Shab reflect direct effects of defective IA and IK channels, rather than the consequence of developmental regulation in response to a chronic deficiency in IA or IK.
These long-lasting giant ejps are consistent with the previous report of plateaued ejps in WT neuromuscular junctions treated with quinidine and dendrotoxin, a Kv1 and Sh channel blocker (Wu et al., 1989). With K+ channels compromised by mutations or drug treatments, the enhanced Na+ influx on the arrival of an orthodromic motor neuron spike may be sufficient to elicit a regenerative Ca2+ spike in the nerve terminal that is enriched in Ca2+ channels. The slower inactivation of Ca2+ channels can outlast the refractory period of Na+ spikes and can thus trigger another Na+ spike once the Na+ channels in the adjacent axonal patch recover from inactivation (Fig. 8). These supernumerary Na+ spikes, thus, propagate antidromically but each contributes to the maintenance of the regenerative Ca2+ potentials for another round of interaction between Ca2+ and Na+ currents (cf. Ganetzky and Wu, 1982). With insufficient IK in Shab mutants, cumulative inactivation of the Sh channel during repetitive stimulation may well decrease the repolarizing force sufficiently to trigger supernumerary spikes in the nerve terminal, leading to sustained transmitter release. Similar explosive events of prolonged neurotransmission have been described in the squid giant synapse (Katz and Miledi, 1969a) and the frog neuromuscular junction (Katz and Miledi, 1969b) after treatment with the K+ channel blocker tetraethylammonium.
Expression of inactivating IA and sustained IK in different presynaptic terminals
Both rapidly inactivating IA (presumably mediated by Kv1.1/1.4) and noninactivating delayed rectifier IK (presumably Kv1.2) have recently been detected in presynaptic terminals of vertebrate species and their functional significance requires further exploration (Dodson and Forsythe, 2004). The proportion of these two types of K+ currents varies among different preparations and such variability may contribute to the distinct temporal characteristics of synaptic transmission typical of individual preparations. Synapses predominantly expressing IA over IK (e.g., hippocampal mossy fibers and pituitary neurohypophysis) (Thorn et al., 1991; Geiger and Jonas, 2000) are known to be modulated by repetitive input for cumulatively enhanced transmission. Such synapses may be more susceptible to high-frequency excitation and could produce explosive output under some diseased conditions. However, synapses predominantly expressing IK over IA (e.g., the calyx of Held in the auditory pathway) (presumably mediated by Kv1.2) (Dodson et al., 2003), could display a high following frequency, as required for high-frequency transmission. In contrast, synapses known to express both IA and IK (e.g., the lobster neuromuscular junction) (French et al., 2004), can exhibit several patterns of transmission, as a substrate for both short-term and long-term synaptic plasticity (Atwood and Wojtowicz, 1986; Zucker and Regehr, 2002).
Contributions of other K+ channels in the regulation of transmitter release
Additional non-Sh IA channels may also participate in the regulation of nerve excitability and synaptic transmission in different sets of motor neurons of Drosophila. Inactivating and noninactivating K+ currents have been directly recorded from nerve terminals by patch clamping from type III terminals that are preferentially enlarged by an ecdysone mutation (Martinez-Padron and Ferrus, 1997). The inactivating IA, however, is still present in Sh-null mutants, suggesting a contribution from the Shal channel that also mediates inactivating K+ currents (Covarrubias et al., 1991). Type III terminals are thought to be responsible for peptidergic rather than glutamatergic synaptic transmission and, thus, may require regulation by IA of different properties. In addition, it has been shown that neuromuscular junctions in eag Sh double mutants or in eag mutants treated with 4-AP can produce plateaued ejps, which are maintained by continuous transmitter release and are associated with spike trains in motor axons (Ganetzky and Wu, 1982, 1983). Even in the absence of stimulation, these double mutants display plateau ejps spontaneously, with durations and amplitudes even greater than those reported here. This is consistent with the previous proposal that the eag K+ channel subunits are capable of interacting with several other K+ channel subunits to affect both inactivating and noninactivating K+ currents (Zhong and Wu, 1993).
Nerve terminal excitability and synaptic plasticity
The major focus in the study of activity-dependent synaptic plasticity has been directed to the regulation of transmitter release machinery, including the studies using the Drosophila neuromuscular junction preparation (Kuromi and Kidokoro, 2000; Renger et al., 2000). Less attention has been directed to the control of presynaptic terminal excitability in this process. In principle, modulation of inactivating IA and sustained IK may lead to important consequences on synaptic efficacy. In Drosophila, differential regulation of membrane repolarization and, thus, neuronal firing properties by Sh and Shab currents have been documented in cultured Drosophila central neurons. Sh current is important for the regularity of spike firing (Yao and Wu, 2001) and Shab is important for maintaining spike repolarization during repetitive firing (I-F. Peng and C.-F. Wu, unpublished observation). Further, both IA and IK channels are the known targets for cAMP- and cGMP-dependent modulation in Drosophila muscle fibers (Zhong and Wu, 1993). In cultured neurons, Sh IA is modulated by cAMP-and cGMP-dependent pathways (Renger et al., 1999; Yao and Wu, 2001) and Shab IK by CaM-dependent kinases (Yao and Wu, 2001). Therefore, modulation of K+ currents in nerve terminals may play an essential role in the activity-dependent plasticity of synaptic transmission. The Drosophila neuromuscular junction expresses both IA and IK with a number of known structural genes as well as auxiliary proteins that modify the channel properties. This model system thus provides a rich source for genetic interaction analysis and easy manipulation of genes and proteins important in the control of presynaptic terminal excitability and activity-dependent modification of synaptic efficacy.
Footnotes
This work was supported by National Institutes of Health Grants NS26528 and HD18577. We thank Drs. Satpal Singh, Jing Wang, Yalin Wang, and Yi Zhong for providing fly stocks. We also thank Ms. Jennifer Heacock for assistance in preparing the manuscript.
References
- Atwood HL, Wojtowicz JM (1986). Short-term and long-term plasticity and physiological differentiation of crustacean motor synapses. Int Rev Neurobiol 28:275–362. [DOI] [PubMed] [Google Scholar]
- Augustine GJ (1990). Regulation of transmitter release at the squid giant synapse by presynaptic delayed rectifier potassium current. J Physiol (Lond) 431:343–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K, Salkoff L (1990). The Drosophila Shaker gene codes for a distinctive K+ current in a subset of neurons. Neuron 4:129–140. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G (1995). Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron 15:951–960. [DOI] [PubMed] [Google Scholar]
- Blaine JT, Ribera AB (2001). Kv2 channels form delayed-rectifier potassium channels in situ. J Neurosci 21:1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadie KS, Bate M (1993). Development of the embryonic neuromuscular synapse of Drosophila melanogaster J Neurosci 13:144–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V, Zhong Y, Wu C-F (1990). Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J Neurosci 10:3754–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Wei AG, Baker K, Salkoff L (1989). A family of putative potassium channel genes in Drosophila Science 243:943–947. [DOI] [PubMed] [Google Scholar]
- Chopra M, Gu GG, Singh S (2000). Mutations affecting the delayed rectifier potassium current in Drosophila J Neurogenet 14:107–123. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B (1999). Molecular diversity of K+ channels. Ann NY Acad Sci 868:233–285. [DOI] [PubMed] [Google Scholar]
- Covarrubias M, Wei AA, Salkoff L (1991). Shaker, Shal, Shab, and Shaw express independent K+ current systems. Neuron 7:763–773. [DOI] [PubMed] [Google Scholar]
- Davis GW, Schuster CM, Goodman CS (1996). Genetic dissection of structural and functional components of synaptic plasticity. III. CREB is necessary for presynaptic functional plasticity. Neuron 17:669–679. [DOI] [PubMed] [Google Scholar]
- Dodson PD, Forsythe ID (2004). Presynaptic K+ channels: electrifying regulators of synaptic terminal excitability. Trends Neurosci 27:210–217. [DOI] [PubMed] [Google Scholar]
- Dodson PD, Billups B, Rusznák Z, Szûcs G, Barker MC, Forsythe ID (2003). Presynaptic rat Kv1.2 channels suppress synaptic terminal hyperexcitability following action potential invasion. J Physiol (Lond) 550:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel JE, Wu C-F (1992). Interactions of membrane excitability mutations affecting potassium and sodium currents in the flight and giant fiber escape systems of Drosophila J Comp Physiol A Neuroethol Sens Neural Behav Physiol 171:93–104. [DOI] [PubMed] [Google Scholar]
- Feng Y, Ueda A, Wu C-F (2004). A modified minimal hemolymph-like solution, HL3.1, for physiological recordings at the neuromuscular junctions of normal and mutant Drosophila larvae. J Neurogenet 18:377–402. [DOI] [PubMed] [Google Scholar]
- Forsythe ID (1994). Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS, in vitro. J Physiol (Lond) 479:381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox LE, Soll DR, Wu C-F (2006). Coordination and modulation of locomotion pattern generators in Drosophila larvae: effects of altered biogenic amine levels by the tyramine β hydroxylase mutation. J Neurosci 26:1486–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French LB, Lanning CC, Matly M, Harris-Warrick RM (2004). Cellular localization of Shab and Shaw potassium channels in the lobster stomatogastric ganglion. Neuroscience 123:919–930. [DOI] [PubMed] [Google Scholar]
- Ganetzky B, Wu C-F (1982). Drosophila mutants with opposing effects on nerve excitability: genetic and spatial interactions in repetitive firing. J Neurophysiol 47:501–514. [DOI] [PubMed] [Google Scholar]
- Ganetzky B, Wu C-F (1983). Neurogenetic analysis of potassium currents in Drosophila: synergistic effects on neuromuscular transmission in double mutants. J Neurogenet 1:17–28. [DOI] [PubMed] [Google Scholar]
- Geiger JRP, Jonas P (2000). Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron 28:927–939. [DOI] [PubMed] [Google Scholar]
- Heinemann SH, Rettig J, Graack HR, Pongs O (1996). Functional characterization of Kv channel beta-subunits from rat brain. J Physiol (Lond) 493:625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (2001). In: Ion channels of excitable membranes Sunderland, MA: Sinauer.
- Iverson LE, Tanouye MA, Lester HA, Davidson N, Rudy B (1988). A-type potassium channels expressed from Shaker locus cDNA. Proc Natl Acad Sci USA 85:5723–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan YN, Jan LY, Dennis MJ (1977). Two mutations of synaptic transmission in Drosophila Proc R Soc Lond B Biol Sci 198: pp. 87–108. [DOI] [PubMed] [Google Scholar]
- Kalman K, Nguyen A, Tseng-Crank J, Dukes ID, Chandy G, Hustad CM, Copeland NG, Jenkins NA, Mohrenweiser H, Brandriff B, Cahalan M, Gutman GA, Chandy KG (1998). Genomic organization, chromosomal localization, tissue distribution, and biophysical characterization of a novel mammalian Shaker-related voltage-gated potassium channel, Kv1.7. J Biol Chem 273:5851–5857. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Klein M, Hochner B, Shuster M, Siegelbaum SA, Hawkins RD, Glanzman DL, Castellucci VF, Abrams TW (1987). Synaptic modulation and learning: new insights into synaptic transmission from the study of behavior. In: Synaptic function (Edelman GM, Gall WE, Cowan WM, eds) pp. 471–518. New York: Wiley.
- Kaplan WD, Trout WE III (1969). The behavior of four neurological mutants of Drosophila Genetics 61:339–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R (1969a). Tetrodotoxin-resistant electric activity in presynaptic terminals. J Physiol (Lond) 203:459–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R (1969b). Spontaneous and evoked activity of motor nerve endings in calcium Ringer. J Physiol (Lond) 203:689–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshishian H, Kim YS (2004). Orchestrating development and function: retrograde BMP signaling in the Drosophila nervous system. Trends Neurosci 27:143–147. [DOI] [PubMed] [Google Scholar]
- Kidokoro Y, Nishikawa K (1994). Miniature endplate currents at the newly formed neuromuscular junction in Drosophila embryos and larvae. Neurosci Res 19:143–154. [DOI] [PubMed] [Google Scholar]
- Kidokoro Y, Kuromi H, Delgado R, Maureira C, Oliva C, Labarca P (2004). Synaptic vesicle pools and plasticity of synaptic transmission at the Drosophila synapse. Brain Res Rev 47:18–32. [DOI] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y (2000). Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron 27:133–143. [DOI] [PubMed] [Google Scholar]
- Malin SA, Nerbonne JM (2002). Delayed rectifier K+ currents, IK, are encoded by Kv2 alpha-subunits and regulate tonic firing in mammalian sympathetic neurons. J Neurosci 22:10094–10105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Padron M, Ferrus A (1997). Presynaptic recordings from Drosophila: correlation of macroscopic and single-channel K+ currents. J Neurosci 17:3412–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, Imoto K (2001). A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat Biotech 19:137–141. [DOI] [PubMed] [Google Scholar]
- Paulsen O, Sejnowski TJ (2000). Natural patterns of activity and long-term synaptic plasticity. Curr Opin Neurobiol 10:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami M, Krishnan KS, Kelly RB (1994). Intermediates in synaptic vesicle recycling revealed by optical imaging of Drosophila neuromuscular junctions. Neuron 13:363–375. [DOI] [PubMed] [Google Scholar]
- Reiff DF, Ihring A, Guerrero G, Isacoff EY, Joesch M, Nakai J, Borst J (2005). An in vivo performance of genetically encoded indicators of neural activity in flies. J Neurosci 25:4766–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renger JJ, Yao WD, Sokolowski MB, Wu C-F (1999). Neuronal polymorphism among natural alleles of a cGMP-dependent kinase gene, foraging, in Drosophila J Neurosci 19:RC28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renger JJ, Ueda A, Atwood HL, Govind CK, Wu C-F (2000). Role of cAMP cascade in synaptic stability and plasticity: ultrastructural and physiological analyses of individual synaptic boutons in Drosophila memory mutants. J Neurosci 20:3980–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeper J, Lorra C, Pongs O (1997). Frequency-dependent inactivation of mammalian A-type K+ channel KV1.4 regulated by Ca2+/calmodulin-dependent protein kinase. J Neurosci 17:3379–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salkoff L, Wyman R (1981). Genetic modification of potassium channels in Drosophila Shaker mutants. Nature 293:228–230. [DOI] [PubMed] [Google Scholar]
- Schwarz TL, Papazian DM, Carretto RC, Jan YN, Jan LY (1990). Immunological characterization of K+ channel components from the Shaker locus and differential distribution of splicing variants in Drosophila Neuron 4:119–127. [DOI] [PubMed] [Google Scholar]
- Sheng M, Tsaur ML, Jan YN, Jan LY (1992). Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron 9:271–284. [DOI] [PubMed] [Google Scholar]
- Sigrist SJ, Reiff DF, Thiel PR, Steinert JR, Schuster CM (2003). Experience-dependent strengthening of Drosophila neuromuscular junctions. J Neurosci 23:6546–6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Singh S (1999). Unmasking of a novel potassium current in Drosophila by a mutation and drugs. J Neurosci 19:6838–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, Tempel BL (1998). Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron 20:809–819. [DOI] [PubMed] [Google Scholar]
- Song W, Ranjan R, Dawson-Scully K, Bronk P, Marin L, Seroude L, Lin YJ, Nie Z, Atwood HL, Benzer S, Zinsmaier KE (2002). Presynaptic regulation of neurotransmission in Drosophila by the G-protein-coupled receptor methuselah. Neuron 36:105–319. [DOI] [PubMed] [Google Scholar]
- Stewart BA, Atwood HL, Renger JJ, Wang J, Wu C-F (1994). Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol A Neuroethol Sens Neural Behav Physiol 175:179–191. [DOI] [PubMed] [Google Scholar]
- Stuhmer W, Ruppersberg JP, Schroter KH, Sakmann B, Stocker M, Giese KP, Perschke A, Baumann A, Pongs O (1989). Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. EMBO J 8:3235–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejedor FJ, Bokhari A, Rogero O, Gorczyca M, Zhang J, Kim E, Sheng M, Budnik V (1997). Essential role for dlg in synaptic clustering of Shaker K+ channels in vivo. J Neurosci 17:152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn PJ, Wang XM, Lemos JR (1991). A fast, transient K+ current in neurohypophysial nerve terminals of the rat. J Physiol (Lond) 432:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpe LC, Schwarz TL, Tempel BL, Papazian DM, Jan YN, Jan LY (1988a). Expression of functional potassium channels from Shaker cDNA in Xenopus oocytes. Nature 331:143–145. [DOI] [PubMed] [Google Scholar]
- Timpe LC, Jan YN, Jan LY (1988b). Four cDNA clones from the Shaker locus of Drosophila induce kinetically distinct A-type potassium currents in Xenopus oocytes. Neuron 1:659–667. [DOI] [PubMed] [Google Scholar]
- Torroja L, Packard M, Gorczyca M, White K, Budnik V (1999). The Drosophila β-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J Neurosci 19:7793–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS (1991). Immunological identification and characterization of a delayed rectifier K+ channel polypeptide in rat brain. Proc Natl Acad Sci USA 88:10764–10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda S, Salkoff L (1995). The major delayed rectifier in both Drosophila neurons and muscle is encoded by Shab. J Neurosci 15:5209–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Sylwester AW, Reed D, Wu DA, Soll DR, Wu C-F (1997). Morphometric description of the wandering behavior in Drosophila larvae: aberrant locomotion in Na+ and K+ channel mutants revealed by computer-assisted motion analysis. J Neurogenet 11:231–254. [DOI] [PubMed] [Google Scholar]
- Wang JW, Soll DR, Wu C-F (2002). Morphometric description of the wandering behavior in Drosophila larvae: a phenotypic analysis of K+ channel mutants. J Neurogenet 16:45–63. [DOI] [PubMed] [Google Scholar]
- Wang JW, Wong AM, Flores J, Vosshall LB, Axel R (2003). Two-photon calcium imaging reveals an odor-evoked map of activity in the fly brain. Cell 112:271–282. [DOI] [PubMed] [Google Scholar]
- Wang Y, Guo HF, Pologruto TA, Hannan F, Hakker I, Svoboda K, Zhong Y (2004). Stereotyped odor-evoked activity in the mushroom body of Drosophila revealed by green fluorescent protein-based Ca2+ imaging. J Neurosci 24:6507–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei A, Covarrubias M, Butler A, Baker K, Pak M, Salkoff L (1990). K+ current diversity is produced by an extended gene family conserved in Drosophila and mouse. Science 248:599–603. [DOI] [PubMed] [Google Scholar]
- Wu C-F, Haugland FN (1985). Voltage-clamp analysis of membrane currents in larval muscle fibers of Drosophila: alteration of potassium currents in Shaker mutants. J Neurosci 5:2626–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-F, Ganetzky B, Jan LY, Jan YN, Benzer S (1978). A Drosophila mutant with a temperature-sensitive block in nerve conduction. Proc Natl Acad Sci USA 75:4047–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-F, Tsai MC, Chen ML, Zhong Y, Singh S, Lee CY (1989). Actions of dendrotoxin on K+ channels and neuromuscular transmission in Drosophila melanogaster, and its effects in synergy with K+ channel-specific drugs and mutations. J Exp Biol 147:21–41. [DOI] [PubMed] [Google Scholar]
- Yao WD, Wu C-F (2001). Distinct roles of CaMKII and PKA in regulation of firing patterns and K+ currents in Drosophila neurons. J Neurophysiol 85:1384–1394. [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Rheuben MB, Kidokoro Y (1997). Transition from growth cone to functional motor nerve terminal in Drosophila embryos. J Neurosci 17:8408–8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Adolfsen B, Galle KT, Littleton JT (2005). Retrograde signaling by Syt 4 induces presynaptic release and synapse-specific growth. Science 310:858–863. [DOI] [PubMed] [Google Scholar]
- Zhao M-L, Sable EO, Iverson LE, Wu C-F (1995). Functional expression of Shaker K+ channels in cultured Drosophila“giant” neurons derived from Sh cDNA transformants: distinct properties, distribution, and turnover. J Neurosci 15:1406–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wu C-F (1991). Altered synaptic plasticity in Drosophila memory mutants with a defective cyclic AMP cascade. Science 251:198–201. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Wu C-F (1993). Modulation of different K+ currents in Drosophila: a hypothetical role for the Eag subunit in multimeric K+ channels. J Neurosci 13:4669–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Budnik V, Wu C-F (1992). Synaptic plasticity in Drosophila memory and hyperexcitable mutants: role of cAMP cascade. J Neurosci 12:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Poo MM (2004). Reversal and consolidation of activity-induced synaptic modifications. Trends Neurosci 27:378–383. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002). Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. [DOI] [PubMed] [Google Scholar]