

Abstract
Cholinergic neurotransmission supports motor, autonomic, and cognitive function and is compromised in myasthenias, cardiovascular diseases, and neurodegenerative disorders. Presynaptic uptake of choline via the sodium-dependent, hemicholinium-3-sensitive choline transporter (CHT) is believed to sustain acetylcholine (ACh) synthesis and release. Analysis of this hypothesis in vivo is limited in mammals because of the toxicity of CHT antagonists and the early postnatal lethality of CHT−/− mice (Ferguson et al., 2004). In Caenorhabditis elegans, in which cholinergic signaling supports motor activity and mutant alleles impacting ACh secretion and response can be propagated, we investigated the contribution of CHT (CHO-1) to facets of cholinergic neurobiology. Using the cho-1 promoter to drive expression of a translational, green fluorescent protein-CHO-1 fusion (CHO-1:GFP) in wild-type and kinesin (unc-104) mutant backgrounds, we establish in the living nematode that the transporter localizes to cholinergic synapses, and likely traffics on synaptic vesicles. Using embryonic primary cultures, we demonstrate that CHO-1 mediates hemicholinium-3-sensitive, high-affinity choline uptake that can be enhanced with depolarization in a Ca2+-dependent manner supporting ACh synthesis. Although homozygous cho-1 null mutants are viable, they possess 40% less ACh than wild-type animals and display stress-dependent defects in motor activity. In a choline-free liquid environment, cho-1 mutants demonstrate premature paralysis relative to wild-type animals. Our findings establish a requirement for presynaptic choline transport activity in vivo in a model amenable to a genetic dissection of CHO-1 regulation.
Keywords: choline, acetylcholine, transporter, C. elegans, CHT, CHO-1
Introduction
Acetylcholine (ACh) is a critical neurotransmitter in both the central and peripheral nervous systems, serving as the primary excitatory neurotransmitter at the vertebrate neuromuscular junction (NMJ), and as a modulator of cognitive function (Sarter and Bruno, 1997). Defects in ACh synthesis or reception at the NMJ result in myasthenias (Engel et al., 2003a,b), and loss of basal forebrain cholinergic neurons contributes to the dementia characteristic of Alzheimer’s disease (Whitehouse et al., 1982).
ACh synthesis occurs in the presynaptic terminal, and is thought to be limited by choline availability, particularly under circumstances of elevated ACh release (Birks and MacIntosh, 1957, 1961; Jope, 1979). Although there is evidence for limited de novo synthesis of choline via membrane phospholipid catabolism in brain (Blusztajn and Wurtman, 1981; Lee et al., 1993), ACh synthesis likely depends on the efficient uptake of choline across the presynaptic membrane (Birks and MacIntosh, 1961) via the high-affinity choline transporter (CHT). CHT is distinguishable from ubiquitous, low-affinity choline uptake mechanisms by its affinity for substrate (Km, ∼1–5 vs 100 μm), its sodium and chloride dependence, and nanomolar sensitivity to the competitive antagonist hemicholinium-3 (HC-3) (Guyenet et al., 1973; Haga and Noda, 1973). Accordingly, ACh synthesis and release is impaired in vitro when CHT is inhibited by HC-3 (Guyenet et al., 1973), and high-affinity choline uptake is modulated to meet the demands of increased ACh synthesis and release (Simon and Kuhar, 1975).
Recent studies have demonstrated that CHT expression is confined in brain to cholinergic neurons (Okuda et al., 2000; Misawa et al., 2001; Kobayashi et al., 2002; Ferguson et al., 2003) and is enriched in cholinergic terminals and at NMJs (Ferguson et al., 2003; Nakata et al., 2004). Murine CHT traffics on small clear synaptic vesicles that contain ACh, the vesicular acetylcholine transporter (VAChT), and other synaptic vesicle markers, and exhibits depolarization-triggered changes in cell surface density, consistent with a physical coupling between ACh release and CHT surface trafficking (Ferguson et al., 2003).
To date, vertebrate model systems have not been amenable to the study of the in vivo effects of a complete loss of CHT function on ACh homeostasis and animal behavior. Acute pharmacological blockage of CHT leads to rapid respiratory failure (Schueler, 1955), and mouse homozygous CHT knock-outs are neonatal lethal (Ferguson et al., 2004). Like the vertebrate nervous system, ACh is the principal excitatory transmitter at the Caenorhabditis elegans NMJ (Richmond and Jorgensen, 1999), and importantly, the core synaptic machinery is well conserved (Nonet, 1999). Unlike vertebrate models, homozygous null alleles of C. elegans CHT (CHO-1) are viable. Because of the simplicity of this genetically tractable model system, the C. elegans model offers a unique opportunity to investigate the consequences of a loss of high-affinity choline uptake on ACh homeostasis, release, and behavior.
We report that green fluorescent protein (GFP)-tagged CHO-1 localizes to cholinergic synapses in vivo in a manner consistent with trafficking via synaptic vesicle precursors. Endogenous CHO-1 mediates high-affinity choline uptake that supports ACh biosynthesis, and displays many of the same properties characteristic of its mammalian ortholog CHT. Loss of CHO-1-mediated choline uptake results in both a loss of normal ACh synthesis, and an activity-dependent fatigue phenotype.
Materials and Methods
C. elegans strains and husbandry.
All strains were derived from the wild-type (WT) strain (Bristol N2) and maintained at 20°C using standard methods (Brenner, 1974), except that in most cases animals were grown on thin lawns of Escherichia coli strain HB101 rather than OP50. The strains used in this study were as follows: N2 (wild type), cho-1(tm373) (Mitani Laboratory, National BioResource Project for the Nematode, Tokyo, Japan), pcho-1:CHO-1:GFP (BY503(vtIs16); lin-15 (n765ts)), pcho-1::GFP (BY505(vtEx14); lin-15 (n765ts)), pcho-1::CHO-1:GFP; cho-1(tm373) (BY506(vtEx15); cho-1(tm373); lin-15 (n765ts)), pcho-1::GFP; cho-1(tm373) (BY507(vtEx14); pcho-1:CHO-1:GFP; pric-1:VAMP:mRFP1 (BY508(vtIs16; vtEx16; lin-15(n765ts)), unc-104(e1265, pld-1(ok986), cha-1(cn101), BZ722 (snf-6 (eg38); cho-1 (tm373)) (Kim et al., 2004), ric-1(md226) (Jorgensen Laboratory, University of Utah, Salt Lake City, UT), acr-16(ok789) (Miller Laboratory, Vanderbilt University, Nashville, TN), unc-18(md299), unc-29(e1072), snt-1(md290). cho-1(tm373) was outcrossed three times to N2 before its use in any experiment.
Creation of transgenic animals and microscopy.
To create pcho-1:CHO-1:GFP (BY503(vtIs16)), genomic sequence representing the cho-1 gene and 5.1 kb of upstream promoter sequence was PCR-amplified from genomic DNA using standard methods. This promoter length was chosen because it had been identified by Okuda et al. (2000) as sufficient to drive GFP in the cholinergic nervous system. Amplification was performed using the upper primer: 5′-(PstI)-ctgcagtatacacgagaagctgctc-3′ and the lower primer: 5′-(KpnI)-ggtaccgttcgtctcttgtaattgcta-3′, and then ligated into Fire Vector pPD95.75 (gift from A. Fire, Stanford University School of Medicine, Stanford, CA) in-frame with the GFP gene. The fully sequenced construct (5 ng/μl) was coinjected with the lin-15 rescuing plasmid pJM23 (50 ng/μl) into lin-15 (n765ts) animals using methods described previously (Jin, 1999). For BY503, genomic integration of the pcho-1:CHO-1:GFP extrachromosomal array (vtEx13) was achieved using an ultraviolet/trimethylpsoralen protocol (Clark and Chiu, 2003) followed by outcrossing four times to N2. After characterization of the expression of this transgene, it was determined that the original promoter identified by Okuda et al. (2000) did not drive expression in all cholinergic neurons. Thus, for rescue experiments the initial promoter was extended an additional 2.5 kb. The additional promoter sequence was PCR amplified, ligated into the original pcho-1:GFP construct and coinjected (20 ng/μl) with pJM23 (50 ng/μl) into lin-15(n765ts) animals to create pcho-1::GFP (BY505(vtEx14)). The extended promoter was ligated into the original pcho-1:CHO-1:GFP construct and coinjected (5 ng/μl) with pJM23 (50 ng/μl) into lin-15(n765ts); cho-1(tm373) to create pcho-1:CHO-1:GFP; cho-1(tm373) (BY506(vtEx15); cho-1(tm373)) used in rescue experiments. For neuroanatomy examination experiments, BY505 males were crossed with cho-1(tm373) hermaphrodites, and GFP-positive homozygous cho-1(tm373) animals isolated creating BY507 (vtEx14; cho-1(tm373)).
For CHO-1:GFP, VAMP:mRFP1 colocalization studies, the mRFP1 gene was amplified from mRFP1/pRSETB (gift from R. Y. Tsien, University of California, San Diego, CA) and subcloned into pJL4 (pacr-5:VAMP:GFP) construct (gift from E. M. Jorgensen, University of Utah, Salt Lake City, UT), using AgeI and SpeI restriction sites to replace GFP with mRFP1. The VAMP:mRFP1 BamH1/ApaI fragment was subcloned into pDM1 (pric-1::GFP), replacing GFP and creating pric-1:VAMP:mRFP1 for pan-neuronal expression. This construct (5 ng/μl) was coinjected with pRF4 (rol-6 dominant marker; 60 ng/μl) into BY503 animals creating BY508(vtIs16, vtEx16; lin-15(n765ts)). Animals positive for both roll and CHO-1:GFP were propagated and imaged using an LSM510-Meta confocal (Carl Zeiss, Thornwood, NY). For synapse development/integrity experiments, pric-1:VAMP:mRFP1 was coinjected (5 ng/μl) with pRF4 (60 ng/μl) into N2 (BY509(vtEx17)) and cho-1(tm373) (BY510(vtEx18; cho-1(tm373)) animals, and roll positive animals were examined and imaged. For unc-104 localization experiments, BY503 (pcho-1:CHO-1:GFP) males were crossed to unc-104 (e1265) hermaphrodites, and unc-104−/−; GFP-positive animals (vtIs16; unc-104(e1265)) were selected for confocal imaging.
Aldicarb and levamisole tests.
Acute aldicarb treatment was performed essentially as described previously (Nonet et al., 1997). For each test, 20–30 young adult animals were picked onto plates containing aldicarb at a final concentration of 0.0–1.0 mm, and scored for paralysis after 2 h of treatment. For levamisole sensitivity, 20–30 young adult animals were placed onto plates containing 0.1 mm levamisole and scored for paralysis at 30, 60, and 90 min after plating.
C. elegans cell culture.
Primary cultures were prepared from BY503 (pcho-1:CHO-1:GFP), wild-type (N2), cho-1 mutant (cho-1 (tm373)), pld-1(ok986), cha-1(cn101), and BY506 (pcho-1:CHO-1:GFP; cho-1(tm373)) animals as described previously (Christensen et al., 2002; Carvelli et al., 2004), and incubated at 18°C. Approximately 25 × 106 cells per well (24-well plate) were plated for uptake assays, and 104 cells per well for imaging.
HPLC analysis of ACh and choline content.
To determine total choline and ACh levels in primary cultures, two wells per sample were lysed and processed for neurochemistry using methods described by Liberato et al. (1985) with modifications. Specifically, cells were extracted with 300 μl/well acetonitrile for 5 min, and then cleared by centrifugation at 10,000 × g for 10 min at 4°C. The supernatant was extracted twice with 0.5 vol of heptane, and the remaining acetonitrile evaporated using a DNA120 SpeedVac (Thermo Savant, Waltham, MA). The choline/ACh pellet was resuspended in 50 mm H2PO4, and the choline/ACh was separated using HPLC. The ACh was detected by electrochemical methods using a reaction with acetylcholinesterase and choline oxidase, both immobilized on a post-column enzyme reactor (immobilized enzyme reactor) as described previously (Damsma et al., 1985) (Vanderbilt Neurochemistry Core Resource). Samples were normalized to total protein. For whole-worm analysis, animals were removed from plates with M9 buffer, and washed three times by centrifugation at 1500 × g for 4 min to remove bacteria. The animals were pelleted, and the M9 buffer was removed using a syringe and 27-gauge needle. Animals were lysed in acetonitrile (1 ml per 100 g) by grinding with a mortar/pestle in liquid nitrogen, and the lysate was cleared by centrifugation at 10,000 × g for 10 min at 4°C. After clearing, the lysate was processed as described above. For protein determination and sample normalization, cell debris from the clearing step was dissolved in 0.1 m NaOH and subjected to protein determination. To determine endogenous free choline levels in bacterial strains, bacteria were washed off of nematode growth medium (NGM) plates in water, and adjusted to equal density based on OD600. Aliquots of pelleted bacteria (100 mg per strain) were resuspended in acetonitrile and sonicated using a probe sonicator. Samples were processed for neurochemistry as described above, and normalized to total protein.
Transport assays.
Choline transport assays were performed on primary cultures of C. elegans embryonic cells 3 d after plating. All transport experiments were performed in triplicate unless otherwise noted, and the data were analyzed using GraphPad (San Diego, CA) Prism software (Student’s t test or one- or two-way ANOVA as noted in the text). For standard transport assays, cells were washed twice in uptake buffer (130 mm NaCl, 10 mm HEPES, 1.5 mm CaCl2, 0.5 mm MgCl2, 1.3 mm KH2PO4, 34 mm dextrose; pH 7.35, 345–350 mOsm), and incubated in 0.1 μm [3H]choline (choline chloride, [methyl-3H]ethanol; New England Nuclear, Boston, MA; 86 Ci/mmol) for 5 min at room temperature. Cells were washed three times in 4°C uptake buffer and lysed in 1% SDS for 20 min at room temperature, and the lysate was analyzed by liquid scintillation spectrometry. For normalization, 50 μl aliquots of lysate per well were used for total protein determination using a BCA (bicinchoninic acid) protein assay (Sigma-Aldrich, St. Louis, MO). Nonspecific uptake was determined either by uptake in cho-1 null mutant cells, uptake at 4°C, or uptake in the presence of 1 or 10 μm HC-3 (as noted in the text). For saturation assays, six concentrations of [3H]choline were used (0.1–6.0 μm). For these studies, unlabeled choline was mixed with [3H]choline in uptake buffer to yield a specific activity of 2.5 Ci/mmol. Km and Vmax values were calculated using nonlinear regression analysis with GraphPad Prism software. For HC-3 competition experiments, six concentrations of HC-3 were used (0.001–9.0 μm), and uptake was performed using 0.1 μm [3H]choline. Data were fit using GraphPad Prism software (nonlinear regression, one site competition). To examine the ion dependency of high-affinity choline transport, [3H]choline uptake was performed under standard conditions except for that 130 mm N-methyl-d-glucamine (NMDG) was used in place of NaCl (for sodium-free uptake), and 130 mm sodium-gluconate was used to substitute for NaCl (Cl-free uptake). For the analysis of activity-dependent changes in CHO-1-mediated transport, cells were washed twice in uptake buffer, and then incubated for 5 min at room temperature in uptake buffer plus 30 mm KCl (depolarized; equimolar reductions in NaCl made to maintain osmolarity) or standard uptake buffer (control). The cells were placed back into standard uptake buffer, and uptake was determined using the above-described methods and 0.1 μm [3H]choline. In some experiments, 100 μm CdCl2 was added to the initial washes and depolarization buffers to block voltage-gated Ca2+ channels. To investigate the time course of recovery after depolarization, cells were incubated for 5 min at room temperature in 30 mm KCl (initial) or standard uptake buffer (control), and then placed in standard uptake buffer for 2, 5, 10, 20, and 30 min at room temperature, followed by standard choline uptake experiments using 0.1 μm [3H]choline.
ACh incorporation studies.
To monitor the incorporation of labeled choline into ACh, cells were subjected to a pulse labeling protocol 3 d after plating. For high-affinity choline uptake and incorporation, cells were washed twice in standard uptake buffer, and then incubated for 5, 10, 20, or 30 min at room temperature in uptake buffer containing 0.1 μm [3H]choline and the acetylcholinesterase inhibitor aldicarb (20 μm; Sigma-Aldrich). Cells were washed three times in 4°C uptake buffer plus 20 μm aldicarb, and either processed for neurochemistry or lysed in 1% SDS and processed as normal uptake samples. To determine the conversion of [3H]choline into ACh, peak fractions for choline and ACh were isolated after HPLC separation, and the accumulated 3H label was determined by scintillation counting.
For low-affinity choline uptake and incorporation, cells were washed twice in uptake buffer, and then incubated for 1 h at room temperature in uptake buffer plus 20 μm aldicarb. The buffer was removed, and the cells were incubated for 20 min at room temperature in 0.1–100 μm [14C]choline ([methyl-14C]choline chloride, 9.25 m; Amersham Biosciences, Piscataway, NJ; 56 mCi/mmol) in uptake buffer containing 20 μm aldicarb. [14C]Choline was used because the low specific activity of [3H]choline precludes its use in experiments in which high choline concentrations are required. Cells were then processed for neurochemistry, or total uptake was determined as described above for [3H]choline incorporation. Nonspecific uptake was defined by the extent of choline uptake at 4°C.
Choline acetyltransferase assays.
Cells were harvested 3 d after plating, and choline acetyltransferase (ChAT) activity was determined using methods described by Fonnum (1975) with modifications. Cultured cells were solubilized with 50 mm Tris-Cl, pH 7.5, 0.1% Triton X-100 by probe sonication, and the lysates were cleared by centrifugation for 10 min, at 4°C, and at 10,000 × g. Duplicate aliquots per well (minimum of three wells per genotype per experiment) were removed and incubated with reaction buffer [300 mm NaCl, 50 mm NaH2PO4, pH 7.4, 10 mm EDTA, 0.1 mm eserine, 20 μm aldicarb, 0.05 mg/ml BSA, 8 mm choline, and 0.2 mm [14C]acetyl-coenzyme A (New England Nuclear; 51.4 mCi/mmol)] for 20 min at room temperature. Control samples were incubated at 4°C. Samples were extracted with tetraphenylborate (15 mg/ml)/heptanone-3, and the organic layer was collected after centrifugation at 5000 × g for 5 min at 4°C. ChAT activity is represented by the incorporation of 14C label into ACh, as determined by scintillation counting. All samples were normalized to total protein.
Assays of nematode motor activity.
Locomotory behavior was examined in wild-type and mutant animals using visual (individual and group assays) and automated methods (individual assays only). In individual visual assays, single worms were picked off of NGM agar plates spread with a thin lawn of HB101 bacteria, and placed into M9 buffer or water (no difference in results between the two media). Swimming behavior was monitored, and the number of body bends per minute was quantitated manually. For group visual assays, 10–15 worms were placed into water in a single well of a 96-well microtiter plate, and a videotape recording was made of thrashing behavior (2 h) using a ZVS-3C75DE analog camera (Carl Zeiss) mounted on a stereo dissecting microscope (Stemi 2000-C; Carl Zeiss). The animal movements were tracked visually/manually, and the number of worms paralyzed or immobile at every minute throughout the 120 min assay was quantitated. For automated analysis, single worms were placed in 50 μl of water in a single well of a 96-well microtiter plate, and audio, video, still image (AVI) movies of their swimming behavior created through the conversion of analog to digital data using a frame grabber [Piccolo graphics card (Ingenieur Helfrich) and VidCap32 AVI capture application (Microsoft, Redmond, CA)]. The resulting AVI movies (30 min each; a total of 2 h per worm) were analyzed using a script written in MatLab 7.0.1 (MathWorks, Natick, MA), which discerns the position of the worm in every frame using motion detection and the selection of a pixel location that designates the worm centroid (available on request). The x and y pixel locations of the worm’s centroid are placed in arrays across time and separated into segments, or windows. For each window, the signal is detrended to remove first-order motion (constant and linear trends as opposed to oscillations), and then a fast Fourier transform (FFT) is taken to determine the frequency spectrum for each window. The Fourier spectra for corresponding x and y dimensions are summed, and the frequency having the largest amplitude in the combined spectrum is selected. Finally, the calculated frequencies for each window are placed in an array so that the frequency of the worm oscillation over time can be displayed. Average frequencies are determined for 6 s windows and plotted for each 30 min video segment.
Results
CHO-1:GFP localizes to synapses in vivo in an UNC-104-dependent manner
Studies on the distribution of mammalian CHT in the CNS and spinal cord using HC-3 binding and CHT immunocytochemistry have revealed that the transporter is expressed in cholinergic terminals (Rainbow et al., 1984; Quirion, 1987; Misawa et al., 2001; Kobayashi et al., 2002; Ferguson et al., 2003; Kus et al., 2003). In an effort to characterize CHO-1 function in the C. elegans model, we first addressed its localization in vivo. Okuda et al. (2000) demonstrated that the cho-1 promoter drives expression of a cytosolic GFP reporter exclusively in cholinergic neurons; however, the subcellular localization of the CHO-1 protein is uncharacterized to date. To this end, we created transgenic animals (BY503) expressing a CHO-1:GFP fusion protein under the control of the endogenous cho-1 promoter (Fig. 1A). CHO-1:GFP was expressed exclusively in cholinergic neurons, including motor neurons of the nerve cord and central neurons of the nerve ring, consistent with the aforementioned GFP reporter studies (Okuda et al., 2000). More importantly, we found that CHO-1:GFP localized to axonal varicosities and colocalized with a fluorescent reporter of the synaptic vesicle marker synaptobrevin [vesicle-associated membrane protein (VAMP)] (Fig. 1A).
Figure 1.

CHO-1:GFP localization in vivo and its UNC-104-dependent trafficking to cholinergic synapses. A, A CHO-1:GFP fusion protein localizes to punctate regions of cholinergic nerve processes and colabels with VAMP:mRFP1. Top panel, Transgenic C. elegans expressing a CHO-1:GFP fusion protein under the control of the endogenous cho-1 promoter in a wild-type background (BY503). The arrows indicate en passant synapses of a cholinergic sublateral neuron. Bottom panels, Transgenic animals coexpressing CHO-1:GFP (BY503) and VAMP:mRFP1 (BY508) in a cholinergic sublateral neuron. Scale bars, 5 μm. B, CHO-1:GFP traffics on synaptic vesicles. Shown are head neurons expressing the CHO-1:GFP fusion protein in the wild-type (BY503) and unc-104 (vtIs16; unc-104(e1265)) mutant backgrounds. The arrows indicate normal synaptic localization in the wild-type background and the loss of synaptic localization in the unc-104 mutant background. The asterisks indicate where the fusion protein is trapped in the cell bodies of these neurons.
Murine CHT resides on small clear vesicles within cholinergic terminals in the brain, and immunoisolates with vesicles that contain VAChT and other synaptic vesicle resident proteins (Ferguson et al., 2003). We sought evidence that CHO-1:GFP also traffics on synaptic vesicles by determining the localization of CHO-1:GFP in mutant animals that fail to transport synaptic vesicles to their nerve terminals. UNC-104 is the kinesin responsible for the transport of synaptic vesicle precursors to the presynaptic terminal (Fig. 1B) (Hall and Hedgecock, 1991; Otsuka et al., 1991). Synaptic vesicle resident proteins such as VAChT, VMAT (vesicular monoamine transporter), and VAMP are selectively retained in the cell bodies of unc-104 mutant neurons, whereas presynaptic membrane proteins are unaffected (Nonet et al., 1993; Rand et al., 2000; Koushika et al., 2001). Thus, retention of GFP-tagged proteins in the cell bodies of neurons when expressed in the unc-104 mutant background is expected if the protein normally localizes to synaptic vesicles. When CHO-1:GFP is expressed in the wild-type background, we observed normal localization to cholinergic synapses of the dorsal nerve cord and nerve ring/head neurons (Fig. 1B). In contrast, when expressed in the unc-104 (e1265) mutant background, CHO-1:GFP failed to localize synaptically, and was retained in the neuronal cell bodies (Fig. 1B). These data provide evidence that CHO-1, like mouse CHT, uses axonal transport mechanisms common to other synaptic vesicle proteins (Ferguson et al., 2003).
CHO-1 supports high-affinity, HC-3-sensitive choline transport activity
The study of choline transport per se is not possible in the intact nematode. However, recently described techniques for preparing C. elegans primary cultures can be exploited for this purpose (Christensen et al., 2002; Carvelli et al., 2004). We used this preparation to verify the functional integrity of the CHO-1:GFP fusion protein; specifically, we examined the localization and choline uptake capabilities of CHO-1:GFP in primary cultures prepared from the BY503 strain (wild-type animals expressing CHO-1:GFP) (Fig. 2A,B). In these cultures, CHO-1:GFP-positive cells represent ∼8% of the population, which is in agreement with estimates placing the total number of cholinergic neurons in the adult animal at ∼10% of the total cell number. We observed prominent CHO-1:GFP expression in the cell soma, likely reflective of biosynthetic structures; however, we also observed localization of CHO-1:GFP at the ends of neuritic projections where contacts were made with other cells (Fig. 2A), possibly reflecting an in vitro synaptic localization. The CHO-1:GFP fusion protein expressed in this model is functional because we could demonstrate an enhanced capacity for high-affinity choline uptake in these cultures relative to nontransgenic cultures (Fig. 2B). Kinetic studies revealed that transgenic cultures, relative to wild-type controls, display an increase in their choline transport Vmax with no apparent change in substrate Km (Fig. 2B), consistent with gene-dosage driven enhancement of endogenous, high-affinity choline transport. This is most likely attributable to increased density at the plasma membrane, indicating that plasma membrane sites are not normally saturated for CHO-1. This finding has possible therapeutic implications, because it suggests that choline uptake and possibly ACh synthesis can be augmented by elevating the amount of CHT expression.
Figure 2.
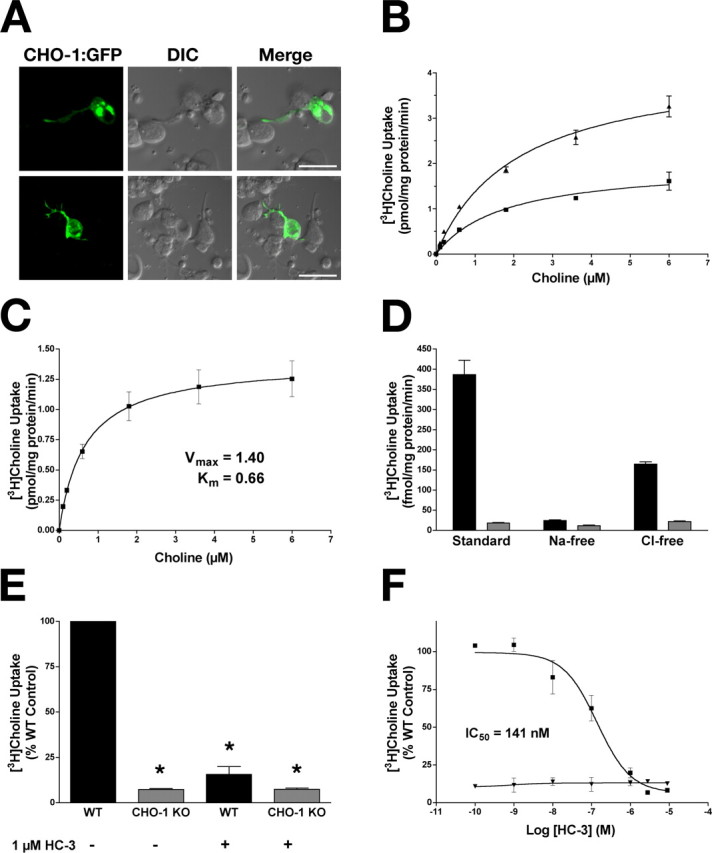
Characterization of high-affinity choline uptake in C. elegans primary cultures. A, Primary cultures prepared from animals expressing the CHO-1:GFP fusion protein (BY503). The arrows indicate the ends of neurite projections contacting adjacent cells. DIC, Differential interference contrast. Scale bars, 10 μm. B, Saturation kinetics for primary cultures derived from BY503 (filled triangles) and WT (filled squares) animals. Overexpression of CHO-1:GFP in wild-type cells increased the Vmax of [3H]choline transport (BY503, 3.73 ± 0.180 pmol · mg protein−1 · min−1; WT, 1.69 ± 0.049 pmol · mg protein−1 · min−1) with no change in apparent Km (BY503, 1.083 ± 0.22 μm choline; WT, 1.014 ± 0.12 μm choline) relative to nonexpressing controls. [3H]Choline uptake from cho-1 knock-out cultures was defined as nonspecific uptake. Data are from two independent experiments performed in triplicate and are plotted as mean ± SEM. C–F, [3H]Choline uptake monitored from primary cultures prepared from WT and cho-1 mutant animals. C, Saturation kinetics of [3H]choline uptake in WT cultures. Nonspecific binding was defined by 1 μm HC-3 (mean ± SEM; n = 3 in triplicate). D, Normal [3H]choline uptake is Na+ and Cl− dependent. Uptake measured from WT (black) and cho-1 knock-out (gray) cultures in the presence of Na+ and Cl− (standard), the absence of Na+ (+NMDG), and the absence of Cl− (+Na-gluconate). Nonspecific binding was defined by uptake at 4°C (mean ± SEM; n = 3 in triplicate). E, High-affinity [3H]choline uptake is lost in cho-1 knock-out cultures. [3H]Choline uptake was measured in WT (black) and cho-1 mutant (gray) cultures in the presence and absence of 1 μm HC-3. Data are expressed as percent WT control (mean ± SEM; n = 3 in triplicate), and nonspecific binding was determined by uptake performed at 4°C. Statistical analysis was performed using a Student’s t test; asterisks indicate statistical significance as described in text. F, High-affinity [3H]choline uptake is inhibited by HC-3. [3H]Choline uptake was measured in WT (filled squares) and cho-1 knock-out (filled triangles) cultures in the presence of increasing concentrations of HC-3. Data are plotted as mean ± SEM of two experiments performed in triplicate, and nonspecific uptake is defined by uptake at 4°C.
To further characterize CHO-1 activity, we analyzed the characteristics of wild-type cultures and compared these with cultures prepared from a cho-1 knock-out line. The cho-1(tm373) allele carries a 1.7 kb deletion in the cho-1 gene, removing exons 4–7 (record no. 373; National BioResource Project for the Experimental Animal C. elegans, Tokyo, Japan). Because this lesion removes transmembrane domains 4–13 (there are 13 total predicted transmembrane domains in CHO-1), it would likely result in a complete loss of function.
We found that choline uptake into WT primary cultures was saturable, with an apparent Km for choline of 0.66 ± 0.17 μm (Fig. 2C). This is comparable with Km estimates for both human and rodent CHT (Km, 0.6–2 μm) (Yamamura and Snyder, 1972; Simon and Kuhar, 1976; Apparsundaram et al., 2001; Ferguson et al., 2003). As in mammalian neurons, choline uptake was both sodium and chloride dependent. Wild-type levels of uptake were decreased to 6.4% of normal when sodium was replaced (standard, 386.8 ± 35.53 fmol · mg protein−1 · min−1; Na-free, 24.82 ± 1.73 fmol · mg protein−1 · min−1) and 42.5% of normal when chloride was replaced (standard, 386.8 ± 35.53 fmol · mg protein−1 · min−1; Cl-free, 164.5 ± 5.88 fmol · mg protein−1 · min−1) (Fig. 2D). Importantly, high-affinity uptake was reduced to 8.1 ± 0.77% (t test, p < 0.0001) of wild-type levels in cultures prepared from cho-1 knock-out animals, suggesting that the bulk, if not all, of high-affinity choline transport activity is CHO-1 mediated (Fig. 2E). High-affinity choline uptake was also HC-3 sensitive: wild-type levels of choline uptake were suppressed to 20 ± 4.45% (t test, p < 0.01) of normal when wild-type cells were pretreated with 1 μm HC-3 (Fig. 2E), an effect that proved to be dose dependent, with a nanomolar IC50 evident (IC50, 141 ± 0.169 nm) (Fig. 2F). Wild-type cells treated with concentrations of HC-3 known to completely block all high-affinity choline transport in other systems, resulted in uptake that matched that displayed by cho-1 mutant cells treated or untreated with HC-3 (Fig. 2E and treatment with 10 μm HC-3, data not shown), suggesting that high-affinity choline uptake is completely lost in cho-1(tm373) mutants. Importantly, high-affinity, HC-3-sensitive choline uptake was rescued to ∼73% of normal in cho-1 mutant cells with the transgenic expression of a CHO-1:GFP fusion protein driven by the endogenous cho-1 promoter (BY506) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Together, these data indicate that C. elegans CHO-1 shares many of the biochemical and pharmacological properties displayed by its mammalian orthologs.
CHO-1 supports depolarization triggered elevations in choline transport activity in vitro
Electrical and pharmacological stimulation of cholinergic neurons leading to increases in ACh release results in enhanced capacity for high-affinity choline uptake (for review, see Ferguson and Blakely, 2004). Treatments that enhance cholinergic neurotransmission and/or ACh release support increases in the CHT Vmax, consistent with an activity-dependent regulation of CHT surface expression. To see whether these properties are evident in the nematode ortholog, we compared choline uptake under standard and depolarizing conditions using wild-type primary cultures. We found that choline uptake could be significantly stimulated (274 ± 2.52% of control; t test, p < 0.0001) when cells were depolarized by elevated potassium (Fig. 3A), an enhancement we found to be dependent on voltage-gated Ca2+ channels as uptake enhancements could be completely blocked by coincubation with the Ca2+ channel blocker Cd2+ during high K+ treatments (Fig. 3A) (t test, p > 0.05) (Lonart et al., 1998). Furthermore, depolarization-elicited effects on choline uptake were persistent: when cells were depolarized, and then placed back into standard uptake buffer and assayed for uptake at 5, 10, and 30 min after depolarization, choline uptake was significantly enhanced relative to control cells at 5 and 10 min after depolarization (ANOVA, p < 0.0001 and p < 0.001, respectively), and did not reach pre-depolarization levels after 30 min (p = 0.05), indicative of sustained plasticity in this paradigm (Fig. 3B).
Figure 3.

High-affinity choline uptake is enhanced with activity in C. elegans primary cultures. A, [3H]Choline uptake is enhanced with depolarization and requires active calcium channels. Uptake was measured from WT cultures under normal conditions (Control), and under depolarizing conditions (+30 mm KCl). This activity-dependent enhancement of [3H]choline uptake was abolished when voltage-gated calcium channels were blocked with 100 μm CdCl2. Data shown are from a representative experiment performed in triplicate, plotting mean + SEM as percent control (n = 3). Nonspecific uptake was defined by uptake in the presence of 2 μm HC-3. B, Activity-dependent enhancement of [3H]choline uptake is persistent. WT cultures were briefly depolarized with 30 mm KCl, and then assayed immediately for [3H]choline uptake (Initial) or placed into standard uptake buffer for 5, 10, or 30 min before measuring uptake [shown as 5, 10, and 30′ post dep(olarization)]. Data are shown as percentage control (control treated identically in parallel with “Initial” cells but without depolarization) and are mean ± SEM of a representative experiment performed in triplicate (n = 2). Uptake at 4°C was subtracted from these data sets as nonspecific uptake. Asterisks designate statistical significance as described in text.
CHO-1 is the only source of high-affinity choline transport used for ACh synthesis
Previous studies have indicated that high-affinity choline uptake is necessary to sustain ACh production and release in cat sympathetic superior cervical ganglia and at vertebrate NMJs (Ferguson and Blakely, 2004; Ferguson et al., 2004). Most previous studies have relied on HC-3 inhibition to establish a role of CHT in ACh production and release in which interpretations are impacted by issues of HC-3 specificity as well as the general toxicity of this agent. Similarly, the early postnatal death of CHT−/− mice precludes identification of possible redundant, high-affinity choline transporters operating to sustain ACh synthesis in the adult. Because CHO-1-deficient animals are viable and fertile, the C. elegans system allows a unique opportunity to study ACh homeostasis in the context of a complete loss of CHT function. We first used HPLC analysis to determine the steady-state levels of choline and ACh in primary cultures prepared from wild-type and mutant animals. Although total choline levels were normal (data not shown), cho-1 knock-out cultures yielded ACh concentrations at ∼43% of wild-type levels (Fig. 4A) (wild type, 1.884 ± 0.08 μm; cho-1 knock-out, 0.8128 ± 0.12 μm; t test, p < 0.0001). Next, we used a pulse-labeling protocol coupled with HPLC determination of total ACh content to track the incorporation of [3H]choline into ACh (Fig. 4B,C). The concentration of [3H]choline used (100 nm) was chosen because it is predicted to lie within the linear range of high-affinity choline transport by CHO-1. As expected, both wild-type and knock-out cultures displayed time-dependent increases in total choline uptake (Fig. 4B), but knock-out cultures consistently displayed a 90–95% reduction in total uptake relative to wild-type cultures (Fig. 4B) [ANOVA test; 5.5% of wild type at 10 min (p < 0.001); 9.1% of wild type at 20 min (p < 0.001); 8.3% of wild type at 40 min (p < 0.001)]. Although there was detectable choline uptake in cho-1 knock-out cultures at [3H]choline concentrations selective for high-affinity transport, there was no measurable incorporation of this choline into total ACh stores (Fig. 4C). In contrast, [3H]choline brought into wild-type cultures was readily incorporated in a time-dependent manner (0.045 ± 0.005% of total ACh was labeled in wild-type cells at 10 min; 0.095 ± 0.005% at 20 min; and 0.1605 ± 0.005% at 40 min). Furthermore, this [3H]ACh was releasable with high potassium depolarization (data not shown), indicating that the ACh synthesized under these conditions supplies a readily releasable pool of vesicles. These data suggest that CHO-1 is the only high-affinity transporter capable of supporting ACh synthesis and release in vitro and that the choline used to synthesize ACh in cho-1 knock-out cultures is derived from a source other than high-affinity transport.
Figure 4.

Effects of a loss of high-affinity choline transport on ACh synthesis in vitro. Results of a combined pulse-labeling/neurochemistry approach reveal that CHO-1 is the only source of high-affinity transport supporting ACh synthesis in primary cultures. A, Total ACh content of WT (black) and cho-1 knock-out (KO) (gray) cultures at steady state as assayed by HPLC. B, Total [3H]choline uptake in WT (black) and cho-1 mutant (gray) cultures over 10, 20, and 40 min periods assayed. Total uptake (mean ± SEM) were as follows: WT, 0.955 ± 0.009 pmol at 10 min, 2.288 ± 0.013 pmol at 20 min, and 3.440 ± 0.002 at 40 min; cho-1 knock-out, 0.053 ± 0.003 pmol at 10 min, 0.208 ± 0.030 pmol at 20 min, and 0.284 ± 0.017 pmol at 40 min. ANOVA comparison of means, p < 0.0001. Uptake was performed with 100 nm [3H]choline, and nonspecific uptake is defined by uptake at 4°C. C, Incorporation of [3H]choline into ACh. WT (black) and cho-1 knock-out (gray) cultures were incubated with 100 nm [3H]choline for 10, 20, or 40 min, and then processed for neurochemistry. Peak fractions for ACh were collected and analyzed by liquid scintillation spectrometry to analyze the conversion of [3H]choline into [3H]ACh. For all panels, shown are mean ± SEM, n = 3 in triplicate; asterisks indicate statistical significance as described in the text.
Low-affinity choline transport mechanisms support ACh synthesis at high substrate concentrations in vitro
In cho-1 knock-out cultures, total ACh levels are reduced 57% relative to wild-type cultures at steady state (Fig. 4A), yet there does not appear to be redundant high-affinity choline uptake mechanisms operating to supply choline for ACh production (Fig. 4C). There is apparent tight linkage between CHT regulation and cholinergic tone in vivo (Ferguson and Blakely, 2004). However, neuroendocrine cells demonstrate ACh synthesis and vesicular release in the absence of detectable CHT expression (Bauerfeind et al., 1993; Apparsundaram et al., 2001), raising questions as to the precursor support pathways available to sustain cholinergic signaling.
Whereas sodium-dependent high-affinity choline uptake is lost in cho-1 knock-out cultures, possibly sodium-independent, low-affinity choline uptake mechanisms may be operating to supply choline to sustain ACh synthesis. Using the same pulse labeling/neurochemistry approach as described above, we loaded the cells with 0.1, 10, 50, and 100 μm [14C]choline and measured its incorporation into ACh (Fig. 5). With the exception of 0.1 μm, these concentrations are predicted to be saturating for high-affinity CHO-1-mediated choline transport. Total ACh content was again found to be reduced relative to wild-type cultures (Fig. 5A) [ANOVA test; 29.40 ± 1.50% of wild-type levels at 0.1 μm [14C]choline (p < 0.01); 34.15 ± 6.55% at 10 μm (p < 0.01); 30.60 ± 3.10% at 50 μm (p < 0.01); and 43.95 ± 16.35% at 100 μm (p < 0.05)], but no significant difference in total ACh levels was detected between cho-1 mutant cultures loaded with the different concentrations of choline (p > 0.05). Although uptake was severely diminished in cho-1 knock-out cultures at low choline concentrations (Fig. 5B) [ANOVA test; 15.50 ± 0.50% of wild type at 0.1 μm [14C]choline (p < 0.001); 37.00 ± 3.0% at 10 μm [14C]choline (p < 0.01)], it was not significantly different from wild-type uptake at higher choline concentrations [96.50 ± 0.50% of wild type at 50 μm [14C]choline (p > 0.05); 113.50 ± 10.50% of wild type at 100 μm [14C]choline (p > 0.05)]. Presumably cho-1 mutant cells have wild-type levels of choline uptake at high precursor concentrations because choline uptake is now occurring through low-affinity mechanisms. As described earlier, there was no measurable incorporation of labeled choline into ACh at 0.1 μm [3H]choline in cho-1 mutant cells. At higher precursor concentrations, we detected ACh production that increased as the concentration of labeled choline was increased, but was still significantly lower than wild type (Fig. 5C). Specifically, there was low, but detectable incorporation of label into ACh at 10 μm [14C]choline [0.017 ± 0.017% of ACh is 14C-labeled in knock-out cultures, as opposed to 0.585 ± 0.035% in wild-type cultures (p < 0.01)]. Furthermore, we observed increasing levels of incorporation at 50 and 100 μm [14C]choline [knock-out, 0.033 ± 0.028%; wild type, 0.585 ± 0.105% (p < 0.01); knock-out, 0.145 ± 0.045%; wild type, 0.940 ± 0.150 (p < 0.001), respectively]. In wild-type cells, the ACh incorporation at high concentrations of [14C]choline (50 and 100 μm) is likely the sum of the activities of both the high- and low-affinity choline transport systems.
Figure 5.
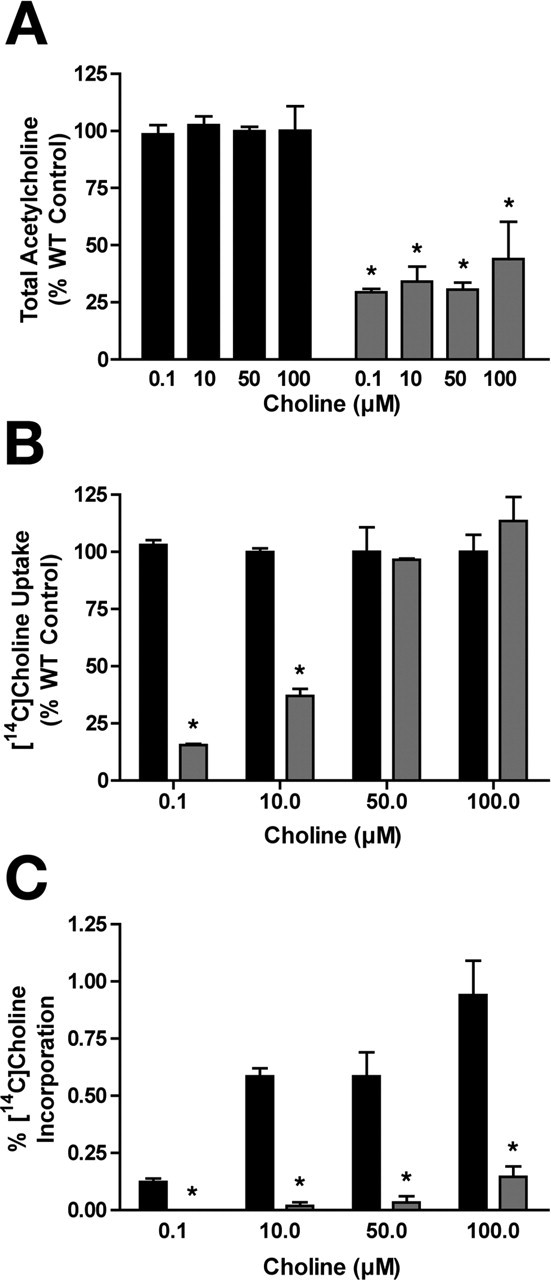
The low-affinity system supports ACh synthesis only at high substrate concentrations. Results of a similar pulse-labeling approach as described in Figure 3, except performed with increasing concentrations of [14C]choline. A, Total ACh content in WT (black) and cho-1 mutant (gray) cultures as detected by HPLC. ANOVA comparison of the means: p = 0.0001 (WT vs cho-1 mutant); p > 0.05 (cho-1−/− cells comparing the four choline concentrations). B, Total [14C]choline uptake in WT (black) and cho-1 mutant (gray) cultures, expressed as percentage of WT control. Uptake was performed in parallel for 10 min at 0.1, 10, 50, and 100 μm [14C]choline, and nonspecific binding was defined by uptake at 4°C. C, Incorporation of [14C]choline into ACh. Conversion of [14C]choline into [14C]ACh was determined as described above (Fig. 3) for WT (black) and cho-1 mutant (gray) cultures. Data are shown as the percentage of total ACh that is [14C]ACh. For all panels, shown are mean ± SEM, n = 3 in triplicate; asterisks indicate statistical significance as described in the text.
The amount of incorporation of labeled choline into ACh in cho-1 mutant cells at 100 μm choline (favorable for low-affinity mechanisms) was approximately equivalent to the amount of incorporation seen in wild-type cells at 0.1 μm choline (favorable only for high-affinity uptake). This suggests that low-affinity mechanisms may be sufficient for driving ACh synthesis in cho-1 mutants given ample extracellular choline concentrations. The molecular identity of low-affinity choline uptake systems evident in our preparation is unknown; however, recent work in mammalian tissue suggests that sodium-independent choline uptake occurs via two neuronally expressed transporters: choline transporter-like 1 (CTL1) (Inazu et al., 2005; Traiffort et al., 2005) and organic cation transporter type 2 (OCT-2) (Busch et al., 1998; Sweet et al., 2001). Potential C. elegans homologs of CTL1 and OCT-2 exist, and it would be interesting to examine whether mutations in these genes enhance the neurochemical and motor phenotypes seen with cho-1 mutants. Alternatively, a transporter with ACh and choline uptake activity has been recently described in C. elegans [sodium:neurotransmitter symporter family member 6 (SNF-6)] (Kim et al., 2004). However, SNF-6 is not expressed in cholinergic motor neurons (Kim et al., 2004) (D. Miller, personal communication) and transports choline with a Km of ∼190 μm (Kim et al., 2004), which is substantially higher than the low-affinity systems characterized to date. Moreover, cho-1;snf-6 double mutants do not show an enhancement of the motor behavior phenotype when compared with cho-1 single-mutant animals (data not shown), which would be expected if SNF-6 serves to supply choline for ACh synthesis in the absence of high-affinity uptake.
ACh synthesized in cho-1 knock-out cultures is not likely derived from choline liberated by phospholipase D activity
An alternative to exogenous choline supporting ACh synthesis is the de novo production of choline by phospholipid catabolism. In mammalian preparations, choline supplied from membrane phosphatidylcholine by phospholipases can drive the synthesis of ACh in vitro (Blusztajn et al., 1987). The most likely phospholipase catalyzing choline release in this manner is phospholipase D (Blusztajn et al., 1987; Lee et al., 1993). There is only one phospholipase D (PLD) identified in the C. elegans genome (designated pld-1), and it is expressed neuronally (Nakashima et al., 2000). Null mutants in pld-1 exist, and are viable. To investigate whether PLD-1 activity is important for supplying choline for ACh synthesis in the absence of cho-1-mediated high-affinity transport, we created cho-1;pld-1 double mutants and assayed wild-type and mutant cultures for choline uptake and total ACh (Fig. 6A,B). As previously discussed, cho-1 knock-out cultures have greatly diminished capacity for high-affinity choline uptake (Fig. 6A, compare WT, CHO-1 KO). We observed no further reduction in choline uptake in the cho-1;pld-1 double mutant (Fig. 6A). As previously noted, total ACh levels were reduced in cho-1 mutant cultures relative to wild-type levels (Fig. 6B); however, no additional reduction was seen in the cho-1;pld-1 double mutant, indicating that the ACh present in cho-1 knock-out cultures is not synthesized from choline derived by phospholipase D (PLD) activity (Fig. 6B). Similarly, no difference in ACh content was detected between cho-1 and cho-1;pld-1 double mutants when assays were performed with whole-worm extracts (data not shown).
Figure 6.

Phospholipase D contribution to ACh synthesis, ChAT activity in the absence of CHO-1, and high-affinity uptake in ChAT knock-out (KO) cultures. A, [3H]Choline uptake in WT (black), cho-1 mutant (dark gray), and cho-1;pld-1 double-mutant (light gray) cultures. Data are plotted as percentage of WT control. *p < 0.0001; ns, no significant difference (p > 0.05); t test. B, Total ACh concentrations in WT (black), cho-1 mutant (dark gray), and cho-1;pld-1 double-mutant (light gray) cells as determined by HPLC analysis. *p < 0.01; ns, no significant difference (p > 0.05); t test. C, ChAT activity in WT (black) and cho-1 mutant (gray) cultures. Data are expressed as percentage of WT control. For A–C, n = 2 in triplicate. D, Total [3H]choline uptake in WT (black), cho-1 mutant (dark gray), and ChAT mutant (cha-1(cn101); light gray) cultures. Data are expressed as percentage of WT control (n = 3 in triplicate). For all panels, shown are mean ± SEM; uptake was performed using 0.1 μm [3H]choline.
Compensatory mechanisms sustaining ACh signaling in CHO-1-deficient animals
Given that nonstressed, well fed cho-1 mutant animals appear behaviorally normal on casual inspection, we investigated whether compensatory mechanisms may be operating in the cho-1 knock-out background to sustain the synthesis of ACh, or augment postsynaptic responses. The enzyme ChAT is responsible for and essential for ACh production (Erickson et al., 1996). We found that cho-1 mutant cultures have normal levels of ChAT activity relative to wild-type cultures (Fig. 6C). Additionally, we found the sensitivity of NMJ receptor fields to agonist stimulated normally in cho-1 mutant animals, as assayed by the treatment of worms with the specific nicotinic ACh receptor agonist levamisole (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). In contrast, we observed a significant increase in basal high-affinity choline uptake in ChAT mutant cultures (Fig. 6D). Specifically, cultured cells derived from animals homozygous for a hypomorphic ChAT allele (cha-1 (cn101)) exhibited a 2.3-fold increase in basal choline uptake relative to cultures from wild-type animals (Fig. 6D) (226.6 ± 11.53% of wild type; t test, p < 0.0001). These studies indicate that, although ChAT activity does not appear to be altered when CHO-1 function is compromised, CHO-1 activity is modulated to compensate for losses in ChAT activity.
cho-1 mutant animals have normal total choline levels, reduced total ACh levels, and visible defects in locomotory activity relative to wild-type animals
Because null mutations in the C. elegans choline transporter are viable, we have the opportunity to investigate the neurochemical and behavioral consequences of a complete loss of high-affinity choline uptake on the intact animal. These questions have been nearly impossible to address in other animal models to date. We examined total choline and ACh levels from whole-worm extracts and found that, similar to our in vitro studies, choline levels did not differ comparing wild type to cho-1 (Fig. 7A) (wild type, 5.25 ± 0.53 × 10−7 μmol/μg protein; cho-1 knock-out, 5.43 ± 0.93 × 10−7 μmol/μg protein). However, total ACh levels at steady state are reduced to 63.6% of normal in cho-1 mutant animals (Fig. 7B) (wild type, 5.34 ± 0.14 × 10−7 μmol/μg protein; cho-1 knock-out, 3.40 ± 0.11 × 10−7 μmol/μg protein), a deficit that is rescued with the expression of a CHO-1:GFP fusion protein in cho-1 mutant cholinergic neurons (Fig. 7B) (BY506, 5.39 ± 0.58 × 10−7 μmol/μg protein). This ∼36% loss of ACh in cho-1 mutant animals does not overtly effect behavior under normal growth conditions. Indeed, in standard plate assays, cholinergic neurotransmission appeared normal when assayed for sensitivity to the acetylcholinesterase inhibitor aldicarb (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). C. elegans mutants that are defective in ACh release are generally resistant to the paralyzing effects of aldicarb. cho-1 mutants displayed a wild-type aldicarb sensitivity profile when tested in parallel with WT, a synaptotagmin allele (snt-1(md290)), and a strong aldicarb resistant mutant that is WT in behavior (ric-1(md226)) (Nguyen et al., 1995).
Figure 7.

Effects of loss of cho-1 activity on total choline and ACh stores. A, Total choline extracted from adult worms and detected by HPLC. No significant difference in choline levels was detected between WT animals, cho-1 mutants, and cho-1 mutants expressing a CHO-1:GFP fusion protein in the cholinergic nervous system (BY506) (ANOVA, p > 0.05). Results are plotted as mean ± SEM from samples performed in triplicate (n = 4). B, Total ACh in adult worms as detected by HPLC. cho-1 mutants animals had 36% less total ACh relative to WT animals (ANOVA test, p < 0.01; mean ± SEM from samples performed in triplicate; n = 4). Normal ACh levels were restored in BY506 animals (ANOVA test, WT vs BY506, p > 0.05; mean ± SEM). C, cho-1 mutants are visibly impaired in locomotory activity. Adult cho-1 mutant animals placed in a choline-free liquid medium demonstrated a 16% reduction in thrashing behavior relative to WT animals, as defined by the number of body bends per minute (WT, n = 32; cho-1 mutant, n = 36). This thrashing defect was rescued in BY506 animals (n = 32). D, The percentage difference in total ACh between WT and cho-1 mutant animals does not increase when animals are subjected to sustained physical stress. Shown is the total ACh extracted from adult worms and detected by HPLC after allowing animals to thrash for 2 h in a choline-free liquid environment (mean ± SEM; n = 3 in triplicate). KO, Knock-out. Asterisks indicate statistical significance as described in the text.
Under well fed conditions on solid medium, cho-1 mutants exhibit little if any motor slowing. However, cho-1 mutant animals do display visible defects in locomotory activity when stressed (Fig. 7C). When placed in a liquid environment, nematodes increase their locomotor rates substantially or “thrash.” Thrashing presumably imposes a high demand on motor neuron firing. When placed in a choline-free liquid medium, cho-1 mutants demonstrated a significant decrease in thrashing rate relative to wild-type animals (Fig. 7C) (222.30 ± 3.35 body bends per minute for wild type; 186.40 ± 1.66 bends per minute for cho-1 mutants; t test, p < 0.0001). Thrashing was restored to wild-type levels when a CHO-1:GFP fusion protein was expressed in cho-1 mutant cholinergic neurons (BY506, 218.3 ± 4.13 bends per minute; p > 0.05 compared with WT). The locomotor defects displayed by cho-1 mutants are not likely attributable to defects in the development of the cholinergic nervous system or synapses, because both cholinergic neuroanatomy and synapse morphology appear normal (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). We asked whether prolonged thrashing and thus increased motor neuron activity would further deplete ACh stores in cho-1 mutant animals (Fig. 7D). Indeed, after 2 h of forced thrashing, the total ACh content in cho-1 animals was further diminished by ∼50%. Wild-type animals also exhibited a loss of ACh levels, consistent with a need for exogenous choline to sustain ACh production during motor stress. Regardless, after 2 h of thrashing, CHO-1-deficient animals remained more significantly depleted of ACh than wild-type animals (wild type, 2.30 ± 0.22 × 10−7 μmol/μg protein; cho-1 mutants, 1.59 ± 0.13 × 10−7 μmol/μg protein; t test, p < 0.05), possibly a reflection of nonmotor pathways deficient in ACh production.
cho-1−/− animals display a stress-triggered, early fatigue phenotype
cho-1 knock-out animals have only a ∼36% reduction in total ACh at steady state, and this percent reduction does not change relative to wild type when further taxed by forced thrashing for 2 h before analysis. One explanation for this is that cho-1 mutants have developed compensatory mechanisms that allow them to use ACh more efficiently than WT animals. However, because motor neurons represent ∼50% of the full complement of cholinergic neurons present in the adult animal, it is probable that our motor “fatigue” paradigm only impacts a fraction of the total nematode stores of ACh. Thus, with sustained activity, there may actually be a more significant depletion in ACh at the NMJ that cannot be resolved by HPLC analysis of total ACh content. We suspected that such a phenotype would be more readily detected through continuous monitoring of nematode motor behavior. Thus, we first characterized the continuous motor behavior of multiple worms simultaneously in group assays using visual methods (Fig. 8A,B). Wild-type and cho-1 mutant worms (10–15 per assay) were placed in a choline-free liquid medium, and thrashing was observed. The number of worms paralyzed or immobile at every minute during a 120 min period was calculated and plotted as the percentage of total animals (Fig. 8A,B). We first attempted group analyses on worms grown on abundant rich food (8P plates with a thick bed of NA22 bacteria), and, under these growth conditions, there was no obvious gross differences in behavior between wild-type and cho-1 mutant animals in the first 90 min of thrashing (Fig. 8A). Between 90 and 120 min, however, cho-1 mutants displayed an increase in paralysis or inactivity over wild-type animals (Fig. 8A).
Figure 8.
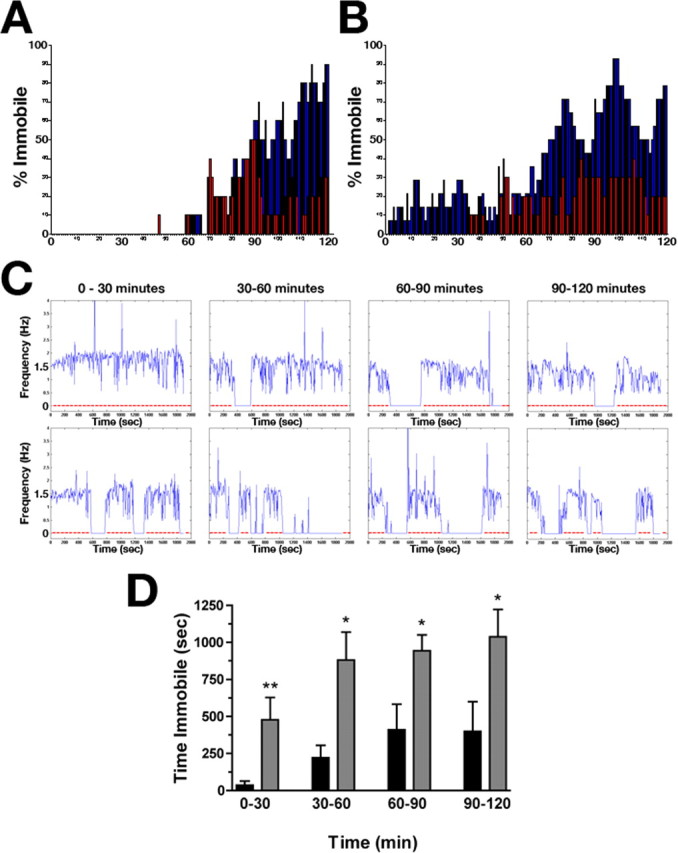
Characterization of cho-1 mutant motor activity. A, B, Group analysis demonstrates an early fatigue phenotype of cho-1 mutants that is influenced by exogenous choline. Shown are representative analyses of 15 worms per genotype (WT, red; cho-1 mutants, blue) in a standard liquid thrashing assay, monitored visually. Plotted are the percentage of total animals immobile per genotype at each minute of a 120 min assay. A, WT and cho-1 mutant animals analyzed after growth on thick lawns of NA22 bacteria. B, WT and cho-1 mutant animals analyzed after growth on sparse lawns of HB101 bacteria. This graph is representative of an extreme example of this phenotype. C, Representative graphs of single-worm automated assays showing animal movement in a choline-free liquid medium over the course of 2 h. Shown are movement traces for WT (top panel) and cho-1 knock-out (bottom panel) animals. Plotted are FFTs of movement frequencies (in hertz) per 6 s window, for a total of 30 min per segment. Shown are 2 h assays broken down into 30 min segments. Periods of inactivity (or fatigue) are shown as a flatline at 0 Hz; the red line is provided as a visual guide to this drop in activity. D, Graph summarizing the amount of time spent immobile (or fatigued) by WT (black) and cho-1 mutant (gray) animals. The data analyzed correspond to the 30 min windows depicted in A but are the combined data of multiple recordings (n = 17 for WT; n = 23 for cho-1 mutants). cho-1 mutant animals displayed a significant increase in fatigue throughout the 2 h assay, with a marked increase in inactivity during the first 30 min relative to WT animals. Error bars indicate SEM; asterisks indicate statitsical significance as described in the text.
Our fatigue results demonstrated significant variability that appeared to relate to growth conditions. We recalled observations by Ghosh and Emmons (2005) who reported that, after ∼90 min of swimming, wild-type animals shift between states of swimming and quiescence in an ultradian manner dependent on growth parameters, primarily temperature and food. Accordingly, we wondered whether restrictions in dietary choline would exacerbate the motor defect displayed by cho-1 mutants. To test this hypothesis, we first analyzed the total choline content in three bacterial strains commonly used for food (OP50, HB101, and NA22). HB101 contained the lowest concentration of total choline of the three strains, and the aforementioned NA22 bacteria had more than five times higher levels (0.533 ± 0.001, 0.930 ± 0.085, 2.664 ± 0.327 × 10−7 μmol choline/μg protein for HB101, OP50, and NA22, respectively). We then analyzed the group behavior of wild-type and cho-1 mutant animals plated on thinly spread lawns of HB101 bacteria (Fig. 8B). Under these conditions, in addition to the increased number of cho-1 animals immobile during later stages of thrashing compared with wild-type animals, cho-1 mutants now displayed an early paralysis phenotype (Fig. 8B). Specifically, cho-1 mutant animals displayed periods of paralysis or inactivity in the first 90 min of the assay that was not exhibited by wild-type animals, sometimes initiating during the first few minutes of the assay (Fig. 8B). Overall, these group assays revealed a trend that when cho-1 mutant animals are surrounded by abundant, rich food, they demonstrate fewer early pauses compared with cho-1 mutants plated on sparse and/or less rich food. Although altogether this trend suggests that restrictions in dietary choline may enhance the fatigue phenotype observed, it should be noted that only free choline measurements were performed on the bacterial strains used as food. There may be additional choline-containing compounds that provide bioavailable choline to these animals in times of stress or high demand. The link between the stress-dependent phenotype and dietary choline availability is attractive, because it indicates that the deleterious effects of a loss of CHO-1-mediated choline uptake can be partially compensated for by provisional supplementation of choline through diet. This is supported by studies performed by Wecker (1988) who showed that, in rat brain slices, ACh synthesis is influenced by the amount of choline consumed in diet.
Individual thrashing behavior can be monitored in worm cultures visually; however, the sensitivity of this method is limited, especially if behavioral deficits are intermittent. To better characterize the early fatigue phenotype exhibited by cho-1 mutants, we designed an automated system for analyzing the movement of individual worms thrashing in a choline-free liquid environment. This method provided a more quantitative assessment of worm motor activity, and removed investigator bias. For all animals, thrashing frequency was monitored for a total of 2 h (4 × 30 min). In agreement with the visual test for thrashing shown in Figure 7C, initial thrash rates differed between wild-type and cho-1 knock-out animals (cho-1 mutants had an ∼15% reduction in thrash rates using both approaches). The mean thrash frequency of wild type in the first 10 min was 1.64 ± 0.04 Hz, compared with 1.40 ± 0.05 Hz by cho-1 mutants (rescued by the expression of CHO-1:GFP in cholinergic neurons; 1.61 ± 0.04 Hz, BY506). More striking was the early fatigue phenotype displayed by cho-1 mutant animals. As indicated by the group behavior assays, both wild-type and cho-1 mutant animals displayed periods of inactivity after time spent thrashing in the choline-free medium. This “resting” period is represented in Figure 8B as a flatline at 0 Hz. In wild-type animals, periods of inactivity typically began after the first 30 min of the assay, and occurred consistently every 20–30 min with an average duration of 4.3 min if they occurred during the first hour of the assay, or 6.2 min if they occurred during the second hour (Fig. 8C). Each period of inactivity was followed by normal vigorous thrashing until the next rest. In contrast, cho-1 mutants demonstrated periods of fatigue as early as the first 2 min of the assay, and the periods of inactivity were more frequent and of longer duration than that of wild-type animals (Fig. 8C,D). Specifically, these events occurred on average every 6 min, and lasted an average of 4.4 min if they occurred during the first hour of the assay, and 8.25 min if they occurred during the second hour of the assay. When the periods of fatigue were plotted as the total time spent immobile during each 30 min window (Fig. 8D), this phenotype was apparent. cho-1 mutants spent significantly more time immobile or in the “resting” state than wild-type animals in every 30 min window (ANOVA; 0–30 min, p < 0.05; 30–60 min, p < 0.01; 60–90 min, p < 0.01; 90–120 min, p < 0.01). This stress-related fatigue phenotype was rescued with the expression of the CHO-1:GFP fusion protein driven by the endogenous cho-1 promoter (BY506) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) (ANOVA test, p > 0.05, WT vs BY506 at every time point). Although we interpret this phenotype as stress-related fatigue, it is important to note that there are other possibilities, including changes in motor control patterns.
We extended our analysis of the motor deficits in cho-1 animals to assess the severity of the phenotype to that displayed by well characterized synaptic mutants (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Specifically, both mild and strong mutants were chosen, which are known to compromise cholinergic neurotransmission, including unc-18 (strong uncoordinated mutant normally required for synaptic vesicle docking) (Weimer et al., 2003); acr-16 [overtly wild-type mutant but measurably reduces evoked cholinergic responses; gene encodes a subunit of the levamisole-resistant nicotinic ACh receptor (AChR) at the NMJ] (Touroutine et al., 2005), and unc-29 (strong uncoordinated mutant; gene encodes an essential subunit of the levamisole-sensitive AChR at the NMJ) (Fleming et al., 1997; Richmond and Jorgensen, 1999). Whereas the two strong uncoordinated mutants (unc-18(md299) and unc-29(e1072)) demonstrated pronounced immobility throughout the 2 h assay, acr-16(ok789) was indistinguishable from WT (ANOVA, p > 0.05 for all time segments). In this comparison, cho-1 mutants displayed an intermediate phenotype (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
Discussion
The use of C. elegans for dissecting molecular aspects of neural signaling is well established (Nonet, 1999). We have adopted this model to study the effects of a loss of presynaptic choline uptake on ACh homeostasis and cholinergic-based behaviors in C. elegans. Multiple studies using pharmacological inhibition of presynaptic choline transport have indicated that the high-affinity, HC-3-sensitive choline transporter is a major regulator of ACh synthesis (for review, see Ferguson and Blakely, 2004). However, subsequent studies in cultured neuroendocrine cells have indicated that ACh synthesis and release can occur in the absence of CHT expression (Bauerfeind et al., 1993; Apparsundaram et al., 2001). To date, the effects of a genetic loss of CHT function on ACh homeostasis and animal behavior has not been established in vivo, because the mouse knock-out displays homozygous neonatal lethality, and heterozygous animals have wild-type levels of high-affinity choline uptake (Ferguson et al., 2004). In contrast, homozygous null alleles of C. elegans CHT (CHO-1) are viable, thus providing an opportunity to study ACh synthesis and cholinergic function in the absence of high-affinity choline uptake.
Using transgenic animals expressing CHO-1:GFP in cholinergic neurons, we found that the transporter localizes to cholinergic synapses, as indicated by its colocalization with synaptic vesicle protein VAMP, and consistent with rodent and primate studies of CHT. Murine CHT resides predominantly on small, clear synaptic vesicles that contain synaptic vesicle markers and VAChT, and demonstrates activity-dependent increases in surface expression, implying a novel mode of regulation for plasma membrane transporters in coupling neurotransmitter precursor uptake to neurotransmitter release (Ferguson et al., 2003). CHO-1 synaptic localization is dependent on UNC-104, a kinesin responsible for the transport of synaptic vesicle precursors to the synapse (Hall and Hedgecock, 1991), suggesting that CHO-1 also traffics on synaptic vesicles.
The transport properties of CHO-1 in primary cultures agree well with data published for vertebrate CHT (Yamamura and Snyder, 1972). CHO-1 mediates high-affinity (Km, ∼0.67 μm) choline uptake that is inhibited by nanomolar concentrations of HC-3, and is both sodium and chloride dependent. CHO-1 appears to be regulated by depolarization, because choline uptake significantly increases when preceded by high potassium depolarization. Similar activity-dependent CHT regulation has been described previously in rodent brain slices and synaptosomes (Ferguson and Blakely, 2004). Together, the UNC-104-dependent synaptic localization and the depolarization-induced enhancement of high-affinity choline uptake suggest that CHO-1 traffics to the presynaptic membrane on synaptic vesicles in response to neuronal activity. The depolarization-induced upregulation of choline uptake requires active calcium channels, and may simply reflect Ca2+-dependent fusion of CHO-1 vesicles. However, this upregulation persists for nearly 30 min after depolarization, indicating a form of short-term plasticity in this system. This may be occurring through increased membrane expression of the transporter, or posttranslational modulation of its activity. The former is more likely based on studies in mouse synaptosomes (Ferguson et al., 2003).
Multiple studies have suggested that, within presynaptic terminals, CHT-mediated choline uptake is rapid and efficient at supplying ChAT with choline to meet the demands for ACh synthesis during periods of high or sustained activity (Ferguson and Blakely, 2004). A critical test of this hypothesis requires studies of ACh production and release in the complete absence of CHT activity. In the mouse model, no difference in hindbrain or forebrain ACh levels was detected in knock-out pups compared with wild-type littermates (Ferguson et al., 2004), possibly because of the immaturity of CNS cholinergic circuits at birth. In contrast, the C. elegans system allows exploration of choline uptake dependence in adult animals. In our studies, we observed that normal, steady-state ACh levels are indeed CHO-1 dependent (∼60% of wild-type levels in cho-1 mutant cells and animals). The presence of ∼40% of normal ACh in these mutants then led us to ask: what is the source of choline maintaining the residual ACh stores in the absence of CHO-1 function? Several possibilities exist: redundant mechanisms of high-affinity uptake, uptake through sodium-independent low-affinity mechanisms, and the liberation of free choline through membrane phospholipases.
We demonstrated that only minimal uptake occurs in cho-1 knock-out cultures over 30 min at conditions favorable for high-affinity transport. This uptake represents <9% of wild-type uptake and does not contribute to detectable ACh synthesis. Thus, we conclude that no redundant high-affinity transport mechanism is operating to sustain ACh production. In contrast, choline uptake in cho-1 mutant cultures was equivalent to wild-type levels at choline concentrations saturated for high-affinity transport and therefore amenable to low-affinity mechanisms. Here, we detected low, but measurable levels of incorporation of choline into ACh. In fact, at 100 μm choline, the amount of labeled choline incorporated into ACh in mutant cells approaches that seen in wild-type cells at 0.1 μm choline. These data indicate that, given sufficiently low demand, low-affinity mechanisms could provide choline for conversion to ACh, if synapses have access to ample choline concentrations. Although the local concentration of choline in the synapse are unknown, circulating plasma choline in humans is only 10.9–13.6 μm in blood plasma, and 1.8 – 2.2 μm in the interstitial and cerebrospinal fluid (Aquilonius et al., 1970; Growdon et al., 1977). Unless the low-affinity system is localized synaptically, and synaptic concentrations approach 100 μm, it seems unlikely that this mechanism contributes significantly to ACh synthesis in the absence of CHO-1. Indeed, previous studies have indicated that low-affinity mechanisms are inefficient for providing synaptic choline for ACh production (Haga and Noda, 1973).
Evidence suggests that membrane phosphatidylcholine may serve as a reservoir of choline under conditions in which external free choline is limited (Maire and Wurtman, 1985; Blusztajn et al., 1987). Phosphatidylethanolamine can be methylated to form phosphatidylcholine via the enzyme PeMT (phosphatidylethanolamine N-methyltransferase) (Blusztajn et al., 1979, 1987b), and free choline liberated from this pathway supports ACh synthesis (Blusztajn et al., 1987a,b). The generation of free choline appears to occur through the direct hydrolysis of phosphatidylcholine by phospholipase D, rather than by phospholipase C or multienzyme pathways consisting of phospholipase A, lysophospholipase, and glycerophosphocholine (Lee et al., 1993). If intracellular phosphatidylcholine is the source of choline used in the absence of high-affinity transport (and low-affinity mechanisms are not contributing measurably to ACh production) (Haga and Noda, 1973; present study), then we would expect to see an additional reduction in total ACh in cells/animals doubly mutant for cho-1 and phospholipase D. Rather, we observed that, in both primary cultures and whole-animal extracts, ACh content remained at cho-1 mutant levels even when phosholipase D activity is absent. Moreover, cho-1;pld-1 double mutants are healthy, and behaviorally indistinguishable from cho-1 mutants, which would not be expected if a membrane cannibalism was a major source of choline in the absence of CHO-1 function (Blusztajn et al., 1987a,b). However, these findings do not invalidate the possibility that choline used for ACh synthesis in cho-1 mutants may be derived from phospholipids catabolized in PLD-independent pathways.
One way that cho-1 mutant animals could offset the effects of a loss in high-affinity choline uptake is through compensatory mechanisms that increase the efficiency of ACh production or the sensitivity of ACh reception. It has been suggested that high-affinity choline transport and ChAT are tightly coupled systems (Barker and Mittag, 1975; Barker et al., 1975). Thus, we tested whether ChAT activity is upregulated in cho-1 mutant cells. We found no difference in ChAT activity between cho-1 and wild-type animals, similar to findings of unaltered ChAT activity in the brains of CHT knock-out mice (Ferguson et al., 2004). Interestingly, basal choline uptake is significantly higher in C. elegans cells carrying a hypomorphic ChAT mutation, a phenomenon that is also seen in mice heterozygous for a lethal mutation in ChAT (Brandon et al., 2004). The combination of these data support the model that choline uptake is limiting for ChAT function: ChAT is already in kinetic excess, and cannot be further upregulated in the absence of high-affinity choline uptake, but when ChAT function is compromised, choline transport can be increased to compensate. Relocation of a portion of vesicular stores of CHT may support this compensatory response.
Despite the fact that cho-1 knock-out animals are capable of synthesizing ∼60% of normal levels of ACh, they do demonstrate observable defects in motor activity. When placed in conditions that require sustained neuronal activity, a stress-related fatigue phenotype is apparent, indicating impairment in ACh release at the NMJ. This may be attributable to a localized depletion of ACh at the NMJ. If this depletion is occurring selectively in motor neurons, then a complete loss of NMJ ACh may not be detected biochemically (in total ACh measurements) because of the background of unaffected ACh levels in cholinergic nonmotor neurons. However, such a localized loss should be detected behaviorally. We suspect that, in times of low ACh demand, for example, crawling on bed of choline-rich bacteria, low-affinity uptake mechanisms likely have time to provide cho-1 mutant cells with sufficient choline to sustain cholinergic signaling. In contrast, when the cho-1 mutant system is taxed during intense, sustained motor activity, depletion of ACh at the NMJ may be seen if synaptic vesicles are not filled, because the demand exceeds the kinetic limitations of alternative pathways providing free choline to the nerve terminal.
Our studies demonstrate an essential role of high-affinity transport for normal ACh synthesis and motor activity in adult animals. CHO-1 appears to be the sole presynaptic transporter capable of sustaining ACh synthesis at low levels of extracellular choline. The stress-dependent motor phenotype displayed by cho-1 mutant animals can be exploited in future genetic screens for the additional characterization of CHT regulation. Furthermore, studies of the regulation and trafficking of CHO-1 should aid in our understanding of the processes sustaining cholinergic tone in disorders in which cholinergic deficits emerge.
Footnotes
This work was supported by American Cancer Society Postdoctoral Fellowship PF-01-113-01-DDC (D.S.M.) and National Institutes of Health Grant MH073159-02 (to R.D.B.). The Neurochemistry Core Facility of the Center for Molecular Neuroscience of Vanderbilt University Medical Center is supported by National Institutes of Health Grant P30 HD15052, and the Vanderbilt University Medical Center Cell Imaging Core Resource is supported by National Institutes of Health Grants CA68485 and DK20593. We greatly appreciate the helpful discussions with Shawn Ferguson (Yale University, New Haven, CT), especially during the initial stages of this project, and the support of Tammy Jessen (Vanderbilt University, Nashville, TN) for lab organization and C. elegans husbandry. We appreciate the useful discussions with David Miller III and members of his laboratory (Vanderbilt University) and are indebted to Professor Andrew Dozier (Engineering, Vanderbilt University) and his engineering students Mohd Fakhrurrazi Mohd Salleh, Suhaili Harun, Saiful Azlan Adanan, and Amani Rafie for the generation of the first working prototype of our worm tracking system. We gratefully acknowledge the use of the Neurochemistry Core Facility of the Center for Molecular Neuroscience (Vanderbilt University Medical Center) and the Vanderbilt University Medical Center Cell Imaging Core Resource.
References
- Apparsundaram S, Ferguson SM, Blakely RD (2001). Molecular cloning and characterization of a murine hemicholinium-3-sensitive choline transporter. Biochem Soc Trans 29:711–716. [DOI] [PubMed] [Google Scholar]
- Aquilonius SM, Schuberth J, Sundwall A (1970). Studies on choline in cerebrospinal fluid. Acta Pharmacol Toxicol (Copenh) 28:35. [PubMed] [Google Scholar]
- Barker LA, Mittag TW (1975). Comparative studies of substrates and inhibitors of choline transport and choline acetyltransferase. J Pharmacol Exp Ther 192:86–94. [PubMed] [Google Scholar]
- Barker LA, Dowdall MJ, Mittag TW (1975). Comparative studies on synaptosomes: high-affinity uptake and acetylation of N-[Me-3H]choline and N-[Me-3H]N-hydroxyethylpyrrolidinium. Brain Res 86:343–348. [DOI] [PubMed] [Google Scholar]
- Bauerfeind R, Regnier-Vigouroux A, Flatmark T, Huttner WB (1993). Selective storage of acetylcholine, but not catecholamines, in neuroendocrine synaptic-like microvesicles of early endosomal origin. Neuron 11:105–121. [DOI] [PubMed] [Google Scholar]
- Birks RI, MacIntosh FC (1957). Acetylcholine metabolism at nerve-endings. Br Med Bull 13:157–161. [DOI] [PubMed] [Google Scholar]
- Birks RI, MacIntosh FC (1961). Acetylcholine metabolism of a sympathetic ganglion. Can J Biochem Physiol 39:787–827. [Google Scholar]
- Blusztajn JK, Wurtman RJ (1981). Choline biosynthesis by a preparation enriched in synaptosomes from rat brain. Nature 290:417–418. [DOI] [PubMed] [Google Scholar]
- Blusztajn JK, Zeisel SH, Wurtman RJ (1979). Synthesis of lecithin (phosphatidylcholine) from phosphatidylethanolamine in bovine brain. Brain Res 179:319–327. [DOI] [PubMed] [Google Scholar]
- Blusztajn JK, Liscovitch M, Mauron C, Richardson UI, Wurtman RJ (1987a). Phosphatidylcholine as a precursor of choline for acetylcholine synthesis. J Neural Transm Suppl 24:247–259. [PubMed] [Google Scholar]
- Blusztajn JK, Liscovitch M, Richardson UI (1987b). Synthesis of acetylcholine from choline derived from phosphatidylcholine in a human neuronal cell line. Proc Natl Acad Sci USA 84:5474–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Mellott T, Pizzo DP, Coufal N, D’Amour KA, Gobeske K, Lortie M, Lopez-Coviella I, Berse B, Thal LJ, Gage FH, Blusztajn JK (2004). Choline transporter 1 maintains cholinergic function in choline acetyltransferase haploinsufficiency. J Neurosci 24:5459–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch AE, Karbach U, Miska D, Gorboulev V, Akhoundova A, Volk C, Arndt P, Ulzheimer JC, Sonders MS, Baumann C, Waldegger S, Lang F, Koepsell H (1998). Human neurons express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine, and memantine. Mol Pharmacol 54:342–352. [DOI] [PubMed] [Google Scholar]
- Carvelli L, McDonald PW, Blakely RD, Defelice LJ (2004). Dopamine transporters depolarize neurons by a channel mechanism. Proc Natl Acad Sci USA 101:16046–16051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen M, Estevez A, Yin X, Fox R, Morrison R, McDonnell M, Gleason C, Miller DM III, Strange K (2002). A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron 33:503–514. [DOI] [PubMed] [Google Scholar]
- Clark SG, Chiu C (2003). C. elegans ZAG-1, a Zn-finger-homeodomain protein, regulates axonal development and neuronal differentiation. Development 130:3781–3794. [DOI] [PubMed] [Google Scholar]
- Damsma G, Westerink BHC, Horn AS (1985). A simple, sensitive, and economic assay for choline and acetylcholine using HPLC, an enzyme reactor, and an electrochemical detector. J Neurochem 45:1649. [DOI] [PubMed] [Google Scholar]
- Engel AG, Ohno K, Sine SM (2003a). Sleuthing molecular targets for neurological diseases at the neuromuscular junction. Nat Rev Neurosci 4:339–352. [DOI] [PubMed] [Google Scholar]
- Engel AG, Ohno K, Shen XM, Sine SM (2003b). Congenital myasthenic syndromes: multiple molecular targets at the neuromuscular junction. Ann NY Acad Sci 998:138–160. [DOI] [PubMed] [Google Scholar]
- Erickson JD, Weihe E, Schafer MK, Neale E, Williamson L, Bonner TI, Tao-Cheng JH, Eiden LE (1996). The VAChT/ChAT “cholinergic gene locus”: new aspects of genetic and vesicular regulation of cholinergic function. Prog Brain Res 109:69–82. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Blakely RD (2004). The choline transporter resurfaces: new roles for synaptic vesicles? Mol Interv 4:22–37. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Savchenko V, Apparsundaram S, Zwick M, Wright J, Heilman CJ, Yi H, Levey AI, Blakely RD (2003). Vesicular localization and activity-dependent trafficking of presynaptic choline transporters. J Neurosci 23:9697–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Bazalakova M, Savchenko V, Tapia JC, Wright J, Blakely RD (2004). Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc Natl Acad Sci USA 101:8762–8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JT, Squire MD, Barnes TM, Tornoe C, Matsuda K, Ahnn J, Fire A, Sulston JE, Barnard EA, Sattelle DB, Lewis JA (1997). Caenorhabditis elegans levamisole resistance genes lev-1, unc-29, and unc-38 encode functional nicotinic acetylcholine receptor subunits. J Neurosci 17:5843–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonnum F (1975). A rapid radiochemical method for the determination of choline acetyltransferase. J Neurochem 24:407–409. [DOI] [PubMed] [Google Scholar]
- Ghosh R, Emmons S (2005). Swimming in C. elegans: molecular and cellular basis of an ultradian temporal pattern. C. elegans International Worm Meeting 2005:31. [Google Scholar]
- Growdon JH, Cohen EL, Wurtman RJ (1977). Effects of oral choline administration on serum and CSF choline levels in patients with Huntington’s disease. J Neurochem 28:229–231. [DOI] [PubMed] [Google Scholar]
- Guyenet P, Lefresne P, Rossier J, Beaujouan JC, Glowinski J. (1973). Inhibition by hemicholinium-3 of [14C]acetylcholine synthesis and [3H]choline high-affinity uptake in rat striatal synaptosomes. Mol Pharmacol 9:630–639. [PubMed] [Google Scholar]
- Haga T, Noda H (1973). Choline uptake systems of rat brain synaptosomes. Biochim Biophys Acta 291:564–575. [DOI] [PubMed] [Google Scholar]
- Hall DH, Hedgecock EM (1991). Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell 65:837–847. [DOI] [PubMed] [Google Scholar]
- Inazu M, Takeda H, Matsumiya T (2005). Molecular and functional characterization of an Na+-independent choline transporter in rat astrocytes. J Neurochem 94:1427–1437. [DOI] [PubMed] [Google Scholar]
- Jin Y (1999). Transformation. In: C. elegans: a practical approach (Hope IA, ed.) pp. 69–95. New York: Oxford UP.
- Jope RS (1979). High-affinity choline transport and acetylCoA production in brain and their roles in the regulation of acetylcholine synthesis. Brain Res 180:313–344. [DOI] [PubMed] [Google Scholar]
- Kim H, Rogers MJ, Richmond JE, McIntire SL (2004). SNF-6 is an acetylcholine transporter interacting with the dystrophin complex in Caenorhabditis elegans. Nature 430:891–896. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Okuda T, Fujioka Y, Matsumura G, Nishimura Y, Haga T (2002). Distribution of the high-affinity choline transporter in the human and macaque monkey spinal cord. Neurosci Lett 317:25–28. [DOI] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML (2001). A post-docking role for active zone protein RIM. Nat Neurosci 4:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kus L, Borys E, Ping Chu Y, Ferguson SM, Blakely RD, Emborg ME, Kordower JH, Levey AI, Mufson EJ (2003). Distribution of high-affinity choline transporter immunoreactivity in the primate central nervous system. J Comp Neurol 463:341–357. [DOI] [PubMed] [Google Scholar]
- Lee HC, Fellenz-Maloney MP, Liscovitch M, Blusztajn JK (1993). Phospholipase D-catalyzed hydrolysis of phosphatidylcholine provides the choline precursor for acetylcholine synthesis in a human neuronal cell line. Proc Natl Acad Sci USA 90:10086–10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberato DJ, Weintraub ST, Yergey AL (1985). Separation and quantification of choline and acetylcholine by thermospray liquid chromatography/mass spectrometry. Biomed Environ Mass Spectrom 13:171–174. [DOI] [PubMed] [Google Scholar]
- Lonart G, Janz R, Johnson KM, Sudhof TC (1998). Mechanism of action of Rab3A in mossy fiber LTP. Neuron 21:1141–1150. [DOI] [PubMed] [Google Scholar]
- Maire JC, Wurtman RJ (1985). Effects of electrical stimulation and choline availability on the release and contents of acetylcholine and choline in superfused slices from rat striatum. J Physiol (Paris) 80:189–195. [PubMed] [Google Scholar]
- Misawa H, Nakata K, Matsuura J, Nagao M, Okuda T, Haga T (2001). Distribution of the high-affinity choline transporter in the central nervous system of the rat. Neuroscience 105:87–98. [DOI] [PubMed] [Google Scholar]
- Nakashima S, Nozawa Y, Hisamoto N, Matsumoto K, Banno Y (2000). Molecular cloning and expression pattern of C. elegans phospholipase D. Worm Breeder’s Gazette 16:27. [Google Scholar]
- Nakata K, Okuda T, Misawa H (2004). Ultrastructural localization of high-affinity choline transporter in the rat neuromuscular junction: enrichment on synaptic vesicles. Synapse 53:53–56. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Alfonso A, Johnson CD, Rand JB (1995). Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics 140:527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet M (1999). Studying mutations that effect neurotransmitter release in C. elegans. In: Frontiers in molecular biology: neurotransmitter release (Bellen H, ed.) Oxford: Oxford UP.
- Nonet ML, Grundahl K, Meyer BJ, Rand JB (1993). Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell 73:1291–1305. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ (1997). Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J Neurosci 17:8061–8073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, Haga T, Kanai Y, Endou H, Ishihara T, Katsura I (2000). Identification and characterization of the high-affinity choline transporter. Nat Neurosci 3:120–125. [DOI] [PubMed] [Google Scholar]
- Otsuka AJ, Jeyaprakash A, Garcia-Anoveros J, Tang Garcia-Anoveros, Fisk G, Harshorne T, Franco R, Born T (1991). The C. elegans unc-104 gene encodes a putative kinesin heavy chain-like protein. Neuron 6:113–122. [DOI] [PubMed] [Google Scholar]
- Quirion R (1987). Characterization and autoradiographic distribution of hemicholinium-3 high-affinity choline uptake sites in mammalian brain. Synapse 1:293–303. [DOI] [PubMed] [Google Scholar]
- Rainbow TC, Parsons B, Wieczorek CM (1984). Quantitative autoradiography of [3H]-hemicholinium-3 binding sites in rat brain. Eur J Pharmacol 102:195–196. [DOI] [PubMed] [Google Scholar]
- Rand JB, Duerr JS, Frisby DL (2000). Neurogenetics of vesicular transporters in C. elegans. FASEB J 14:2414–2422. [DOI] [PubMed] [Google Scholar]
- Richmond JE, Jorgensen EM (1999). One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci 2:791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Bruno JP (1997). Cognitive functions of cortical acetylcholine: toward a unifying hypothesis. Brain Res Rev 23:28–46. [DOI] [PubMed] [Google Scholar]
- Schueler FW (1955). A new group of respiratory paralyzants. I. The “hemicholiniums.”. J Pharmacol Exp Ther 115:127–143. [PubMed] [Google Scholar]
- Simon JR, Kuhar MG (1975). Impulse-flow regulation of high-affinity choline uptake in brain cholinergic nerve terminals. Nature 255:162–163. [DOI] [PubMed] [Google Scholar]
- Simon JR, Kuhar MG (1976). High-affinity choline uptake: ionic and energy requirements. J Neurochem 27:93–99. [DOI] [PubMed] [Google Scholar]
- Sweet DH, Miller DS, Pritchard JB (2001). Ventricular choline transport: a role for organic cation transporter 2 expressed in choroid plexus. J Biol Chem 276:41611–41619. [DOI] [PubMed] [Google Scholar]
- Touroutine D, Fox RM, Von Stetina SE, Burdina A, Miller DM III, Richmond JE (2005). acr-16 encodes an essential subunit of the levamisole-resistant nicotinic receptor at the Caenorhabditis elegans neuromuscular junction. J Biol Chem 280:27013–27021. [DOI] [PubMed] [Google Scholar]
- Traiffort E, Ruat M, O’Regan S, Meunier FM (2005). Molecular characterization of the family of choline transporter-like proteins and their splice variants. J Neurochem 92:1116–1125. [DOI] [PubMed] [Google Scholar]
- Wecker L (1988). Influence of dietary choline availability and neuronal demand on acetylcholine synthesis by rat brain. J Neurochem 51:497–504. [DOI] [PubMed] [Google Scholar]
- Weimer RM, Richmond JE, Davis WS, Hadwiger G, Nonet ML, Jorgensen EM (2003). Defects in synaptic vesicle docking in unc-18 mutants. Nat Neurosci 6:1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delong MR (1982). Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215:1237–1239. [DOI] [PubMed] [Google Scholar]
- Yamamura HI, Snyder SH (1972). Choline: high-affinity uptake by rat brain synaptosomes. Science 178:626–628. [DOI] [PubMed] [Google Scholar]