

Abstract
We show that desert hedgehog (dhh), a signaling molecule expressed by Schwann cells, is essential for the structural and functional integrity of the peripheral nerve. Dhh-null nerves display multiple abnormalities that affect myelinating and nonmyelinating Schwann cells, axons, and vasculature and immune cells. Myelinated fibers of these mice have a significantly increased (more than two times) number of Schmidt-Lanterman incisures (SLIs), and connexin 29, a molecular component of SLIs, is strongly upregulated. Crossing Dhh-null mice with myelin basic protein (MBP)-deficient shiverer mice, which also have increased SLI numbers, results in further increased SLIs, suggesting that Dhh and MBP control SLIs by different mechanisms. Unmyelinated fibers are also affected, containing many fewer axons per Schwann cell in transverse profiles, whereas the total number of unmyelinated axons is reduced by approximately one-third. In Dhh-null mice, the blood–nerve barrier is permeable and neutrophils and macrophage numbers are elevated, even in uninjured nerves. Dhh-null nerves also lack the largest-diameter myelinated fibers, have elevated numbers of degenerating myelinated axons, and contain regenerating fibers. Transected dhh nerves degenerate faster than wild-type controls. This demonstrates that a single identified glial signal, Dhh, plays a critical role in controlling the integrity of peripheral nervous tissue, in line with its critical role in nerve sheath development (Parmantier et al., 1999). The complexity of the defects raises a number of important questions about the Dhh-dependent cell–cell signaling network in peripheral nerves.
Keywords: glia, degeneration, regeneration, hedgehog, PNS, electron microscopy, neuropathy
Introduction
It is commonly believed that signals from glial cells play an essential part, directly and indirectly, in establishing and maintaining the structural and functional integrity of the nervous system. Nevertheless, it has proved hard to identify particular signaling molecules that perform such functions in vivo and to define the consequences of their absence. Here, we have addressed this issue by examining the signaling molecule desert hedgehog (Dhh) in peripheral nerves, where this gene is expressed exclusively by myelinating Schwann cells.
Hedgehogs (Hhs) are intercellular signaling proteins that are best known for controlling tissue patterning during development. In mammals, the Hh family consists of three members: Sonic hedgehog (Shh), Indian hedgehog (Ihh), and Dhh (Hammerschmidt et al., 1997). They bind to the transmembrane receptors patched to activate complex intracellular signaling cascades involving smoothened (smo) and the three gli transcription factors (Ruiz i Altaba et al., 2003). We have shown previously that during development, Dhh secreted by Schwann cells is required for the correct formation of the perineurial tissue sheath around peripheral nerves (Parmantier et al., 1999). In humans, a null mutation in dhh also leads to peripheral neuropathy with mini-fascicle formation and gonadal dysgenesis (Umehara et al., 2000).
There is increasing recognition that long- and short-term term signaling from glial cells has an essential role in the nervous system. Glial cells are a key source not only of developmental signals and trophic factors but also of signals that regulate blood flow, neuronal survival after injury, and pain pathways (Jessen, 2004; Newman, 2004; Schipke and Kettenmann, 2004; Ullian et al., 2004; Tsuda et al., 2005). They also secrete gliotransmitters that take part in the short-term regulation of synaptic function and neuronal excitability (Zhang and Haydon, 2005).
A number of observations indicates the importance of glial-derived signals in peripheral nerves. Schwann cell precursors and immature Schwann cells are likely to provide indispensable survival support for embryonic motor and sensory neurons, because removal of these glial cells through genetic manipulations results in extensive neuronal death (Garratt et al., 2000; Jessen and Mirsky, 2005). In mature nerves also, disruption of the relationship between unmyelinated axons and nonmyelinating Schwann cells leads to death of sensory neurons perhaps as a result of decreased glial cell line-derived neurotrophic factor expression (Chen et al., 2003). In myelinated fibers, axonal death accompanies inherited Schwann cell disorders, pointing to a disruption of as yet unidentified signaling pathways between axons and Schwann cells, whereas at the neuromuscular junction, unknown signals from perisynaptic glia modulate synaptic activity (Colomar and Robitaille, 2004).
In the present study, we find that removal of Dhh, a signaling molecule selectively expressed in myelinating Schwann cells in adult nerves, has wide-ranging adverse consequences for every tissue component of peripheral nerves. This demonstrates an important signaling function for myelinating Schwann cells and identifies a glial signal that is essential for tissue integrity.
Materials and Methods
Animals
Dhh-null mice were obtained by homologous recombination as described previously (Bitgood et al., 1996) and were made available by Prof. A. P. McMahon (Harvard University, Boston, MA). Animals were genotyped as dhh+/+, dhh+/−, or dhh−/− by PCR on genomic DNA (Parmantier et al., 1999). The Dhh-null mutant and wild-type mice were on a mixed CD1/C57BL6 genetic background. Myelin basic protein (MBP)-deficient shiverer (shi) mice were obtained from Prof. I. Griffiths (University of Glasgow Veterinary School, Glasgow, UK). They are on a C3H/101 background and were genotyped as shi+/+, shi+/−, and shi−/− by PCR (Gomez et al., 1990). All conclusions are based on observations on a minimum of three animals per genotype. The number of animals in each particular test is indicated in the text.
In situ hybridization
Digoxygenin-labeled cRNA antisense probes were generated as described by Parmantier et al. (1999). Postnatal day 10 (P10) mice were perfused with 4% phosphate-buffered parafomaldehyde for 30 min. The nerves were then removed and fixed for an additional 3 h with fresh 4% paraformaldehyde at 4°C. They were rinsed in PBS and protected in 30% sucrose in PBS overnight before mounting in Tissue Tek (Sakura Finetek, Tokyo, Japan). Frozen sections (20 μm) were mounted on Superfrost slides before in situ hybridization overnight at 65°C with a digoxygenin-labeled antisense probe to dhh as described previously (Parmantier et al., 1999). Subsequently, slides were incubated overnight with antibody to human smooth muscle actin (clone 1A4; 1:70; a gift from L. Morgan, EISAI London Research, London, UK) (DakoCytomation, Carpinteria, CA) and with goat anti-mouse Ig rhodamine for 1 h (MP Biomedicals, Irvine, CA), followed by mouse anti-MBP (1:200; Sternberger Monoclonals, Baltimore, MD) overnight and by goat anti-mouse Ig fluorescein (Jackson ImmunoResearch, West Grove, PA) for 1 h before being mounted in Citifluor (London, UK) anti-fade mounting medium before examination in an Eclipse 800 Nikon (Tokyo, Japan) fluorescence microscope.
Labeling and analysis of Schmidt-Lanterman incisures
Nerve fixation and immunolabeling were as described by Gould et al. (1995). Immediately after the mice were killed, the sciatic nerves were exposed from the straightened thigh and fixed in situ with 4% phosphate-buffered paraformaldehyde for 30 min. The nerves were then removed and fixed for an additional 3 h with fresh 4% paraformaldehyde at 4°C. They were rinsed in PBS and stored in newly made PBS at 4°C. On the next day, the nerves were desheathed and cut into smaller pieces. They were teased individually into small bundles, transferred into a 96-well plate containing rhodamine-conjugated phalloidin (1:100; Sigma, St. Louis, MO), and kept overnight at 4°C. They were then rinsed three times in PBS and transferred individually to Superfrost-coated slides, where they were teased further into single fibers and smaller bundles. Slides were then either mounted in Citifluor anti-fade mounting medium or immunolabeled with antibodies to MUPP1 (Poliak et al., 2002) (a gift from E. Peles, The Weizmann Institute of Science, Rehovot, Israel) or neurofascin (NF) 155 (Chang et al., 2000) (a gift from P. Brophy, University of Edinburgh, Edinburgh, UK) and goat anti-rabbit FITC (MP Biomedicals) before being mounted in Citifluor anti-fade mounting medium before examination in an Nikon Eclipse 800 fluorescence microscope. Connexin 29 (cx29) labeling was performed on ethanol-fixed nerves as described by Li et al. (2002), and KV1.2 antibody (Alomone Laboratories, Jerusalem, Israel) labeling was performed after phalloidin labeling as described by Altevogt et al. (2002). Images were captured using a digital camera at 40× magnification with Lucia acquisition software, which was also used for measurements of Schmidt-Lanterman incisures (SLIs) and fiber width. In randomly chosen fibers, the inter-SLI distance was measured from the center of one SLI to the center of the next adjacent SLI, and the fiber width was measured at three positions along the internode as described by Gould et al. (1995). The numbers of SLIs were counted along measured distances of randomly selected individual teased fibers. Measurements were taken from fibers from two animals of each genotype at each time point. A total of 1311 SLI intervals was measured in dhh−/− fibers, 689 in dhh+/− fibers, and 221 in dhh+/+ fibers (P10 only). For counts of the number of SLIs, a total of 133 fibers was analyzed in dhh−/− mice, 138 in dhh+/− mice, and 51 in dhh+/+ mice (P10 only).
Electron microscopic analysis of tissue sections
P30 dhh+/+, dhh+/−, and dhh−/− mice (n = 3 per genotype) were anesthetized and perfused with 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 m phosphate buffer.
Analysis of nerve fiber diameter.
Sciatic nerves were removed and postfixed in the same solution at 4°C overnight. They were then processed for electron microscopy as described previously (Parmantier et al., 1999). Ultrathin transverse sections were viewed and photographed using a JEOL (Peabody, MA) 1010 electron microscope. Pictures were taken from the lower right quadrant of transverse sections at 2000× magnification, covering in total one-fourth of the entire nerve area. They were scanned and imported into Open Lab software, where the diameter of myelinated axons and of myelin plus axons was measured. From these measurements, the g-ratio (axon diameter/axon plus myelin sheath diameter) was calculated. To study the size distribution of axons and of axons plus myelin, the measurements were grouped into increasing size categories separated by 0.1 μm, and the number in each category was determined.
Nonmyelinating Schwann cells.
Using the sections and micrographs taken for the previous analysis, the number of unmyelinated axons/Schwann cell unit was counted in two 2-month-old and one 4-month-old animal of each genotype, and the number of axons in a 1:1, 2:1, etc., ratio was expressed as a percentage of the total number of unmyelinated axons.
Axon numbers in the tibial branch.
The total number of myelinated and unmyelinated axons in the entire tibial branch of the sciatic nerve was counted directly in the electron microscope using sections mounted on membrane-coated grids without grid bars. Nerves from 2-, 4-, and 5.5-month-old dhh+/+ and dhh−/− littermates were counted (n = 3 nerves for each genotype).
Degeneration and regeneration in uninjured sciatic nerve.
dhh+/+ and dhh−/− mice aged 2–6 months (two mice per genotype) and 21 months (two mice per genotype) were perfused with 2.5% paraformaldehyde and 2.5% glutaraldehyde in 0.1 m phosphate buffer. Sciatic nerves were dissected and processed for electron microscopy as before. Photographic (500×) montages covering a transverse section of the entire tibial and common peroneal branches at the mid-thigh level from each animal were analyzed. Degenerating fibers were identified as profiles either without an axon or with a highly reduced axon caliber in association with obviously broken myelin. The regenerating myelinated fibers were identified by the presence of loose, redundant basal lamina and a reduced number of myelin lamellae (usually <10 lamellae) relative to axon diameter. The sections were also observed at higher magnification to confirm the data.
The number of compact and interrupted myelinated fibers.
Photographic montages were made as described above, including the tibial and common peroneal branch of the sciatic nerve, and the entire nerve profiles were analyzed (three animals per genotype).
Degeneration after nerve cut.
The left sciatic nerve from 2-month-old dhh+/− and dhh−/− mice was transected (n = 2 per genotype per time point). At 2 and 7 d after operation, mice were perfused and the nerves were processed for electron microscopy. The right sciatic nerve from each animal was used as a control. Sections were taken at approximately mid-thigh level, and photographic montages of transverse sections were made as described above. Fibers in both uncut and 2 d postcut nerve were categorized into (1) normal-looking fibers with intact myelin, (2) fibers with collapsed axons, and (3) fibers with collapsed myelin. The entire section of each nerve was counted. In parallel experiments, nerves were processed for immunoblotting (see below).
Measurement of motor neuron cell body size
P30 dhh+/+, dhh+/−, and dhh−/− mice were perfused with 4% paraformaldehyde, 4% picric acid, and 0.15 m phosphate buffer. The spinal cord was exposed, and the tissue from lumbar (L) levels L4–L5 was further dissected and postfixed for 1 h. Segments were washed in 0.1 m phosphate buffer before overnight incubation in 30% sucrose/PBS at 4°C. Forty-micrometer sections were cut transversely on a freezing microtome and transferred individually into wells of a 96-well plate containing 0.1 m phosphate buffer, 5% sucrose, and 0.02% sodium azide. The sections were transferred onto gelatinized slides and allowed to dry for 24 h before 4–6 min immersion in Neutral Red solution (0.5% in 1% acetic acid). Slides were dehydrated, mounted in DPX mounting medium (BDH Chemicals, Poole, UK), and observed at 20× in a Nikon Eclipse 800 microscope. Images of sections were captured using a digital camera and AKT software (Nikon), and the area of the cell soma of individual motor neurons was measured using Open Lab software.
Immunoblotting
Proteins were extracted from sciatic nerves of dhh+/− and dhh−/− mice as described by Wrabetz et al. (2000). Samples were denatured and resolved on either 12% or 8% SDS-polyacrylamide gels and transferred onto Hybond-ECL nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ). The blot was blocked with 0.05% Tween and 5% dry milk in PBS overnight at 4°C, followed by incubation in appropriate antibody in 0.05% Tween and 1% dry milk in PBS overnight at 4°C. Rabbit polyclonal P0 antibodies (Morgan et al., 1991) or mouse monoclonal antibodies recognizing P0 (Archelos et al., 1993) (a gift from J. J. Archelos, Karl-Franzens University, Graz, Austria), MBP, MAG (Roche Products, Welwyn Garden City, UK), β-catenin, E-cadherin (BD Biosciences, Franklin Lakes, NJ), and β-tubulin (Sigma) were used. Goat anti-mouse Ig peroxidase or goat anti-rabbit Ig peroxidase (Promega, Madison, WI) were used as second layers.
Specific protein complexes were developed using ECL-plus chemiluminescent reagent and x-ray film (Amersham Biosciences). In some experiments, uncut and transected nerves, 2 d after transection, were immunoblotted with antibodies to TUJ1 (Covance, Princeton, NJ) or P0. Antibodies to glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Abcam, Cambridge, MA) were used to control for equal loading of samples.
Semiquantitative reverse transcription-PCR
Total RNA was extracted from freshly dissected sciatic nerves from dhh+/− and dhh−/− mice of different ages, or from purified Schwann cells or fibroblasts, with Trizol reagent (Invitrogen, San Diego, CA) according to the manufacturer's instructions. The RNA was quantified, and 500 ng of each was reverse transcribed into cDNA, as described previously (Parmantier et al., 1999). To check that the same quantity of cDNA was used in the PCRs, the relative amount of cDNA synthesized from each sample was assessed by PCR amplification using specific primers for GAPDH. One microliter of cDNA was used in each 50 μl PCR. For the sciatic nerve extracts, reactions were cycled through one cycle at 94°C for 3 min, followed by 30 rounds (27 for GAPDH) at 94°C for 30 s, 62°C (MBP, MAG, cx32, S100) for 30 s, 55°C (cx29 and JAM) for 45 s, 63°C (GAPDH) for 30 s, 72°C for 30 s, and one final extension at 72°C for 5 min. The primer pairs were as follows: MBP: forward primer 5′ACTCACACACGAGAACTACCCA3′, reverse primer 5′CCAGCTAAATCTGCTGAGGG 3′ (169 bp) (Nicholson et al., 2001); MAG: forward primer 5′GGAGACCTGGGCCTACGAAA3′, reverse primer 5′GACGTCCAAACTGGCGTAAC3′ (442 bp) (Nicholson et al., 2001); cx32: forward primer 5′TACGTGGCGTGAATCGGCAC3′, reverse primer 5′GTTGGTGAGCTACGTGCATT3′ (266 bp) (Nicholson et al., 2001); cx29: forward primer 5′ATGTGCGGCAGGTTCCTGAGACA3′, reverse primer 5′TCAAAATGGCTCTTTTGCCTCCA3′ (777 bp) (Sohl et al., 2001); S100: forward primer 5′GGTGACAAGCACAAGCTGAA3′, reverse primer 5′CTGGAAGTCACACTCCCCA3′ (150 bp) (Nicholson et al., 2001); JAM2: forward primer 5′GTCGCACAGATGTGTTTGG3′, reverse primer 5′GAGTTCACATGGAAAGAGG3′ (327 bp); JAM3: forward primer 5′TTGGTCTACTACCAACAGG3′, reverse primer 5′TTTCGTGTTCATTGTGTACG3′ (369 bp). For Ptc1 reverse transcription (RT)-PCR of Schwann cells, 1 μl of cDNA was used in a 50 μl PCR mixture that was subjected to one cycle at 94°C for 3 min, followed by 35 cycles at 94°C for 30 s, 55°C for 1 min, 72°C for 30 s, and one final extension at 72°C for 5 min [Ptc1: forward primer 5′AACAAAAATTCAACCAAACCTC3′, reverse primer 5′TGTCTTCATTCCAGTTGATGTG3′ (244 bp) (Takabatake et al., 1997); Cld5: forward primer 5′GAAGGGGCTGTGGATGTC3′, reverse primer 5′ACCGTCGGATCATAGAAC3′ (314 bp) (Poliak et al., 2002)].
Preparation of purified neonatal mouse Schwann cells
P3 nerve Schwann cells from ICR mice were purified by immunopanning using antibodies to Thy-1 as described previously (Dong et al., 1999). Schwann cells were plated on to 35 mm poly-l-lysine- and laminin-coated tissue culture dishes in defined medium with low insulin (Dong et al., 1999), d-glucose, and 10 ng/ml neuregulin-β1 (R & D Systems, Minneapolis, MN). Twenty-four hours before the experiment, the Schwann cell medium was changed to DMEM and 10% fetal calf serum (Invitrogen). Cells were then treated with the Hh pathway agonist Cur-0199567 (C67) (40 nm; a gift from Dr. L. Rubin, Curis, Cambridge, MA) (Frank-Kamenetsky et al., 2002). Fibroblasts were treated with recombinant human Dhh protein (a gift from Biogen, Cambridge, MA). Three and 24 h later, RNA was extracted as described above.
Labeling of macrophages and neutrophils
The left sciatic nerves from 2-month-old dhh+/− and dhh−/− mice were transected. The mice were killed at 2 d after operation, and the distal stumps of cut nerve and the uncut sciatic nerve from each animal were fresh frozen in Tissue Tek (Sakura Finetek). Frozen sections (10 μm) of dhh+/− and dhh−/− nerves were fixed with 4% paraformaldehyde and labeled with F4/80 antibody (Serotec, Indianapolis, IN) (a gift from S. Davies, University College London, London, UK) to identify macrophages (Starkey et al., 1987) and Ly-6G (GR1)-FITC antibody (BD Biosciences) (a gift from J. Roes, University College London, London, UK) to identify neutrophils (Parmantier et al., 1999).
Endoneurial blood vessel permeability
Five percent bovine albumin (Cohn fraction V) was mixed with 1% Evans blue albumin (EBA) in HBSS (Sigma) and dialyzed against HBSS overnight at 4°C (Kristensson and Olsson, 1971). The following day, the tracer was filtered, and 1 ml/kg body weight of mice (P30) was injected into the jugular vein of anesthetized mice (n = 3 per genotype). The tracer was left to circulate for 3 h, 30 min before the mice were killed, and the sciatic nerve and the brain from each animal were fixed with 4% paraformaldehyde, sectioned, and viewed under the fluorescence microscope.
Results
The number of SLI is elevated in dhh−/− Schwann cells
We previously used teased nerve fiber preparations to show that in postnatal nerves dhh is selectively expressed by myelinating Schwann cells (Parmantier et al., 1999). We confirmed this finding using in situ hybridization on transverse sections of P10 sciatic nerve because this allows visualization of other cellular components of the nerve, including blood vesselsand the perineurium, both of which were essentially negative (Fig. 1).
Figure 1.
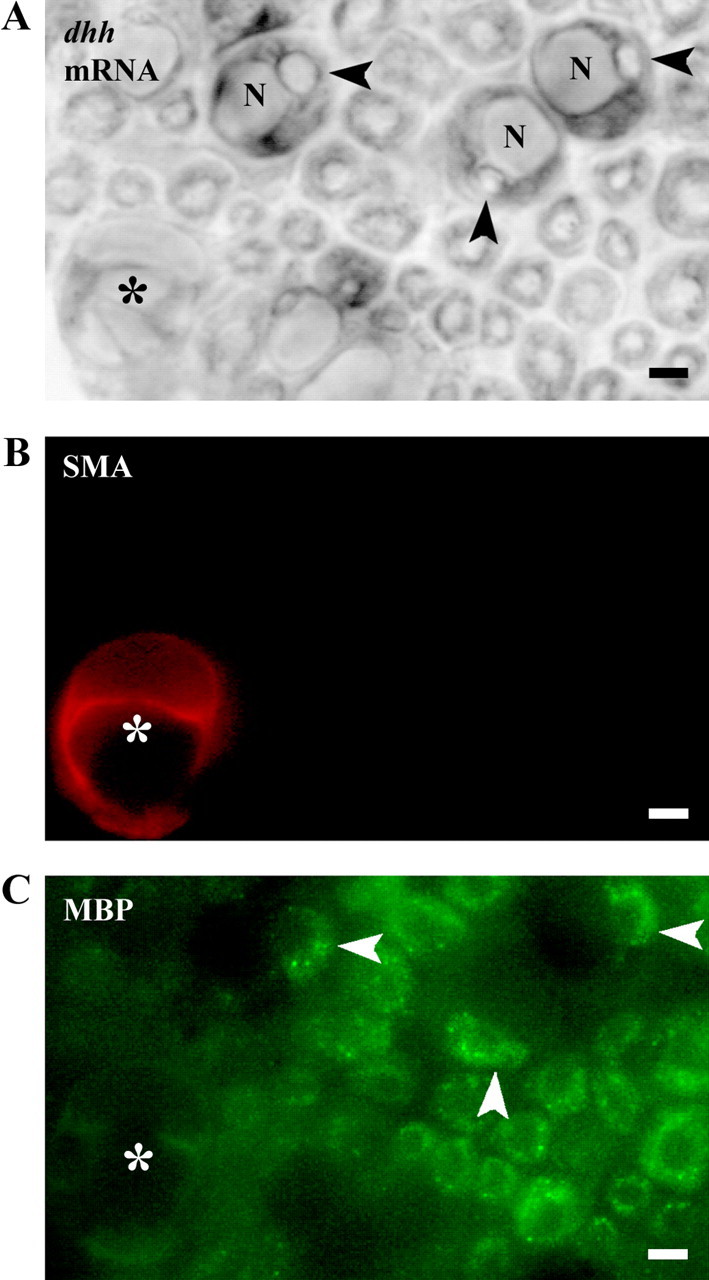
Dhh mRNA is selectively expressed in myelinating Schwann cells. The same field from a transverse section of a P10 sciatic nerve is shown in A (bright field to visualize dhh mRNA by in situ hybridization), B (immunofluorescence to visualize blood vessels), and C (immunofluorescence to visualize myelin). A, Unambiguous Dhh signal is only seen where the section cuts through the nuclei (N) of myelinating cells displacing the myelin sheath (arrowheads) to an eccentric position. B, The field contains one capillary (asterisk) visualized by smooth muscle actin (SMA) antibodies. C, Myelin sheaths shown by MBP antibodies. Myelin sheaths associated with Schwann cell nuclei that are labeled for dhh mRNA (arrowheads). Asterisk in A and C indicates a capillary. Scale bars, 4 μm.
To investigate the effect of Dhh on the general architecture of myelinated fibers, nerves were taken from dhh+/− and dhh−/− mice at P10, P23, P30, and 8 months (n = 2 for each genotype at each age), fixed, teased, and stained with fluorescein-conjugated phalloidin, which binds to F-actin and clearly demarcates nodes of Ranvier, paranodal loops, and SLIs, which are cytoplasmic channels within compact myelin that are considered to facilitate communication between the cytoplasmic compartments on abaxonal and adaxonal sides of the myelin sheath via gap junctions (Hall and Williams, 1970; Ghabriel and Allt, 1981; Trapp et al., 1989; Gould et al., 1995; Balice-Gordon et al., 1998; Poliak and Peles, 2003). Examination of dhh+/+, dhh+/−, and dhh−/− fibers revealed an obvious increase in SLI numbers in the dhh−/− animals (Fig. 2; see Fig. 5). This was quantified by measuring distances between SLIs (Gould et al., 1995). This showed that the distance between SLIs in dhh−/− fibers was about one-half of that in dhh+/+ or dhh+/− fibers (Table 1). We also counted the number of SLIs along internodes at P10, P23, and P30 and found that the number of SLIs per 100 μm in dhh−/− fibers was about twice that seen in controls (Table 1). The number of SLIs in dhh+/+ and dhh+/− mice was similar showing that a single copy of dhh is sufficient to maintain the normal SLI phenotype. Because the number of SLIs was increased, the distance between SLIs was decreased (Table 1). When visualized by phalloidin labeling, the gross morphology of SLIs in dhh−/− nerves differed from that of normal nerves. Whereas the majority of SLIs in normal fibers were relatively simple structures, those in dhh−/− fibers were frequently broader and more complex, often consisting of closely spaced doublets (Fig. 2). Similar observations were made in shi mice, where the number of SLIs is also increased as a result of lack of MBP (Gould et al., 1995).
Figure 2.

Teased nerve fibers of dhh−/− mice, labeled with rhodamine-conjugated phalloidin, show increased numbers of SLIs in a dhh+/− nerve fiber (A) and in a dhh−/− nerve fiber (B). Note also the increased size and complexity of the SLI in B. Scale bar, 10 μm.
Figure 5.

cx29, NF155, and MUPP1 are localized to SLI in nerves of both dhh+/− and dhh−/− mice. A, Sciatic nerves were teased and immunolabeled with antibodies to cx29, revealing the increased number of SLIs in the dhh−/− fibers. B, Teased nerves were double labeled with phalloidin and antibodies to NF155, showing the increased number of SLIs in the dhh−/− fibers. Note the co-incidence of actin and NF155 labeling. C, Teased nerves were double labeled with phalloidin and antibodies to MUPP1. Note the co-incidence of actin and MUPP1 labeling. Scale bar, 33 μm.
Table 1.
The number of SLI in dhh−/− and control nerves
| P10 |
P23 |
P30 |
|||||
|---|---|---|---|---|---|---|---|
| dhh+/+ | dhh+/− | dhh−/− | dhh+/− | dhh−/− | dhh+/− | dhh−/− | |
| Fiber width | 5.76 ± 0.92 | 5.62 ± 0.93 | 5.14 ± 0.77 | 7.48 ± 0.11 | 7.42 ± 0.11 | 8.22 ± 0.94 | 6.14 ± 0.14 |
| Distance between SLIs | 37.2 ± 1.70 | 38.3 ± 1.58ns | 20.12 ± 0.61*** | 39.69 ± 1.91 | 20.30 ± 0.56* | 38.29 ± 1.09 | 16.88 ± 0.39* |
| SLI/100 μm | 2.38 ± 0.11 | 2.39 ± 0.14ns | 4.74 ± 0.20*** | 2.54 ± 0.14 | 5.5 ± 0.51* | 2.79 ± 0.12 | 6.38 ± 0.39* |
At all ages, the distance between SLIs in dhh−/− fibers is approximately one-half that seen in dhh+/+or dhh+/− nerves. Fiber width and distance is expressed in micrometers. The mean value ± SEM is shown. Superscripts indicate significant difference from same age control:
*p < 0.001 versus dhh+/−;
**p < 0.001 versus dhh+/+; nsNot significant versus dhh+/+ (Mann–Whitney test).
Electron microscopic analysis shows a decreased number of large fibers in dhh−/− mice
To test whether the absence of Dhh changed other structural features of myelinated fibers, electron micrographs from the sciatic nerves of dhh+/+, dhh+/−, and dhh−/− mice (n = 3 per genotype) were scanned, and the diameter of axons and axons plus myelin of >1000 fibers for each genotype was measured. We found that the number of larger-diameter myelinated axons was reduced in dhh−/− mice compared with dhh+/− mice (Fig. 3A). In these nerves, the number of axons between 6.0 and 7.5 μm in diameter was significantly reduced (p < 0.05; Kolmogorov–Smirnov Z test) and larger axons were absent. We did not find a statistically significant difference between any of the genotypes in the average diameter of axons or in the diameter of axons plus myelin (Fig. 3B), although there was a trend toward smaller diameters in dhh−/− nerves. This is in line with our previous light microscopic observations on myelinated units in dhh−/− and dhh+/+ nerves (Bajestan et al., 2006). There were no significant differences between dhh+/+, dhh+/−, and dhh−/− nerves in the g-ratios of myelinated axons at any diameter This accords with previous light-microscopic measurements of average g-ratios in dhh+/+ and dhh−/− mice (Bajestan et al., 2006), indicating that although there are more incisures in dhh−/− nerves, the myelin thickness in most individual axons remains unaltered (Fig. 3C). To determine whether the total number of myelinated axons was altered in the absence of Dhh, we used the electron microscope to count the myelinated axons in the tibial branch of the sciatic nerve in three pairs of dhh+/+ and dhh−/− 2- to 6-month-old littermates. In dhh+/+ mice, there were 2765 ± 39.15 myelinated axons, and in dhh−/−, mice there were 2344.67 ± 33.33 myelinated axons. Comparison revealed that there was a small but significant (p < 0.05; Student's t test) drop in the number of myelinated axons in dhh−/− animals.
Figure 3.
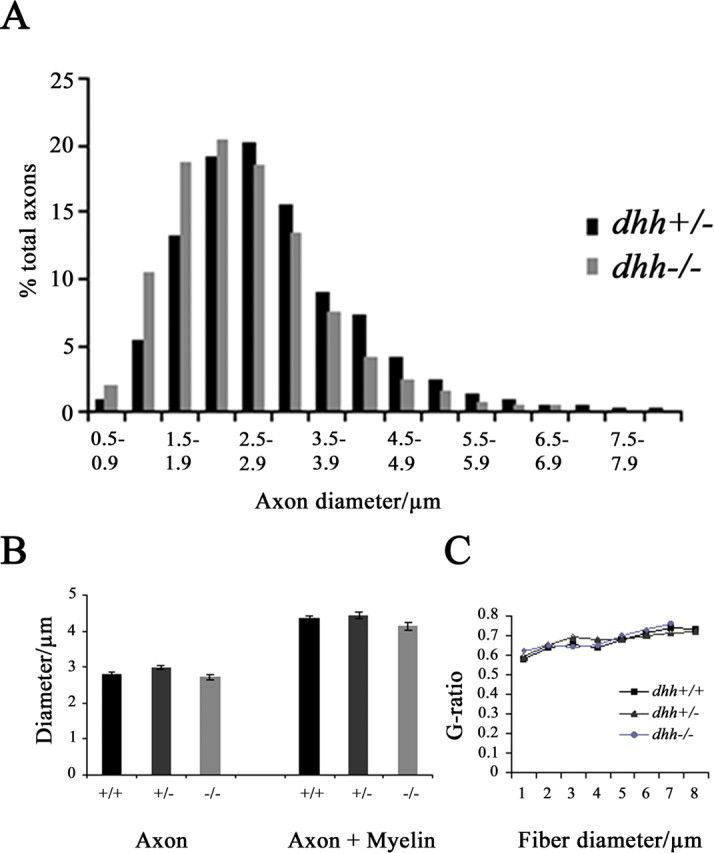
Analysis of myelinated axon and fiber diameters. A, Axon diameters in dhh+/− and dhh−/− nerves. Note that myelinated axons of the largest diameter are absent in dhh−/− nerves. B, Average diameters of dhh+/+, dhh+/−, and dhh−/− axons and myelinated profiles are similar, although the dhh−/− myelinated profiles are slightly but not significantly smaller. C, The g-ratio for each fiber size is similar in all three genotypes.
Motor neuron number and cell body size are unchanged in dhh−/− mice
The largest-diameter axons within the sciatic nerve include those of motor neurons. We therefore investigated whether the observed loss of large-diameter axons in dhh−/− nerves was attributable to a loss or change in the size of the motor neurons.
We compared the number and average area of the cell body of motor neurons, as judged by Neutral Red staining, in the L4/5 region of the spinal cord where the largest motor neurons are located. There was no significant difference in the average size of the motor neurons between dhh−/− and dhh+/− mice (2.9 ± 0.12 μm in dhh+/+ and 3.2 ± 0.1 μm in dhh−/−). The average number of motor neurons in the two genotypes was also similar (data not shown).
cx29 is strongly upregulated in dhh−/− nerves
Because the number of SLIs was at least double in dhh−/− mice at all ages, we investigated whether this increase in number had any effect on the expression levels of myelin proteins (P0 and MBP) or proteins that are expressed in the incisures (MAG, E-cadherin, β-catenin, and cx29) (Gould et al., 1995; Smith-Slatas and Barbarese, 2000; Meier et al., 2004). Protein extracts were prepared from dhh+/− and dhh−/− mouse sciatic nerves at different ages (P10, P21, P30, and 3 months) and analyzed by immunoblotting (Fig. 4A). In dhh−/− nerves, the level of MBP was somewhat lower at P30 and 3 months than in dhh+/− nerves, but there was no apparent difference at earlier ages even though the difference in SLI number is already established by P10 (Table 1). There were minimal differences in the expression levels of P0, MAG, E-cadherin, and β-catenin protein at any of the ages tested. Western blotting of cx29, however, revealed that this protein was markedly upregulated at P21 (Fig. 4B) and other ages (data not shown). In agreement with others, we found that cx29 protein displayed anomalous behavior on Western blotting (Altevogt et al., 2002; Li et al., 2002), showing a strong band at ∼60 kDa and a weaker band at ∼30 kDa, both of which were upregulated in the dhh−/− nerve.
Figure 4.

Western blot and RT-PCR analysis of dhh+/− and dhh−/− sciatic nerves. A, Western blot analysis of MBP, P0, β-catenin, MAG, and E-cadherin in sciatic nerves from dhh+/− and dhh−/− mice at P10, P21, P30, and 3 months. β-Tubulin is included to show that samples are equally loaded. Note that MBP levels in dhh−/− nerves are lower at 3 months than in dhh+/− nerves, whereas other proteins are unchanged. B, Western blot analysis of cx29 at P21. Note the upregulation of a strong band at ∼60 kDa and a less prominent band at ∼29 kDa in the dhh−/− nerves. C, RT-PCR of mRNA in sciatic nerves from dhh+/− and dhh−/− mice at P10, P21, P30, and 3 months of molecules that are expressed either in SLI and/or endothelial junctions. GAPDH is included to show that samples are equally loaded. Note that cx29 mRNA levels are increased in dhh−/− nerve at 3 months.
In dhh−/− mice, many SLI-associated mRNAs are unchanged, whereas cx29 mRNA levels are elevated in older nerves
RT-PCR was performed on sciatic nerve extracts from dhh+/− and dhh−/− mice aged P10, P23, P30, and 3 months to survey the relative mRNA levels of the proteins analyzed by immunoblotting. We also investigated the mRNA levels of other proteins such as cx32, cx29, S100, and claudin5, which are associated with SLIs (Gould et al., 1995; Altevogt et al., 2002; Li et al., 2002; Poliak et al., 2002; Poliak and Peles, 2003; Meier et al., 2004), and of JAM2 and JAM3, which are associated with tight junctions and with MUPP1 (Ebnet et al., 2004). After standardization with the housekeeping gene GAPDH, samples were run separately with primers against MBP, MAG, cx32, cx29, S100, E-cadherin, claudin5, JAM2, and JAM3 (Fig. 4C). The results showed a clear increase in cx29 mRNA expression in dhh−/− mice at 3 months with smaller increases detectable at all ages. The expression level of mRNAs for MBP, cx32, S100, JAM2, and JAM3 remained unchanged. MAG and E-cadherin expression levels appeared marginally higher in dhh−/− than dhh+/− nerves at P13 and P23 but did not appear to be different at older ages.
The SLI-associated proteins MUPP1, NF155, and cx29 are expressed in the additional SLIs of dhh−/− Schwann cells
To test whether the additional SLIs present in dhh−/− fibers had the same molecular composition as SLIs in normal fibers, sciatic nerves from adult dhh+/− and dhh−/− animals were teased and immunolabeled with antibodies against a number of SLI proteins. The localization of cx29, NF155, and MUPP1 is shown in Figure 5. MUPP1 is primarily confined to SLIs (Poliak et al., 2002), whereas cx29 and NF155 are also present in paranodal loops adjacent to the node of Ranvier. We found that NF155 and MUPP1 were seen in essentially all phalloidin-labeled SLIs in both dhh+/+ and dhh−/− nerve fibers, and immunolabeling for cx29 showed a similar distribution (Fig. 5). Comparable results were obtained with S100 and MAG antibodies (data not shown). Thus, the new incisures that are generated in the absence of Dhh signaling appear to have an essentially normal molecular composition.
It has been suggested that there is an association between cx29 in myelinating Schwann cells and KV1.2 potassium channels in axonal membranes (Arroyo et al., 1999; Altevogt et al., 2002). We therefore used antibodies and immunolabeling to examine KV1.2 channels in dhh−/− teased nerves (data not shown). In three separate experiments, we found that axonal KV1.2 immunolabeling was generally weaker in dhh−/− fibers than in dhh+/− ones. This may not be quantitatively significant. On the other hand, it is clear that KV1.2 expression in axons is not increased commensurately with Schwann cell cx29 expression in dhh−/− fibers.
Dhh/shi double null mice have increased numbers of SLIs compared with single null mice
The fact that SLI numbers are increased in both dhh and shi (MBP) mutants led us to ask whether these conditions were linked.
To investigate this genetically, we crossed dhh null mice with shi mice to obtain mice that were null for both dhh and MBP. We reasoned that if Dhh and MBP acted along an identical pathway to control SLIs, there should be little or no increase in the number of SLIs in the double mutants compared with the single mutant mice. In contrast, if Dhh and MBP were components of two relatively independent and parallel mechanisms, the number of SLIs should be further increased. Eleven double null mice were obtained. Five were used to make teased nerve preparations and for electron microscopy. It was found that the distance between SLIs was substantially smaller in dhh/shi double null nerve fibers than in either of the single null mice both at 3 and 15 weeks (n = 2 for each genotype at each time point) (Fig. 6C, Table 2). We also counted the number of SLIs along internodes and found that the number of SLIs per 100 μm was elevated in the double mutant compared with the shi−/− at both time points (Table 2). As an additional measure of myelin disruption, we used electron micrographs of transversely sectioned nerves to compare the number of continuous, undisrupted compact myelin profiles (sheaths) in normal, dhh−/−, and double null mice (n = 3 per genotype) with the total number of myelinated fibers, which included profiles that contained incisures or nodal structures. We found that 92.3 ± 1.20% of the total myelinated axon profiles in normal mice had intact sheaths, whereas the corresponding figure was 75.7 ± 3.93% in dhh−/− mice. In the double mutants, however, only 23 ± 2.52% of total myelinated profiles had intact sheaths. This measure of myelin compactness therefore confirms the difference between single and dhh/shi double mutant nerves revealed by measuring SLI intervals or counting SLI numbers.
Figure 6.

Dhh−/−/shi−/− double null sciatic nerves have increased numbers of SLI. A, B, Semithin toluidine blue-stained sections of wild-type (A) and dhh−/−/shi−/− (B) nerves. Note the arrangement of the double null nerve into mini-fascicles, a phenotype typical of dhh−/− null nerves. C, Teased preparations of wild-type, shi, and double null nerves from 3.5-month-old mice, using phalloidin to label SLIs. The double null nerve fibers have many more SLIs than the wild-type ones. D, Electron micrograph showing a complex SLI in a transverse section of double null nerve. E, Electron micrograph of multiple SLIs (arrows) in a double mutant nerve shown in a longitudinal section. Scale bars: A, B, 70 μm; C, 66 μm; D, 0.9 μm; E, 0.7 μm.
Table 2.
The number of SLIs in double mutants
| Dhh+/+/shi+/+ | dhh+/−/shi+/− | shi+/− | shi−/− | dhh−/− | Dhh−/−/ shi−/− | |
|---|---|---|---|---|---|---|
| Fiber diameter | 7.42 ± 0.10 | 6.94 ± 0.84 | 6.98 ± 0.93 | 7.04 ± 0.93 | 6.86 ± 0.13 | |
| 8.69 ± 0.22 | 9.03 ± 0.11 | 8.41 ± 0.16 | 9.19 ± 0.16 | 7.96 ± 0.14 | ||
| Distance between SLIs | 40.64 ± 1.66 | 27.47 ± 0.95 | 32.44 ± 1.43 | 16.73 ± 0.37 | 13.92 ± 0.38 * | |
| 41.53 ± 2.04 | 31.64 ± 0.74 | 19.51 ± 0.44 | 23.70 ± 0.73 | 12.90 ± 0.21*** | ||
| SLI/100 μm | 2.02 ± 0.11 | 2.89 ± 0.12 | 2.21 ± 0.12 | 4.77 ± 0.17 | 6.42 ± 0.31 * | |
| 2.36 ± 0.18 | 3.04 ± 0.18 | 4.79 ± 0.20 | 4.17 ± 0.26 | 7.87 ± 0.2*** |
Nerve fiber analysis of the sciatic nerves of littermates of the indicated genotypes at 3 weeks (bold letters) and 15 weeks of age. Note that double null nerves have shorter SLI distances and higher numbers of SLIs per 100 μm than the single mutants.
*p < 0.001 versus shi−/−;
**p < 0.001 versus shi−/− (Mann–Whitney test).
Together, these results suggest that Dhh and MBP control SLI numbers by distinct and relatively independent mechanisms. We also confirmed that the number of SLIs in shi+/− mice was intermediate between the wild-type and shi−/− mice (data not shown) (Smith-Slatas and Barbarese, 2000). Toluidine blue semithin sections revealed that the nerves of the double null mice, like those of dhh−/− mice, were arranged in mini-fascicles (Fig. 6A,B).
Nonmyelinating Schwann cells and unmyelinated axons are affected in dhh−/− nerves
In transverse sections of dhh−/− nerves, we noted a large number of unmyelinated axons in a 1:1 ratio with a single Schwann cell, whereas nonmyelinating cells with large numbers of axons were seldom seen (Fig. 7A). This was confirmed by using electron microscopy to count the number of unmyelinated axons in a 1:1, 1:2, etc., ratio with Schwann cells in an entire transverse section of sciatic nerve from 2- to 4-month-old dhh−/− and dhh+/− littermates (n = 3 per genotype) (Fig. 7B). This showed that the percentage of axons in a 1:1 ratio was highly elevated in dhh−/− nerves. In line with this, the number of axons found in groups of 10 or larger was lower in the dhh−/− nerves. Axon/Schwann cell units containing two to nine axons were present in similar numbers and accommodated 40–45% of axons in both genotypes (Fig. 7A,B). To determine whether the total number of unmyelinated axons was altered in the absence of Dhh, we used the electron microscope to count the total number of unmyelinated axons in the tibial branch of the sciatic nerve at 2, 4, and 5.5 months of age. In dhh+/+ mice, there were 4335.33 ± 47.88 unmyelinated axons, and in dhh−/− mice, there were 2807.67 ± 138.81 unmyelinated axons. Comparison revealed a statistically significant difference (p < 0.001; Student's t test) in the number of unmyelinated axons in dhh−/− nerves.
Figure 7.
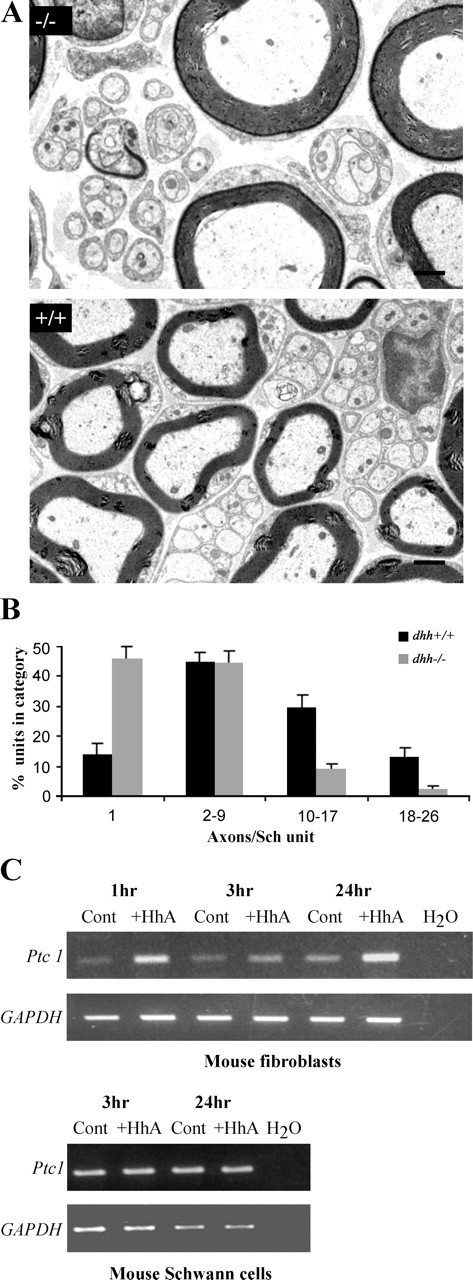
A, Electron micrographs showing transverse sections of the sciatic nerve of 2-month-old mice, illustrating the typical difference in appearance of nonmyelinating Schwann cells in dhh−/− and dhh+/+ animals. Scale bar, 1 μm. B, The percentage of the total number of small unmyelinated axons that is found in a 1:1 ratio and ratios of 1:2–9, etc. with nonmyelinating Schwann (Sch) cells. Note the marked increase in the percentage of axons in a 1:1 ratio with a Schwann cell in dhh−/− nerves and the decrease in the Schwann cell units containing large numbers of axons. C, RT-PCR analysis showing that ptc1 mRNA in cultured Schwann cells is not upregulated in response to hedgehog ligands (HhA) in contrast to that seen in fibroblasts. Dhh protein (40 nm) was applied to Schwann cells, and the Hh agonist Cur-019956767 (40 nm) was applied to fibroblasts. GAPDH is shown to indicate that samples are equally loaded. Cont, Control. Mean values ± SEM are shown.
Thus, in addition to the changes in myelinating Schwann cells (see above), the nonmyelinating Schwann cells are also altered in the absence of Dhh, a fact that is likely to be related to the expression of Ptc2 receptors on these cells (Bajestan et al., 2006).
In Schwann cells, ptc1 is not regulated in response to Hh signaling
We showed previously that in developing peripheral nerve, the Hh receptor ptc1 was localized at highest levels in the perineurial cells surrounding nerve fibers. Furthermore, its expression level in these cells of fibroblastic origin was upregulated by Dhh, as would be expected, because Hhs normally upregulate ptc1 expression in target cells (Parmantier et al., 1999). In view of the Schwann cell phenotype reported here, we wanted to determine whether Schwann cells, in addition to expressing dhh, also expressed ptc1 receptors that were responsive to Hh signaling.
Purified neonatal mouse Schwann cells or nerve-derived fibroblasts were cultured with Hh ligands or agonists (Frank-Kamenetsky et al., 2002) for 3 and 24 h. RT-PCR revealed that Schwann cells expressed detectable levels of ptc1, but unlike nerve fibroblastic cells, they did not upregulate ptc1 expression in response to Hh signals (Fig. 7C). The reason for this is unknown. Ptc2 receptors are also expressed in peripheral nerve where they are found selectively on nonmyelinating Schwann cells in teased preparations of normal adult nerves (Bajestan et al., 2006).
In so far as the absence of upregulation of ptc1 can be taken to indicate nonfunctionality of the ptc1 signaling pathway and the fact that ptc1 mRNA levels are very low in adult nerves (Parmantier et al., 1999), these observations suggest that the effects of Dhh on myelinating Schwann cells are most likely to be indirect, perhaps via Dhh-induced signals from perineurial cells, rather than autocrine, whereas the effects of Dhh on nonmyelinating cells are likely to be via Ptc2.
Blood vessels in peripheral nerves of dhh−/− mice are abnormally permeable
In dhh−/− nerves, the molecular and cellular composition of the endoneurium is likely to be altered because of the breakdown of the tissue–nerve barrier (Parmantier et al., 1999). The endoneurial capillary–nerve barrier (blood–nerve barrier) constitutes another protective mechanism for endoneurial nerve fibers. We therefore tested whether the blood vessels in dhh−/− mice were abnormally leaky, using permeability to EBA as an indicator (Kristensson and Olsson, 1971).
EBA was injected into the jugular vein of dhh+/+ and dhh−/− mice under general anesthesia and allowed to circulate for 30 min or 3 h. The mice were then killed, and the sciatic nerves and the brain from each animal were fixed, sectioned, and viewed under the fluorescence microscope (Fig. 8). In the brain, EBA was confined to blood vessels at both time points, and no differences were seen between dhh+/+ and dhh−/− animals. In the sciatic nerve of control mice, EBA was associated with the perineurium/epineurium, presumably reflecting the normal permeability of epineurial vessels. In the endoneurium, EBA was, however, confined to blood vessels at both time points, demonstrating the existence of the blood–nerve barrier in dhh+/+ nerves. But in dhh−/− nerves, EBA distribution was markedly different. In this case, EBA was present throughout the nerve at both time points.
Figure 8.

Endoneurial blood vessels in dhh−/− nerves are permeable to dyes. Comparison of permeability to EBA in dhh+/− and dhh−/− mouse tissues is shown. Left column, Sciatic nerve. Note the labeling throughout the nerve in the dhh−/− nerve (bottom), whereas in the dhh+/− nerve, EBA is confined to the epineurium and three capillaries within the endoneurium. Middle and right columns, Labeling is confined to capillaries in both the cortex and cerebellum and does not differ between dhh+/− and dhh−/− mice. Scale bars, 50 μm.
Thus, the double protection offered to endoneurial nerve fibers by the perineurial tissue–nerve barrier and the blood–nerve barrier is absent in dhh−/− mice. The permeability of the blood–brain barrier is, however, independent of Dhh signaling. Although this is likely to have adverse effects on peripheral nerve fibers, it is probably not a significant cause of the elevated SLI numbers in dhh−/− fibers, because SLI numbers are elevated before the perineurial and vascular barriers develop fully (Kristensson and Olsson, 1971; Weerasuriya et al., 1990). Schwann cell-derived Dhh could also be involved in promoting vascularization of peripheral nerves (Kusano et al., 2004), although we did not find any obvious reduction in the number of blood vessels in dhh−/− nerves (data not shown).
Uninjured dhh−/− nerves show age-dependent axonal degeneration and myelin breakdown
The loss of myelinated axons indicated that these uninjured dhh−/− nerves suffered spontaneous axonal degeneration, perhaps accompanied by regeneration.
To investigate this for myelinated fibers, nerves from 2- to 6- and 21-month-old dhh−/− mice were examined by electron microscopy (n = 2 per genotype per age group) (Fig. 9A). This revealed that 33 ± 9.5 (1.1 ± 0.1%) of the myelinated axons (total axons counted, 2864 ± 561) were degenerating at any one time in mutant nerves from 2- to 6-month-old mice, whereas no degenerating profiles (total axons counted, 4020 ± 156) were found in littermates. By 21 months, 12 ± 1 (0.3 ± 0%) degenerating fibers were seen in wild-type controls (total axons counted, 4399 ± 913), but they were still approximately four times more common, 50 ± 2 (1.2 ± 0.2%), in dhh−/− nerves (total axons counted, 4444 ± 844). In dhh−/− nerves, the number of degenerating profiles increased markedly from 2–6 months to 21 months.
Figure 9.
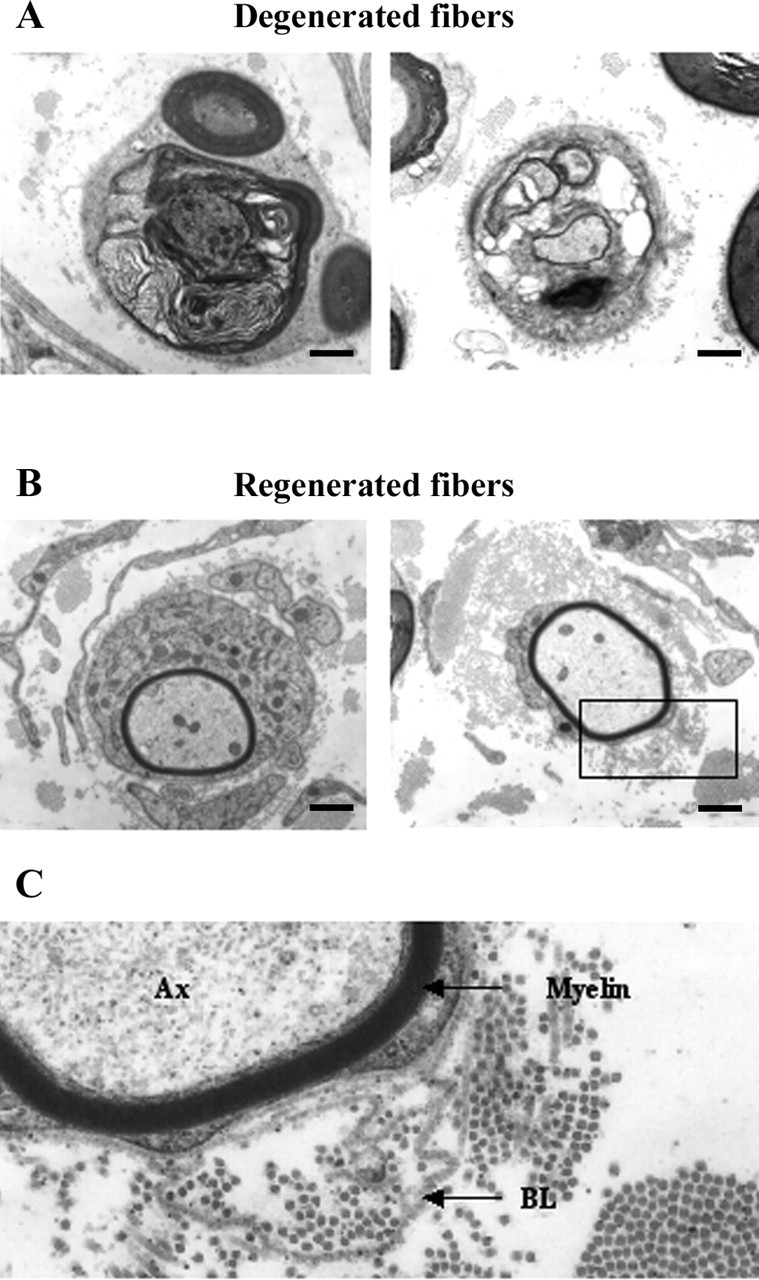
Dhh−/− nerves contain degenerating and regenerating nerve fibers. A, Electron micrographs showing two profiles of degenerating fibers. Left, Shrunken axon surrounded by myelin debris. Right, Shrunken axon surrounded by a vacuolated Schwann cell. B, Two profiles of regenerating axons with thin myelin sheaths relative to the axon diameter. C, Enlargement of the boxed in area in B. Note the redundant basal lamina (BL) associated with a thinly myelinated fiber. Ax, Axon. Scale bars: A, B, 1.8 μm.
In the dhh−/− nerves, we also noted a number of axons that had thin myelin sheaths for their size (Fig. 9B). These were often associated with loops of redundant basal lamina (Fig. 9C). These features strongly indicate that these profiles represent regenerating axons in the early stages of myelination.
Dhh−/− nerves contain high numbers of macrophages and neutrophils
Significant, ongoing axonal degeneration and myelin breakdown, as seen in dhh−/− nerves, could be expected to be accompanied by elevated numbers of neutrophils and macrophages. To test this, frozen sections of dhh−/− and dhh+/+ nerves (n = 2 per genotype) were labeled with F4/80 antibody to reveal macrophages and with Gr-1 antibody to reveal neutrophils. This showed that in dhh−/− nerves, the number of macrophages was elevated approximately fivefold and the number of neutrophils was elevated approximately fourfold compared with wild-type controls (Fig. 10A). By electron microscopy, macrophages and neutrophils were easily found in dhh−/− nerves, but these cells were rarely seen in dhh+/+ nerves (data not shown).
Figure 10.

Normal and transected dhh−/− nerves contain increased numbers of macrophages and neutrophils. A, Comparison of macrophage and neutrophil numbers in intact dhh−/− and null nerves. Note the significant increase in the number of macrophages (p < 0.004) and neutrophils (p < 0.005) in the dhh−/− nerves. B, Comparison of macrophage and neutrophil numbers in the distal stump of dhh+/− and null nerves 2 d after transection. Both normal and mutant nerves show increased macrophage numbers. Transected normal nerves show a substantial increase in neutrophil numbers as expected, but neutrophil numbers in cut dhh−/− nerves are little changed. Mean values ± SEM are shown.
These cell counts are likely to reflect a chronic inflammatory cellular response to the ongoing degeneration in dhh−/− nerves.
Degeneration and inflammatory cell recruitment in transected dhh−/− nerves
The elevated number of inflammatory cells, described above, is likely to be a part of the answer to why dhh−/− nerves degenerate rapidly after transection (Bajestan et al., 2006), because for myelin breakdown, these nerves would be less reliant on the macrophage and neutrophil recruitment seen in normal nerves during the first few days after injury.
To examine macrophage and neutrophil recruitment, sciatic nerves of dhh−/− and dhh+/+ mice were transected and processed for cell counts, whereas other nerves were fixed for electron microscopy or extracted for Western blotting to monitor nerve degeneration.
Accelerated degeneration of the distal stump was clearly evident by electron microscopy at 2 and 7 d after transection (n = 2 per genotype per time point) (Fig. 11A). Two days after injury, the percentage of all myelinated fibers that were intact had collapsed axons, or collapsed axons and myelin was 45, 47, and 7%, respectively, in dhh+/+ mice, whereas the corresponding figures in dhh−/− nerve were 4, 75, and 20%. Western blotting using TUJ1 antibody to detect neuron-specific β-tubulin showed that 2 d after transection, the levels of β-tubulin were markedly lower in the dhh−/− nerves than in wild-type nerves, although at this early time point, P0 levels were still unchanged (Fig. 11B). This confirms the rapid degeneration of axons in the mutants and agrees with previous observations that axonal degeneration precedes loss of myelin (Koeppen, 2004).
Figure 11.
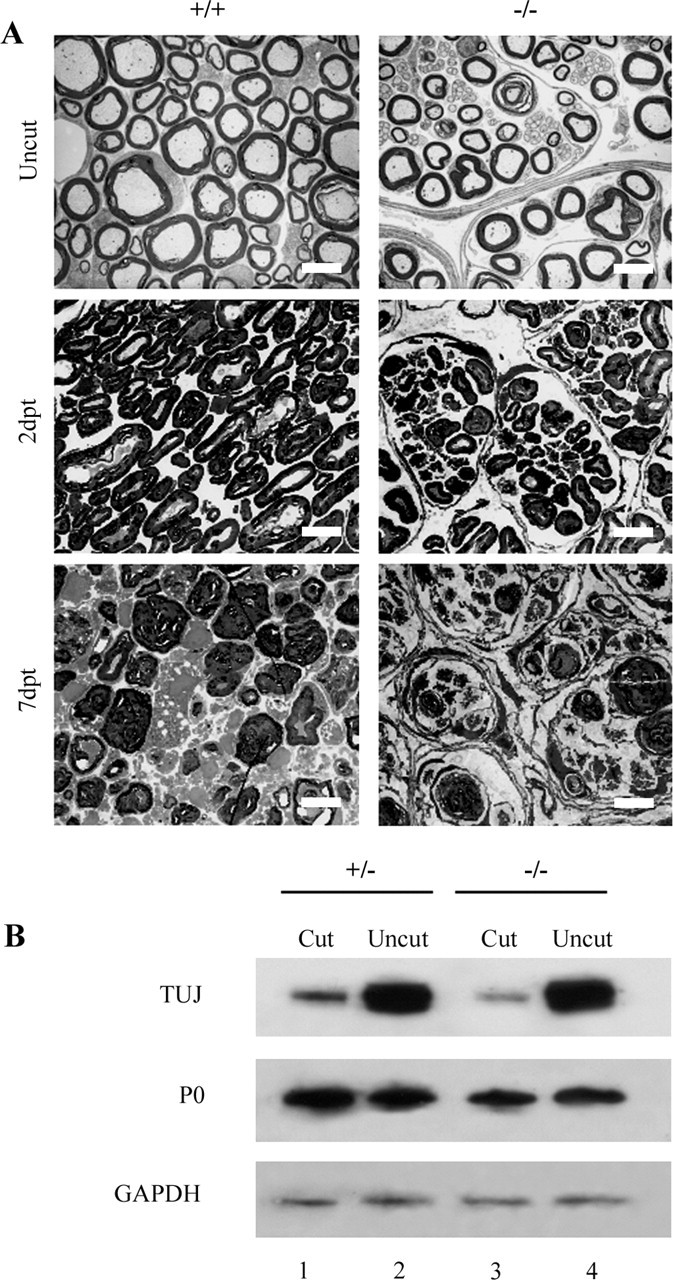
Dhh−/− fibers degenerate rapidly after injury, and the number of mini-fascicles increases. A, Low-power view of nerves 2 and 7 d post-transection (2dpt and 7dpt). At both time points, the dhh−/− nerve shows more advanced degeneration, although the difference is more pronounced at 2 d. Note that an increased number of mini-fascicles is present in the dhh−/− nerve at 2 and 7 d. Scale bars, 5.5 μm. B, TUJ1 (TUJ) immunoblotting reveals that TUJ1 levels, an indicator of axon integrity, show a more marked decrease in dhh−/− nerves than in dhh+/− nerve at 2 d after transection. P0 levels are not markedly altered at this time.
Counts of macrophages using F4/80 antibodies showed that in dhh+/+ nerves, their number had increased sevenfold to eightfold at 2 d after cut, in line with previous observations (Perry et al., 1987; Clemence et al., 1989), whereas in mutant nerves, macrophage numbers increased only approximately twofold to reach comparable numbers to those seen in cut dhh+/+ nerve (Fig. 10B). Neutrophil invasion is one of the earliest responses to nerve injury and is followed slightly later by the arrival of macrophages (Perkins and Tracey, 2000). Counts of neutrophils using Gr-1 antibodies revealed that in dhh+/+ nerves, their number had risen sevenfold to eightfold 2 d after transection. In dhh−/− nerves, however, neutrophil numbers remained unaltered.
Uninjured dhh−/− nerves are organized into mini-fascicles, not found in controls (Parmantier et al., 1999). Surprisingly, in injured dhh−/− nerves, the number of mini-fascicles increased rapidly, even from this high baseline, and was elevated by almost 50% on day 2 after transection (Fig. 11A). Thus, the number of mini-fascicles in entire transverse sections of dhh−/− uninjured nerves was 140, in 2 d transected nerves (Fig. 11A) was 222, and in 7 d transected nerves was 260 (n = 2). Wild-type nerves show slow reorganization into mini-fascicles associated with nerve transection (Bradley et al., 1998).
Discussion
This study shows compound tissue abnormalities in nerves that lack the signaling protein Dhh. This indicates that the cell–cell signaling networks that control tissue maturation and homeostasis in peripheral nerves include, and are crucially dependent on, Dhh. Because Dhh is derived from glial cells, these observations also make the general point that in the peripheral nervous system, glial signals are essential for broad-spectrum tissue integrity. This is likely to apply to glial cells more generally, although, in vivo, signaling molecules with this function in mammalian glia do not appear to have been identified previously.
We found that myelinated Schwann cells in dhh−/− mice have more than double the normal number of SLIs. Although SLI numbers are also increased in MBP-deficient shi mice (Gould et al., 1995), we found that the number of SLIs is substantially increased again in dhh/shi double mutants, which suggests that Dhh and MBP are likely to control SLI numbers through distinct mechanisms. One of the molecular components of SLI, cx29, is strikingly elevated in dhh−/− nerves. Nonmyelinating Schwann cells (in Remak fibers) are also affected, because Remak fibers with large numbers of axons are reduced in number, whereas cells that envelop a single axon only are more numerous. The total number of unmyelinated axons is also reduced by one-third, whereas myelinated axons are lost, albeit to a lesser extent. We show that the blood–nerve barrier is permeable in dhh−/− mice, a finding that parallels the breakdown of the perineurial diffusion barrier in these animals (Parmantier et al., 1999). In dhh−/− nerves, the number of resident macrophages and neutrophils is also elevated. Significantly, uninjured dhh−/− nerves contain a substantial number of degenerating myelinated fibers, which increases with age. They also contain regenerating axons. In addition to this age-dependent neuropathy, degeneration of injured nerves is more rapid in dhh animals than in controls.
Although Dhh is expressed in myelinating Schwann cells, Hh receptors are mainly found on the perineurial cells (ptc1) and nonmyelinating Schwann cells (ptc2). Very low levels of ptc1 are seen on Schwann cells in tissue sections, although they can be detected on neonatal cells in culture by RT-PCR. Even in this case, however, they are not upregulated in response to Hh agonists as expected for ptc1 and as seen for ptc1 in fibrobasts. This argues against an autocrine Dhh circuit in myelinating Schwann cells. A more likely mechanism is indicated by the action of Ihh in cartilage. In this case, Ihh from chondrocytes acts on ptc1 receptors in the perichondrium that surrounds the cartilage and induces the expression of a second signal that acts back on the chondrocytes (Vortkamp et al., 1996). It is therefore possible that Schwann cell-derived Dhh acts on the perineurium that surrounds the nerve, inducing signals that, in turn, act back on Schwann cells and perhaps other elements in the nerve.
It will be a complex task to untangle Dhh-dependent cell–cell signaling networks in peripheral nerves and to define the causal sequences that translate the absence of Dhh in myelinating Schwann cells into the set of tissue changes we have observed. At this point, we can only speculate. The breakdown of the tissue–nerve and blood–nerve barriers is likely to alter the chemical composition of the endoneurial space and therefore the microenvironment of the axon–Schwann cell units (Olsson, 1990; Beamish et al., 1991; Sinnreich et al., 2005). This, however, is unlikely to be the cause of the adverse changes in myelinating Schwann cells, the increase in SLI, because this is already evident at P10, which is before these barriers are effective (Kristensson and Olsson, 1971; Weerasuriya et al., 1990). Similarly, the abnormalities seen in nonmyelinating fibers are likely to relate to the striking and selective expression of ptc2 Hh receptors on these cells (Bajestan et al., 2006). Schwann cell defects, in turn, are likely to contribute to the axon loss seen in dhh−/− nerves. In the case of myelinating axons, this is accompanied by regeneration as we have observed. But it is also plausible to suggest that the large reduction in unmyelinated axons, likely caused by a lack of ptc2 activation in these cells, leads to the observed remodeling of Remak fibers to accommodate fewer axons per Schwann cell.
It is unclear to what extent the elevated number of neutrophils and macrophages in uncut dhh−/− nerves is attributable to the absence of vascular and perineurial barriers to their entry, or to what degree they are attracted by the ongoing fiber degeneration. The presence of these cells could contribute to fiber instability, because in another model of demyelinating peripheral neuropathy (i.e., P0+/− heterozygote mice) there are increased numbers of macrophages within the endoneurium and both resident and blood-borne macrophages play an active role in the demyelination (Carenini et al., 2001; Maurer et al., 2003). In cut dhh−/− nerves, the relatively rapid disappearance of TUJ1 only 2 d after injury indicates abnormally fast axonal degeneration, and there is a concomitant increase in myelin degradation. This accelerated deterioration in injured nerves may be caused partly by the same factors that cause ongoing axon loss in intact dhh−/− nerves. But it is also likely that the increased numbers of resident macrophages and neutrophils play a part, because these cells are known to be crucial to the degenerative response (Scheidt et al., 1986; Coleman and Perry, 2002). The effect is the converse of that observed in the C57BL/Wld mouse in which a chromosome translocation results in delayed axonal degeneration and myelin degradation (Lunn et al., 1989; Coleman and Perry, 2002).
Extensive investigation of molecules known to be associated with SLI revealed that cx29 was upregulated at both the mRNA and protein level in dhh−/− nerves. This is intriguing because in adult nerves, cx29 is localized to the innermost layers of the myelin sheath, showing a striking coincidence with KV1.2 potassium channels in the underlying axonal membrane (Altevogt et al., 2002; Li et al., 2002). Our immunohistochemical experiments failed, however, to show any increase in KV1.2 channels corresponding to the strong cx29 upregulation. A small decrease was observed in MBP (but not mRNA) levels at older ages only, with minimal changes in the MAG protein level. This contrasts with the findings of Smith-Slatas and Barbarese (2000) in shi mice, in which increased MAG and cx32 protein levels were found in shi nerves relative to wild-type nerves, although mRNA levels were unaltered. We did not see an increase in cx32 mRNA levels or in protein levels (data not shown). The SLI increase in dhh−/− mice therefore seems to be mechanistically different from that in the shi mice, a conclusion we reinforced by the elevated SLI numbers in shh/shi double mutants. Increased SLIs are also seen in Schwann cells in Caspr/paranodin null mice (Bhat et al., 2001) and in the CNS in ceramide galactosyl transferase null mice (Dupree et al., 1998).
It has been reported recently that E-cadherin is required to stabilize SLIs and that dominant-negative mutations in E-cadherin that interfere with binding to p120 catenin cause destabilization and disappearance of SLIs (Tricaud et al., 2005). Because we did not find an increase in E-cadherin levels on Western blot, we think it unlikely that alterations in E-cadherin levels are responsible for the increased number of SLIs.
It is of great interest that Dhh levels are reduced in experimental diabetic neuropathy (Calcutt et al., 2003). The present finding that loss of Dhh leads to a broad-spectrum neuropathy indicates that the Dhh reduction in experimental diabetic neuropathy may be a significant cause of the pathological changes seen in that model. This interpretation is also strongly supported by the finding that external Shh, which activates similar signaling pathways to Dhh, restores nerve function to nondiabetic levels and enhances nerve regeneration after injury (Pepinsky et al., 2002; Calcutt et al., 2003; Kusano et al., 2004).
Furthermore, it is noteworthy that a number of the faults caused by Dhh deficiency and described here have been identified previously as age-related changes. These include axonal loss that involves substantially greater reduction in the number of unmyelinated axons than myelinated ones, the presence of regenerating myelinated fibers, increased numbers of resident macrophages, widening of individual SLIs, increase in SLI number, and a shift toward fewer axons per nonmyelinating Schwann cell in Remak fibers (Kanda et al., 1991; Ceballos et al., 1999; Tabata et al., 2000).
Together, these observations lend strong support to the conclusion drawn from the present study that glial-derived Dhh is an essential developmental and trophic signal in peripheral nerves. They also raise the possibility that a failure of this system contributes to nerve pathology and the nerve deterioration that accompanies aging.
Footnotes
This work was supported by a program grant from the Wellcome Trust to K.R.J. and R.M. and by a grant from The Ministry of Education, Culture, Sports, Science, and Technology of Japan to F.U. We thank P. Brophy, S. Davies, L. Morgan, E. Peles, J. Roes, and J. Archelos for the gift of antibodies, A. P. McMahon and I. Griffiths for provision of mutant mice, L. Rubin for the gift of the hedgehog pathway agonist C67, Biogen for Desert hedgehog protein, J. Golding for discussions about statistics, Yuko Shirahama for technical assistance, Jane Pendjiky for photographic assistance, and Debbie Bartram for editing this manuscript.
References
- Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL (2002). Connexin 29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci 22:6458–6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archelos JJ, Roggenbuck K, Schneider-Schaulies J, Linington C, Toyka KV, Hartung HP (1993). Production and characterization of monoclonal antibodies to the extracellular domain of P0. J Neurosci Res 35:46–53. [DOI] [PubMed] [Google Scholar]
- Arroyo EJ, Xu YT, Zhou L, Messing A, Peles E, Chiu SY, Scherer SS (1999). Myelinating Schwann cells determine the internodal localization of Kv1.1, Kv1.2 Kvbeta2 and Caspr. J Neurocytol 28:333–347. [DOI] [PubMed] [Google Scholar]
- Bajestan SN, Umehara F, Shirahama Y, Itoh K, Sharghi-Namini S, Jessen KR, Mirsky R, Osame O (2006). Desert hedgehog-patched 2 expression in peripheral nerves during Wallerian degeneration and regeneration. J Neurobiol 66:243–255. [DOI] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Bone LJ, Scherer SS (1998). Functional gap junctions in the Schwann cell myelin sheath. J Cell Biol 142:1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beamish NG, Stolinski C, Thomas PK, King RH (1991). Freeze-fracture observations on normal and abnormal perineurial tight junctions: alterations in diabetic polyneuropathy. Acta Neuropathol (Berl) 81:269–279. [DOI] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ (2001). Axon-glia interactions and the domain organisation of myelinated axons requires neurexinIV/Caspr/paranodin. Neuron 30:369–383. [DOI] [PubMed] [Google Scholar]
- Bitgood MJ, Shen L, McMahon AP (1996). Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr Biol 6:298–304. [DOI] [PubMed] [Google Scholar]
- Bradley JL, Abernethy DA, King RH, Muddle JR, Thomas PK (1998). Neural architecture in transected rabbit sciatic nerve after prolonged nonreinnervation. J Anat 192:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcutt NA, Allendoerfer KL, Mizisin AP, Middlemas A, Freshwater JD, Burgers M, Ranciato R, Delcroix JD, Taylor FR, Shapiro R, Strauch K, Dudek H, Engber TM, Galdes A, Rubin LL, Tomlinson DR (2003). Therapeutic efficacy of sonic hedgehog protein in experimental diabetic neuropathy. J Clin Invest 111:507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carenini S, Maurer M, Werner A, Blazyca H, Toyka KV, Schmid CD, Raivich G, Martini R (2001). The role of macrophages in demyelinating peripheral nervous system of mice heterozygously deficient in P0 J Cell Biol 152:301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos D, Cuadras J, Verdu E, Navarro X (1999). Morphometric and ultrastructural changes with ageing in mouse peripheral nerve. J Anat 195:563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Cho I, Brophy PJ (2000). A study on the immunocytochemical localization of neurofascin in rat sciatic nerve. J Vet Sci 1:67–71. [PubMed] [Google Scholar]
- Chen S, Rio C, Ji RR, Dikkes P, Coggeshall RE, Woolf CJ, Corfas G (2003). Disruption of ErbB receptor signaling in adult non-myelinating Schwann cells causes progressive sensory loss. Nat Neurosci 6:1186–1193. [DOI] [PubMed] [Google Scholar]
- Clemence AM, Mirsky R, Jessen KR (1989). Non-myelin-forming Schwann cells proliferate rapidly during Wallerian degeneration in the rat sciatic nerve. J Neurocytol 18:185–192. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Perry VH (2002). Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci 25:532–537. [DOI] [PubMed] [Google Scholar]
- Colomar A, Robitaille R (2004). Glial modulation of synaptic transmission at the neuromuscular junction. Glia 47:284–289. [DOI] [PubMed] [Google Scholar]
- Dong Z, Sinanan A, Parkinson D, Parmantier E, Mirsky R, Jessen KR (1999). Schwann cell development in embryonic mouse nerves. J Neurosci Res 56:334–348. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Suzuki K, Popko B (1998). Myelin abnormalities in mice deficient in galactocerebroside and sulfatide. J Neurocytol 27:649–659. [DOI] [PubMed] [Google Scholar]
- Ebnet K, Suzuki A, Ohno S, Vestweber D (2004). Junctional adhesion molecules (JAMs): more molecules with dual functions? J Cell Sci 117:19–29. [DOI] [PubMed] [Google Scholar]
- Frank-Kamenetsky M, Zhang XM, Bottega S, Guicherit O, Wichterle H, Dudek H, Bumcrot D, Wang FY, Jones S, Shulok J, Rubin LL, Porter JA (2002). Small-molecule modulators of Hedghog signaling: identification and characterization of Smoothened agonists and antagonists. J Biol 1:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garratt AN, Britsch S, Birchmeier C (2000). Neuregulin, a factor with many functions in the life of a Schwann cell. BioEssays 22:987–996. [DOI] [PubMed] [Google Scholar]
- Ghabriel MM, Allt G (1981). Incisures of Schmidt-Lanterman. Prog Neurobiol 17:25–28. [DOI] [PubMed] [Google Scholar]
- Gomez CM, Muggleton-Harris AL, Whittingham DG, Hood LE, Readhead C (1990). Rapid preimplantation detection of mutant (shiverer) and normal alleles of the mouse myelin basic protein gene allowing selective implantation and birth of live young. Proc Natl Acad Sci USA 87:4481–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould RM, Byrd AL, Barbarese E (1995). The number of Schmidt-Lanterman incisures is more than doubled in shiverer PNS myelin sheaths. J Neurocytol 24:85–98. [DOI] [PubMed] [Google Scholar]
- Hall SM, Williams PL (1970). Studies on the “incisures” of Schmidt and Lanterman. J Cell Sci 6:767–791. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M, Brook A, McMahon AP (1997). The world according to hedgehog. Trends Genet 13:14–21. [DOI] [PubMed] [Google Scholar]
- Jessen KR (2004). Glial cells. Int J Biochem Cell Biol 36:1862–1867. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R (2005). The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci 6:671–682. [DOI] [PubMed] [Google Scholar]
- Kanda T, Tsukagoshi H, Oda M, Miyamoto K, Tanabe H (1991). Morphological changes in unmyelinated nerve fibres in the sural nerve with age. Brain 114:585–599. [DOI] [PubMed] [Google Scholar]
- Koeppen AH (2004). Wallerian degeneration: history and clinical significance. J Neurol Sci 220:115–117. [DOI] [PubMed] [Google Scholar]
- Kristensson K, Olsson Y (1971). The perineurium as a diffusion barrier to protein tracers. Acta Neuropathol (Berl) 17:127–138. [DOI] [PubMed] [Google Scholar]
- Kusano KF, Allendoerfer KL, Munger W, Pola R, Bosch-Marce M, Kirchmair R, Yoon YS, Curry C, Silver M, Kearney M, Asahara T, Losordo DW (2004). Sonic hedgehog induces arteriogenesis in diabetic vasa nervorum and restores function in diabetic neuropathy. Arterioscler Thromb Vasc Biol 24:2102–2107. [DOI] [PubMed] [Google Scholar]
- Li X, Lynn BD, Olson C, Meier C, Davidson KG, Yasamura T, Rash JE, Nagy JI (2002). Connexin 29 expression, immunocytochemistry and freeze-fracture replica immunogold labelling (FRIL) in sciatic nerve. Eur J Neurosci 16:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S (1989). Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci 1:27–33. [DOI] [PubMed] [Google Scholar]
- Maurer M, Muller M, Kobsar I, Leonhard C, Martini R, Kiefer R (2003). Origin of pathogenic macrophages and endoneurial fibroblast-like cells in an animal model of inherited neuropathy. Mol Cell Neurosci 23:351–359. [DOI] [PubMed] [Google Scholar]
- Meier C, Dermietzel R, Davidson KG, Yasumura T, Rash JE (2004). Connexin32-containing gap junctions in Schwann cells at the internodal zone of partial myelin compaction and in Schmidt-Lanterman incisures. J Neurosci 24:3186–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan L, Jessen KR, Mirsky R (1991). The effects of cAMP on differentiation of cultured Schwann cells: progression from an early phenotype (04+) to a myelin phenotype (P0+, GFAP−, N-CAM−, NGF-receptor−) depends on growth inhibition. J Cell Biol 112:457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA (2004). Glial modulation of synaptic transmission in the retina. Glia 47:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson SM, Gomes D, de Nechaud B, Bruzzone R (2001). Altered gene expression in Schwann cells of connexin32 knockout animals. J Neurosci Res 66:23–36. [DOI] [PubMed] [Google Scholar]
- Olsson Y (1990). Microenvironment of the peripheral nervous system under normal and pathological conditions. Crit Rev Neurobiol 5:265–311. [PubMed] [Google Scholar]
- Parmantier E, Lynn B, Lawson D, Turmaine M, Sharghi Namini S, Chakrabarti L, McMahon AP, Jessen KR, Mirsky R (1999). Schwann cell-derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron 23:713–724. [DOI] [PubMed] [Google Scholar]
- Pepinsky RB, Shapiro RI, Wang S, Chakraborty A, Gill A, Lepage DJ, Wen D, Rayhorn P, Horan GSB, Taylor FR, Garber EA, Galdes A, Engber TM (2002). Long-acting forms of Sonic hedgehog with improved pharmacokinetic and pharmacodynamic properties are efficacious in a nerve injury model. J Pharm Sci 91:371–387. [DOI] [PubMed] [Google Scholar]
- Perkins NM, Tracey DJ (2000). Hyperalgesia due to nerve injury: role of neutrophils. Neuroscience 101:745–757. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Gordon S (1987). The macrophage response to central and peripheral nerve injury. A possible role for macrophages in regeneration. J Exp Med 165:1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Peles E (2003). The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci 4:968–980. [DOI] [PubMed] [Google Scholar]
- Poliak S, Matlis S, Ullmer C, Scherer SS, Peles E (2002). Distinct claudins and associated PDZ domain proteins form autotypic tight junctions in myelinating Schwann cells. J Cell Biol 159:361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz i Altaba A, Nguyen V, Palma V (2003). The emergent design of the neural tube: prepattern, SHH morphogen and GLI code. Curr Opin Genet Dev 13:513–521. [DOI] [PubMed] [Google Scholar]
- Scheidt P, Waehneldt TV, Beuche W, Friede RL (1986). Changes of myelin proteins during Wallerian degeneration in situ and in Millipore diffusion chambers preventing active phagocytosis. Brain Res 379:380–384. [DOI] [PubMed] [Google Scholar]
- Schipke CG, Kettenmann H (2004). Astrocyte responses to neuronal activity. Glia 47:226–232. [DOI] [PubMed] [Google Scholar]
- Sinnreich M, Taylor BV, Dyck PJ (2005). Diabetic neuropathies. Classification, clinical features and pathophysiological basis. Neurologist 11:63–79. [DOI] [PubMed] [Google Scholar]
- Smith-Slatas C, Barbarese E (2000). Myelin basic protein gene dosage effects in the PNS. Mol Cell Neurosci 15:343–354. [DOI] [PubMed] [Google Scholar]
- Sohl G, Eiberger J, Jung YT, Kozak CA, Willecke K (2001). The mouse gap junction gene connexin29 is highly expressed in sciatic nerve and regulated during brain development. Biol Chem 382:973–978. [DOI] [PubMed] [Google Scholar]
- Starkey PM, Turley L, Gordon S (1987). The mouse macrophage-specific glycoprotein defined by monoclonal antibody F4/80: characterization, biosynthesis and demonstration of a rat analogue. Immunology 60:117–122. [PMC free article] [PubMed] [Google Scholar]
- Tabata H, Ikegami H, Kariya K (2000). Spontaneous age-related peripheral neuropathy in B6C3F1 mice. J Toxicol Sci 25:95–104. [DOI] [PubMed] [Google Scholar]
- Takabatake T, Ogawa M, Takahashi TC, Mizuno M, Okamoto M, Takeshima K (1997). Hedgehog and patched gene expression in adult ocular tissues. FEBS Lett 410:485–489. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Andrews SB, Wong A, O'Connell M, Griffin JW (1989). Co-localization of the myelin-associated glycoprotein and the microfilament components, F-actin and spectrin, in Schwann cells of myelinated nerve fibers. J Neurocytol 18:47–60. [DOI] [PubMed] [Google Scholar]
- Tricaud N, Perrin-Tricaud C, Bruses JL, Rutishauser U (2005). Adherens junctions in myelinating Schwann cells stabilize Schmidt-Lanterman incisures via recruitment of p120 catenin to E-cadherin. J Neurosci 25:3259–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW (2005). Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci 28:101–107. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Christopherson KS, Barres BA (2004). Role for glia in synaptogenesis. Glia 15:209–216. [DOI] [PubMed] [Google Scholar]
- Umehara F, Tate G, Itoh K, Yamaguchi N, Douchi T, Mitsuya T, Osame M (2000). A novel mutation of desert hedgehog in a patient with 46,XY partial gonadal dysgenesis accompanied by minifascicular neuropathy. Am J Hum Genet 67:1302–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ (1996). Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 273:613–622. [DOI] [PubMed] [Google Scholar]
- Weerasuriya A, Curran GL, Poduslo JF (1990). Developmental changes in blood-nerve transfer of albumin and endoneurial albumin content in rat sciatic nerve. Brain Res 521:40–46. [DOI] [PubMed] [Google Scholar]
- Wrabetz L, Feltri ML, Quattrini A, Imperiale D, Previtali S, D'Antonio M, Martini R, Yin X, Trapp BD, Zhou L, Chiu SY, Messing A (2000). P0 glycoprotein overexpression causes congenital hypomyelination of peripheral nerves. J Cell Biol 148:1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Haydon PG (2005). Roles for gliotransmission in the nervous system. J Neural Transm 112:121–125. [DOI] [PubMed] [Google Scholar]