

Abstract
Characterization of soluble oligomeric amyloid β (Aβ) species in the brains of Alzheimer's disease (AD) patients and transgenic models has raised the possibility that different conformations of Aβ may contribute to AD pathology via different mechanisms. To characterize the toxic effect of different Aβ conformations, we tested side by side the effect of well characterized Aβ oligomers (AβOs), Aβ-derived diffusible ligands (ADDLs), and fibrillar Aβ (Aβf) preparations in human cortical neurons (HCNs). Both AβOs and ADDLs bind rapidly and with high affinity to synaptic contacts and cellular membranes. AβOs (5 μm) induced rapid and massive neuronal death. Calcium influx accelerated, but was not required for, AβO toxicity. AβOs elicited a stereotyped succession of cellular changes consistent with the activation of a mitochondrial death apoptotic pathway. At low concentrations AβOs caused chronic and subtler mitochondrial alterations but minimal cell death. ADDLs induced similar toxic changes as AβOs but on a fivefold longer time scale. Higher concentrations of Aβf and longer incubation times were required to produce widespread neuritic dystrophy but modest HCN cell death. Thus various Aβ species may play relevant roles in AD, causing neurotoxicity by distinct non-overlapping mechanisms affecting neuronal function and viability over multiple time courses.
Keywords: amyloid β, oligomers, Alzheimer's disease, apoptosis, mitochondria, synapses
Introduction
Typical neuropathological features of Alzheimer's disease (AD) include the presence of senile plaques, neurofibrillary tangles, and neuronal loss in the cortex and hippocampal formation of affected individuals. Senile plaques are composed mainly of amyloid β (Aβ) peptide in its fibrillar form. Fibrillar Aβ (Aβf) triggers a variety of pathological changes including tau hyperphosphorylation, leading to neuronal dysfunction and degeneration (Busciglio et al., 1992, 1995; Pike et al., 1992; Geula et al., 1998). In culture, the chronic exposure of neurons to Aβf induces aberrant activation of adhesion signaling pathways, neuritic dystrophy, and synaptic loss (Grace et al., 2002; Grace et al., 2003). In vivo imaging in transgenic models show neuritic dystrophy and distortion in direct apposition with Aβf deposits (Tsai et al., 2004; Spires et al., 2005), which cause alterations in neocortical synaptic responses (Stern et al., 2004). Fibrillization of Aβ is preceded by multiple conformational changes including trimer, pentamer, or higher molecular weight complex formation, also known as Aβ-derived diffusible ligands (ADDLs) (Lambert et al., 1998), oligomers composed of 15–20 monomers (AβOs) (Kayed et al., 2003), protofibrils (string of oligomers) (Nguyen and Hall, 2004), and dodecameric oligomers Aβ*56 (Lesné et al., 2006). These intermediate Aβ species are designated collectively as “soluble Aβ” (Glabe, 2004). Soluble Aβ species are found in cerebrospinal fluid of AD patients (Kuo et al., 1996; Georganopoulou et al., 2005), can be neurotoxic at low concentrations, and induce inhibition of long-term potentiation and cognitive dysfunction in rodents (Lambert et al., 1998; Hartley et al., 1999; Dahlgren et al., 2002; Walsh et al., 2002b; Lesné et al., 2006). Most relevant, the brain levels of soluble Aβ species appear to correlate better than density of plaque deposition with severity of cognitive impairment (Lue et al., 1999; Naslund et al., 2000). In this regard, oligomers derived from different amyloidogenic proteins have been proposed as primary toxic species in several neurodegenerative diseases, including prion in spongiform encephalopathies, α-synuclein in Parkinson's disease, and polyglutamine in Huntington's disease (Lashuel et al., 2002b; Caughey and Lansbury, 2003; Kayed et al., 2003; Sanchez et al., 2003). Oligomer toxicity appears to be related to a common conformational state, because various oligomers react with the conformation-dependent antibody A11 and oligomeric forms of nondisease-related proteins and peptides are equally toxic (Bucciantini et al., 2002; Kayed et al., 2003, 2004; Demuro et al., 2005). These results raise the possibility that different conformations of Aβ, including soluble and fibrillar Aβ species, may contribute to AD pathology via different mechanisms. To address this issue, we tested side by side the effect of well characterized AβO, ADDL, and Aβf preparations on neuronal function and viability. We used cultured human cortical neurons (HCNs), one of the principal cell types affected in AD. The results show that Aβ species cause neurotoxicity by multiple mechanisms, affecting neuronal function and viability at various concentrations and over different time courses. Thus several Aβ species may play relevant roles in AD by exerting distinct deleterious effects on neuronal cells during the disease process.
Materials and Methods
Neuronal cultures.
Neuronal cultures were established from 16-to 21-week-old human fetal brain tissue as described (Busciglio et al., 2002; Pelsman et al., 2003). The protocols for tissue procurement complied with federal and institutional guidelines. Briefly, the cells were dissociated into single-cell suspensions by incubation with 0.25% trypsin/ PBS at 37°C for 30 min and mechanically dissociated by using a fire-polished glass Pasteur pipette. Cells were plated at a density of 20,000 cells/cm2 in 24- or 96-well plates and 35 and 60 mm dishes. At 2 h after plating the medium was changed to Neurobasal medium plus N2 and B27 supplements (Invitrogen, Grand Island, NY). Partial medium changes (50%) were performed every 5 d. For low calcium experiments, after the initial 5 d, 11 consecutive daily medium changes were performed with calcium-free DMEM plus N2 and B27 supplements to reach a 50 nm calcium concentration in the culture medium. Control cultures received similar medium changes, but normal calcium levels were preserved in the culture. Alternatively, magnesium concentration was increased to 400 μm while calcium was decreased simultaneously to 50 nm on 15 d in vitro (15 DIV). Both protocols worked well, yielding equivalent results.
Aβ treatments.
AβO preparations have been characterized in previous studies (Kayed et al., 2003). A detailed protocol for the preparation of AβOs is included by Demuro and colleagues (2005). AβO preparations have an approximate molecular mass of 90 kDa and contain very little material of lower molecular mass; the ultrastructural analysis indicates that they are composed of spherical vesicles with diameters of 2–5 nm (Demuro et al., 2005). Comprehensive descriptions of ADDL preparations also have been published previously (Lambert et al., 1998; Chromy et al., 2003). ADDL solutions have a heterogeneous composition and show a major component migrating at ∼17 kDa in addition to several minor faster and slower migrating bands. The molecular weights of ADDLs formed in vitro are consistent with Aβ trimer, tetramer, pentamer, and higher molecular weights up to 24-mer (Lambert et al., 1998; Chromy et al., 2003). Fibrillar and oligomeric Aβ1–42 was prepared as described (Demuro et al., 2005). ADDLs were prepared with Aβ1–42 peptide provided by Dr. W. Klein (Northwestern University, Evanston, IL) and followed the standardized protocol (Lambert et al., 1998; Lacor et al., 2004). For some experiments a kit was used to label Aβf with Alexa Fluor 350 (Invitrogen Molecular Probes, Carlsbad, CA), following the manufacturer's instructions. Aβ preparations were added to the cultures at 18 DIV, and viability was assessed at the indicated time points.
Immunofluorescence.
Neurons were fixed in 4% paraformaldehyde and 0.12 m sucrose in PBS for 30 min at 37°C, permeabilized with 0.2% Triton X-100 in PBS, and blocked for 1 h in 5% bovine serum albumin/PBS. Then the cultures were incubated with mouse anti-synaptophysin (1:500; Calbiochem, La Jolla, CA), rabbit anti-tau (1:1000; Dako, Carpinteria, CA), anti-oligomer A11 (1:2500) (Kayed et al., 2003), anti-Aβ 6E10 (1:1000; Signet, Dedham, MA), mouse anti-PSD-95 (1:1000), a gift from Dr. Matthew Rasband (University of Connecticut Health Center, Farmington, CT), mouse anti-cytochrome c (cyt c; 1:600; BD Transduction Laboratories, San Diego, CA), or rabbit anti-apoptosis-inducing factor (AIF; 1:500; Chemicon, Temecula, CA) for 24 h at 4°C, followed by incubation in fluorescent-conjugated secondary antibodies (Alexa, Invitrogen Molecular Probes). Triple labeling was performed by sequential incubation with the appropriate antibodies. Competition with antigenic peptide, use of non-immune IgG instead of primary antibody, or omission of primary antibody resulted in complete elimination of specific labeling. An Axiovert 200 inverted microscope (Zeiss, Jena, Germany) was used for specimen examination and imaging. Fluorescent images were captured with a digital camera (Zeiss) at a final magnification of 630× and processed by using AxioVision (Zeiss). To assess colocalization of synaptic markers, we used an apotome device (Zeiss) for z-sectioning. To quantify the frequency of colocalization of fluorescent signals, we captured at least 20 fields per section of each individual case at a final magnification of 630× and analyzed them with AxioVision software (Zeiss). To avoid bias, we performed colocalization assessments in coded preparations. To confirm triple colocalization of fluorescent markers, we used TetraSpec beads (Invitrogen Molecular Probes) as a positive control. Each 500 nm bead is labeled with four different fluorophores, enabling us to correct for potential plane shifts associated with each individual fluorescent filter (TetraSpeck Fluorescent Microspheres Sampler kit; Invitrogen Molecular Probes).
Time-lapse imaging.
HCNs grown on 35 mm glass-bottom culture dishes treated with Aβ preparations or vehicle alone were placed in a temperature- and CO2-controlled incubator chamber mounted on the microscope stage. Time-lapse images that used differential interference contrast (DIC) optics were taken every 30 min for 24 h at 400× final magnification. During experiments the temperature was maintained at 37°C, and the CO2 concentration was held at 5%. For quantitative image analysis, random microscopic fields were selected (at least 15 microscopic fields per culture dish in at least triplicate dishes). Microscopic fields were selected randomly, with care taken to select a similar number of fields per quadrant of the 12 mm glass area (seven to eight fields/quadrant) of glass-bottom 35 mm culture dishes. The culture dish remained in the culture chamber on the stage of the automated microscope at all times during the experiments. Selection of fields for analysis was performed by using the “mark and find” function immediately before the initiation of the treatments. Then the image acquisition software automatically returned to the selected fields to capture images at each predetermined time point. Once the experiment was finished, the image analysis software marked each scored object (e.g., condensed nucleus) and then added up the number of scored objects per field for each time point, ruling out the possibility of scoring the same object more than once.
Cell death and mitochondrial assays.
Cell death was assessed by quantifying lactate dehydrogenase release to the cultured medium with the use of a commercial kit (CytoTox 96; Promega, Madison, WI) and by counting the number of condensed nuclei per microscopic field at 400× final magnification. At least 10 fields per culture in triplicate cultures were analyzed per individual experiment. Caspase 3 and 7 activities were quantified by using the Caspase-Glo 3/7 kit (Promega). Mitochondrial oxidoreductase activity was determined by analyzing the ability of mitochondria to convert a tetrazolium compound to formazan. The reagent 3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium (MTS) is reduced to formazan by mitochondrial succinate dehydrogenase in complex II (succinate/ubiquinone oxidoreductase complex) and possibly other complexes of the electron transport chain (CellTiter 96 AQueous; Promega) (Busciglio et al., 2002). ATP level was quantified by using a luminescence assay (CellTiter Glo, Promega). Mitochondrial membrane potential (MMP) was determined by using the fluorescent probe JC1 as described previously (Busciglio et al., 2002). Briefly, HCNs cultured in black-wall clear bottom plates were labeled with JC1 (1 μg/ml) at 37°C for 20 min and washed twice with medium. JC1 fluorescence was measured on a 96-well fluorescence reader (Gemini Spectra XPS, Molecular Devices, Union City, CA), and relative changes in MMP were assessed as the change in JC1 590/525 fluorescence ratio. Similar studies also were performed with two other commonly used mitochondrial and cell membrane potential indicators: tetramethylrhodamine ethyl ester (TMRE) at 100 nm for 10 min and bis-(1,3-dibutylbarbituric acid)-trimethine oxonol [DiBAC4(3)] at 100 nm for 20 min.
Statistical analysis.
All experiments were repeated at least three times in cultures derived from at least three different fetal specimens. Each individual experiment was performed in triplicate cultures. Data were analyzed by unpaired Student's t test and expressed as the mean ± SEM. Significance was assessed at p < 0.05. All reported results correspond to single representative experiments.
Results
Synaptic localization of AβOs and ADDLs
Initial tests were performed to validate the experimental model and the effectiveness of Aβ preparations to induce cellular responses. By 15–20 DIV the HCN cultures developed a dense network of neuronal processes, compartmentalized MAP2 (mitogen-activated protein 2) and tau in somatodendritic and axonal compartments, respectively, and established numerous active synapses (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Kerkovich et al., 1999). To study the effects that soluble and fibrillar Aβ species have in differentiated neurons, we initiated treatments at 18 DIV. Previous work has shown a very specific synaptic targeting of ADDLs in rat hippocampal neurons in culture (Lacor et al., 2004). To determine whether AβOs also were localized to synaptic sites, we fixed HCN cultures treated with 5 μm AβOs at 30 min and 1 h after initiation of the incubation, and we performed triple immunofluorescence with anti-synaptophysin, anti-postsynaptic density-95 (anti-PSD-95), and anti-oligomer (A11) or anti-Aβ (6E10) antibodies. Synapses were defined by the colocalization of the presynaptic and postsynaptic markers synaptophysin and PSD-95 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We found a clear time-dependent increase in the number of AβOs bound to cells in cultures fixed at 30 min and 1 h (data not shown). Multiple fluorescence labeling demonstrated a striking colocalization of oligomers with presynaptic and postsynaptic markers (Fig. 1A). Approximately 43 ± 13% of synapses labeled positive for AβOs with the anti-oligomeric specific antibody A11 (Fig. 1B). We also investigated the localization of ADDLs (5 μm) in HCNs at similar time points. We found that ADDLs have a slightly higher preference for synaptic localization (54 ± 11%) than AβOs (Fig. 1B). Similar results were obtained by using anti-Aβ 6E10 antibody (data not shown). Thus both AβOs and ADDLs bind rapidly and with remarkable specificity to synaptic sites in HCN cultures.
Figure 1.
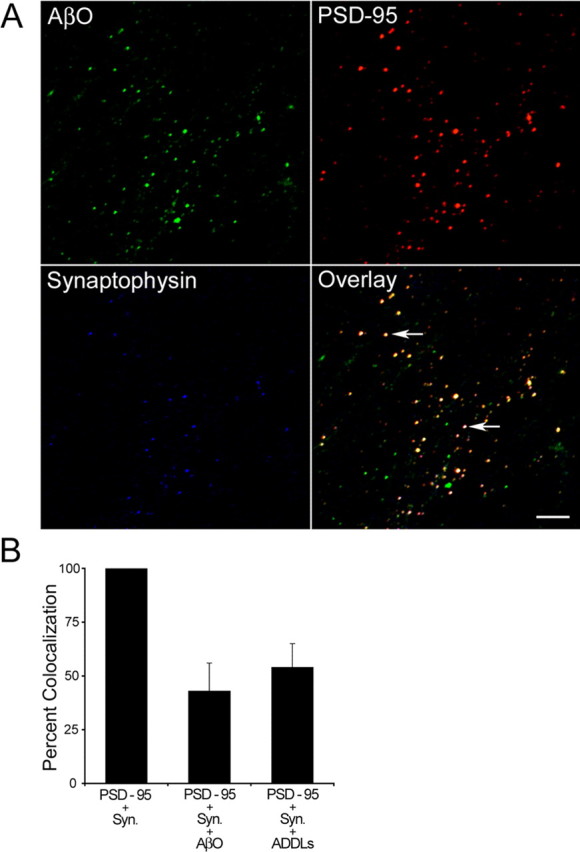
AβOs colocalize with synaptic markers in HCNs. A, Triple immunofluorescence of 18 DIV HCNs with AβOs (A11, green; 1:2500), anti-PSD-95 (red; 1:1000), and anti-synaptophysin (blue; 1:500). Cultures were incubated with 5 μm AβOs for 1 h before fixation. The overlay image shows the colocalization of the three antibodies. Triple colocalization is observed as light yellow fluorescent spots (arrows). Scale bar, 5 μm. B, Quantification of the frequency of colocalization of AβOs and ADDLs (antibody A11) with the synaptic markers PSD-95 and synaptophysin (Syn.). Error bars indicate the mean ± SEM.
Aβ-soluble species cause a fast and massive toxic effect, whereas Aβ fibrils induce progressive dystrophy and modest cell death in HCNs
Earlier experiments characterized the dystrophic effect of Aβf on rat cortical and hippocampal neurons (Pike et al., 1992; Grace et al., 2002; Grace and Busciglio, 2003). A similar dystrophic-inducing effect of Aβf was observed on HCNs. Chronic exposure of HCNs to 20 μm Aβf for 10 d induced progressive dystrophy, including wavy shapes, sharp angles, tortuosity, and formation of loops in neuronal processes (Fig. 2B,C). No such changes were observed in control neurons treated with vehicle (Fig. 2A). Despite these dramatic changes in neuronal morphology and consistent with previous results in rat cortical neurons (Grace et al., 2002), only modest cell death (20 ± 7%) was observed after 10 d of treatment with Aβf. In sharp contrast, 5 μm of AβOs induced rapid and extensive cell death in HCNs (Fig. 2D–G) (see also the supplemental movie, available at www.jneurosci.org as supplemental material). HCNs treated with AβOs showed rapid changes in morphology as compared with those treated with Aβf. In the presence of AβOs neuritic retraction was observed as early as 3 h after initiation of the treatment, followed by progressive apoptotic features, including disintegration of neuronal processes, swelling of cell bodies, and nuclear condensation (Fig. 2E,F). The supplemental movie (available at www.jneurosci.org as supplemental material) contains a complete time-lapse sequence starting at time point 0 (Fig. 1D) and finishing 24 h later (Fig. 1E). By 12 h 30 ± 8% of the neurons were lost, and by 24 h ∼73 ± 5% of the cells were dead (Fig. 2H). The effect of AβOs was dose-dependent. Incubation with 2 μm AβOs required 96 h to induce massive death, and incremental concentrations (5, 10, and 20 μm) showed faster and higher toxic rates (Fig. 2H). The toxic effect was reduced markedly by the preincubation of AβO preparations with anti-oligomer A11 or anti-Aβ 6E10 antibodies before their addition to the cultures or by simultaneous addition of Aβ and A11 antibodies to the cultures (data not shown).
Figure 2.

AβOs cause rapid and massive neuronal death. Shown is double immunofluorescence of 18 DIV HCNs with anti-tau (red; 1:500) and anti-Aβ (blue). A, Neuronal processes in vehicle-treated HCNs exhibit considerable development and a normal, smooth appearance (Ctrl). B, After 10 d of treatment with 20 μm Aβf, neuronal processes display aberrant morphologies, including tortuosity and irregular neuritic caliber. C, Combined DIC/fluorescence at higher magnification illustrates a neuronal process exhibiting loops and sharp turns close to Aβf deposits (blue). D–F, DIC images of living HCN cultures treated with 5 μm AβOs. Immediately after AβO addition, the culture exhibits a normal appearance (D); after 24 h, extensive cell death is indicated by retraction and disintegration of processes and nuclear condensation (E, arrows). At higher magnification the beading and disintegration of neuronal processes are observed clearly (F, arrows). The supplemental movie (available at www.jneurosci.org as supplemental material) contains a complete time-lapse sequence of this experiment. Scale bars, 20 μm. G, Quantification of condensed nuclei/field over the 24 h period of AβO treatment. The graph illustrates one representative field. At least 20 different fields were analyzed with similar results. H, Bar graph illustrating the increase in condensed nuclei in HCN cultures treated with increasing concentrations of AβOs. The number of condensed nuclei was scored at the indicated time points. Similar results were obtained in five independent experiments. Error bars indicate the mean ± SEM.
To characterize the death process further, we analyzed three parameters associated with apoptosis and loss of cell viability after 24 h of AβO treatment (5 μm): mitochondrial metabolic activity, caspase activation, and lactate dehydrogenase (LDH) release. Mitochondrial activity was studied by using an oxidoreductase assay, which measures the amount of formazan salt converted to a soluble-colored product by mitochondria (MTS assay). By 24 h a significant drop of 61 ± 11% in mitochondrial oxidoreductase activity was observed in HCNs treated with AβOs (Fig. 3D). We also analyzed the levels of activated caspases 3 and 7, which are critical mediators of the execution phase of apoptosis (Earnshaw et al., 1999; Slee et al., 1999; Fan et al., 2005). We found a 224 ± 24% increase in caspase activity as compared with control or Aβf-treated cells (Fig. 3E). Similarly, there was a major increase in LDH level (257 ± 18%) in the medium of AβO-treated cultures after 24 h (Fig. 3F), indicating massive cell death.
Figure 3.
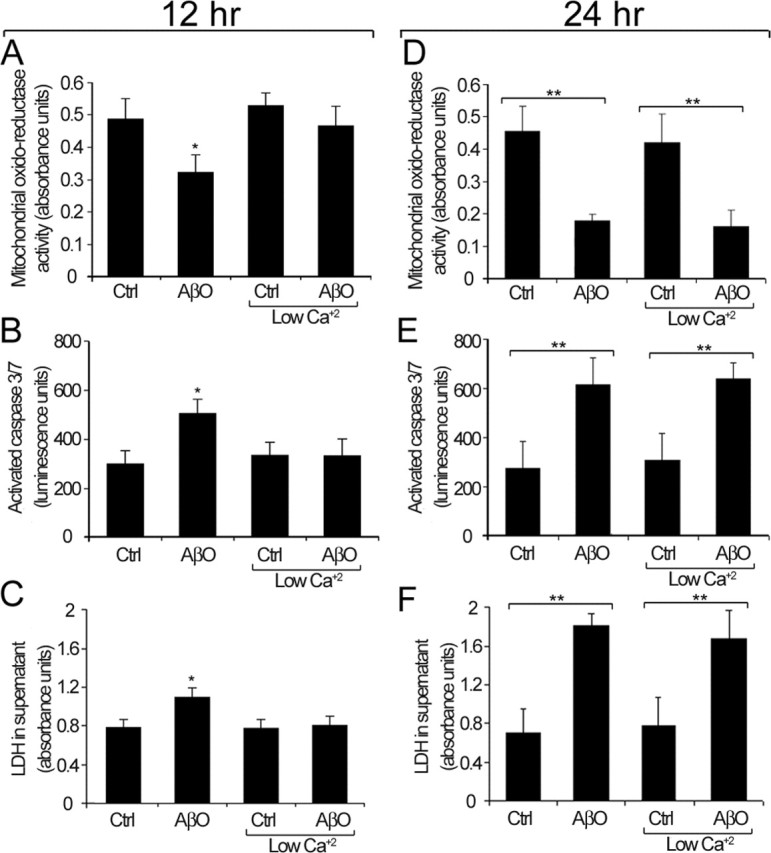
AβO toxic effect under normal and low extracellular calcium. A, D, Mitochondrial oxidoreductase activity at 12 h (A) and 24 h (D). B, E, Caspase 3 and 7 activation at 12 h (B) and 24 h (E). C, F, LDH levels in the culture medium at 12 h (C) and 24 h (F). Under normal culture conditions, these three parameters are significantly different from controls (Ctrl) after 12 h of AβO treatment (A–C). Under low calcium conditions, no significant differences in any of these parameters are observed between Ctrl and AβO-treated cultures at 12 h (A–C). By 24 h, there is a significant enhancement of neurotoxic parameters in AβO-treated cultures under both normal and low calcium conditions (D–F). Similar results were obtained in four independent experiments. Values are expressed as the mean ± SEM by unpaired Student's t test; ∗p < 0.05; ∗∗p < 0.01.
At a similar concentration the ADDLs induced neurotoxicity on a much slower time course. Only after 5–7 d of continuous ADDL exposure did changes in mitochondrial oxidoreductase activity reach a similar level to that of AβO-treated cells for 24 h. After 7 d of ADDL treatment the caspase activation was elevated to a lesser but significant extent when compared with that of AβO-treated cultures for 24 h. Consistent with these results, the LDH level was elevated only slightly by 7–8 d, indicating only moderate cell death in ADDL-treated cells (data not shown). Thus AβOs induced rapid apoptotic changes on HCNs, whereas similar concentrations of ADDLs resulted in comparable changes but on a markedly slower time course. No significant modifications in mitochondrial activity, caspase activation, or LDH release were present in HCNs treated with Aβf for 24 h (data not shown). Thus soluble (AβOs and ADDLs), but not fibrillar, Aβ species are associated with HCN apoptotic cell death in culture.
Previous studies indicate that Aβ-soluble species are toxic at very low concentrations in different experimental paradigms (Hartley et al., 1999; Walsh et al., 22217; Kayed et al., 2003; Demuro et al., 2005). To assess the effect of Aβ species at lower doses, we analyzed the effect of AβOs at 10 nm, 100 nm, and 1 μm. At 1 μm and 100 nm the AβOs decreased MMP significantly within 6 h (35.7 ± 7 and 54.2 ± 13%, respectively) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). At 10 nm AβOs the MMP did not change as compared with control cells. At 1 μm and 100 nm AβOs the ATP levels dropped to ∼50%, in both cases by day 3 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). LDH levels in the supernatant increased by only 10% in HCNs treated with 1 μm and 100 nm AβOs at the end of day 3 (data not shown). These results indicate that, although low concentrations of AβOs do not cause significant neuronal loss, they induce early and chronic alterations in mitochondrial function that may impact overall neuronal homeostasis.
Calcium influx accelerates, but is not essential for, AβO-induced neurotoxicity
Recent experiments demonstrated that AβOs induce a fast calcium influx in neuroblastoma cells (Demuro et al., 2005), and numerous studies have implicated increased intracellular calcium as a central event in apoptotic processes in various cell types (Duchen, 2000; Polster and Fiskum, 2004). To establish the role of calcium in AβO neurotoxicity, we grew HCNs under normal or low calcium concentration conditions (see Materials and Methods) and treated them with 5 μm AβOs. Data collected at 12 h indicated that, in the presence of normal calcium levels, there was a significant decrease of 41 ± 25% in mitochondrial oxidoreductase activity (Fig. 3A), a 40 ± 11% increase in caspase activity (Fig. 3B), and 26 ± 6% higher LDH levels in the culture medium (Fig. 3C). These changes were not observed in neurons treated for 12 h with AβOs under low calcium conditions (Fig. 3A–C). However, by 24 h the levels of oxidoreductase activity, caspase activation, and LDH release were similar to those in HCN cultures exposed to AβOs in the presence of calcium (Fig. 3A–C). These results suggest that, although calcium influx can accelerate AβO toxicity markedly, it is not an absolute requirement for the toxic effect of soluble Aβ species. There were no differences in the dystrophic effect induced by Aβf in normal and low calcium conditions (data not shown).
AβOs activate a mitochondrial death pathway
In many paradigms of apoptosis a mitochondrial death pathway is activated by signaling molecules recruited to mitochondria, leading to a rapid drop of MMP and ATP levels, opening of the mitochondrial transition pore (MTP), translocation of cyt c and AIF from the mitochondrial matrix to the cytoplasm, and activation of caspases in charge of the “execution” phase of the death process (Johnstone et al., 2002; Danial and Korsmeyer, 2004; Helguera et al., 2005). To delineate further the chain of events triggered by Aβ species, we analyzed mitochondrial changes at early time points after Aβ treatment, before the occurrence of massive cell death. Mitochondrial membrane potential, measured as the emission ratio of red over green fluorescence (590/525 nm) of the fluorescent probe JC-1, dropped drastically within the first 2 h of exposure to AβOs (89 ± 15%) (Fig. 4A). Comparable results were observed by using the fluorescent probes TMRE and DiBAC4(3), two other commonly used mitochondrial and cell membrane potential indicators, respectively (data not shown). Similarly, ATP levels dropped 70 ± 10% by 2 h and decreased steadily thereafter (Fig. 4B). Caspase activation and LDH release increased 35 ± 6 and 23 ± 8%, respectively, by 8 h (Fig. 4C,D). Treatment with 5 μm ADDLs decreased MMP and ATP levels progressively but to a lesser extent as compared with AβOs at similar concentrations (data not shown). Changes in mitochondrial membrane potential were observed only after 5 d of treatment with 20 μm Aβf, because it decreased 34 ± 4% as compared with controls, whereas ATP levels decreased 33 ± 9%, indicating that dystrophic neurons also exhibit mitochondrial impairment.
Figure 4.

AβOs induce rapid mitochondrial alterations in HCNs. A–D, MMP, ATP levels, caspase activity, and LDH release were quantified in control (Ctrl) and AβO-treated cultures (5 μm) at the indicated time points. Significant changes in MMP and ATP levels can be observed in AβO-treated cultures within 2 h of treatment (A, B). Significant changes in caspase activity and LDH release are detected by 8 h (C, D). Values are expressed as the mean ± SEM by unpaired Student's t test; ∗p < 0.05; ∗∗p < 0.01. Similar results were obtained in four independent experiments.
Mitochondrial release of cyt c and AIF is a direct consequence of increased permeability of the mitochondrial membrane mediated by the mitochondrial permeability transition pore (Danial and Korsmeyer, 2004). Immunofluorescence analysis revealed that both cyt c and AIF showed diffuse cytoplasmic localization in cell bodies and processes of HCNs treated with AβOs for 8 h, which were devoid of obvious degenerative morphology (Fig. 5). In contrast, discrete labeling and restricted mitochondrial localization of cyt c and AIF were observed in cell bodies and processes of HCNs treated with vehicle (Fig. 5, Ctrl). Similarly, HCNs treated with ADDLs for 96 h showed diffuse cytoplasmic localization of cyt c and AIF (Fig. 5). Thus early mitochondrial changes consistent with activation of a mitochondrial death pathway are present early on during soluble Aβ toxicity.
Figure 5.

Soluble AβOs induce translocation of cyt c and AIF from mitochondria to the cytoplasmic compartment. Shown is immunofluorescence of HCNs treated with vehicle (Control), AβOs, or ADDLs for the indicated times. Control cultures incubated with vehicle for 96 h exhibit mitochondrial localization of cyt c and AIF in cell bodies and processes (green fluorescence). In contrast, AβO- and ADDL-treated cultures exhibit diffuse cyt c and AIF localization in both cell bodies and neuronal processes (green fluorescence). The bottom panels are higher magnifications of the boxed areas. Scale bars, 10 μm. Neurons were double-labeled with the neuronal-specific marker anti-tubulin Class III (red fluorescence). Nuclei were counterstained with Hoechst (blue fluorescence).
Discussion
To understand the pathophysiology of AD, it is critical to elucidate the role of Aβ and its different conformations in the disease process. We used primary HCNs, the target cells in AD, as an experimental paradigm to show that Aβf, AβO, and ADDL preparations affect neuronal function and viability by distinct toxic mechanisms, suggesting that Aβ neurotoxicity in AD is a complex and multifaceted phenomenon.
The presence of Aβf in the AD brain is well established as the major component of senile plaques. Previous work demonstrates that Aβf is toxic to primary neurons (Busciglio et al., 1992, 1995; Pike et al., 1992; Geula et al., 1998). Chronic exposure to Aβf induces abnormal activation of adhesion signaling pathways, neuritic dystrophy, and synaptic loss (Grace et al., 2002; Grace and Busciglio, 2003). In vivo experiments in transgenic models show the presence of widespread dystrophy in neuronal processes in contact with Aβf deposits (Tsai et al., 2004; Spires et al., 2005), which contributes to major alterations in neocortical synaptic responses (Stern et al., 2004), yet the apparent lack of correlation between the amount of Aβf deposition and the severity of dementia strengthened the proposition that soluble, and not fibrillar, forms of Aβ may be responsible for the alterations in neuronal function and viability in AD (Hardy and Selkoe, 2002; Klein, 2002; Tanzi, 2005). This view is supported by experiments showing that soluble Aβ species are present in AD brains and transgenic models (Gong et al., 2003; Kayed et al., 2003; Takahashi et al., 2004; Oddo et al., 2006, Lesné et al., 2006) and that their levels correlate better than plaque density with cognitive impairment (Lue et al., 1999; Naslund et al., 2000). In fact, previous studies indicate that Aβ-soluble species are toxic at low concentrations, reduce synaptic density, induce cognitive dysfunction, and inhibit long-term potentiation (Lambert et al., 1998; Hartley et al., 1999; Dahlgren et al., 2002; Walsh et al., 2002b; Wang et al., 2002).
To perform a side by side comparison of the effect of Aβ species in HCNs, we used homogeneous preparations of AβOs, ADDLs, and Aβf that have been well characterized by EM, HPLC, and immunoreactivity to conformation-dependent antibodies (Lacor et al., 2004; Demuro et al., 2005). We found a very specific synaptic localization of AβOs and ADDLs in HCNs. The targeting of AβOs and ADDLs to synaptic contacts was fast and accounted for ∼45–50% of AβO total deposits present on HCN cultures after 1 h of incubation. Similar results were obtained with ADDLs, which also show remarkable colocalization with synaptic sites in rat hippocampal neurons (Lacor et al., 2004).
The mechanism by which AβOs and ADDLs are targeted to synaptic sites may be a crucial step in the pathological cascade. One possibility is that the high concentration of metal ions at synapses, particularly Cu+2 and Zn+2 (Frederickson and Bush, 2001; Mocchegiani et al., 2005; Mathie et al., 2006), “attracts” Aβ oligomers, given the high binding affinity of Aβ for Cu+2 and Zn+2 (Bush et al., 1994; Atwood et al., 2000). Then metal–Aβ complexes in the synaptic area may initiate a toxic cascade (Cherny et al., 2001; Frederickson and Bush, 2001), consistent with the synaptic alterations linked to soluble forms of Aβ (Hartley et al., 1999; Walsh et al., 22217). Recent results suggest that ADDLs bind to and reduce the number of NMDA receptors, providing an alternative pathway by which soluble forms of Aβ may cause synaptic failure (Lacor et al., 2005). Notably, most AβOs and ADDLs not associated with synapses were found in close contact with cellular membranes, suggesting that soluble Aβ may initiate toxicity not only at synapses but also at multiple cellular locations.
The fast and highly toxic effect of AβOs was correlated with a sequence of cellular alterations, which is consistent with the activation of a mitochondrial death pathway, including early changes in MMP and ATP production, gradual decrease in mitochondrial oxidoreductase activity, cytoplasmic translocation of cyt c and AIF, caspase activation, and nuclear condensation (Walker et al., 1988; Cai et al., 1998; Joza et al., 2001; van Loo et al., 2002). These toxic changes were concentration-dependent because lower AβO concentrations induced chronic mitochondrial alterations, but not cell death (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In this regard, chronic and subtle impairment of mitochondrial function by low concentrations of Aβ-soluble species may underlie defective synaptic activity and cognitive impairment in AD patients. Furthermore, transport and localization of mitochondria into neuronal processes, an energy-dependent event, is necessary for the development and maintenance of both spines and synaptic plasticity (Li et al., 2004).
Despite inducing similar changes, the time course of ADDL toxicity was longer. ADDLs required approximately five times longer than AβOs to cause similar effects. Structural differences between AβOs and ADDLs influencing pore-forming or receptor-binding activities are likely to account for this disparity. An alternative possibility is that the toxic effect of ADDLs could be receptor-mediated, leading to mitochondrial changes by downstream signals over a longer time course.
We found that lowering extracellular calcium delayed significantly the AβO toxic process. However, after 24 h AβO toxicity in low calcium approached that of AβOs in normal calcium. One interpretation is that the rapid toxic response induced by AβOs is accelerated by calcium influx (Mattson, 1992; Mattson and Rydel, 1992; Li et al., 1996; Berridge et al., 1998; LaFerla, 2002; Pierrot et al., 2004), but in the absence of calcium the pore-forming properties of Aβ-soluble species (Kagan et al., 2002; Lashuel et al., 22217; Arispe, 2004; Kayed et al., 2004; Quist et al., 2005; Thundimadathil et al., 2005) still trigger the apoptotic process. In this scenario, regardless of calcium influx the progressive formation of Aβ pores in the plasma membrane and internal endomembrane system leads to a general destabilization of ion homeostasis and activation of the apoptotic process.
Supplemental Figure 3A (available at www.jneurosci.org as supplemental material) summarizes the time line of degenerative changes induced by AβOs, ADDLs, and Aβf. AβOs and ADDLs at micromolar concentrations result in similar morphological and biochemical changes. AβOs exert a faster effect, killing most neurons by 24 h. Five to seven days of incubation are required to generate a similar extent of cell death with ADDLs. Chronic incubation for 10 d and higher concentrations of Aβf are required to produce generalized dystrophic changes but only modest cell death, whereas nanomolar concentrations of AβOs and ADDLs result in chronic mitochondrial dysfunction and minor changes in cell viability.
Supplemental Figure 3B (available at www.jneurosci.org as supplemental material) illustrates a model of the toxic pathway(s) triggered by Aβ-soluble species. AβOs and ADDLs are recruited to synapses, possibly by metal ions enriched at synaptic sites and/or synaptic receptors. Another possibility is that Aβ is released at synapses, where it may undergo oligomerization (Gylys et al., 2004). Regardless, the pore-forming activity of Aβ-soluble species could channel a rapid influx of calcium inside cells at synaptic sites and other cellular locations. Mitochondria act as a major buffering system for calcium, but extreme calcium loads lead to the opening of the MTP (Korge et al., 2002; Scorrano et al., 2003; Miyamoto et al., 2005), causing the collapse of the MMP and the release of cyt c and AIF from the mitochondrial matrix to the cytosol where caspases are activated for the execution phase of the apoptotic process. The end stage is illustrated by the complete loss of cell membrane integrity and the release of LDH.
In conclusion, the emerging view of multiple Aβ species capable of deleterious effects at multiple levels coexisting in AD will require a refined therapeutic strategy to address Aβ-mediated neurotoxicity. Additional studies in AD brains will be necessary to assess the synaptic localization of Aβ-soluble species, to establish how the load of Aβ-soluble species changes during disease progression, to determine whether there is a precursor–product relationship among oligomeric and/or fibrillar species, and to understand how each of these factors contributes to neurodegeneration.
Footnotes
This work was supported by grants from the Alzheimer's Association (J.B.), the National Institutes of Health (NS21230 to C.G.), and The Larry L. Hillblom Foundation (C.G.). We express our gratitude to Dr. Pascale N. Lacor (Northwestern University, Evanston, IL) for synthetic peptides and Drs. Mary P. Lambert (Northwestern University), William L. Klein (Northwestern University), Ian Parker (University of California, Irvine), and Angelo Demuro (University of California, Irvine) for helpful advice.
References
- Arispe N (2004). Architecture of the Alzheimer's AβP ion channel pore. J Membr Biol 197:33–48. [DOI] [PubMed] [Google Scholar]
- Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP, Tanzi RE, Bush AI (2000). Characterization of copper interactions with Alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid β1–42 J Neurochem 75:1219–1233. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Lipp P (1998). Calcium—a life and death signal. Nature 395:645–648. [DOI] [PubMed] [Google Scholar]
- Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M (2002). Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416:507–511. [DOI] [PubMed] [Google Scholar]
- Busciglio J, Lorenzo A, Yankner BA (1992). Methodological variables in the assessment of beta amyloid neurotoxicity. Neurobiol Aging 13:609–612. [DOI] [PubMed] [Google Scholar]
- Busciglio J, Lorenzo A, Yeh J, Yankner BA (1995). β-Amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 14:879–888. [DOI] [PubMed] [Google Scholar]
- Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, Yankner BA (2002). Altered metabolism of the amyloid β precursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron 33:677–688. [DOI] [PubMed] [Google Scholar]
- Bush AI, Pettingell WH Jr, Paradis MD, Tanzi RE (1994). Modulation of Aβ adhesiveness and secretase site cleavage by zinc. J Biol Chem 269:12152–12158. [PubMed] [Google Scholar]
- Cai J, Yang J, Jones DP (1998). Mitochondrial control of apoptosis: the role of cytochrome c. Biochim Biophys Acta 1366:139–149. [DOI] [PubMed] [Google Scholar]
- Caughey B, Lansbury PT (2003). Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26:267–298. [DOI] [PubMed] [Google Scholar]
- Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI (2001). Treatment with a copper–zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30:665–676. [DOI] [PubMed] [Google Scholar]
- Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL (2003). Self-assembly of Aβ1–42 into globular neurotoxins. Biochemistry 42:12749–12760. [DOI] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB Jr, Baker LK, Krafft GA, LaDu MJ (2002). Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J Biol Chem 277:32046–32053. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ (2004). Cell death: critical control points. Cell 116:205–219. [DOI] [PubMed] [Google Scholar]
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG (2005). Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280:17294–17300. [DOI] [PubMed] [Google Scholar]
- Duchen MR (2000). Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium 28:339–348. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Martins LM, Kaufmann SH (1999). Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem 68:383–424. [DOI] [PubMed] [Google Scholar]
- Fan TJ, Han LH, Cong RS, Liang J (2005). Caspase family proteases and apoptosis. Acta Biochim Biophys Sin (Shanghai) 37:719–727. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Bush AI (2001). Synaptically released zinc: physiological functions and pathological effects. Biometals 14:353–366. [DOI] [PubMed] [Google Scholar]
- Georganopoulou DG, Chang L, Nam JM, Thaxton CS, Mufson EJ, Klein WL, Mirkin CA (2005). Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer's disease. Proc Natl Acad Sci USA 102:2273–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA (1998). Aging renders the brain vulnerable to amyloid β-protein neurotoxicity. Nat Med 4:827–831. [DOI] [PubMed] [Google Scholar]
- Glabe CG (2004). Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem Sci 29:542–547. [DOI] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL (2003). Alzheimer's disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA 100:10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace EA, Busciglio J (2003). Aberrant activation of focal adhesion proteins mediates fibrillar amyloid β-induced neuronal dystrophy. J Neurosci 23:493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace EA, Rabiner CA, Busciglio J (2002). Characterization of neuronal dystrophy induced by fibrillar amyloid β: implications for Alzheimer's disease. Neuroscience 114:265–273. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM (2004). Synaptic changes in Alzheimer's disease: increased amyloid-β and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol 165:1809–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ (1999). Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19:8876–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helguera P, Pelsman A, Pigino G, Wolvetang E, Head E, Busciglio J (2005). ets-2 promotes the activation of a mitochondrial death pathway in Down's syndrome neurons. J Neurosci 25:2295–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW (2002). Apoptosis: a link between cancer genetics and chemotherapy. Cell 108:153–164. [DOI] [PubMed] [Google Scholar]
- Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zuniga-Pflucker JC, Kroemer G, Penninger JM (2001). Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410:549–554. [DOI] [PubMed] [Google Scholar]
- Kagan BL, Hirakura Y, Azimov R, Azimova R, Lin MC (2002). The channel hypothesis of Alzheimer's disease: current status. Peptides 23:1311–1315. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486–489. [DOI] [PubMed] [Google Scholar]
- Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG (2004). Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem 279:46363–46366. [DOI] [PubMed] [Google Scholar]
- Kerkovich DM, Sapp D, Weidenheim K, Brosnan CF, Pfeiffer SE, Yeh HH, Busciglio J (1999). Fetal human cortical neurons grown in culture: morphological differentiation, biochemical correlates, and development of electrical activity. Int J Dev Neurosci 17:347–356. [DOI] [PubMed] [Google Scholar]
- Klein WL (2002). Aβ toxicity in Alzheimer's disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int 41:345–352. [DOI] [PubMed] [Google Scholar]
- Korge P, Honda HM, Weiss JN (2002). Protection of cardiac mitochondria by diazoxide and protein kinase C: implications for ischemic preconditioning. Proc Natl Acad Sci USA 99:3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE (1996). Water-soluble AβN-40, N-42 oligomers in normal and Alzheimer disease brains. J Biol Chem 271:4077–4081. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL (2004). Synaptic targeting by Alzheimer's-related amyloid β oligomers. J Neurosci 24:10191–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Sanz-Clemente A, Viola KL, Klein WL (2005). Changes in NMDA receptor subunit 1 and 2B expression in ADDL-treated hippocampal neurons. Soc Neurosci Abstr 31:786.17. [Google Scholar]
- LaFerla FM (2002). Calcium dyshomeostasis and intracellular signaling in Alzheimer's disease. Nat Rev Neurosci 3:862–872. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL (1998). Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95:6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT Jr (22217). α-Synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol 322:1089–1102. [DOI] [PubMed] [Google Scholar]
- Lashuel HA, Hartley DM, Balakhaneh D, Aggarwal A, Teichberg S, Callaway DJ (2002b). New class of inhibitors of amyloid-β fibril formation. Implications for the mechanism of pathogenesis in Alzheimer's disease. J Biol Chem 277:42881–42890. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH (2006). A specific amyloid-β protein assembly in the brain impairs memory. Nature 440:352–357. [DOI] [PubMed] [Google Scholar]
- Li YP, Bushnell AF, Lee CM, Perlmutter LS, Wong SK (1996). β-Amyloid induces apoptosis in human-derived neurotypic SH-SY5Y cells. Brain Res 738:196–204. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119:873–887. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J (1999). Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155:853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A, Sutton GL, Clarke CE, Veale EL (2006). Zinc and copper: pharmacological probes and endogenous modulators of neuronal excitability. Pharmacol Ther in press. [DOI] [PubMed]
- Mattson MP (1992). Calcium as sculptor and destroyer of neural circuitry. Exp Gerontol 27:29–49. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Rydel RE (1992). β-Amyloid precursor protein and Alzheimer's disease: the peptide plot thickens. Neurobiol Aging 13:617–621. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Howes AL, Adams JW, Dorn II GW, Brown JH (2005). Ca2+ dysregulation induces mitochondrial depolarization and apoptosis: role of Na+/Ca2+ exchanger and AKT. J Biol Chem 280:38505–38512. [DOI] [PubMed] [Google Scholar]
- Mocchegiani E, Bertoni-Freddari C, Marcellini F, Malavolta M (2005). Brain, aging, and neurodegeneration: role of zinc ion availability. Prog Neurobiol 75:367–390. [DOI] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD (2000). Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA 283:1571–1577. [DOI] [PubMed] [Google Scholar]
- Nguyen HD, Hall CK (2004). Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc Natl Acad Sci USA 101:16180–16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM (2006). Temporal profile of amyloid-beta (Aβ) oligomerization in an in vivo model of Alzheimer disease. A link between Aβ and tau pathology. J Biol Chem 281:1599–1604. [DOI] [PubMed] [Google Scholar]
- Pelsman A, Hoyo-Vadillo C, Gudasheva TA, Seredenin SB, Ostrovskaya RU, Busciglio J (2003). GVS-111 prevents oxidative damage and apoptosis in normal and Down's syndrome human cortical neurons. Int J Dev Neurosci 21:117–124. [DOI] [PubMed] [Google Scholar]
- Pierrot N, Ghisdal P, Caumont AS, Octave JN (2004). Intraneuronal amyloid-β1–42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem 88:1140–1150. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Cummings BJ, Cotman CW (1992). β-Amyloid induces neuritic dystrophy in vitro: similarities with Alzheimer pathology. NeuroReport 3:769–772. [DOI] [PubMed] [Google Scholar]
- Polster BM, Fiskum G (2004). Mitochondrial mechanisms of neural cell apoptosis. J Neurochem 90:1281–1289. [DOI] [PubMed] [Google Scholar]
- Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R (2005). Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci USA 102:10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez I, Mahlke C, Yuan J (2003). Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 421:373–379. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003). BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300:135–139. [DOI] [PubMed] [Google Scholar]
- Slee EA, Adrain C, Martin SJ (1999). Serial killers: ordering caspase activation events in apoptosis. Cell Death Differ 6:1067–1074. [DOI] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT (2005). Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci 25:7278–7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern EA, Bacskai BJ, Hickey GA, Attenello FJ, Lombardo JA, Hyman BT (2004). Cortical synaptic integration in vivo is disrupted by amyloid-β plaques. J Neurosci 24:4535–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK (2004). Oligomerization of Alzheimer's β-amyloid within processes and synapses of cultured neurons and brain. J Neurosci 24:3592–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE (2005). The synaptic Aβ hypothesis of Alzheimer disease. Nat Neurosci 8:977–979. [DOI] [PubMed] [Google Scholar]
- Thundimadathil J, Roeske RW, Jiang HY, Guo L (2005). Aggregation and porin-like channel activity of a beta sheet peptide. Biochemistry 44:10259–10270. [DOI] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB (2004). Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci 7:1181–1183. [DOI] [PubMed] [Google Scholar]
- van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P (2002). The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ 9:1031–1042. [DOI] [PubMed] [Google Scholar]
- Walker NI, Harmon BV, Gobe GC, Kerr JF (1988). Patterns of cell death. Methods Achiev Exp Pathol 13:18–54. [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (22217). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo Nature 416:535–539. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ (2002b). Amyloid-β oligomers: their production, toxicity, and therapeutic inhibition. Biochem Soc Trans 30:552–557. [DOI] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL (2002). Soluble oligomers of β amyloid1–42 inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res 924:133–140. [DOI] [PubMed] [Google Scholar]