

Abstract
Mutant huntingtin can affect vesicular and receptor trafficking via its abnormal protein interactions, suggesting that impairment of intracellular trafficking may contribute to Huntington's disease. There is growing evidence that huntingtin-associated protein-1 (HAP1) also interacts with microtubule-dependent transporters and is involved in intracellular trafficking. However, it remains unclear how the trafficking of HAP1 is regulated and contributes to neuronal function. Here we report that phosphorylation of HAP1 decreases its association with microtubule-dependent transport proteins dynactin p150 and kinesin light chain and reduces its localization in neurite tips. Suppressing HAP1 expression by RNA interference reduces neurite outgrowth and the level of tropomyosin-related kinase A receptor tyrosine kinase (TrkA), a nerve growth factor receptor whose internalization and trafficking are required for neurite outgrowth. HAP1 maintains the normal level of membrane TrkA by preventing the degradation of internalized TrkA. Mutant huntingtin also reduces the association of HAP1 with dynactin p150 and kinesin light chain and thereby decreases the intracellular level of TrkA. These findings suggest that HAP1 trafficking is critical for the stability of TrkA and neurite function, both of which can be attenuated by mutant huntingtin.
Keywords: huntingtin, neuropathology, TrkA, polyglutamine, neurite, trafficking
Introduction
Huntington's disease (HD) is caused by an expansion of a polyglutamine tract (>37 glutamines) in huntingtin (htt). The gain of toxic function of mutant htt is thought to critically contribute to its neuropathology. Evidence to support this theory includes the findings that mutant htt abnormally interacts with other proteins and affects intracellular trafficking (Gunawardena and Goldstein, 2005; McGuire et al., 2005; Morfini et al., 2005). Thus, proteins that bind mutant htt and are also enriched in neurons are likely to contribute to the selective neuropathology of HD. Growing evidence has shown that Huntingtin associated protein-1 (HAP1) is a strong candidate for being involved in HD pathology, because HAP1 is a brain-enriched protein and participates in intracellular trafficking in neurons (Li et al., 1995; Li and Li, 2005).
The role of HAP1 in intracellular trafficking is supported by the following findings. First, HAP1 interacts with dynactin p150Glued (Engelender et al., 1997; Li et al., 1998) and kinesin light chain (KLC) (McGuire et al., 2006). Kinesin light chain and p150Glued associate with microtubule motors kinesin and dynein, which mediate intracellular anterograde and retrograde transport, respectively. Consistently, HAP1 and htt are cotransported bidirectionally in axons (Block-Galarza et al., 1997). Second, HAP1 is localized to microtubules and various vesicles in the brain (Gutekunst et al., 1998; Martin et al., 1999). Third, HAP1 interacts with hepatocyte growth factor-regulated tyrosine kinase substrate, a protein critical for endocytic trafficking of membrane proteins (Li et al., 2002). Fourth, HAP1 is able to stabilize the internalized membrane receptors (R) for epidermal growth factor (EGF) and GABAA (Li et al., 2002; Kittler et al., 2004). Furthermore, recent studies show that mutant htt affects intracellular trafficking via its abnormal interaction with HAP1. For example, htt-mediated transport of BDNF vesicles involves HAP1 and dynactin p150, and mutant htt impairs this transport via its increased binding to HAP1 (Gauthier et al., 2004). HAP1 interacts with the type 1 inositol (1,4,5)-triphosphate receptor (IP3R1), forming an IP3R1–HAP1A–htt ternary complex in which mutant htt enhances the sensitivity of IP3R1 to IP3 (Tang et al., 2003). In addition, the critical role of HAP1 for neuronal function has been demonstrated in HAP1 knock-out mice, which show a feeding defect and postnatal death (Chan et al., 2002; Dragatsis et al., 2004) possibly attributable to the selective degeneration of hypothalamic neurons (Li et al., 2003).
The interactions of HAP1 with various partners suggest that HAP1 may be a trafficking protein or an adaptor to link cargos to intracellular transporters. However, little is known about cellular regulation of the interactions of HAP1 with trafficking proteins. Here we report that phosphorylation of HAP1 regulates its association with kinesin light chain and dynactin p150. Reducing HAP1 expression or decreasing its association with microtubule-dependent transporters in the presence of mutant htt diminishes the intracellular level of tropomyosin-related kinase A receptor tyrosine kinase (TrkA), a nerve growth factor receptor whose intracellular trafficking is required for neurite outgrowth. These findings suggest that the trafficking function of HAP1 is regulated by phosphorylation and important for stabilizing the internalized TrkA.
Materials and Methods
Antibodies and reagents.
Rabbit antibodies to glutathione S-transferase (GST), HAP1A or HAP1B isoform, and dynactin p150 (Li et al., 1998), mouse monoclonal antibody (mEM48) to htt, and guinea pig antibody (EM78) to HAP1 (Li et al., 2000) were generated in our previous studies. Rabbit antibody to kinesin light chain-2 was generated using a GST fusion protein encoding KLC-2 [amino acids (aa) 124–411] (McGuire et al., 2006). Synthesized peptides CGHPPASGTSYRSS(pT)L were used as antigen to generate rabbit antibody against phosphorylated T598 in HAP1A by AnaSpec (San Jose, CA). The antibody (p1A) was then purified with the phosphorylated peptides using affinity column. Other antibodies used are those against γ-tubulin (Sigma, St. Louis, MO), TrkA (Upstate, Charlottesville, VA; and a gift from Dr. Louis Reichardt, University of California San Francisco, San Francisco, CA), the p75 neurotrophin receptor p75NTR (a gift from Dr. Moses V. Chao, New York University Medical Center, New York, NY), HAP1 (mouse anti-HAP; Transduction Laboratories, Lexington, KY), early endosomal antigen EEA1 (Transduction Laboratories), phospho-(Ser/Thr) protein kinase A (PKA) substrate (Cell Signaling Technology, Beverly, MA), kinesin heavy chain (Chemicon, Temecula, CA), the hemagglutinin (HA) epitope (Cell Signaling Technology), neuronal-specific nuclear protein (NeuN) (Chemicon), dynactin p150 (mouse antibody 610473; BD Biosciences PharMingen, San Diego, CA), and Akt and phospho-Akt (Ser473) (Cell Signaling Technology). Alexa 488-conjugated secondary antibodies were purchased from Invitrogen (Carlsbad, CA). Rhodamine-conjugated secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). Protein A-agarose beads were obtained from Sigma. Okadaic acid, staurosporine, and NGF were purchased from Calbiochem (La Jolla, CA). Sulfo-NHS-SS-biotin and UltraLink Neutravidin beads were from Pierce (Rockford, IL). PKA (P-2645) was obtained from Sigma, and protein kinase B (PKB) (9274) was from Cell Signaling Technology. HAP1 knock-out mice were established and described previously (Li et al., 2003). Other mice include HD transgenic mice (N171-82Q) that express the N-terminal 171 amino acids with an 82-glutamine repeat and HD CAG150 knock-in mice that express full-length mutant huntingtin.
Immunoprecipitation.
For immunoprecipitation of HAP1A or phosphorylated HAP1A from adult rat brain, hypothalami were dissected and homogenized in 0.2% Triton X-100 in PBS buffer containing protease inhibitor cocktail (Sigma) and phosphatase inhibitors (5 mm NaF and 2 mm Na3VO4) at a concentration of 0.1 g/ml. Samples were gently rocked for 1 h at 4°C, followed by centrifugation at 16,000 × g for 15 min at 4°C. One milligram of the clarified supernatant was preincubated with protein A-agarose beads (30 μl of 1:1 slurry) for 1 h at 4°C. After pelleting the beads, the supernatant was incubated with 10 μl of anti-HAP1A or 20 μl of anti-phosphorylated HAP1A (anti-p1A) antibody overnight at 4°C. Protein A-agarose beads (30 μl) were added to the mixture for an additional 1 h incubation. Immunoprecipitations with preimmune serum served as a control. The beads containing immunocomplexes were pelleted and washed three times with homogenization buffer. Precipitated proteins were resuspended in 80 μl of SDS-PAGE sample buffer and boiled for 5 min, resolved by 4–12% SDS-PAGE, and detected with antibodies and the ECL plus kit (Amersham Biosciences, Arlington Heights, IL). For immunoprecipitation of HAP1A from PC12 cells, PC12 cells in a 10 cm plate were lysed in 1 ml of 0.2% Triton X-100 in PBS containing protease inhibitor cocktail and phosphatase inhibitors by gentle rocking for 1 h at 4°C. After centrifugation at 16,000 × g for 15 min at 4°C, 500 μg of supernatant was subjected to immunoprecipitation with anti-HAP1A as described above.
Phosphorylation studies.
PC12 cells that had been incubated with serum-free medium overnight were treated with NGF (100 ng/ml) for 15 or 60 min to investigate the effect of NGF on HAP1A phosphorylation. The serum-starved PC12 cells were also treated without or with 2 μm okadaic acid (OA) for 1 h and subjected to immunoprecipitation with anti-HAP1A. Transfected HEK293 cells were also incubated with serum-free medium and treated with OA (0–2 μm for 60 min) to increase phosphorylation or staurosporine (0–1 μm for 60 min) to inhibit phosphorylation. The cells were lysed in buffer containing 1% NP-40, 10 mm HEPES, 150 mm NaCl, pH 7.4, protease inhibitor cocktail, and phosphatase inhibitors (5 mm NaF and 2 mm Na3VO4) and subjected to Western blotting. For in vitro phosphorylation with purified PKA or PKB, 0.5 μg of purified GST fusion proteins containing C-terminal HAP1A (amino acids 293–599) or full-length HAP1A were incubated without or with 20 U of PKA or PKB at 30°C for 30 min in kinase buffer containing 25 mm Tris, pH 7.5, 5 mm β-glycerolphosphate, 2 mm DTT, 0.1 mm Na3VO4, 10 mm MgCl2, and 400 μm ATP. Reactions were stopped with SDS sample buffer and analyzed by Western blotting. Plasmid encoding the HA-tagged catalytic subunit of PKA (Moore et al., 2002) provided by Haian Fu (Emory University, Atlanta, GA) was also used for cotransfection with HAP1A in HEK293 cells.
Adenoviral infection experiments.
To generate adenoviral vectors expressing green fluorescent protein (GFP)-htt-23Q and 130Q, human htt cDNAs encoding the first 208 amino acids plus an additional 23 or 130 glutamine repeat were fused in-frame to the GFP C-terminal cDNA, resulting in GFP-htt fusion proteins containing a 23 (htt-23Q) or 130 (htt-130Q) glutamine repeat that was expressed under the control of the cytomegalovirus (CMV) promoter. Recombinant adenoviruses that express GFP-htt were prepared in our previous study (Shin et al., 2005). To generate adenoviral HAP1 small interfering RNA (siRNA), we tested a number of siRNA sequences and found that one siRNA (1695–1713, GAAGTATGTCCTCCAGCAA) was able to effectively inhibit the expression of HAP1 (McGuire et al., 2006). This siRNA was inserted into an adenoviral vector with the mouse U6 promoter, which also independently expresses GFP driven by the CMV promoter. A vector that expresses GFP alone or a scramble siRNA (GCGCGCTTTGTAGGATTCG) served as controls. Adenoviruses for siRNA experiments were generated and purified by Welgen (Worcester, MA). The viral titer was determined by measuring the number of infected HEK293 cells expressing GFP or showing cytopathic effect. All viral stocks were adjusted to 1 × 108 pfu/μl before their use. PC12 cells were incubated with adenovirus at a multiplicity of infection of 25–50 for 2 h and cultured for an additional 24–96 h in the absence of adenovirus. Cultured sympathetic neurons were infected with 0.5 μl of adenoviral HAP1 siRNA or 1 μl of scramble siRNA and GFP (at ratio of 1:1) for 24 h and cultured for an additional 72 h before immunostaining.
Primary neuronal culture.
Hypothalamic neurons were isolated from the hypothalamus of embryonic day 14–16 rats. Dissected tissue was treated with 0.0625 mg/ml trypsin and 0.0625 mg/ml DNase in 1× HBSS buffer without calcium and magnesium for 10 min at 37°C, followed by triturating with a 1 ml pipette tip 20 times. Cells were then washed once with the tissue culture medium and spun down at 1500 × g for 3 min. Cells were plated on the top of a layer of astrocytes and grown initially in a 50% glial conditioned medium [DMEM containing 0.25% glucose, 2 mm glutamate, 10% FCS, 500 nm insulin, 1× vitamin mixture (M6895; Sigma) and 1% antibiotic–antimytotic (Invitrogen)]. The cells were then cultured in neurobasal/B27 medium following the method used in our previous study (Li et al., 2000). For culturing sympathetic neurons, superior cervical ganglia were dissected from 3- to 4-week-old rats using the method as described previously (Orike et al., 2001) with some modifications. The ganglia were desheathed, minced into 8–10 pieces, and treated enzymatically in 1× HBSS buffer containing 1.0 mg/ml collagenase, 0.0625 mg/ml trypsin, and 0.0625 mg/ml DNase at 37°C for 30 min. Neurons were dissociated by gentle trituration through a 1 ml pipette tip and were then plated onto glass coverslips that had been precoated with 0.1 mg/ml poly-d-lysine and 1 μg/ml laminin. Neurons were cultured in neurobasal-A/B27 medium (Invitrogen) supplemented with 2 mm glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10 ng/ml NGF. To reduce glial proliferation, 5 μm cytosine arabinoside was added 12–16 h after plating.
Immunofluorescence, light microscopy, and confocal microscopy.
Cells were fixed in 4% paraformaldehyde in PBS for 10 min at room temperature. After three rinses with PBS, cells were permeabilized and blocked with 0.2% Triton X-100/3% BSA/PBS for 30 min at room temperature. Cells were then incubated with primary antibodies diluted in 3% BSA/PBS overnight at 4°C. Cells were washed three times with PBS and then incubated with Alexa 488- or rhodamine-conjugated secondary antibodies and nuclear dye Hoechst diluted in 3% BSA/PBS for 30 min at 4°C. Immunostaining of brain sections was performed as described previously (Shin et al., 2005). Light micrographs were taken using a Zeiss (Oberkochen, Germany) microscope (Axiovert 200 MOT) and a 20× or 63× lens (LD-Achroplan 20×/0.4 or 63×/0.75) with a digital camera (Orca-100; Hamamatsu, Bridgewater, NJ) and the Openlab software (Improvision, Lexington, MA). Confocal imaging was performed using the 63× oil immersion objective lens and a Zeiss LSM 510 confocal microscope system.
Imaging of transfected cells and quantification of HAP1A signal in growth cones.
HAP1A tagged with red fluorescent protein (RFP) in the PRK vector (McGuire et al., 2006) was transfected into PC12 cells that had been plated on cover glass in two-well chamber slides (155379; Lab-Tek, Naperville, IL). We also generated GFP-HAP1A containing the T598A mutation (GFP-T598A) for transfection. Cells were then immediately infected by adenoviral GFP-htt-23Q or 130Q for 16 h. After transfection of RFP-HAP1A for 5 h, NGF was added to 100 ng/ml for 36 h. Cells were plated on cover glass in two-well chamber slides (155379; Lab-Tek) and incubated in CO2-independent medium (18045-088; Invitrogen) containing 100 ng/ml NGF. Cells were maintained at 37°C on a heated stage and imaged using a 63× objective lens. Fifty scans were taken over the course of ∼20 min. The captured images were used to create a movie file using Windows Movie Maker software (Microsoft, Seattle, WA). For quantifying HAP1 signals in neurites, cultured cells were fixed, and their images were captured using a 63× objective lens. The exact same settings were used to image all cells examined. Three regions of interest (ROIs) were created for each cell: the soma, the growth cone farthest from the soma, and a region of the plate with no cells to correct for background. Cells were selected for measurement based on the following criteria: cells must express both RFP-HAP1A and GFP-htt, have a cell body GFP signal >600 (arbitrary units for average pixel intensity within the ROI) over the background, and have a discernible growth cone. Two scans were taken for each cell, and their ROI signals were averaged. RFP-HAP1A signal values were then corrected for background. The data for the relative level of RFP-HAP1A at the growth cone were represented as the ratio of growth cone RFP signal to cell body RFP signal. A total of 30 cells from two independent experiments were measured. A similar method was used to quantify the ratio of growth cone signal to cell body signal of transfected GFP-HAP1A or GFP-T598A in ∼120 PC12 cells in two independent experiments.
Biotinylation assays.
Biotinylation assays were performed as described previously (Kuruvilla et al., 2004) with some modifications. For biotinylation of membrane proteins, PC12 cells were rinsed twice with PBS containing 1 mm CaCl2 and 0.5 mm MgCl2 (PBS-Ca-Mg) and then incubated with sulfo-NHS-SS-biotin (1.0 mg/ml in PBS-Ca-Mg) for 30 min at 4°C. Cells were then quenched with PBS-Ca-Mg containing 100 mm glycine for 15 min at 4°C and then washed twice with PBS before precipitation. To measure internalized membrane TrkA, serum-starved PC12 cells were subjected to biotinylation as described above. To prevent lysosomal degradation of internalized TrkA, cells were treated with 100 μg/ml of the lysosomal protease inhibitor leupeptin for 30 min before biotinylation. Receptor internalization was then allowed to occur by incubation of cells with 50 ng/ml NGF at 37°C for 30 min. Remaining biotin at the cell surface was removed with cleaving buffer (50 mm glutathione, 75 mm NaCl, 10 mm EDTA, 1% BSA, and 0.075N NaOH) before precipitation. To study NGF-mediated degradation of membrane TrkA, PC12 cells that had been incubated with serum-free medium overnight were biotinylated as described above. Cells were then treated with NGF for different lengths of time (0, 75, and 150 min) and rinsed with PBS. The remaining TrkA after NGF treatment was determined by precipitation and Western blotting in which PC12 cells were lysed with immunoprecipitation assay buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 0.1% SDS, 0.5% deoxycholate, and 1% Triton X-100). Biotinylated proteins were precipitated by 40 μl (1:1 slurry) of UltraLink Neutravidin beads, resuspended in 80 μl of SDS-PAGE sample buffer, and analyzed by Western blotting with anti-TrkA. Supernatant after immunoprecipitation was also probed with different antibodies for controls.
Statistical analysis.
Statistical significance (p < 0.05) was assessed by using Student's t test. Calculations were performed by SigmaPlot 4.11 (SPSS, Chicago, IL) and Prism (version 4) software (GraphPad Software, San Diego, CA).
Results
Phosphorylation of HAP1A
In rodents, HAP1 consists of two HAP1 isoforms, HAP1A and HAP1B, which only differ in their short C-terminal sequences (579–599 aa of HAP1A vs 579–629 aa of HAP1B) (Fig. 1A). Both isoforms form homodimers and heterodimers and interact with htt, but only HAP1A is able to promote neurite outgrowth in PC12 cells (Li et al., 2000). Examination of differentiated PC12 cells, which have been treated with NGF and stained with antibodies specific to HAP1A or HAP1B, revealed that HAP1A is enriched in neurite tips, whereas HAP1B remains in the bodies of PC12 cells (Fig. 1B). The result suggests that C-terminal sequences of HAP1A are important for its localization in neurite tips and that this neuritic localization promotes neurite outgrowth.
Figure 1.

Comparison of HAP1A with HAP1B. A, Different C-terminal sequences of rat HAP1A and HAP1B. The PKA phosphorylation site is indicated in gray. The htt binding region is indicated in the black box. B, Distribution of HAP1A and HAP1B in PC12 cells treated with NGF (50 ng/ml) for 3 d. HAP1A (top) is localized in neurite tips, whereas HAP1B (bottom) is predominantly in the cell body. PC12 cells were labeled by antibodies against the specific C-terminal sequences of HAP1A and HAP1B as underlined in A.
We found that the C-terminal sequence 595RSST598 of HAP1A fits well with the phosphorylation motif (RXXpS/T) for PKA (Pearson and Kemp, 1991). We therefore examined whether transfected HAP1A is phosphorylated using Western blotting with the rabbit phospho-(Ser/Thr) PKA substrate antibody, which recognizes the RXXpS/T motif. Indeed, transfected HAP1A, but not HAP1B (data not shown), was recognized by this antibody (Fig. 2A). To confirm the idea that threonine (T598) of HAP1A is a phosphorylated residue, we replaced T598 with alanine in HAP1A and expressed this mutated HAP1A (T598A) in transfected HEK293 cells. Consistent with our prediction, transfected T598A did not react with the PKA substrate antibody (Fig. 2A). Furthermore, the HAP1A immunoreaction signal was increased by the phosphatase inhibitor okadaic acid, which increases protein phosphorylation, and decreased by staurosporine, which inhibits protein phosphorylation (Fig. 2B). All of these results suggest that the residue T598 of HAP1A is phosphorylated by PKA.
Figure 2.

Phosphorylation of HAP1A. A, B, Western blotting of transfected HAP1A in HEK293 cells with anti-phospho-(Ser/Thr) PKA substrate antibody. In A, substitution of threonine 598 by alanine (T598A) eliminates the immunoreactivity of HAP1A with this antibody (right). The blots were also probed with anti-HAP1A antibody (left). Vector is PRK vector transfection alone. In B, cells were treated with okadaic acid or staurosporine and then subjected to Western blotting with the anti-phospho-(Ser/Thr) PKA substrate antibody (top). Increasing phosphorylation by okadaic acid (1–2 μm) or inhibiting phosphorylation by staurosporine (0.5–1 μm) alters the immunoreactivity of HAP1A to this antibody (top). p1A indicates the phosphorylated HAP1A. The same blots were also probed with guinea pig anti-HAP1 antibody (bottom). C–E, Western blotting with the anti-p1A antibody specific to the phosphorylated residue of T598 in HAP1A. In C, transfected HAP1A and mutated HAP1A (T598A) in HEK293 cells reacted with anti-HAP1A (left), but T598A was not recognized by anti-p1A (right). In D, OA treatment (1 μm for 1 h) increased the immunoreaction of transfected HAP1A with anti-p1A (top). The blot was also probed with anti-HAP1A (bottom) to show the equal expression of transfected proteins. In E, purified GST fusion proteins containing C-terminal HAP1A (amino acids 293–599) or full-length HAP1A were incubated with purified PKA or PKB in vitro. Both GST-HAP1A fusion proteins were selectively phosphorylated by PKA, which is reflected by the increased anti-p1A signal (top). GST-HAP1A fusion proteins were also revealed by anti-GST antibody. F, Western blotting of cell lysates of HEK293 cells transfected with HAP1A/HAP1B (1:3) with (+) or without (−) HA-tagged PKA. The blots were probed with antibodies to p1A, HAP1A, and the HA epitope.
Identification of the phosphorylation of HAP1A led us to generate a phosphor-specific antibody against a C-terminal HAP1A sequences (585–599 aa) containing phosphorylated T598 antigen. This antibody only recognized wild-type HAP1A, but not mutant HAP1A (T598A), in transfected cells (Fig. 2C). Furthermore, increased protein phosphorylation by okadaic acid enhanced the immunoreaction signals of anti-p1A (Fig. 2D). To provide direct evidence that PKA phosphorylates HAP1A, we generated GST fusion proteins containing either C-terminal (amino acids 293–599) or full-length (amino acids 1–599) HAP1A and incubated these fusion proteins in vitro with purified PKA. As a control, we used PKB for incubation. PKA, but not PKB, increased the immunoreaction of anti-p1A with both C-terminal and full-length HAP1A (Fig. 2E). We also examined whether overexpression of the PKA catalytic subunit can increase the phosphorylation of HAP1A in transfected HEK293 cells. Although overexpressed PKA also increased HAP1A phosphorylation in transfected cells (Fig. 2F), this increase appeared to be less than that of GST-HAP1 in vitro. It is possible that endogenous PKA is sufficient to phosphorylate HAP1A. Another possibility is that other regulatory subunits are required to significantly augment or stabilize the phosphorylation of HAP1A by overexpressed PKA in HEK293 cells. Nevertheless, all of these results verify that anti-p1A specifically recognizes phosphorylated HAP1A.
Anti-p1A allowed us to examine the expression of phosphorylated HAP1A in the mouse brain (Fig. 3A). The expression levels of HAP1 (HAP1A and HAP1B) vary in different brain regions, with the high levels in the hypothalamus, amygdala, and brainstem. The brain regional distribution of p1A is almost identical to that of HAP1, suggesting that the phosphorylation of HAP1A correlates with the total amount of synthesized HAP1.
Figure 3.

Subcellular distribution of HAP1A and its phosphorylated form (p1A). A, Western blotting of tissues from different mouse brain regions. The blots were probed with anti-p1A for phosphorylated HAP1A or anti-HAP1 (EM78) for both HAP1A and HAP1B. Antibody to tubulin was used to reflect the loading amount of proteins in each lane. B, Immunostaining of the dorsomedial nucleus of the mouse hypothalamus showing that antibody to HAP1A labels the cell body, processes, and cytoplasmic puncta, whereas antibody to phosphorylated HAP1A (p1A) labels the cell body and cytoplasmic puncta. C, Immunofluorescent double labeling of mouse hypothalamus with antibodies to HAP1A or p1A and NeuN, a neuronal protein marker. Arrows indicate neuronal processes labeled by anti-HAP1A. Glial nuclei are indicated by arrowheads. D, E, Immunofluorescent double labeling of PC12 cells (D) and confocal imaging of cultured rat hypothalamic neurons (E). Cells were double labeled with mouse anti-HAP1 (red) and rabbit anti-p1A (green). Note that p1A is only seen in the cell body. Arrows indicate HAP1-positive neurite tips that lack p1A staining. F, NGF (100 ng/ml) treatment of PC12 cells for 15 or 60 min reduces p1A signal. Control is PC12 cells treated with vehicle. The blots contain samples from three treatments and were probed with antibodies to p1A, HAP1A, Akt, and phosphorylated Akt (pAkt). Scale bars, 5 μm.
We then examined the cellular distribution of p1A in the mouse hypothalamus, a HAP1-enriched brain region. Because we do not have an antibody specific to nonphosphorylated HAP1A, we used the anti-HAP1A antibody that recognized both phosphorylated and nonphosphorylated HAP1A. This antibody stained the cell body and neurites as well as cytoplasmic puncta of HAP1. However, the antibody to phosphorylated HAP1A (p1A) only labeled the cell body and HAP1 puncta (Fig. 3B). Immunofluorescent double labeling of mouse hypothalamus confirmed that HAP1A is expressed in the bodies and processes of neurons, whereas p1A staining is mostly restricted to the cell body (Fig. 3C). All of these data suggest that phosphorylated HAP1A is enriched in the cell body and that nonphosphorylated HAP1A preferentially localizes to neurite tips. To definitively determine HAP1 distribution in the body and neurites in the same cells, we examined cultured neuronal cells. PC12 cells were labeled with rabbit anti-p1A and a mouse antibody to HAP1, which reacts with the internal region (100–289 aa) of both HAP1A and HAP1B. Similarly, anti-p1A only stained the cell body without obvious signals in neurite tips, whereas the mouse anti-HAP1 labeled both the cell body and neurite tips (Fig. 3D). To examine whether this different labeling also occurs in primary neurons, we performed confocal imaging of cultured rat hypothalamic neurons (Fig. 3E). As expected, the result showed that phosphorylated HAP1A (p1A) was mainly distributed in the cell body, whereas neuritic HAP1 was only labeled by the mouse antibody to HAP1, suggesting that nonphosphorylated HAP1A localizes to neurites.
We have shown that NGF stimulation can enrich HAP1A in neurites (Fig. 1). Treatment of PC12 cells with NGF (100 ng/ml) for 15 and 60 min increased the phosphorylation of protein kinase B (Akt) or activated PKB. This treatment, however, reduced the p1A signal, which reflects the decreased phosphorylation of HAP1A (Fig. 3F). This effect is similar to NGF-induced dephosphorylation of p38 mitogen-activated protein kinase (Torcia et al., 2001; Rosini et al., 2004) and other trafficking proteins such as actin depolymerizing factor (Meberg et al., 1998) and myosin light chain (Fujita et al., 2001). Together, the results demonstrate that HAP1A is phosphorylated in neurons and suggest that the dephosphorylation of HAP1A by NGF relocates it to neurites and their tips.
Phosphorylation of HAP1A reduces its association with dynactin p150 and kinesin
HAP1 has been found to interact with the microtubule motor-associated protein dynactin p150Glued (p150) (Engelender et al., 1997; Li et al., 1998). Our recent study demonstrates that HAP1 also interacts with KLC-2 (McGuire et al., 2006), which is highly homologous to KLC-1 and forms dimers with KLC-1 in vivo (Rahman et al., 1998). These findings prompted us to examine whether phosphorylation of HAP1A regulates its interactions with microtubule transporters. Although the in vivo interactions of HAP1 with trafficking proteins are dynamic, we were able to show the association of endogenous HAP1 with dynactin p150 and kinesin light chains via immunoprecipitation (Fig. 4A). Antibody specific to HAP1A (both nonphosphorylated and phosphorylated HAP1A) could precipitate dynactin p150 and kinesin light chain (Fig. 4B). Antibody specific to phosphorylated HAP1A (p1A), however, was unable to coprecipitate a significant amount of dynactin p150 and kinesin light chain, although it precipitated the similar amount of p1A compared with anti-HAP1A (Fig. 4B). More importantly, when PC12 cells were treated with okadaic acids to increase protein phosphorylation, the amount of p1A was increased in the precipitates, but the amount of coprecipitated dynactin p150 and kinesin light chain was reduced (Fig. 4C). To rule out the influence of the different amounts of the lysate input used in immunoprecipitation, we used densitometry to quantify the proteins that were precipitated with the equivalent inputs in different treatments. We measured the ratios of associated proteins to the precipitated HAP1A (bound/precipitated HAP1A) and compared the relative amount of proteins precipitated by anti-p1A with those precipitated by anti-HAP1A. The densitometry data also support the idea that anti-p1A precipitates less dynactin p150 and KLC or that phosphorylation of HAP1A diminishes the association of HAP1A with trafficking proteins (Fig. 4D). We also generated GST fusion proteins containing full-length KLC-2 (GST-KLC) or the HAP1 binding region in p150 dynactin (GST-p150). These fusion proteins were used to pull down transfected HAP1A and T598A in HEK293 cells. More T598A than HAP1A was pulled down by both of these GST fusion proteins in vitro (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), also supporting the idea that dephosphorylation of T598 in HAP1A can increase its association with dynactin p150 and KLC.
Figure 4.

Phosphorylation decreases the association of HAP1 with dynactin p150 and kinesin as well as neuritic distribution of HAP1A in PC12 cells. A, Immunoprecipitation of endogenous HAP1 (bottom) in PC12 cells coprecipitated endogenous KLC (middle) and dynactin p150 (top). Preimmune (Pre) serum served as a control. B, Comparison of immunoprecipitation (IP) of HAP1 and its associated proteins from rat hypothalamus. The immunoprecipitates were probed with antibodies to HAP1A, p1A, KLC, and dynactin p150. Note that anti-HAP1A precipitated more dynactin p150 and KLC than did anti-p1A. C, PC12 cells were treated with (+) or without (−) OA (2 μm for 1 h) and then immunoprecipitated by anti-HAP1A. Increased phosphorylation by okadaic acid reduced the association of HAP1A with KLC and dynactin p150, although it increased the precipitated amount of phosphorylated HAP1A (p1A). D, Densitometry analysis of immunoprecipitated p1A, dynactin p150, and KLC of rat hypothalamus by anti-p1A or anti-HAP1A in B (top). Densitometry analysis of immunoprecipitated p1A, dynactin p150, and KLC by anti-HAP1A of PC12 cells in the presence (+) or absence (−) of OA in C (bottom). The relative immunoprecipitated proteins (mean ± SE; n = 3) were quantified as the ratio of the bound to the precipitated HAP1A. The ratios of the bound to precipitated HAP1A for anti-HAP1A immunoprecipitation or HAP1A immunoprecipitation in the absence of OA were normalized to 1 to reduce variations caused by immunoblotting or protein loading amounts when compared with p1A immunoprecipitation results. E, PC12 cells were transfected with GFP-tagged HAP1A or T598A and then treated with NGF for 2 d. F, PC12 cells were treated with 50 nm staurosporine or okadaic acid for 48 h and stained with rabbit anti-HAP1A (red) and mouse anti-kinesin heavy chain (green in top) or mouse anti-p150 dynactin (green in middle). Cell nuclei were stained with Hoechst dye (blue). Reduced phosphorylation by staurosporine increased the distribution of HAP1A in the neurite tips, whereas increased phosphorylation by okadaic acid reduces this distribution (bottom). Neurite tips are indicated by arrows. G, The percentage of PC12 cells in F showing HAP1 in neurite tips (top) or elongated neurites (bottom). Cells containing neurites longer than two cell bodies were counted as positive cells with elongated neurites. Data are expressed as mean ± SE (n = 3). ∗∗p < 0.01 compared with control cells without drug treatment.
If dephosphorylation of HAP1A regulates its trafficking in neurites, mutated HAP1A (T598A), which lacks T598 phosphorylation, may be more likely to localize in the neurites than wild-type HAP1A. We therefore transfected GFP-tagged HAP1A and T598A into cultured PC12 cells and treated these cells with NGF (100 ng/ml) to examine their neuritic localization. To minimize the influence by various levels of transfected proteins in individual cells, we measured the ratio of HAP1 fluorescent signal in growth cone to HAP1 signal in the cell body. Transfected T598A showed an increased neuritic distribution compared with wild-type HAP1A (Fig. 4E). By quantifying HAP1 fluorescent signal ratios (growth cone/cell body), we also observed an increase in this ratio in T598A-transfected cells (0.55 ± 0.02; n = 111) compared with HAP1A-transfected cells (0.43 ± 0.02, mean ± SE; n = 122; p < 0.01). We then examined whether altering phosphorylation by okadaic acid or staurosporine could change the distribution of endogenous HAP1A in neurites. Using mouse anti-kinesin heavy chain and rabbit anti-HAP1A, we were able to examine the distribution of HAP1A and kinesin in the same cells in a double-immunofluorescent assay. It is known that staurosporine can promote neurite extension of PC12 cells (Tischler et al., 1991; Rasouly et al., 1992). In those cells that develop long neurites induced by staurosporine (50 nm for 48 h), both HAP1A and kinesin accumulated in neurite tips (Fig. 4F, top). Similarly, p150 dynactin and HAP1A were also enriched in the neurites of these cells (Fig. 4F, middle). Conversely, okadaic acid (50 nm for 48 h) reduces neurite outgrowth and distribution of HAP1 in neurites, even in cells that still possess long neurites (Fig. 4F, bottom). Counting cells with neurites longer than two cell bodies and those containing neuritic HAP1 confirmed that HAP1 distribution in neurite tips is increased in parallel with the increased neurite outgrowth after staurosporine treatment (Fig. 4G). All of these results suggest that phosphorylation of HAP1A reduces its interaction with microtubule transporters, whereas nonphosphorylated HAP1A is able to move to neurite tips in association with neurite outgrowth.
HAP1 stabilizes intracellular TrkA
Because HAP1 is found to stabilize the internalized receptors EGFR and GABAAR by preventing their degradation in lysosomes (Li et al., 2002; Kittler et al., 2004), we asked whether HAP1 is involved in stabilizing proteins that are important for neurite outgrowth. To address this issue, we used siRNA approach to inhibit the expression of HAP1. Because both HAP1A and HAP1B are generated from the same gene, it is difficult to use siRNA to specifically interfere with the expression of HAP1A without affecting HAP1B. Nevertheless, we identified an siRNA that could effectively reduce the expression of both endogenous HAP1A and HAP1B in PC12 cells (Fig. 5A). This siRNA was expressed via an adenoviral vector that independently expressed GFP, allowing us to identify cells that also express HAP1 siRNA. For the control, we infected PC12 cells with adenoviral GFP or a control siRNA of a scramble sequence with nucleic acid composition similar to that of HAP1 siRNA. Adenoviral HAP1 siRNA-infected PC12 cells showed much shorter neurites in the presence of NGF than control cells that were infected with adenoviral GFP (Fig. 5B,C) or the control siRNA (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). In those PC12 cells with short neurites, we did not see nuclear DNA fragmentation and obvious cell body degeneration, suggesting that reduced neurite outgrowth in HAP1 siRNA-infected PC12 cells is not caused by cell death.
Figure 5.

Suppression of HAP1 inhibits neurite outgrowth and the level of TrkA. A, Expression of HAP1 siRNA in PC12 cells via an adenoviral vector that also expresses GFP. Note that endogenous HAP1 was reduced in a time-dependent manner. Control is adenoviral GFP infection. B, PC12 cells were infected with adenoviral GFP or HAP1 siRNA and treated with NGF (50 ng/ml) for 2 d. Cells were then stained with anti-HAP1A (red). Note that cells infected by adenoviral HAP1 siRNA have lost their HAP1 staining and show reduced neurite extension compared with cells infected by adenoviral GFP. C, Percentage of viral-infected PC12 cells containing neurites longer than two cell bodies (mean ± SE; n = 3; ∗∗p < 0.01). D, Western blotting of PC12 cells that had been infected with adenoviral HAP1 siRNA or adenoviral GFP for ∼60 h. Note that the levels of HAP1 and TrkA were decreased, but other proteins [dynactin p150, KLC, EEA1, p75NTR (p75), and tubulin] did not show significant changes. E, Densitometry analysis of the ratios (mean ± SE; n = 3 experiments; ∗∗p < 0.01 compared with the GFP control) of proteins (HAP1, TrkA, p150, and KLC) to tubulin in adenoviral-infected PC12 cells. The ratios in the control group were adjusted to 1. F, PC12 cells infected by adenovirus for ∼60 h were biotinylated and precipitated by Neutravidin beads to examine the membrane and internalized TrkA. Note that both cell membrane TrkA and internalized TrkA were decreased when HAP1 expression was suppressed by siRNA compared with adenoviral GFP infection. The supernatant after precipitation (soluble fraction) was also immunoblotted with antibodies to HAP1, KLC, dynactin p150, and tubulin. G, Verification of the specificity of adenoviral HAP1 siRNA to suppress TrkA in PC12 cells compared with the control scramble siRNA in three experiments. The Western blots were probed with antibodies to TrkA, HAP1, p75NTR, and tubulin (for densitometry data, see supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). H, Western blot analysis of NGF-mediated degradation of membrane TrkA in PC12 cells treated with scramble or HAP1 siRNA for 48 h. The remaining biotinylated TrkA in PC12 cells after NGF treatment for 0, 75, and 150 min was precipitated and subjected to Western blotting. I, Densitometry of the relative change of remaining biotinylated TrkA after NGF treatment compared with the pretreatment value, which serves as 100%. The data (mean ± SE) were obtained from three experiments. ∗∗p < 0.01.
Having found that the expression of HAP1 and its related function can be suppressed by siRNA, we then asked whether any trafficking proteins or receptors important for neurite outgrowth could be affected by reducing the expression of HAP1. By examining a number of proteins, we found that TrkA was specifically reduced concomitant with the decreased level of HAP1 (Fig. 5D,E). TrkA is the major type of membrane receptors that, during activation by NGF, is internalized and retrogradely transported to the cell body to mediate its signaling pathways. The internalized receptors are also recycled back to the plasma membrane or degraded by lysosomes. Thus, like other membrane receptors, the endocytosis of TrkA is regulated through the endosome–lysosomal pathway and is important for its intracellular signaling (Kuruvilla et al., 2004; Reichardt and Mobley, 2004; Saxena et al., 2005). The decrease of TrkA by HAP1 siRNA was specific, because HAP1 siRNA did not affect the expression of dynactin p150, KLC, EEA1, an early endosomal protein, or the p75 neurotrophin receptor p75NTR, another NGF receptor in PC12 cells. It is conceivable that reducing the expression of HAP1 impairs the recycling of internalized TrkA to the plasma membrane, causing more internalized TrkA to be localized to lysosomes for degradation. Indeed, the membrane pool of TrkA as determined by precipitation of cell surface biotinylated proteins was decreased (Fig. 5F). Consequently, less internalized TrkA was found after NGF treatment (Fig. 5F). To rule out nonspecific effects of small RNA interference, we used a scramble siRNA as a control. Compared with this control, HAP1 siRNA significantly reduced the expression of TrkA in PC12 cells, which is reflected by Western blotting (Fig. 5G) and densitometry analysis (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Furthermore, NGF-mediated TrkA degradation in PC12 cells, which normally occurs 2–3 h after NGF treatment (Jullien et al., 2002), was augmented when the expression of HAP1 is suppressed (Fig. 5H). This observation was verified by densitometry analysis of the remaining biotinylated TrkA in PC12 cells after NGF treatment for 150 min (Fig. 5I). As a result, the total TrkA level was also decreased because of the increased degradation of internalized TrkA. Together, all of these results demonstrate that decreasing cellular level of HAP1 specifically alters the stability of the internalized TrkA, leading to a reduced level of TrkA available for mediating its signaling pathway during NGF treatment, thereby resulting in inhibition of neurite outgrowth.
We have shown that HAP1 is abundant in PC12 cells, a cell line that highly expresses TrkA. To investigate whether HAP1 is also present in TrkA-containing primary neurons, we used anti-HAP1 to stain cultured sympathetic neurons isolated from adult rat superior cervical ganglia, a cell model that has been widely used to study TrkA (Orike et al., 2001; Kuruvilla et al., 2004). Both HAP1 and TrkA are specifically enriched in sympathetic neurons and their neurites (Fig. 6A). Similar to PC12 cells, sympathetic neurons treated with HAP1 siRNA show reduced neurite outgrowth compared with those treated with the control siRNA (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Although these data provide strong evidence for the coexpression of HAP1A and TrkA in the same cultured neurons, it is important to know whether this coexpression also occurs in the brain. Immunohistochemistry on slices from adult mouse hypothalamus revealed that some HAP1-containing neurons showed strong TrkA labeling (Fig. 6B) and some TrkA-positive neurons showed weak HAP1 staining. This suggests that HAP1 expression may be important for TrkA function in a subtype of neurons in the adult brain. We then used Western blotting, which is more quantitative than immunofluorescent staining, to analyze the expression of TrkA in the hypothalamus of HAP1 knock-out mouse pups [postnatal day 1 (P1) to P2]. Consistent with the results obtained from PC12 cells (Fig. 5), the absence of HAP1 in HAP1 knock-out mice also leads to reduced levels of TrkA in the hypothalamic tissue (Fig. 6C), which was confirmed by densitometry analysis of the ratio of HAP1 to tubulin in mouse hypothalamic tissue (1.1 ± 0.15 for wild type vs 0.54 ± 0.09 for knock-out, mean ± SE; n = 3; p < 0.05).
Figure 6.
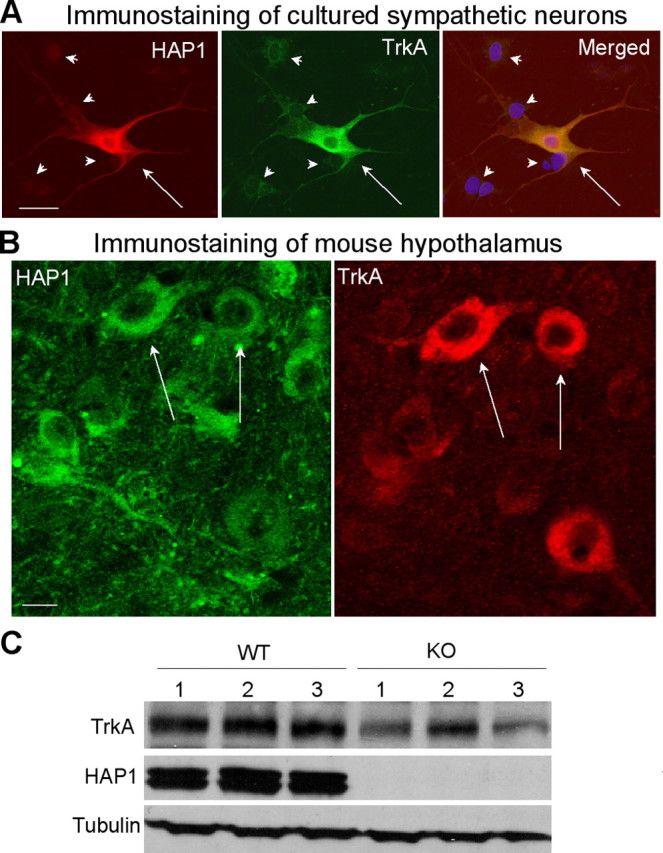
TrkA is colocalized with HAP1 in neurons and is reduced in HAP1 knock-out mouse brain tissue. A, Double-immunofluorescent staining of cultured sympathetic neurons showing that HAP1 (red) and TrkA (green) are coexpressed in the same neurons (arrows). Note that adjacent cells (arrowheads) are negative to HAP1 and TrkA staining. Nuclei (blue) were also shown in the merged image. HAP1 siRNA also reduces neurite extension of sympathetic neurons (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). B, Immunofluorescent double labeling of lateral hypothalamic region of mice. Note that some HAP1-positive cells (arrows) were also labeled by anti-TrkA and some were not. C, Western blotting analysis of the expression of TrkA in the hypothalamus of wild-type (WT; n = 3) and HAP1 knock-out (KO; n = 3) mice at postnatal age (P1–P2). TrkA was decreased in hypothalamic tissue of HAP1 knock-out mice. Scale bars: A, 10 μm; B, 5 μm.
Mutant htt inhibits the intracellular trafficking function of HAP1
The association of HAP1 with trafficking proteins seems important for level of the intracellular TrkA. It is known that mutant htt binds HAP1 avidly (Li et al., 1995; Gauthier et al., 2004). The increased binding of mutant htt to HAP1 could reduce the association of HAP1 with other proteins by competing for or masking the binding sites in HAP1. Another possibility is that mutant htt affects the phosphorylation of HAP1. Examining adenoviral GFP-htt-infected PC12 cells and HD mouse brains showed no difference in HAP1A phosphorylation between wild-type and HD cells or tissues (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). We therefore favor a model in which mutant htt perturbs HAP1 association with trafficking proteins or impairs its ability to stabilize intracellular TrkA. To test this possibility, we expressed N-terminal 208 amino acids of htt in cultured cells. Mutant htt with130Q colocalized with HAP1A puncta in transfected HEK293 cells (Fig. 7A), suggesting that this mutant htt fragment binds soluble HAP1. We therefore expressed GFP fusion protein containing this N-terminal htt in PC12 cells via adenoviral vector infection (Shin et al., 2005). As expected, mutant htt inhibits NGF-induced neurite outgrowth of PC12 cells compared with the control htt infection (Fig. 7B,C). Using fluorescence imaging, we have shown recently that HAP1A can move within neurites of live PC12 cells (McGuire et al., 2006), presumably via its association with kinesin and dynactin p150. With the same technique, we observed the distribution of RFP-HAP1A in the neurite tips, which also formed moving vesicles or vesicular structures (supplemental movies, available at www.jneurosci.org as supplemental material). The movement of RFP-HAP1A is consistent with the trafficking function that may be critical for recycling membrane receptors to the plasma membrane. We then examined mutant htt-infected cells that still showed neurites to evaluate the distribution and movement of RFP-HAP1A in these cells (Fig. 7C). Quantitative analysis of the ratio of RFP-HAP1 signal in neurites to that in the cell body suggests that the neuritic distribution of RFP-HAP1A was significantly decreased (p < 0.05) in PC12 cells expressing GFP-htt-130Q (0.375 ± 0.038, mean ± SE; n = 30) compared with those expressing GFP-htt-23Q (0.260 ± 0.028, mean ± SE; n = 30). Moreover, PC12 cells expressing mutant htt showed the reduced movement of HAP1A and the dynamic recruitment of HAP1A into htt inclusions (supplemental movie for htt-130Q-transfected cells, available at www.jneurosci.org as supplemental material).
Figure 7.
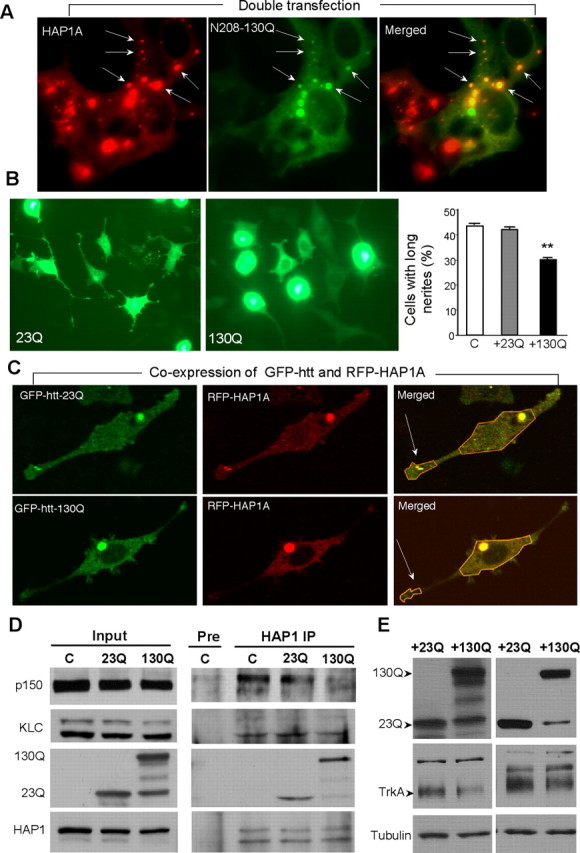
Mutant htt inhibits the association of HAP1 with trafficking proteins and the level of TrkA. A, HEK293 cells were cotransfected with HAP1A and N-terminal htt (1–208 aa) containing 130Q (130Q). Transfected mutant htt (130Q) is colocalized with HAP1A in some cytoplasmic puncta (arrows). Note that HAP1A itself also forms cytoplasmic puncta. B, PC12 cells were infected with adenoviral GFP-htt (1–208 aa) containing 23Q or 130Q and were treated with NGF (50 ng/ml for 48 h). Mutant htt (+130Q) inhibits neurite outgrowth compared with normal htt (+23Q). The percentage of PC12 cells containing neurites longer than two cell bodies was determined (right). C, Noninfected PC12 cells. Data (mean ± SE) were obtained from three experiments. ∗∗p < 0.01. C, Expression of GFP-htt-130Q reduced the distribution of RFP-HAP1A in neurite tips (arrows) of PC12 cells. Merged images show colocalized RFP-HAP1A and GFP-htt (yellow signal). Also see live-cell imaging in supplemental movies (available at www.jneurosci.org as supplemental material). D, Immunoprecipitation (IP) of HAP1A and its associated proteins in PC12 cells infected with adenoviral GFP-htt for 48 h. Mutant N-terminal htt (130Q) bound more HAP1 than did control htt (23Q) and resulted in decreased association of endogenous HAP1 with dynactin p150 and kinesin light chain in PC12 cells. Note that small or degraded protein products in the 130Q sample did not bind HAP1. The same HAP1 immunoprecipitates and input were probed with a panel of different antibodies as indicated. E, PC12 cells infected with adenoviral htt-23Q or 130Q were subjected to Western blotting. Mutant htt (130Q) reduced the level of TrkA in PC12 cells compared with wild-type htt (23Q). Results from two independent experiments are shown.
Although the above findings support the idea that mutant htt abnormally binds HAP1 and affects its trafficking within neurites, it is important to know whether mutant htt reduces the association of HAP1 with dynactin p150 and/or kinesin to affect the trafficking of HAP1 or its function for maintaining the internalized TrkA. We therefore infected PC12 cells with adenoviral htt and then immunoprecipitated endogenous HAP1A. Mutant htt (130Q) was expressed as the intact form and smaller fragments (Fig. 7D, Input), which were not present in control PC12 cells. Immunoprecipitation of HAP1A only coprecipitated the intact form of mutant htt (Fig. 7D, HAP1 IP). Densitometry showed that the ratio of precipitated mutant htt (130Q) to input (0.68 ± 0.03) was significantly greater than the ratio of precipitated normal htt (23Q) to input (0.40 ± 0.05, mean ± SE; n = 3; p < 0.01). Thus, compared with normal N-terminal htt (23Q), more mutant htt (130Q) was coprecipitated with HAP1A. Meanwhile, the association of HAP1A with dynactin p150 and KLC was reduced in the presence of mutant htt (Fig. 7D). This result is also supported by densitometry analysis of the ratios of precipitated proteins to their input, which were normalized by the ratio from the uninfected control cells and show significantly less dynactin p150 (0.90 ± 0.04 for 23Q vs 0.70 ± 0.03 for 130Q, mean ± SE; n = 3; p < 0.05) and KLC (1.21 ± 0.17 for 23Q vs 0.73 ± 0.09 for 130Q, mean ± SE; n = 3; p < 0.05) in the immunoprecipitates with mutant htt (130Q) compared with control htt (23Q). It is possible that mutant htt reduces the association of HAP1 with these trafficking proteins by competing with them for binding HAP1 or by sequestering kinesin and dynactin complexes as reported in other studies (Gunawardena et al., 2003; Trushina et al., 2004). Although these possibilities remain to be verified, it is important to know whether mutant htt can also affect TrkA. Indeed, expression of mutant htt in PC12 cells decreased the level of TrkA (Fig. 7E). To verify that the difference seen was not attributable to different loading amounts of protein samples, we measured the relative change of tubulin and TrkA in the presence of 130Q htt compared with 23Q htt (the band density in 130Q cells/the density of the corresponding band in 23Q cells × 100). Mutant htt (130Q) caused a significant reduction of TrkA (61 ± 0.82%) but not tubulin (92 ± 0.13%, mean ± SE; n = 3; p < 0.05). These results also support the idea that mutant htt binds HAP1A and affects the trafficking of HAP1, which may in turn impair its function to stabilize the internalized TrkA and to promote neurite outgrowth.
Discussion
Several lines of strong evidence indicate that phosphorylation of a C-terminal site of HAP1A occurs in vivo. First, the antibody specific to the phosphorylated T598 can recognize endogenous HAP1 in the mouse brain and PC12 cells, and this immunoreaction in PC12 cells is enhanced by conditions that increase phosphorylation. Second, replacing the T598 residue with alanine diminishes the phosphorylation of HAP1A. Third, phosphorylated HAP1A is primarily confined to the cell body, whereas HAP1A in neurite tips is not labeled by the antibody to phosphorylated HAP1A. Immunoprecipitation experiments using rat brain lysates show that phosphorylated HAP1A binds less dynactin p150 and kinesin than nonphosphorylated HAP1A. These findings suggest that phosphorylation of HAP1A alters its subcellular localization and that nonphosphorylated HAP1 is more likely to favor the binding of other trafficking proteins. Although we provide convincing in vitro evidence for the phosphorylation of the amino acid residue T598 in HAP1A by PKA, this residue could also be a substrate of other kinases in cells. Also, there are multiple regulatory and catalytic subunits of PKA, which are expressed at different levels across tissues and various types of cells (Brandon et al., 1997; Taylor et al., 2004). Thus, it remains to be investigated how HAP1A phosphorylation is regulated in different types of neurons.
Dynactin p150 and kinesin are microtubule-dependent transporters that are primarily involved in retrograde and anterograde transport, respectively. These transport processes are required for a variety of cellular functions, including endocytosis of membrane receptors (Aniento et al., 1993; Lin et al., 2002). In nerve terminals, endocytosis and trafficking of membrane receptors are essential for synaptic transmission and plasticity. In cultured neurons, the function of intracellular trafficking is reflected by neurite outgrowth that requires active vesicular transport as well as the endocytosis and trafficking of membrane receptors. Cultured PC12 cells are a widely used cell model to examine the function of neurotrophic factor receptors and their endocytosis and trafficking. In PC12 cells, abundant TrkA provides a good target to assess the function and trafficking of membrane receptors. Although little is known about the involvement of kinesin and dynactin p150 in TrkA trafficking, it is evident that microtubule-dependent transporters participate in receptor internalization at nerve terminals (Feiguin et al., 1994; Apodaca, 2001; Lin et al., 2002; Bananis et al., 2004).
In the present study, we provide clear evidence that HAP1 deficiency can reduce the level of TrkA. This effect is likely attributable to the action of HAP1 on the protein level, rather than transcripts, of TrkA. First, HAP1 is not a nuclear protein and is unlikely to act on nuclear transcription. Second, decreasing internalized TrkA levels by HAP1 siRNA occurs in a short time period (75–150 min) during NGF stimulation, also suggesting that it acts on the protein level. Third, HAP1 is found to prevent degradation of other membrane receptors such as EGFR and GABAAR (Li et al., 2002; Kittler et al., 2004). Given a large body of evidence that TrkA is endocytosed and degraded by lysosomes, we believe that HAP1 prevents the degradation of internalized TrkA.
Studies of different cellular and animal systems have shown that mutant htt can affect kinesin- or dynactin-associated transport (Gunawardena et al., 2003; Szebenyi et al., 2003; Gauthier et al., 2004; Trushina et al., 2004) and inhibit neurite outgrowth (Li et al., 2000; Wyttenbach et al., 2001; Song et al., 2002). Because mutant htt in the nucleus can interfere with gene transcription, leading to altered signaling that may also affect neuronal function (Apostol et al., 2005), an important issue to be resolved is whether mutant htt in the cytoplasm causes defective neuronal differentiation and neurite outgrowth by altering the trafficking of growth factor receptors. In the current study, we focused on receptors important for neurite extension of PC12 cells and provided evidence that mutant htt, which is predominantly present in the cytoplasm (Fig. 7), could reduce the level of TrkA in neuronal cells.
The effect of mutant htt on TrkA seen in our study is consistent with the previous report that cytoplasmic mutant htt reduces the level of TrkA and affects its signaling in PC12 cells (Song et al., 2002). Our findings, however, offer mechanistic insight into this phenomenon by showing that mutant htt decreases the interaction of HAP1 with kinesin and dynactin. We also provided direct evidence that decreasing the expression of HAP1 affects TrkA level and neurite outgrowth. This strong evidence supports the idea that the dysfunction of HAP1, which can be caused by mutant htt, could also lead to impairment in the trafficking of TrkA or the stability of internalized TrkA. Because HAP1 is found to transport membrane receptors between the endosomes and lysosomes (Li et al., 2002), lack of HAP1 or inhibition of its function may cause more receptors to be degraded in lysosomes, reducing the amount of TrkA that is able to be recycled to the plasma membrane and therefore impairing cellular function related to TrkA.
Although we also provide strong evidence for the coexpression of HAP1 and TrkA in PC12 cells and cultured sympathetic neurons, it should be pointed out that the expression of HAP1 varies in different types of neurons in the brain. The fact that HAP1 is most abundant in hypothalamic neurons suggests that the functions of HAP1, such as stabilizing the internalized membrane receptors, may be cell-type specific. We have shown that some hypothalamic neurons express both HAP1 and TrkA and some do not in adult mouse brain. It remains to be investigated how HAP1 and TrkA are expressed in embryonic brains or during early development. In those cells that express both HAP1 and TrkA, HAP1 may play an important role in the intracellular trafficking of TrkA. Although this possibility remains to be further investigated in vivo using the gene targeting knock-out technique to selectively eliminate HAP1 in specific subtypes of neurons, it is clear that reducing the expression of HAP1 in cultured neuronal cells affects the level of TrkA and impairs neurite outgrowth.
Based on the present findings, we propose a model for the regulation of HAP1 trafficking (Fig. 8). HAP1 interacts with microtubule-dependent transporters such as dynactin p150 and kinesin. The interactions of HAP1 with these microtubule-dependent transporters allow HAP1 to localize in neurite tips and to participate in intracellular trafficking of membrane receptors. The phosphorylation of the C terminus of HAP1A reduces its interactions with these transporters and leads to the retention of HAP1A in the cell body. Similarly, when HAP1 is bound to mutant htt, the interaction of HAP1 with microtubule-dependent transporters is diminished. As a result, membrane receptors, such as TrkA, may not be efficiently transported to the appropriate cellular locations and are instead targeted to the lysosomes for degradation, leading to the decreased level of TrkA and the inhibition of neurite outgrowth. Although this proposed model is based on the findings from cells that highly express TrkA, it would also provide a basis for additional investigation of the effect of mutant htt on the intracellular trafficking of other membrane receptors in different types of neurons.
Figure 8.

Model of the regulation of trafficking of HAP1. PKA phosphorylation of C-terminal HAP1A reduces its binding to dynactin p150 and kinesin. Dephosphorylation by an okadaic acid-sensitive phosphatase(s) increases this binding, which may allow HAP1A to be transported to neurites in which it stabilizes the internalized TrkA and promotes neurite outgrowth. Mutant htt may reduce the association of HAP1 with these trafficking proteins to affect the function of HAP1 (see also the description in Discussion).
Footnotes
This work was supported by National Institutes of Health Grants NS36232 and AG41669. We thank Louis Reichardt for providing antibody to TrkA, Moses V. Chao for antibody to p75NTR receptor, Frank Gordon for help with sympathetic neuronal culture, and Haian Fu for advice and PKA plasmid.
References
- Aniento F, Emans N, Griffiths G, Gruenberg J (1993). Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J Cell Biol 123:1373–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apodaca G (2001). Endocytic traffic in polarized epithelial cells: role of the actin and microtubule cytoskeleton. Traffic 2:149–159. [DOI] [PubMed] [Google Scholar]
- Apostol BL, Illes K, Pallos J, Bodai L, Wu J, Strand A, Schweitzer E, Olson JM, Kazantsev A, Marsh JL, Thompson LM (2005). Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum Mol Genet 15:273–285. [DOI] [PubMed] [Google Scholar]
- Bananis E, Nath S, Gordon K, Satir P, Stockert RJ, Murray JW, Wolkoff AW (2004). Microtubule-dependent movement of late endocytic vesicles in vitro: requirements for Dynein and Kinesin. Mol Biol Cell 15:3688–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block-Galarza J, Chase KO, Sapp E, Vaughn KT, Vallee RB, DiFiglia M, Aronin N (1997). Fast transport and retrograde movement of huntingtin and HAP 1 in axons. NeuroReport 8:2247–2251. [DOI] [PubMed] [Google Scholar]
- Brandon EP, Idzerda RL, McKnight GS (1997). PKA isoforms, neural pathways, and behaviour: making the connection. Curr Opin Neurobiol 7:397–403. [DOI] [PubMed] [Google Scholar]
- Chan EY, Nasir J, Gutekunst CA, Coleman S, Maclean A, Maas A, Metzler M, Gertsenstein M, Ross CA, Nagy A, Hayden MR (2002). Targeted disruption of Huntingtin-associated protein-1 (Hap1) results in postnatal death due to depressed feeding behavior. Hum Mol Genet 11:945–959. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Zeitlin S, Dietrich P (2004). Huntingtin-associated protein 1 (Hap1) mutant mice bypassing the early postnatal lethality are neuroanatomically normal and fertile but display growth retardation. Hum Mol Genet 13:3115–3125. [DOI] [PubMed] [Google Scholar]
- Engelender S, Sharp AH, Colomer V, Tokito MK, Lanahan A, Worley P, Holzbaur EL, Ross CA (1997). Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum Mol Genet 6:2205–2212. [DOI] [PubMed] [Google Scholar]
- Feiguin F, Ferreira A, Kosik KS, Caceres A (1994). Kinesin-mediated organelle translocation revealed by specific cellular manipulations. J Cell Biol 127:1021–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita A, Hattori Y, Takeuchi T, Kamata Y, Hata F (2001). NGF induces neurite outgrowth via a decrease in phosphorylation of myosin light chain in PC12 cells. NeuroReport 12:3599–3602. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118:127–138. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS (2005). Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch Neurol 62:46–51. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS (2003). Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40:25–40. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Ferrante RJ, Li XJ, Hersch SM (1998). The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): comparison with huntingtin in rat and human. J Neurosci 18:7674–7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullien J, Guili V, Reichardt LF, Rudkin BB (2002). Molecular kinetics of nerve growth factor receptor trafficking and activation. J Biol Chem 277:38700–38708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Thomas P, Tretter V, Bogdanov YD, Haucke V, Smart TG, Moss SJ (2004). Huntingtin-associated protein 1 regulates inhibitory synaptic transmission by modulating γ-aminobutyric acid type A receptor membrane trafficking. Proc Natl Acad Sci USA 101:12736–12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruvilla R, Zweifel LS, Glebova NO, Lonze BE, Valdez G, Ye H, Ginty DD (2004). A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell 118:243–255. [DOI] [PubMed] [Google Scholar]
- Li SH, Gutekunst CA, Hersch SM, Li XJ (1998). Interaction of huntingtin-associated protein with dynactin P150Glued. J Neurosci 18:1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Li H, Torre ER, Li XJ (2000). Expression of huntingtin-associated protein-1 in neuronal cells implicates a role in neuritic growth. Mol Cell Neurosci 16:168–183. [DOI] [PubMed] [Google Scholar]
- Li SH, Yu ZX, Li CL, Nguyen HP, Zhou YX, Deng C, Li XJ (2003). Lack of huntingtin-associated protein-1 causes neuronal death resembling hypothalamic degeneration in Huntington's disease. J Neurosci 23:6956–6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XJ, Li SH (2005). HAP1 and intracellular trafficking. Trends Pharmacol Sci 26:1–3. [DOI] [PubMed] [Google Scholar]
- Li XJ, Li SH, Sharp AH, Nucifora FC Jr, Schilling G, Lanahan A, Worley P, Snyder SH, Ross CA (1995). A huntingtin-associated protein enriched in brain with implications for pathology. Nature 378:398–402. [DOI] [PubMed] [Google Scholar]
- Li Y, Chin LS, Levey AI, Li L (2002). Huntingtin-associated protein 1 interacts with hepatocyte growth factor-regulated tyrosine kinase substrate and functions in endosomal trafficking. J Biol Chem 277:28212–28221. [DOI] [PubMed] [Google Scholar]
- Lin SX, Gundersen GG, Maxfield FR (2002). Export from pericentriolar endocytic recycling compartment to cell surface depends on stable, detyrosinated (glu) microtubules and kinesin. Mol Biol Cell 13:96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EJ, Kim M, Velier J, Sapp E, Lee HS, Laforet G, Won L, Chase K, Bhide PG, Heller A, Aronin N, Difiglia M (1999). Analysis of Huntingtin-associated protein 1 in mouse brain and immortalized striatal neurons. J Comp Neurol 403:421–430. [PubMed] [Google Scholar]
- McGuire JR, Rong J, Li SH, Li XJ (2005). Polyglutamine protein trafficking and neurodegeneration. Curr Genomics 6:189–194. [Google Scholar]
- McGuire JR, Rong J, Li SH, Li XJ (2006). Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J Biol Chem 281:3552–3559. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Ono S, Minamide LS, Takahashi M, Bamburg JR (1998). Actin depolymerizing factor and cofilin phosphorylation dynamics: response to signals that regulate neurite extension. Cell Motil Cytoskeleton 39:172–190. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Kanter JR, Jones KC, Taylor SS (2002). Phosphorylation of the catalytic subunit of protein kinase A. Autophosphorylation versus phosphorylation by phosphoinositide-dependent kinase-1. J Biol Chem 277:47878–47884. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Brady ST (2005). Polyglutamine expansion diseases: failing to deliver. Trends Mol Med 11:64–70. [DOI] [PubMed] [Google Scholar]
- Orike N, Thrasivoulou C, Wrigley A, Cowen T (2001). Differential regulation of survival and growth in adult sympathetic neurons: an in vitro study of neurotrophin responsiveness. J Neurobiol 47:295–305. [DOI] [PubMed] [Google Scholar]
- Pearson RB, Kemp BE (1991). Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations. Methods Enzymol 200:62–81. [DOI] [PubMed] [Google Scholar]
- Rahman A, Friedman DS, Goldstein LS (1998). Two kinesin light chain genes in mice. Identification and characterization of the encoded proteins. J Biol Chem 273:15395–15403. [DOI] [PubMed] [Google Scholar]
- Rasouly D, Rahamim E, Lester D, Matsuda Y, Lazarovici P (1992). Staurosporine-induced neurite outgrowth in PC12 cells is independent of protein kinase C inhibition. Mol Pharmacol 42:35–43. [PubMed] [Google Scholar]
- Reichardt LF, Mobley WC (2004). Going the distance, or not, with neurotrophin signals. Cell 118:141–143. [DOI] [PubMed] [Google Scholar]
- Rosini P, De Chiara G, Bonini P, Lucibello M, Marcocci ME, Garaci E, Cozzolino F, Torcia M (2004). Nerve growth factor-dependent survival of CESS B cell line is mediated by increased expression and decreased degradation of MAPK phosphatase 1. J Biol Chem 279:14016–14023. [DOI] [PubMed] [Google Scholar]
- Saxena S, Howe CL, Cosgaya JM, Steiner P, Hirling H, Chan JR, Weis J, Kruttgen A (2005). Differential endocytic sorting of p75NTR and TrkA in response to NGF: a role for late endosomes in TrkA trafficking. Mol Cell Neurosci 28:571–587. [DOI] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ (2005). Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol 171:1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Perides G, Liu YF (2002). Expression of full-length polyglutamine-expanded Huntingtin disrupts growth factor receptor signaling in rat pheochromocytoma (PC12) cells. J Biol Chem 277:6703–6707. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST (2003). Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 40:41–52. [DOI] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I (2003). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Yang J, Wu J, Haste NM, Radzio-Andzelm E, Anand G (2004). PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta 1697:259–269. [DOI] [PubMed] [Google Scholar]
- Tischler AS, Ruzicka LA, Dobner PR (1991). A protein kinase inhibitor, staurosporine, mimics nerve growth factor induction of neurotensin/neuromedin N gene expression. J Biol Chem 266:1141–1146. [PubMed] [Google Scholar]
- Torcia M, De Chiara G, Nencioni L, Ammendola S, Labardi D, Lucibello M, Rosini P, Marlier LN, Bonini P, Dello Sbarba P, Palamara AT, Zambrano N, Russo T, Garaci E, Cozzolino F (2001). Nerve growth factor inhibits apoptosis in memory B lymphocytes via inactivation of p38 MAPK, prevention of Bcl-2 phosphorylation, and cytochrome c release. J Biol Chem 276:39027–39036. [DOI] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger II JD, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, Chacko C, McMurray CT (2004). Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol 24:8195–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, Bradley J, Brown R, Maxwell M, Schapira A, Orntoft TF, Kato K, Rubinsztein DC (2001). Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum Mol Genet 10:1829–1845. [DOI] [PubMed] [Google Scholar]