

Abstract
Alzheimer's disease (AD) is characterized by progressive loss of neurons in selected brain regions, extracellular accumulations of amyloid β, and intracellular fibrils containing hyperphosphorylated tau. Tau mutations in familial tauopathies confirmed a central role of tau pathology; however, the role of tau alteration and the sequence of tau-dependent neurodegeneration in AD remain elusive. Using Sindbis virus-mediated expression of AD-relevant tau constructs in hippocampal slices, we show that disease-like tau modifications affect tau phosphorylation at selected sites, induce Alz50/MC1-reactive pathological tau conformation, cause accumulation of insoluble tau, and induce region-specific neurodegeneration. Live imaging demonstrates that tau-dependent degeneration is associated with the development of a “ballooned” phenotype, a distinct feature of cell death. Spine density and morphology is not altered as judged from algorithm-based evaluation of dendritic spines, suggesting that synaptic integrity is remarkably stable against tau-dependent degeneration. The data provide evidence that tau-induced cell death involves apoptotic as well as nonapoptotic mechanisms. Furthermore, they demonstrate that targeted expression of tau in hippocampal slices provides a novel model to analyze tau modification and spatiotemporal dynamics of tau-dependent neurodegeneration in an authentic CNS environment.
Keywords: Sindbis virus, tauopathy, hippocampus, neurodegeneration, spines, phosphorylation
Introduction
Tau alteration and dysfunction and extensive neuron loss has long been associated with several neurodegenerative diseases now collectively called tauopathies. In tauopathies such as Alzheimer's disease (AD) and frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), tau is abnormally and hyperphosphorylated and is accumulated as intracellular neurofibrillary tangles (NFTs) (Lee et al., 2001; Shahani and Brandt, 2002; Brandt et al., 2005). In AD, the tau pathology is joined by the accumulation of extracellular amyloid plaques formed from amyloid β (Aβ) peptides. The identification of mutations in the tau gene in FTDP-17 has confirmed that dysfunction of tau protein is central to the neurodegenerative process and that changes in tau suffice to induce neuronal loss.
Abnormal phosphorylation of tau is considered one of the earliest signs of neuronal degeneration and appears to precede tau aggregation or amyloid formation (Braak et al., 1994). A good correlation between the amount of hyperphosphorylated tau and the clinical severity of AD patients has been demonstrated (Holzer et al., 1994). Site-specific phosphorylation of tau can be regulated by concerted and sequential action of many protein kinases and phosphatases (Iqbal et al., 2005; Stoothoff and Johnson, 2005). These findings, and the changes in kinases and phosphatases observed in AD (Iqbal et al., 2005), suggest that tau hyperphosphorylation is likely to be caused by an imbalance of the complex protein phosphorylation and dephosphorylation systems.
Glutamate is commonly used as a mimic of phosphoserine or phosphothreonine because of similar steric and electrostatic properties (Huffine and Scholtz, 1996; Davis, 2004) and imitates the effect of phosphorylation on the function of many proteins (Maciejewski et al., 1995; Wang et al., 1998). We showed previously that tau constructs in which serine/threonine residues (which are phosphorylated to a high extent in AD tau) were substituted with glutamate to create a pseudohyperphosphorylation mimic key structural and functional aspects of hyperphosphorylated tau protein (Eidenmüller et al., 2000, 2001; Maas et al., 2000), thus providing a model to test the effect of a disease-like permanent and high stoichiometric tau phosphorylation. Interestingly, pseudohyperphosphorylated tau (PHP tau) exerted a neurotoxic effect when expressed in rat pheochromocytoma 12 (PC12) cells and human model neurons (Fath et al., 2002). It should, however, be noted that mimicry of phosphoserine or phosphothreonine with glutamate is limited to certain aspects, because the mutated residues are usually not detected by antibodies that are specific for phosphorylated serine or threonine (Eidenmüller et al., 2000).
Slice cultures combine the accessibility and maintenance of in vitro culture systems while preserving intact the hippocampal synaptic circuitry and anatomy. Here we show that tau constructs are efficiently expressed in individual neurons of hippocampal slices using recombinant Sindbis virus. The enhanced green fluorescent protein (EGFP) tag allows an excellent visualization of all cellular compartments down to single dendritic spines of proximal and distal dendrites. We show that tau modifications induce region-specific neurodegeneration, cause accumulation of insoluble tau, induce distinct pathological tau conformations, and affect tau phosphorylation at selected sites. Three-dimensional reconstructions of individual neurons suggest that synaptic integrity is remarkably stable against tau-dependent degeneration.
Materials and Methods
Materials.
Chemicals were purchased from Sigma (Deisenhofen, Germany). Cell culture media and supplements were obtained from Sigma and Invitrogen (Gaithersburg, MD), and culture flasks, plates, and dishes were obtained from Nunc (Roskilde, Denmark), unless stated otherwise.
Construction of Sindbis virus expression vectors.
Eukaryotic expression plasmids for adult [441 amino acids (aa)] and fetal (352 aa) human tau with N-terminally fused EGFP were constructed in pHSV expression vectors. PHP tau was constructed by changing the codons for S198, S199, S202, T231, S235, S396, S404, S409, S413, and S422 to glutamate as described previously (Eidenmüller et al., 2000). EGFP–tau [wild type (wt) and PHP; adult and fetal isoform] cDNA was first subcloned into pcDNA3.1 vector by EcoRI–XhoI sites. For recloning into pSinRep5 vector (a generous gift from Sondra Schlesinger, Washington University School of Medicine, St. Louis, MO), EGFP–tau constructs were excised from pcDNA3.1 vector using EcoRI, blunt-ended with Klenow fragment, then cut with XbaI, and cloned into pSinRep5 plasmid, which was cut previously with XbaI and PmlI. As a control, an expression vector was constructed with EGFP alone. EGFP was excised from pEGFP-C1 (Clontech, Heidelberg, Germany) with BglII, blunt-ended with Klenow fragment, then cut with NheI, and cloned into pSinRep5 plasmid, which was cut previously with XbaI and PmlI.
pSinRep5–EGFP–tau constructs, pSinRep5–EGFP, and helper DH (26S) DNA (which encodes the structural proteins of the Sindbis virus; S. Schlesinger) were then transcribed in vitro (mMessage Machine SP6 kit; Ambion, Huntingdon, UK). The recombinant RNA and helper DH (26S) RNA were coelectroporated into baby hamster kidney (BHK) cells using the Gene Pulser II electroporation system (Bio-Rad, München, Germany). The recombinant RNA encoding EGFP–tau or EGFP alone was packaged and enveloped by the structural proteins produced by the helper RNA. The replication-incompetent pseudovirions thus produced and released into the extracellular medium were harvested after 24 h and filtered and stored at −80°C. The titer tested on BHK cells ranged from 1.7 to 3.0 × 108 infectious particles per milliliter for different constructs.
Organotypic hippocampal slice cultures and Sindbis virus infections.
Organotypic slice cultures from hippocampus were prepared according to the technique described by Stoppini et al. (1991). Six-day-old Naval Medical Research Institute mice were decapitated, the brains were removed, and the hippocampus of both sides were isolated and cut into transverse slices of 400 μm thickness on a McIlwain tissue chopper (Gabler, Bad Schwalbach, Germany). The slices were then cultured on Millipore (Bedford, MA) membrane culture inserts (Millicell CM; pore size, 0.4 μm) in six-well culture plates with 1 ml of slice culture media [50% minimum essential medium (DMEM with HEPES modification), 25% DMEM, 25% heat-inactivated horse serum, 2 mm glutamine, 0.65% glucose, pH 7.2] per well. The six-well culture plates were kept at 37°C in a humidified atmosphere with 5% CO2. The slice culture media were changed every third day. Slices were kept in culture for 12 d before the experiments. After the first 8 d in culture, the slice culture medium was replaced gradually with serum-free culture medium (Neurobasal medium, pH 7.2, supplemented with N1, 2 mm glutamine, and 0.65% glucose) as follows: (1) 50% serum medium/50% serum-free medium on day 8 in vitro and (2) 100% serum-free medium on day 11. On day 12 in vitro, the slice cultures were infected with the Sindbis virus by the droplet method. For this, the virus stock was diluted (according to the experiment) and was applied as a drop (1 μl) on each hippocampal slice. Infected slices were then used for different experimental purposes described below.
Antibodies.
The following primary antibodies were used, and specificity and sources are given in parentheses: phosphorylation-independent human tau antibodies: Tau-5 (mouse; PharMingen, San Diego, CA), anti-human tau protein antiserum (rabbit; Biosource, Camarillo, CA); phosphorylation-dependent and site-specific tau antibodies: AT270 (Thr-181; mouse; Pierce, Rockford, IL), pT205 (Thr-205; rabbit; Biosource), pT212 (Thr-212; rabbit; Biosource), pS214 (Ser-214; rabbit; Biosource), pS262 (Ser-262; rabbit; Biosource), pS356 (Ser-356; rabbit; Biosource); conformation-dependent tau antibodies: Alz-50 (7–9, 312–342; mouse; a generous gift from Peter Davies, Albert Einstein College of Medicine, Bronx, NY), MC1 (7–9, 312–342; mouse; P. Davies); anti-GFP antibody (rabbit; Invitrogen). As secondary antibodies, cyanine 3 (Cy3)-coupled anti-rabbit antibody (Dianova, Hamburg, Germany) and peroxidase-conjugated anti-mouse and anti-rabbit antibodies (Jackson ImmunoResearch, West Grove, PA) were used.
Immunohistochemistry.
Infected hippocampal slices were left attached on the insert membranes throughout the immunostaining protocol to preserve the hippocampal structure and to ensure even penetration of antibodies inside the tissue. Slices were first washed with PBS and fixed with 4% paraformaldehyde in PBS containing 4% sucrose for 1 h at 4°C. After washing with PBS, slices were treated with 0.4% Triton X-100 in PBS for 90 min and then 50 mm ammonium chloride for 45 min at room temperature and blocked with PBS containing 5% fetal calf serum, 1% BSA, 0.1% Triton X-100, and 0.02% NaN3 at 4°C overnight. Slices were then incubated for 2–3 d at 4°C with primary antibodies diluted in PBS containing 5% fetal calf serum, 1% BSA, 0.1% Triton X-100, and 0.1% NaN3. After washing with PBS, they were incubated with Cy3-coupled anti-rabbit antibody for 2 d at 4°C. The slices were then washed in PBS, mounted in Confocal-Matrix (Micro-Tech-Lab, Graz, Austria) or Vectashield (Vector Laboratories, Burlingame, CA), and coverslipped.
Confocal imaging of fixed and live hippocampal slices.
All images were acquired on Nikon (Tokyo, Japan) laser scanning microscope (Nikon Eclipse TE2000-U inverted), equipped with C1 confocal laser scanning unit, argon (Ar; 488 nm), helium/neon (He/Ne; 543 nm), and blue diode (405 nm) lasers, and EZ-C1 software. Neurons expressing EGFP or EGFP–tau were visualized using the 488 nm argon laser line and 510–540 nm bandpass emission filter. Cy3 was imaged using He/Ne 543 laser excitation and 570–610 nm emission filter. Microscope objectives (Nikon) used were a dry 4× [numerical aperture (NA), 0.13], a dry 20× (NA, 0.45; ELWD), an oil–water–glycerine 20× (NA, 0.75), an oil-immersion 60× (NA, 1.4), and an oil-immersion 100× (NA, 1.3). The microscope was enclosed in an incubation chamber maintained at 37°C and 5% CO2 (Solent Scientific, Sagensworth, UK). For two-channel imaging of EGFP and Cy3, images were acquired via sequential scanning. Image stacks (typically 10–35 optical sections per stack; 1024 × 1024 pixels) were collected for the whole hippocampal slice (30.45 μm z-axis steps) and different hippocampal subfields [CA1, CA3, and dentate gyrus (DG); 0.9–2.5 μm z-axis steps] of the infected hippocampal slice fixed at day 3 postinfection. Maximum projection images were generated from the resulting Z stacks.
Live imaging of infected neurons expressing EGFP alone or EGFP–tau constructs in CA1, CA3, and DG regions of organotypic hippocampal slices was performed over a period of 4 d (day 2 to day 5 postinfection). For these experiments (n = 3), culture inserts were transferred to a glass-bottom culture dish (MatTek, Ashland, MA) after the last change of medium. Image stacks (20–35 optical sections per stack; 1024 × 1024 pixels) were collected at 0.45 μm z-axis steps. Images were taken with lowest practical laser (Ar; 488 nm) intensity and shortest practical illumination time to limit photodynamic damage. By carefully marking the culture plate orientation, the same region was scanned at different days. Two-dimensional maximum projection images were reconstructed for each region. For the quantitation of the extent of degeneration, the number of nondegenerating neurons (i.e., neurons without any swellings having processes without any beading) was determined over time (day 3 and 4) and expressed relative to the number at day 2 postinfection from the same microscopic field in different regions for all constructs. Evaluations were performed blindly by a coworker without knowledge of the identity of the samples.
For morphological analysis of spines, two neurons per construct and per hippocampal subregion were completely imaged at high resolution. Image stacks were captured in a montage manner by translating the x–y stage to adjacent microscopic fields until the cell was completely contained within the stacks (pixel size, 0.086 × 0.086 × 0.25 μm; one channel at 12 bit; 60× oil objective; NA, 1.4). To yield a complete map of the neurons, two-dimensional projections of the image stacks were generated, aligned using Adobe Photoshop (Adobe Systems, San Jose, CA), and dendritic segments assigned to subclasses according to the criteria of Megias et al. (2001). To improve spatial resolution and signal-to-noise ratio, the image stacks were processed using adaptive blind deconvolution (25 iterations; Autoquant, Watervliet, NY). The deconvolved stacks were dissected, and traversing axons and other processes were cropped away and grouped according to their dendritic type. Spine detection and measurements of spine densities, lengths, and volume were performed by algorithm-based evaluation using the automated morphometric software package 3DMA-Neuron (Koh et al., 2002; Weaver et al., 2004) on batches of groups of dendrite segments. For every neuron, all dendritic segments of the respective subclass were combined and spine density calculated by dividing the total number of spines by the total length. Spine length and volume were obtained from a total of 747 dendritic segments from stratum radiatum thick and thin by averaging over the mean spine length and volume of every segment (segment lengths varying between 15 and 150 μm).
Assessment of cell death.
Lactate dehydrogenase (LDH) release into the culture media was determined fluorimetrically using the CytoTox-ONE Homogeneous Membrane Integrity Assay kit (Promega, Madison, WI) according to the manufacturer's protocol. The assay was performed on 96-well microplates and read on Fluostar OPTIMA plate reader (BMG Lab Technologies, Offenburg, Germany) at 544 nm excitation and 590 nm emission. LDH activity was normalized to the EGFP fluorescence, which was determined from the respective fusion protein lysates (from caspase-3 experiments) at different times after infection.
Caspase-3 activity was determined fluorimetrically from the lysates of infected hippocampal slices at different times after infection using EnzChek Caspase-3 assay kit number 1 with N-CBZ-l-aspartyl-l-glutamyl-l-valyl-l-aspartic acid/aminomethyl coumarin (AMC) substrate (Invitrogen) according to the manufacturer's protocol. The assay was performed on 96-well microplates and read on Fluostar OPTIMA plate reader at 355–10 nm excitation, 460 nm emission (for AMC) and 485 nm excitation, and 520 nm emission (for EGFP). Caspase-3 activity was normalized to the EGFP fluorescence of the respective fusion protein lysates.
For determination of DNA fragmentation, DNA was extracted from four to eight hippocampal slices using the TACS apoptotic DNA laddering kit from R & D Systems (Wiesbaden, Germany) according to the manufacturer's protocol. Approximately 1 μg of DNA from each sample was electrophoresed through 1.5% agarose in Tris acetic acid buffer, visualized by ethidium bromide staining, and photographed under UV illumination.
Immunoblot analysis.
Cultured hippocampal slices (at different times after infection) were harvested and sonicated in radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris/HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% NP-40, 0.5% deoxycholate, and 0.1% SDS, pH 8.0) containing protease inhibitors (1 mm PMSF, 10 μg/ml each of leupeptin and pepstatin, 1 mm EGTA) and phosphatase inhibitors (1 mm sodium orthovanadate, 20 mm sodium fluoride, and 1 mm sodium pyrophosphate) and centrifuged for 15 min at 13,000 × g at 4°C. The supernatant (lysate) was collected, frozen, and stored at −80°C. Protein concentration of the samples was determined with BCA (Pierce). Samples (10 μg of protein in lysates) or recombinant tau proteins (0.1 μg of protein) were subjected to SDS-PAGE and transferred to Immobilon-P (Millipore) followed by immunoblotting with various antibodies against tau and GFP. As secondary antibody, peroxidase-coupled anti-mouse or rabbit antiserum was used. Detection used enhanced chemiluminescence using SuperSignal West Dura extended duration substrate (Pierce) and was performed according to the manufacturer's protocol. Quantification of the blots was performed with Gel-Pro Analyzer 4.0 (Media Cybernetics, Silver Spring, MD). To determine quantitatively alterations in EGFP–tau phosphorylation/conformation at different epitopes, all data obtained with phosphorylation-dependent/conformation-dependent antibodies were normalized to total tau levels in the samples, and differences in phosphorylation/conformation between EGFP–PHP tau and EGFP–wt tau were determined.
Sequential extraction of EGFP–tau proteins.
To study the solubility of tau proteins in infected hippocampal slices, we generated a tau solubility profile by applying a sequential extraction approach on hippocampal slices using buffers of increasing stringency (Zhukareva et al., 2004). At each extraction step, samples were sonicated briefly, spun at 45,000 × g for 30 min at 4°C, and supernatants were collected and subjected to immunoblot analysis as described above. The following sequential series of solubilizing buffers with a mixture of protease and phosphatase inhibitors (described above) were used for extraction: (1) buffer A (high salt, 750 mm NaCl, 50 mm Tris buffer, pH 7.2), (2) 1% Triton in buffer A, (3) RIPA buffer (50 mm Tris/HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% NP-40, 0.5% deoxycholate, and 0.1% SDS, pH 8.0), (4) 2% SDS, and (5) 70% formic acid (FA). FA samples were then diluted with distilled water to a 20% final concentration of FA and lyophilized for at least 24 h. Freeze-dried FA samples were reconstituted in the original volume of 0.1 m Tris-HCl, pH 7.5, and adjusted to pH 7.5 with NaOH. The same amount from each extract was loaded per lane (6% of total extract). Quantitative immunoblots were performed using Tau-5 antibody to determine the tau level in each fraction.
Statistical analysis.
Data were expressed as means ± SE (n = 3–5). All experiments were performed in triplicates or duplicates and repeated at least three times. Statistical analysis among experimental groups was performed using paired Student's t test (Origin 7.0; Microcal Software, Northampton, MA). p values are */°p < 0.05, **/°°p < 0.01, and ***/°°°p < 0.001.
Results
Targeted expression of EGFP-tagged human tau in organotypic hippocampal slices permits high-resolution imaging of individual neurons
To analyze the behavior of tau in neurons in an authentic CNS environment, we prepared a panel of EGFP-tagged tau constructs and expressed them in cultured mouse hippocampal slices. The constructs were based on the longest adult (441 aa) and fetal (352 aa) CNS tau isoforms (Fig. 1A). Wild-type tau and PHP tau sequences, which mimic key structural and functional aspects of hyperphosphorylated tau protein (Eidenmüller et al., 2000; Maas et al., 2000; Fath et al., 2002), were prepared and fused amino terminally with EGFP. The constructs were cloned in Sindbis viral vectors, which allows for a targeted transient expression in different types of neurons in hippocampal slice cultures (Ehrengruber et al., 1999; Maletic-Savatic et al., 1999).
Figure 1.
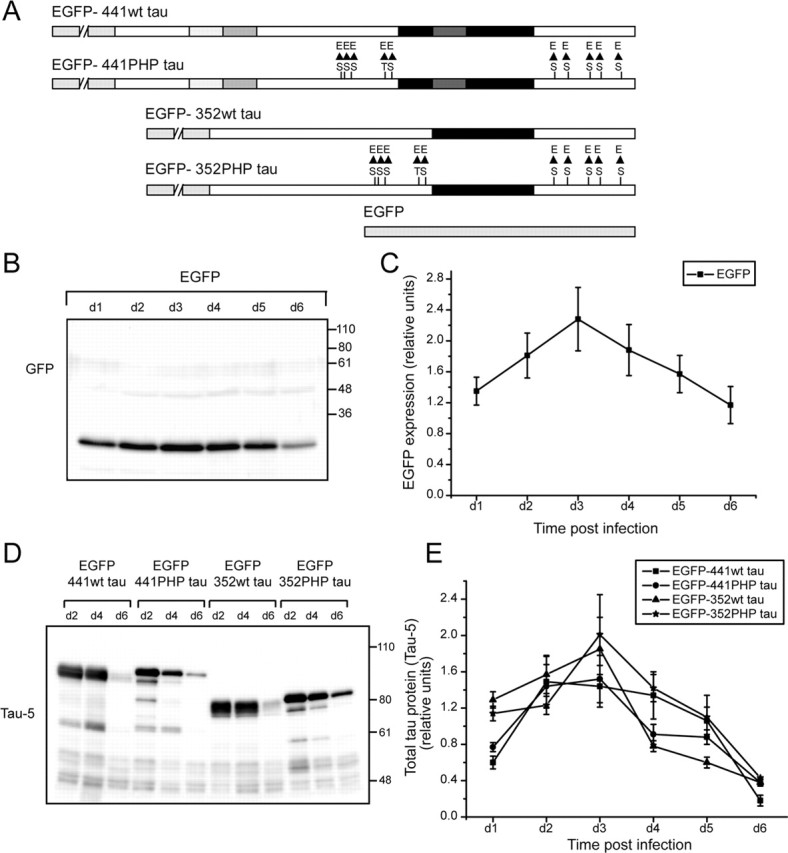
Sindbis-virus-mediated expression of EGFP-tagged human tau constructs in organotypic hippocampal slices. A, Schematic representation of EGFP-tagged human tau constructs. The constructs were based on the longest adult (441 aa) and fetal (352 aa) wild-type tau isoforms with an amino terminally fused EGFP sequence (discontinuous gray box). Serine (S)/threonine (T) residues, which have been substituted with glutamate (E) to create a pseudohyperphosphorylation of these residues, are indicated. As a control, EGFP sequence alone was used. The microtubule-binding repeats of tau are indicated by the thick black box. Adult-specific exons are shaded. B, C, Time course of EGFP expression in infected hippocampal slices. B, Lysates of the slices at different times after infection were prepared and separated by 10% SDS-PAGE and immunoblotted with GFP antibody. C, The graph shows the relative amounts of EGFP expressed at days 1–6 (d1–d6) postinfection. EGFP was detected as early as 7 h postinfection (data not shown), peaked at around day 3, and persisted until 1 week. D, E, Time course of EGFP–tau expression in infected hippocampal slices at d2, d4, and d6 postinfection (D) and the quantitation of results (E). Note that the kinetics of expression of the different tau fusion proteins is very similar to EGFP alone. Lysates of slices at different times after infection were prepared and analyzed by immunoblot using phosphorylation-independent antibody (Tau-5). Data are presented as mean ± SEM from three independent experiments.
To determine the time course of expression, virus was applied onto the hippocampal slices by the droplet method and lysates of the slices at different times after infection were prepared and analyzed by immunoblot. As a control, a construct with EGFP sequence alone was used. EGFP was detected as early as 7 h postinfection (data not shown), peaked at ∼3 d, and persisted until 1 week (Fig. 1B,C). The kinetics of expression of the different tau fusion proteins was very similar to EGFP alone (Fig. 1D,E).
Efficient infection of neurons in all regions of the hippocampal slice was observed 3 d after application of the virus as indicated by the intense EGFP fluorescence (Fig. 2A). At higher magnification, many individual infected CA1 and CA3 pyramidal neurons as well as DG granule neurons were identified by their characteristic morphology and location (Figs. 2B–D). In addition, some interneurons in all regions were also infected. EGFP fluorescence was present in cell bodies, processes, and dendritic spines of the proximal and distal dendrites and allowed high-resolution imaging at the level of individual spines for several days (Fig. 2E–G). At later time points after infection (>5 d), many fluorescent cells appeared to have died in all hippocampal regions (data not shown).
Figure 2.
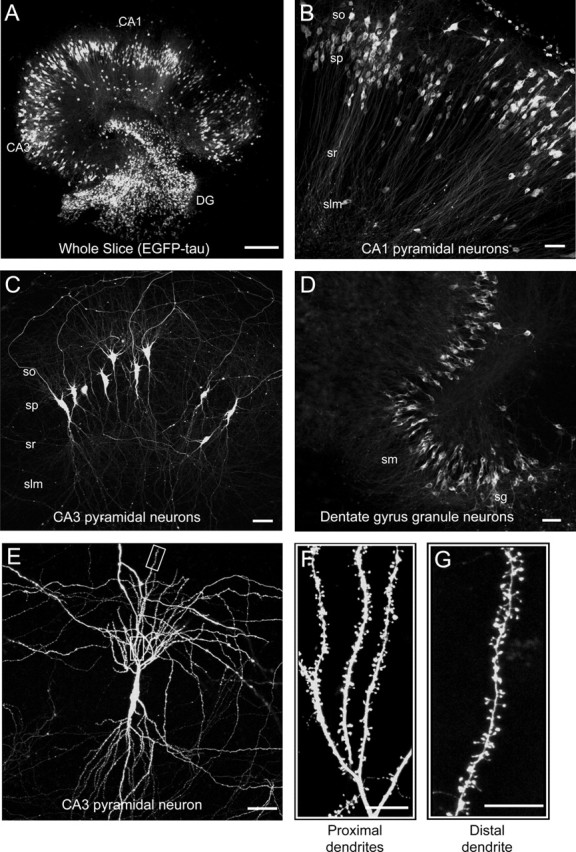
Distribution of infected neurons in cultured hippocampal slices. A–D, Immunofluorescence micrographs of the whole slice (A), CA1 region (B), CA3 region (C), and DG region (D) of EGFP–tau-infected slices 3 d postinfection. Note efficient infection of neurons in all regions. E–G, High-power magnifications of a single CA3 pyramidal neuron (E) and sections of the proximal (F) and distal (G) dendrites. Note intense EGFP fluorescence in all cellular compartments. Slices were fixed and processed for immunofluorescence as described in Materials and Methods. so, Stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; slm, stratum lacunosum-moleculare; sm, stratum moleculare; sg, stratum granulosum. Scale bars: A, 300 μm; B–E, 50 μm; F, G, 10 μm.
Tau-dependent neurodegeneration occurs region specifically in organotypic culture
To determine a potential cytotoxic effect of tau expression in cultured hippocampal slices, release of LDH as a marker for plasma membrane damage was analyzed fluorimetrically at different times after infection. LDH activity was normalized to the EGFP fluorescence, which was determined from the respective lysates. In most experiments, expression of adult or fetal wt tau caused only a small increase in LDH release compared with EGFP alone at 3 and 4 d postinfection (Fig. 3A). In contrast, PHP tau expression resulted in an ∼100% increase in LDH release. Both adult and fetal PHP tau induced a significant increase in comparison to the respective wild-type isoform. We did not observe an obvious difference between the different isoforms (adult wt vs fetal wt tau or adult PHP vs fetal PHP tau) or at different times after infection.
Figure 3.

Effect of tau expression on LDH release, caspase-3 activity, and DNA fragmentation. A, Release of LDH from the infected hippocampal slices at 3 (left) and 4 (right) days postinfection. Both adult (441) and fetal (352) PHP tau induce a significant increase compared with the respective wt isoform or EGFP alone. B, Caspase-3 activity after 3 (left) and 4 (right) days postinfection. Caspase-3 activity is significantly increased for the PHP tau constructs compared with wt tau or EGFP alone in the lysates of infected hippocampal slices at different times after infection (day 3–4). C, DNA fragmentation after 3 d postinfection or treatment with staurosporine (Stp). The pattern of DNA damage for the PHP tau constructs was similar to that induced by the typical apoptosis inducer staurosporine (10 μm; 6 h). LDH and caspase-3 activity were assayed fluorimetrically and normalized to the EGFP fluorescence of the respective fusion protein. All values are shown as the mean ± SEM from at least three independent experiments. Data for EGFP–wt or PHP tau are expressed as percentage of the value for EGFP alone. Data were analyzed using Student's t test and considered to be significantly different from EGFP–wt tau at ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. For the assay for DNA fragmentation, samples were separated by agarose gel electrophoresis on 1.5% agarose. Similar results were obtained by two independent experiments. M, Marker.
To determine the mechanisms that are associated with the PHP tau-induced neurodegeneration, activation of caspase-3 as a marker for apoptotic events was assayed fluorimetrically and normalized to the EGFP fluorescence of the respective fusion protein. Caspase-3 activity was significantly increased for the PHP tau constructs compared with wt tau or EGFP alone in the infected hippocampal slices (Fig. 3B). The pattern of caspase-3 activation for the different isoforms and time points after infection was very similar to that of LDH release, indicating that the neurodegenerative effect of PHP tau is closely associated with apoptotic mechanisms. Support for an apoptotic contribution to PHP tau-induced cell death was provided by the appearance of DNA fragmentation revealed by agarose gel electrophoresis (Fig. 3C). The fragmentation pattern was very similar to the fragmentation induced by the typical apoptosis inducer staurosporine (Gianinazzi et al., 2004).
To determine the spatiotemporal dynamics of tau-induced neurodegeneration, we performed live imaging of infected neurons expressing the EGFP-tagged constructs in the CA1, CA3, and DG regions over a period of 4 d. We observed a time-dependent loss of individual neurons in all hippocampal regions and with all constructs (Fig. 4A,B, asterisks). In addition, in CA3 and DG, many PHP tau-expressing neurons developed a “ballooned” phenotype characterized by a swollen neuronal perikaryon starting at day 3 postinfection for the adult isoform (Fig. 4B, arrowheads) and day 4 for the fetal isoform. Ballooned neurons with and without apparent processes were observed. It was not possible to preserve ballooned neurons by standard fixation procedures, suggesting that they had lost their contacts and cytoskeletal integrity. It should be noted that the development of a ballooned phenotype is considered to be distinct from apoptosis (Gleckman et al., 1999), indicating that tau-induced cell death involves apoptotic as well as nonapoptotic mechanisms.
Figure 4.
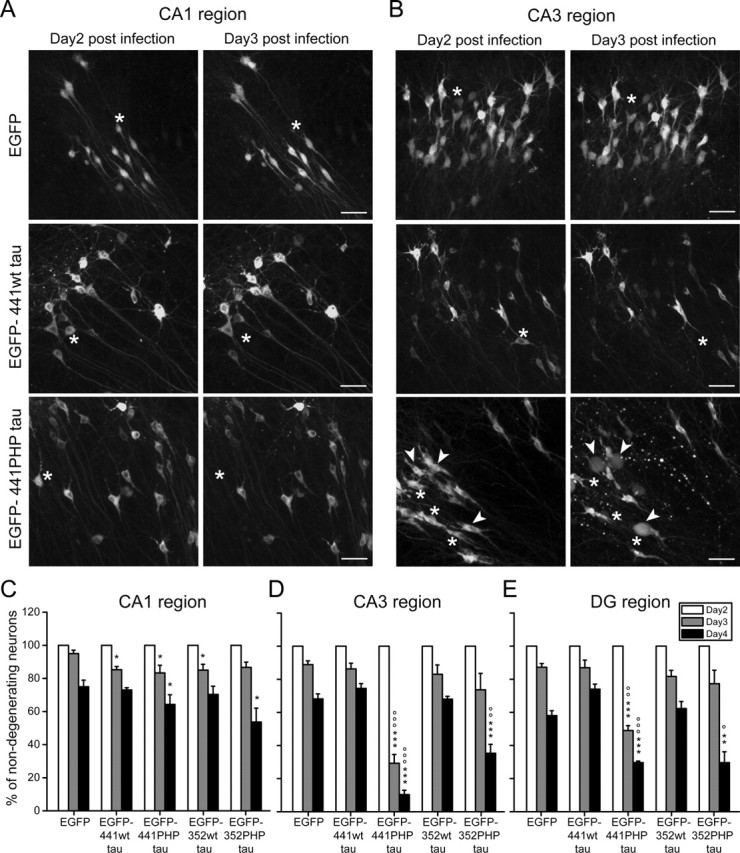
Live imaging of the effect of tau expression on the survival of individual neurons in hippocampal slices. A, B, Fluorescence micrographs of the CA1 region (A) and CA3 region (B) of living hippocampal slices 2 and 3 d postinfection. Time-dependent loss of individual neurons was observed in all hippocampal regions (A, B, asterisks) and with all constructs, which appeared higher in the CA3 region for PHP tau (B). In addition, in CA3 and DG, many PHP tau expressing neurons developed a ballooned phenotype characterized by a swollen neuronal perikaryon with or without processes (arrowheads). Scale bars: A, B, 50 μm. C–E, Percentage of nondegenerating neurons (i.e., neurons with unaffected perikarya and processes) at days 2, 3, and 4 postinfection and expressed relative to the number at day 2 postinfection. In CA3 (D) and DG (E) regions, but not in the CA1 (C) region, the number of nondegenerating neurons expressing the PHP tau isoforms dramatically decreased compared with neurons expressing EGFP alone or wt tau isoforms. All values are shown as the mean ± SEM from at least three independent experiments (C–E). The data for EGFP alone or EGFP–wt or EGFP–PHP tau (at day 3 or day 4) are expressed as percentage of the respective value for day 2 postinfection. Data were analyzed using Student's t test and considered to be significantly different from EGFP alone at ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 or from EGFP–wt tau at °p < 0.05, °°p < 0.01, and °°°p < 0.001.
To quantitate the extent of degeneration, the number of nondegenerating neurons (i.e., neurons without any swelling having processes without beading) was determined over time (day 3 and 4) and expressed relative to the number at day 2 postinfection from the same microscopic field in different regions for all constructs (Fig. 4C–E). In the CA1 region, tau expression resulted in a slight reduction of the number of nondegenerating neurons compared with neurons expressing EGFP alone (Fig. 4C). We did not observe a difference between wt tau- and PHP tau-expressing neurons. In contrast, in CA3 and DG regions, the number of nondegenerating neurons expressing the PHP tau isoforms dramatically decreased compared with neurons expressing EGFP alone or wt tau isoforms (Fig. 4D,E). Neurons expressing the adult isoform of PHP tau degenerated earlier (day 3) than fetal PHP tau-expressing neurons, in which the majority of neurons were degenerated only at day 4.
Collectively, the data indicate that the PHP modification induces a region-specific neurodegeneration in the CA3 and DG subfields of hippocampal slices and that cell death involves both apoptotic and nonapoptotic mechanisms.
Spine density and spine morphology are stable against tau-induced degeneration
In AD, alterations in synaptic integrity and plasticity have been reported as an early event (Selkoe, 2002; Coleman and Yao, 2003); however, it is unclear whether synaptic changes are attributable to the amyloid or the tau pathology. To determine tau-dependent effects on spine density and spine morphology, EGFP–tau-expressing neurons in the CA1 and CA3 regions of organotypic hippocampal slices at day 3 postinfection were analyzed. At these conditions, PHP tau-induced neurodegeneration was evident in the CA3 region but not in CA1 (Fig. 4). We acquired high-resolution images of complete neurons, which allows for detailed spine analysis of the complete dendritic arbors and every type of dendrite. To avoid restricting the evaluation to a too-narrow domain a priori, we included the total dendritic segments of the neuron in the analysis. The segments were assigned to five to seven subclasses to confine the typically high variance of spine densities within parts of the dendritic field (Fig. 5A). Spine-related parameters were determined by automated algorithm-based morphometry as described in Materials and Methods. Spine densities in the different dendritic subclasses ranged between 0.5 (stratum lucidum) and 2.5 per micrometer (stratum radiatum). They were maximal in the proximal stratum radiatum and decreased distally in the CA1 neurons. We did not observe an obvious difference in spine densities between wt and PHP tau-expressing neurons for both isoforms in any hippocampal region or dendritic subclass (Fig. 5B,C). The mean length and volume of spines were 0.94 μm (±0.12 μm; SEM) and 0.41 μm3 (±0.09 μm3; SEM), respectively, for all dendritic segments evaluated and similar for wt and PHP tau-expressing neurons (Fig. 5D).
Figure 5.
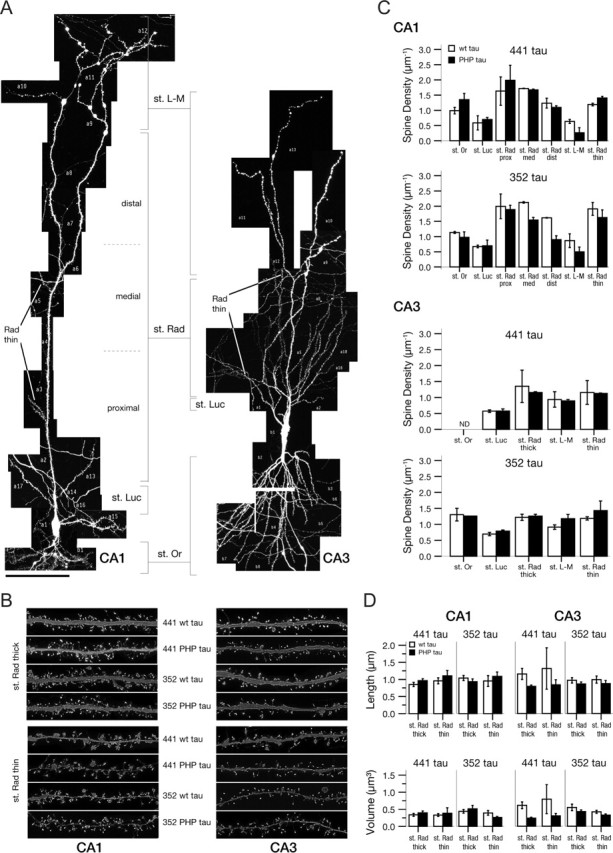
Spine density and spine morphology of tau-expressing neurons in hippocampal slices. A, Complete map of an individual neuron of the CA1 (left) and CA3 (right) region infected with EGFP–tau. Segments are assigned to dendritic subclasses. Scale bar, 100 μm. B, Representative dendritic segments after deconvolution from stratum radiatum thick and thin subclasses from CA1 and CA3 pyramidal neurons expressing tau constructs as indicated. Each frame has a width of 30 μm. C, Spine density in dendritic subclasses of CA1 and CA3 pyramidal neurons expressing adult (441) and fetal (352) wt and PHP tau constructs. Data are based on the analysis of the complete dendritic arbor of two neurons per construct and per hippocampal region. Mean and range are indicated. D, Spine length and volume in stratum radiatum thick and thin subclasses of CA1 and CA3 pyramidal neurons expressing adult (441) and fetal (352) wt and PHP tau constructs. Data were analyzed as described in Materials and Methods from a total of 747 dendritic segments. All values are shown as the mean ± SEM. St. Or, Stratum oriens; st. Luc, stratum lucidum; st. Rad., stratum radiatum; st. L-M, stratum lacunosum-moleculare; prox, proximal; med, medial; dist, distal.
The data suggest that synaptic integrity is remarkably stable against tau-dependent degeneration, providing evidence that alterations in synaptic plasticity in AD are directly caused by the amyloid pathology rather than by tau-mediated mechanisms.
Disease-like tau modifications cause accumulation of insoluble tau, induce pathological tau conformation, and affect tau phosphorylation at selected sites
Filamentous tau aggregates that can be biochemically isolated from diseased brain tissue are hallmarks of tauopathies such as Alzheimer's disease (Selkoe et al., 1982). To determine whether tau becomes insoluble in infected slices, we generated a tau solubility profile by applying a sequential extraction approach on hippocampal slices using buffers of increasing stringency (Fig. 6A) (Zhukareva et al., 2004). In wt tau-infected slices, the majority of tau was soluble and present in the high salt extract (>80%), and almost no insoluble tau (<1%) was detected. In contrast, a significant portion of PHP tau was insoluble (5–6%), indicating the presence of aggregated tau. Tau was not detected in the 70% formic acid fraction implicating the absence of highly insoluble tau species. Interestingly, fetal tau isoforms (wt and PHP) showed a different solubility profile compared with the respective adult isoforms with the fetal tau isoforms generally requiring a higher stringency buffer for extraction.
Figure 6.
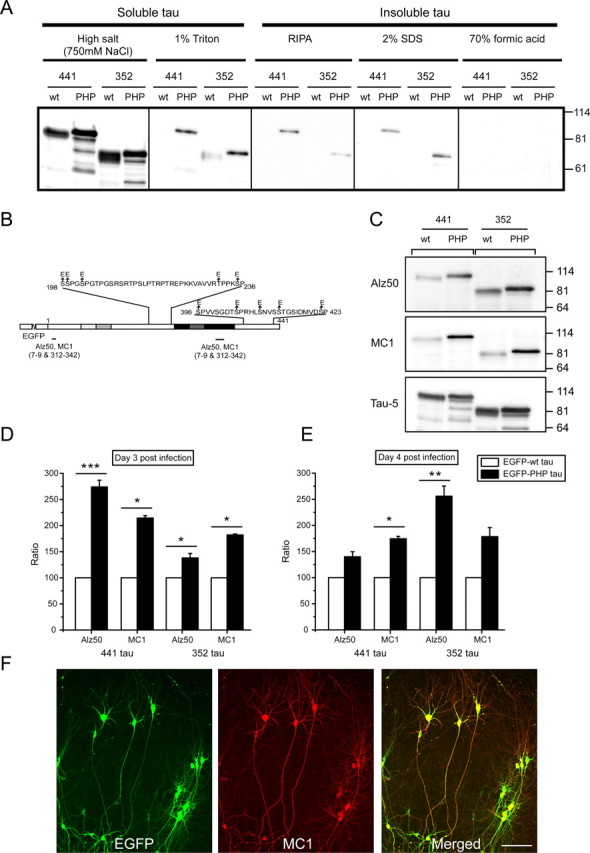
Accumulation of insoluble tau and induction of pathological tau conformation after targeted tau expression. A, Tau solubility profile from lysates of infected hippocampal slices. The profile was generated using buffers of increasing stringency: high salt, 1% Triton, RIPA buffer, 2% SDS, and 70% formic acid. In wt tau (441 and 352 isoform)-infected slices, the majority of tau was soluble and present in the high salt extract with almost no insoluble tau. In contrast, a significant portion of PHP tau isoforms was insoluble. No tau was detected in the 70% formic acid fraction. An equal amount of the material from each extraction step was loaded, and Tau-5 antibody was used for immunoblot analysis. B, Schematic representation that indicates the epitopes of the conformational-dependent antibodies Alz50 and MC1. C, Immunoblot of lysates of infected hippocampal slices. The PHP tau proteins showed a strong increase in immunoreactivity for Alz50 and MC1 antibodies compared with the respective wt proteins. D, E, Quantitation of the immunoblots. Conformational alteration for PHP tau was increased for both adult and fetal tau isoforms at different times after infection (day 3 and day 4). To determine quantitatively the alterations in EGFP–tau conformation, all data obtained with conformation-dependent antibodies were normalized to total tau (Tau-5) levels in the samples, and then the difference in conformation between EGFP–PHP tau and EGFP–wt tau was determined. Data for EGFP–PHP tau are expressed as percentage of the value for EGFP–wt tau (wt tau set as 100%). All values are shown as the mean ± SEM from three independent experiments. Data were analyzed using Student's t test and considered to be significantly different from EGFP-wt tau at ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. F, Double immunofluorescence staining of all infected neurons stained with the MC1 antibody showed a subcellular distribution closely reflecting the EGFP fluorescence. Scale bar, 100 μm.
To test whether the accumulation of insoluble tau is associated with an induction of pathological tau conformation, immunoblots with Alz50 and MC1 antibodies were performed. Both antibodies recognize a conformational epitope in AD brains that depends on two discontinuous portions of the tau sequence as shown in Figure 6B (Jicha et al., 1997) and that appears to be one of the earliest pathological alterations of tau in AD (Wolozin et al., 1986; Uboga and Price, 2000; Weaver et al., 2000). Both antibodies showed slight reactivity against recombinant tau isoforms (data not shown). In lysates of infected hippocampal slices, a strong increase in immunoreactivity for the PHP tau proteins was observed, whereas the signal for the wt proteins remained low (Fig. 6C). Quantitative analysis confirmed the conformational alteration for PHP tau for both adult and fetal tau isoforms at different times after infection (Fig. 6D,E). Practically all infected neurons were stained with the MC1 antibody with a subcellular distribution that closely reflected the EGFP fluorescence (Fig. 6F), indicating that this pathological alteration occurs in all processes and all infected cells.
Abnormal phosphorylation may be involved to generate a conformational state that is typical for the disease. To determine whether pseudohyperphosphorylation of tau induces abnormal phosphorylation in hippocampal slices expressing tau constructs, immunoblot analysis using a panel of phosphorylation-dependent and site-specific antibodies against selected serine and threonine residues was performed (Fig. 7A). None of these antibodies was reactive against recombinant wt or PHP tau with the exception of an antibody against phosphorylated S356, which was less reactive with PHP tau isoforms compared with wild-type tau (10–40% for the fetal and adult isoform, respectively). In lysates of infected hippocampal slices, we observed a change in phosphorylation at several sites compared with the respective wt tau constructs (Fig. 7B). A strong increase in phosphorylation was seen with both adult and fetal PHP tau isoforms at T212, S214, and S262. In contrast, a marked decrease was observed at T205. The amount of total tau as determined using a phosphorylation-independent antibody (Tau-5) was very similar for both isoforms (wt and PHP tau). Quantitative analysis confirmed increased phosphorylation for PHP tau (30–150% compared with the respective wt tau isoform) at T212, S214, and S262 for both adult and fetal tau isoforms at different times after infection (Figs. 7C–F). A significant increase in phosphorylation at S356 (50–60%) was observed for the fetal PHP tau isoform (Fig. 7D,F). A slight increase (10–30%) was also observed for T181. At all conditions, phosphorylation at T205 was strongly decreased (40–70%) for PHP tau.
Figure 7.
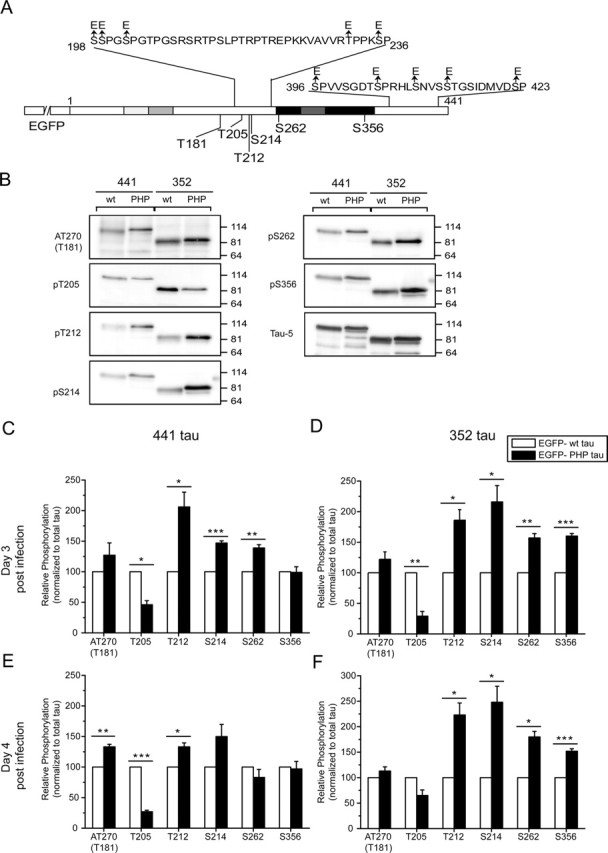
Phosphorylation state of tau in infected neurons. A, Schematic representation that indicates the epitopes of phosphorylation-sensitive antibodies detecting serine (S) and threonine (T) residues that were not mutated in PHP tau. B, Immunoblot of lysates of infected hippocampal slices with different phosphorylation-sensitive antibodies. Note that some antibodies show a strong increase in the staining of the PHP isoforms relative to total tau (Tau-5). C–F, Quantitation of the immunoblots. An increased phosphorylation for PHP tau was detected at T212, S214, and S262 for both adult and fetal tau isoforms compared with the respective wt tau isoform at different times after infection. At all conditions, phosphorylation at T205 was strongly decreased for PHP tau. To determine quantitatively the alterations in EGFP–tau phosphorylation at different epitopes, all data obtained with phosphorylation-dependent antibodies were normalized to total tau (Tau-5) levels in the samples, and then the difference in phosphorylation between EGFP–PHP tau and EGFP–wt tau was determined. The data for EGFP–PHP tau are expressed as percentage of the value for EGFP–wt tau (wt tau set as 100%). All values are shown as the mean ± SEM from three independent experiments. Data were analyzed using Student's t test and considered to be significantly different from EGFP–wt tau at ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
To determine the distribution of exogenously expressed tau and the different phosphoisoforms, immunohistochemistry of infected hippocampal slices was performed. Tau was present in all infected (EGFP-positive) neurons and exhibited a strong staining in processes and cell bodies as detected using a phosphorylation-independent polyclonal antibody against human tau (Fig. 8A). Immunostainings with the phosphorylation-dependent and site-specific antibodies confirmed phosphorylation of the exogenously expressed tau proteins at the respective sites. With all antibodies, only infected neurons in all regions were immunoreactive (Fig. 8B–D and data not shown). Within the population of stained neurons, marked differences in the antibody stainings compared with the EGFP fluorescence were observed. In particular, most interneurons in different zones (e.g., in the CA1 region) showed only weak or no immunoreactivity against selected antibodies (Fig. 8B–D, arrowheads); however, exceptions were also observed (Fig. 8B, asterisk). For some antibodies, staining was prominent only in cell bodies (Fig. 8C), whereas others also showed staining in processes (Fig. 8B,D).
Figure 8.
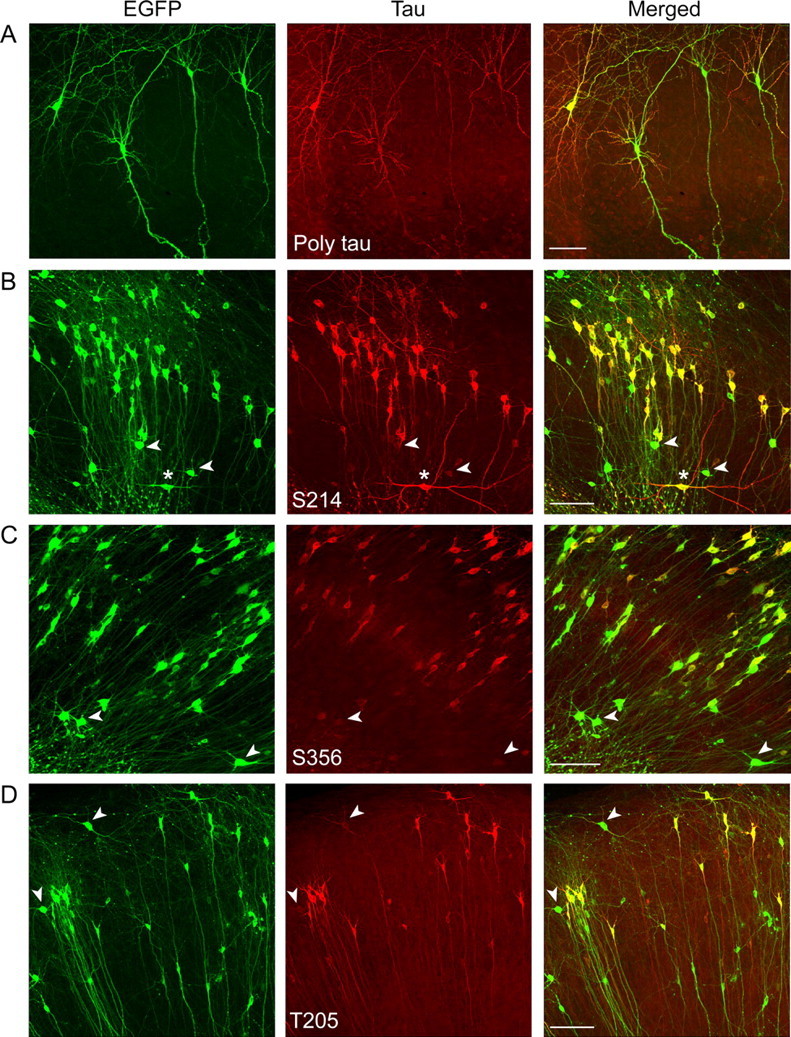
Distribution of the infected neurons that were immunoreactive with different phosphorylation-sensitive antibodies. A–D, Representative images for the CA1 region of hippocampal slices with EGFP fluorescence (left), stained for the respective antibodies (middle) and overlay (right). With all antibodies, only infected neurons were immunoreactive (B–D). Note that most interneurons in different zones in the CA1 region showed only weak or no immunoreactivity against selected antibodies (B–D, arrowheads) with some exceptions (B, asterisk). Some antibodies stained most prominently the cell bodies (C), whereas others showed staining also in processes (B, D). Scale bars, 100 μm.
The data indicate that abnormal phosphorylation at selected sites, induction of pathological tau conformation and formation of tau aggregates are closely associated.
Discussion
Imaging and evaluation of synaptic morphology
We expressed the cytoskeletal protein tau tagged with EGFP using the neurotropic, recombinant Sindbis virus, which provides an efficient, transient expression of the fusion proteins in individual neurons of hippocampal slices. Because EGFP expression revealed the detailed morphology of living neurons, we were able to follow and image structural changes of the same neuron over a time course of several days. EGFP labeling also allowed us a high-resolution imaging and algorithm-based evaluation of the neuronal morphology at the level of individual spines in fixed tissue.
A number of studies have indicated alterations in synaptic integrity and plasticity as an early phenotypic manifestation in the pathogenesis of AD (Selkoe, 2002; Coleman and Yao, 2003). Synapse density is significantly decreased in the AD brain (Terry et al., 1991), and the level of synapse loss provides a better correlate of the pattern and severity of cognitive impairment than amyloid plaques and NFTs (Terry et al., 1991). Also, in the hippocampus, there is a progressive loss of dendritic spines from pyramidal neurons in AD (el Hachimi and Foncin, 1990; Ferrer and Gullotta, 1990). In addition to the loss of synapses, the dendrites of affected neurons in AD show a dystrophic pattern and an accumulation of phosphorylated tau epitopes (McKee et al., 1989; Kowall and McKee, 1993; Su et al., 1994a). By determining spine density and spine morphology of tau-expressing neurons, we demonstrated that PHP tau expression does not induce major alterations in dendritic spines of the infected neurons at the conditions in which neuronal degeneration was evident in the hippocampal slices. The data suggest that in our tau pathology model, synaptic connectivity is remarkably stable against tau-mediated degeneration. Thus, the decrease in synaptic density in AD is probably not caused by a specific tau-dependent mechanism but is caused by the amyloid pathology.
Mechanisms of cell death
AD and other neurodegenerative diseases are characterized by progressive cell loss of specific neuronal populations, but the underlying molecular mechanisms that are responsible for the temporally and spatially defined cell death are still unresolved. Postmortem analysis of human brain has shown apoptotic DNA fragmentation (Su et al., 1994b; Lassmann et al., 1995; Smale et al., 1995) and elevated levels of caspase-3 activity (Gervais et al., 1999; Su et al., 2001) in AD patients. On the other hand, several animal models for tauopathies provided evidence for nonapoptotic neurodegeneration (Allen et al., 2002; Zehr et al., 2004). However, because of the chronic nature of AD and other tauopathies, the relative contribution of apoptosis is difficult to assess and other mechanisms may also contribute to the massive neuronal death. Genetic analyses have established a clear cause-and-effect relationship between tau alteration and neuronal cell death and dementia in FTDP-17, but the mechanisms, which render tau to become a toxic agent, are still unclear.
We observed that disease-like tau modification induced extensive cell death in organotypic hippocampal cultures, which is associated with apoptotic mechanisms as indicated by an increase in caspase-3 activity and typical DNA fragmentation. In addition, many neurons specifically degenerated by establishing a ballooned phenotype, as has also been reported for patients with AD and other tauopathies (Dickson et al., 1986; Fujino et al., 2004). In Pick's disease, no clear association between neuronal DNA fragmentation and the presence of structural abnormalities such as ballooned neurons was observed (Gleckman et al., 1999), indicating that apoptosis and the development of a ballooned phenotype represent distinct mechanisms of cell death. Thus, our data provide evidence that tau-dependent degeneration involves both apoptotic as well as nonapoptotic mechanisms.
We observed that neurodegeneration induced by PHP tau occurs predominantly in the CA3 and DG region of the hippocampal slices. This indicates that disease-relevant tau modification do not cause general toxicity but exert their neurodegenerative effect in a region- and neuron-type specific manner. In AD and after hypoxic/ischemic insults, CA1 region typically demonstrates profound vulnerability to neuronal cell death (Schmidt-Kastner and Freund, 1991; West et al., 1994). Regional susceptibility similar to that seen in our experiments has been reported with human immunodeficiency virus transactivating protein TAT, prenatal stress, and okadaic acid, in which pyramidal cells in the CA3 region and granule cells in DG are more sensitive compared with pyramidal cells in CA1 (Runden et al., 1998; Maragos et al., 2003; Zhu et al., 2004). The mechanism of okadaic acid toxicity is likely attributable to sustained activation of the mitogen-activated protein kinase (MAP kinase) pathway (Runden et al., 1998). Sustained MAP kinase activation is seen after brief ischemic episodes in the CA3 region of the hippocampus (Hu and Wieloch, 1994), and the MAP kinase family is one group of kinases that has been implicated in the phosphorylation of tau in AD (Drewes et al., 1992; Arendt et al., 1995; Pei et al., 2002) and in other neuronal death models (Subramaniam et al., 2004).
The neurotoxic effect of PHP tau may be caused by a selective disturbance of signal transduction cascades in CA3 pyramidal neurons and DG granule neurons; however, the precise molecular events and the relationship to AD remain to be shown. It should be taken into account that our approach results in the presence of tau in all neuronal compartments, whereas endogenous tau is enriched in the axon. This may cause the slight toxicity observed after expression of wt tau but would not explain the obvious difference between wt tau and PHP tau. Furthermore, a relocation of tau from the axonal to the somatodendritic compartment where neurofibrillary tangles become apparent is a characteristic event during the development of tau pathology. Thus, the presence of tau in the “wrong” compartment could well be relevant for disease progression.
Although PHP tau mimics key aspects of hyperphosphorylation, including the induction of structural and functional changes, it does not longer participate in the complex phosphorylation/dephosphorylation events occurring with “normal” tau protein. Whether this is relevant for tau-mediated toxicity in disease in which dephosphorylation of tau is drastically reduced (Matsuo et al., 1994) remains to be shown.
Abnormal phosphorylation and tau aggregation
Constructs with pseudophosphorylated residues provide a tool to determine the effect of disease-like tau modifications on tau phosphorylation at selected sites. Here, we show that pseudohyperphosphorylation increases phosphorylation at several other sites including T181, T212, S214, S262, and S356. Some of these sites are known to be abnormally hyperphosphorylated in AD, constitute recognition sites for some AD diagnostic antibodies, and are also known to correlate with the severity of neuronal pathology (Augustinack et al., 2002; Lauckner et al., 2003). Interestingly, in one case (T205), a decreased phosphorylation was observed. Previously, it has been shown that phosphorylated T205 is completely and specifically dephosphorylated by protein phosphatase 2A as a result of microtubule depolymerization (Merrick et al., 1996). This may suggest that dephosphorylation at this residue is indirectly induced by tau modifications as a result of a disturbed microtubule array in PHP tau-infected neurons. Changed phosphorylation of PHP tau was associated with an induction of a pathological tau conformation as recognized by Alz50 and MC1 antibodies. A conformational change defined by an increased immunoreactivity against these antibodies is considered to be one of the earliest pathological alterations of tau in AD (Wolozin et al., 1986; Uboga and Price, 2000; Weaver et al., 2000).
The contribution of tau modification and tau aggregation to the neurodegenerative process remains elusive. Both neuronal loss and NFTs increase in parallel with the duration and severity of illness, but the amount of neuronal loss exceeds by manyfold the amount of NFTs accumulated (Gomez-Isla et al., 1997). Unfortunately, in the disease and all mouse models, both features are closely interweaved, which precludes analyzing the contribution of each factor. Several cell culture and invertebrate animal models (Drosophila melanogaster and Caenorhabditis elegans) now provide converging evidence that aberrant tau phosphorylation in the absence of large tau aggregates can cause a neurodegenerative phenotype similar to that seen in human tauopathies (for review, see Brandt et al., 2005). In agreement, PHP tau also exerted a neurotoxic effect in neural cells (PC12 cells and human model neurons), with no evidence for the formation of tau aggregates (Fath et al., 2002). When expressed in cultured hippocampal slices, some aggregation of PHP tau was observed. Interestingly, in contrast to cell free assembly reactions in which the PHP modification inhibited aggregation (Eidenmüller et al., 2000), increased aggregation of these constructs was observed in the slices. This indicates that additional modifications (e.g., phosphorylation events as described above) and cellular factors exist that specifically act on PHP tau in the neurons and promote tau aggregation and NFT formation. In fact, data from several mouse models suggest that Aβ42 fibrils or mutant amyloid precursor protein accelerate the formation of NFTs in mouse strains expressing FTDP-17 mutated tau (Götz et al., 2001; Lewis et al., 2001). It will be interesting to systematically analyze the effect of AD-relevant proteins on the aggregation of wt tau by infecting organotypic slices from transgenic animals.
Footnotes
This work was supported by the Ministry for Science and Culture of Lower Saxony. We thank Dr. Sondra Schlesinger (Washington University School of Medicine, St. Louis, MO) for the generous gift of pSinRep5 vector and helper DH (26S) DNA and Dr. Peter Davies (Albert Einstein College of Medicine, Bronx, NY) for Alz-50 and MC1 antibody. We thank Dr. Brent Lindquist (Stony Brook University, Stony Brook, NY) for providing the source code for 3DMA-Neuron and Dr. Christina Weaver (Mount Sinai School of Medicine, NY) for helpful suggestions on the usage of the software. We appreciate the help of Nataliya Golovyashkina, Julian Heidelberg, Ann-Theres Lütkhoff, Jörg Rathert, and Christian Stößel with morphological analyses.
References
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M (2002). Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22:9340–9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Holzer M, Grossmann A, Zedlick D, Bruckner MK (1995). Increased expression and subcellular translocation of the mitogen activated protein kinase kinase and mitogen-activated protein kinase in Alzheimer's disease. Neuroscience 68:5–18. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT (2002). Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol (Berl) 103:26–35. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, Strothjohann M (1994). Abnormally phosphorylated tau protein related to the formation of neurofibrillary tangles and neuropil threads in the cerebral cortex of sheep and goat. Neurosci Lett 171:1–4. [DOI] [PubMed] [Google Scholar]
- Brandt R, Hundelt M, Shahani N (2005). Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta 1739:331–354. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Yao PJ (2003). Synaptic slaughter in Alzheimer's disease. Neurobiol Aging 24:1023–1027. [DOI] [PubMed] [Google Scholar]
- Davis BG (2004). Mimicking posttranslational modifications of proteins. Science 303:480–482. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Yen SH, Suzuki KI, Davies P, Garcia JH, Hirano A (1986). Ballooned neurons in select neurodegenerative diseases contain phosphorylated neurofilament epitopes. Acta Neuropathol (Berl) 71:216–223. [DOI] [PubMed] [Google Scholar]
- Drewes G, Lichtenberg-Kraag B, Doring F, Mandelkow EM, Biernat J, Goris J, Doree M, Mandelkow E (1992). Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J 11:2131–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrengruber MU, Lundstrom K, Schweitzer C, Heuss C, Schlesinger S, Gahwiler BH (1999). Recombinant Semliki Forest virus and Sindbis virus efficiently infect neurons in hippocampal slice cultures. Proc Natl Acad Sci USA 96:7041–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidenmüller J, Fath T, Hellwig A, Reed J, Sontag E, Brandt R (2000). Structural and functional implications of tau hyperphosphorylation: information from phosphorylation-mimicking mutated tau proteins. Biochemistry 39:13166–13175. [DOI] [PubMed] [Google Scholar]
- Eidenmüller J, Fath T, Maas T, Pool M, Sontag E, Brandt R (2001). Phosphorylation-mimicking glutamate clusters in the proline-rich region are sufficient to simulate the functional deficiencies of hyperphosphorylated tau protein. Biochem J 357:759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Hachimi KH, Foncin JF (1990). [Loss of dendritic spines in Alzheimer's disease]. C R Acad Sci III 311:397–402. [PubMed] [Google Scholar]
- Fath T, Eidenmüller J, Brandt R (2002). Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J Neurosci 22:9733–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Gullotta F (1990). Down's syndrome and Alzheimer's disease: dendritic spine counts in the hippocampus. Acta Neuropathol (Berl) 79:680–685. [DOI] [PubMed] [Google Scholar]
- Fujino Y, Delucia MW, Davies P, Dickson DW (2004). Ballooned neurones in the limbic lobe are associated with Alzheimer type pathology and lack diagnostic specificity. Neuropathol Appl Neurobiol 30:676–682. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW (1999). Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 97:395–406. [DOI] [PubMed] [Google Scholar]
- Gianinazzi C, Grandgirard D, Simon F, Imboden H, Joss P, Tauber MG, Leib SL (2004). Apoptosis of hippocampal neurons in organotypic slice culture models: direct effect of bacteria revisited. J Neuropathol Exp Neurol 63:610–617. [DOI] [PubMed] [Google Scholar]
- Gleckman AM, Jiang Z, Liu Y, Smith TW (1999). Neuronal and glial DNA fragmentation in Pick's disease. Acta Neuropathol (Berl) 98:55–61. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol 41:17–24. [DOI] [PubMed] [Google Scholar]
- Götz J, Chen F, van Dorpe J, Nitsch RM (2001). Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293:1491–1495. [DOI] [PubMed] [Google Scholar]
- Holzer M, Holzapfel HP, Zedlick D, Bruckner MK, Arendt T (1994). Abnormally phosphorylated tau protein in Alzheimer's disease: heterogeneity of individual regional distribution and relationship to clinical severity. Neuroscience 63:499–516. [DOI] [PubMed] [Google Scholar]
- Hu BR, Wieloch T (1994). Tyrosine phosphorylation and activation of mitogen-activated protein kinase in the rat brain following transient cerebral ischemia. J Neurochem 62:1357–1367. [DOI] [PubMed] [Google Scholar]
- Huffine ME, Scholtz JM (1996). Energetic implications for protein phosphorylation. Conformational stability of HPr variants that mimic phosphorylated forms. J Biol Chem 271:28898–28902. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I (2005). Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739:198–210. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Bowser R, Kazam IG, Davies P (1997). Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 48:128–132. [DOI] [PubMed] [Google Scholar]
- Koh IY, Lindquist WB, Zito K, Nimchinsky EA, Svoboda K (2002). An image analysis algorithm for dendritic spines. Neural Comput 14:1283–1310. [DOI] [PubMed] [Google Scholar]
- Kowall NW, McKee AC (1993). The histopathology of neuronal degeneration and plasticity in Alzheimer disease. Adv Neurol 59:5–33. [PubMed] [Google Scholar]
- Lassmann H, Bancher C, Breitschopf H, Wegiel J, Bobinski M, Jellinger K, Wisniewski HM (1995). Cell death in Alzheimer's disease evaluated by DNA fragmentation in situ Acta Neuropathol (Berl) 89:35–41. [DOI] [PubMed] [Google Scholar]
- Lauckner J, Frey P, Geula C (2003). Comparative distribution of tau phosphorylated at Ser262 in pre-tangles and tangles. Neurobiol Aging 24:767–776. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ (2001). Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E (2001). Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293:1487–1491. [DOI] [PubMed] [Google Scholar]
- Maas T, Eidenmüller J, Brandt R (2000). Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J Biol Chem 275:15733–15740. [DOI] [PubMed] [Google Scholar]
- Maciejewski PM, Peterson FC, Anderson PJ, Brooks CL (1995). Mutation of serine 90 to glutamic acid mimics phosphorylation of bovine prolactin. J Biol Chem 270:27661–27665. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K (1999). Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283:1923–1927. [DOI] [PubMed] [Google Scholar]
- Maragos WF, Tillman P, Jones M, Bruce-Keller AJ, Roth S, Bell JE, Nath A (2003). Neuronal injury in hippocampus with human immunodeficiency virus transactivating protein, Tat. Neuroscience 117:43–53. [DOI] [PubMed] [Google Scholar]
- Matsuo ES, Shin RW, Billingsley ML, Van de Voorde A, O’Connor M, Trojanowski JQ, Lee VM (1994). Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau. Neuron 13:989–1002. [DOI] [PubMed] [Google Scholar]
- McKee AC, Kowall NW, Kosik KS (1989). Microtubular reorganization and dendritic growth response in Alzheimer's disease. Ann Neurol 26:652–659. [DOI] [PubMed] [Google Scholar]
- Megias M, Emri Z, Freund TF, Gulyas AI (2001). Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 102:527–540. [DOI] [PubMed] [Google Scholar]
- Merrick SE, Demoise DC, Lee VM (1996). Site-specific dephosphorylation of tau protein at Ser202/Thr205 in response to microtubule depolymerization in cultured human neurons involves protein phosphatase 2A. J Biol Chem 271:5589–5594. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak H, An WL, Winblad B, Cowburn RF, Iqbal K, Grundke-Iqbal I (2002). Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer's disease. Brain Res Mol Brain Res 109:45–55. [DOI] [PubMed] [Google Scholar]
- Runden E, Seglen PO, Haug FM, Ottersen OP, Wieloch T, Shamloo M, Laake JH (1998). Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J Neurosci 18:7296–7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Freund TF (1991). Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 40:599–636. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2002). Alzheimer's disease is a synaptic failure. Science 298:789–791. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Ihara Y, Salazar FJ (1982). Alzheimer's disease: insolubility of partially purified paired helical filaments in sodium dodecyl sulfate and urea. Science 215:1243–1245. [DOI] [PubMed] [Google Scholar]
- Shahani N, Brandt R (2002). Functions and malfunctions of the tau proteins. Cell Mol Life Sci 59:1668–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale G, Nichols NR, Brady DR, Finch CE, Horton WE Jr (1995). Evidence for apoptotic cell death in Alzheimer's disease. Exp Neurol 133:225–230. [DOI] [PubMed] [Google Scholar]
- Stoothoff WH, Johnson GV (2005). Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta 1739:280–297. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991). A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37:173–182. [DOI] [PubMed] [Google Scholar]
- Su JH, Cummings BJ, Cotman CW (1994a). Subpopulations of dystrophic neurites [correction of neuritis] in Alzheimer's brain with distinct immunocytochemical and argentophilic characteristics. Brain Res 637:37–44. [DOI] [PubMed] [Google Scholar]
- Su JH, Anderson AJ, Cummings BJ, Cotman CW (1994b). Immunohistochemical evidence for apoptosis in Alzheimer's disease. NeuroReport 5:2529–2533. [DOI] [PubMed] [Google Scholar]
- Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW (2001). Activated caspase-3 expression in Alzheimer's and aged control brain: correlation with Alzheimer pathology. Brain Res 898:350–357. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Zirrgiebel U, von Bohlen Und Halbach O, Strelau J, Laliberte C, Kaplan DR, Unsicker K (2004). ERK activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3. J Cell Biol 165:357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991). Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580. [DOI] [PubMed] [Google Scholar]
- Uboga NV, Price JL (2000). Formation of diffuse and fibrillar tangles in aging and early Alzheimer's disease. Neurobiol Aging 21:1–10. [DOI] [PubMed] [Google Scholar]
- Wang ZY, Wang F, Sellers JR, Korn ED, Hammer III JA (1998). Analysis of the regulatory phosphorylation site in Acanthamoeba myosin IC by using site-directed mutagenesis. Proc Natl Acad Sci USA 95:15200–15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CL, Espinoza M, Kress Y, Davies P (2000). Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging 21:719–727. [DOI] [PubMed] [Google Scholar]
- Weaver CM, Hof PR, Wearne SL, Lindquist WB (2004). Automated algorithms for multiscale morphometry of neuronal dendrites. Neural Comput 16:1353–1383. [DOI] [PubMed] [Google Scholar]
- West MJ, Coleman PD, Flood DG, Troncoso JC (1994). Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet 344:769–772. [DOI] [PubMed] [Google Scholar]
- Wolozin BL, Pruchnicki A, Dickson DW, Davies P (1986). A neuronal antigen in the brains of Alzheimer patients. Science 232:648–650. [DOI] [PubMed] [Google Scholar]
- Zehr C, Lewis J, McGowan E, Crook J, Lin WL, Godwin K, Knight J, Dickson DW, Hutton M (2004). Apoptosis in oligodendrocytes is associated with axonal degeneration in P310L tau mice. Neurobiol Dis 15:553–562. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Li X, Chen W, Zhao Y, Li H, Qing C, Jia N, Bai Z, Liu J (2004). Prenatal stress causes gender-dependent neuronal loss and oxidative stress in rat hippocampus. J Neurosci Res 78:837–844. [DOI] [PubMed] [Google Scholar]
- Zhukareva V, Trojanowski JQ, Lee VM (2004). Assessment of pathological tau proteins in frontotemporal dementias: qualitative and quantitative approaches. Am J Geriatr Psychiatry 12:136–145. [PubMed] [Google Scholar]