

Abstract
Neurotransmitter release in the nucleus accumbens core (NACore) during the acquisition of remifentanil or cocaine reinforcement was determined in an operant runway procedure by simultaneous tandem mass spectrometric analysis of dopamine, acetylcholine, and remifentanil or cocaine itself. Run times for remifentanil or cocaine continually decreased over the five consecutive runs of the experiment. Intra-NACore dopamine, acetylcholine, and drug peaked with each intravenous remifentanil or cocaine self-administration and decreased to pre-run baseline with half-lives of ∼10 min. As expected, remifentanil or cocaine peaks did not vary between the five runs. Surprisingly, however, drug-contingent dopamine peaks also did not change over the five runs, whereas acetylcholine peaks did. Thus, the acquisition of drug reinforcement was paralleled by a continuous increase in acetylcholine overflow in the NACore, whereas the overflow of dopamine, the expected prime neurotransmitter candidate for conditioning in drug reinforcement, did not increase. Local intra-accumbens administration by reverse microdialysis of either atropine or mecamylamine completely and reversibly blocked the acquisition of remifentanil reinforcement. Our findings suggest that activation of muscarinic and nicotinic acetylcholine receptors in the NACore by acetylcholine volume transmission is necessary during the acquisition phase of drug reinforcement conditioning.
Keywords: remifentanil, cocaine, nucleus accumbens core, acetylcholine, dopamine, acquisition, reinforcement, runway, muscarinic, nicotinic
Introduction
Among the brain regions determining an addict's deleterious preference for drug seeking over other behaviors, the nucleus accumbens (NAC) has received enormous attention over the last decades. It has been found that the NAC consists, in fact, of two neuroanatomically and neurochemically distinct regions, a “core” (NACore) and a “shell” (NAShell) (Heimer et al., 1991; Jongen-Relo et al., 1994; Haber et al., 1995). Converging evidence from a number of laboratories suggests that the evaluation of (1) the interoceptive (“direct pharmacological”) stimulus of the drug of abuse itself (Pontieri et al., 1995) and of (2) drug-associated stimuli (discriminative stimuli as well as secondary reinforcing stimuli) (Ito et al., 2000; Ciccocioppo et al., 2001; Ghitza et al., 2003) is performed in the NAShell, whereas the learned (“conditioned”) response to such stimuli (i.e., drug seeking) is executed by the NACore (Shippenberg et al., 1992; Ito et al., 2000; Neisewander et al., 2000; Phillips et al., 2003; Koob et al., 2004; Peoples et al., 2004; Voorn et al., 2004).
This study was originally designed to investigate NACore neurotransmitter changes during the acquisition phase of drug reinforcement by simultaneously quantifying operant behavior and neurotransmitter overflow in a rat runway procedure (Wakonigg et al., 2003b; Geist and Ettenberg, 1990), an operant conditioning paradigm with high face validity that also allows complete experimenter control over the temporal pattern of drug self-administration. To our surprise, acquisition of drug reinforcement was paralleled by an increase in acetylcholine (ACh) overflow in the NACore, whereas the overflow of dopamine (DA) (i.e., the expected prime neurotransmitter candidate for conditioning in drug reinforcement) did not change in the expected way during the development of drug conditioning. The details of these findings and a discussion of their relevance are given below.
Materials and Methods
Subjects.
Male Sprague Dawley rats were obtained from the Research Institute of Laboratory Animal Breeding (Himberg, Austria) weighing 250–300 g on receipt. Before surgery, all animals were housed in groups of six at a constant room temperature of 24°C and had ad libitum access to tap water. Rats were fed ∼15.5 g/d each of pelleted chow (Tagger, Innsbruck, Austria). All rats were tested during the light phase of a 12 h light/dark cycle (lights on at 7:00 A.M.).
Drugs.
Remifentanil HCl (Ultiva; GlaxoSmithKline, Vienna, Austria) was obtained from the Innsbruck Medical University Hospital pharmacy, cocaine (COC) was donated by the National Institute on Drug Abuse, deuterated DA was purchased from Cambridge Isotope Laboratories (Andover, MA), and all other chemicals were purchased from Sigma-Aldrich (Vienna, Austria), unless indicated otherwise. Doses and concentrations refer to pure base.
Surgery.
Male Sprague Dawley rats were implanted under isoflurane (2–4%) anesthesia with self-made jugular vein catheters (Wakonigg et al., 2003b) with the following dimensions: 0.6 mm inner diameter (ID) × 1.2 mm outer diameter (OD) × 0.3 mm silicone tubing thickness. Concentric microdialysis probes (membrane, Filtral AN69; 40,000 molecular cutoff; 0.24 mm ID; 0.31 mm OD; Gambro Hospal, Wiener Neudorf, Austria) were assembled as detailed previously (Zernig et al., 1997; Taber et al., 1998; Zernig and Fibiger, 1998; Acquas et al., 2001). Microdialysis probes were aimed at the NACore region. The coordinates of the probe tip (with a 2 mm active dialysis membrane) relative to bregma were +1.6 mm anteroposterior, −1.6 mm mediolateral, and 8.2 mm dorsoventral (Paxinos and Watson, 1998). Only experiments in which the probe tip location was confirmed by visual inspection of postmortem brain slices were included in the study. Rats were treated with 28 mg of chloramphenicol (Paraxin; Hoffmann-La Roche, Grenzach-Wuehlen, Germany) during the 2 consecutive days after surgery. After the end of each experiment, patency of the catheters was tested by injection of 6 mg/kg methohexital (Brevital; Eli Lilly, Vienna, Austria). All experiments were approved by the Ethics Committee of the Austrian Federal Ministry of Science and performed in accordance with the Guide for the Care and Use of Laboratory Animals.
Runway procedure.
In the operant runway procedure (Geist and Ettenberg, 1990; McFarland and Ettenberg, 1998; Ettenberg, 2004), the time that an animal needs to obtain a stimulus, the “run time,” is thought to be inversely proportional to the reinforcing strength of the stimulus. “Run time” includes both the time the rat spends in the start area before committing to traversing the runway alley and the time it needs to reach the photobeam. Thus, a run time of ∼5 s indicates both that the rat immediately decides to approach the goal area and that it approaches the goal area without engaging in alternative behavior (e.g., rearing or exploring) in the runway alley. Using this procedure, we could reliably demonstrate acquisition of drug reinforcement for opioids and psychostimulants within only five consecutive trials in completely drug- and experiment-naive rats (Wakonigg et al., 2003b). In the present study, 2 d after the implantation of the intravenous catheters and microdialysis probes, rats were given the opportunity to run for an intravenous injection of 0.032 mg/kg remifentanil (RMF) or 0.33 mg/kg COC for five consecutive trials (“runs”). Runs were started by opening a sliding door separating a start area from the main alley (length, 1 m) of the runway and by indicating the availability of RMF or COC with a white cue light in the goal area. The click of a photobeam, the blinking of the cue light, and the start of an automated infusion pump indicated successful completion of the operant response for the (run-contingent) drug. To determine the effect of noncontingent (i.e., experimenter-delivered) drug, rats first passively received the drug within the confines of the start area of the runway, the sliding door was opened, and the rats were then allowed to traverse the alley. The white cue light at the end of the runway was also illuminated during the noncontingent condition. Intertrial intervals of 40 min (RMF) or 60 min (COC) were chosen to guarantee de facto elimination of the drug from the brain (Pan et al., 1991; Haidar et al., 1997; Hemby et al., 1997; Cox et al., 1999) before the start of the next trial. The operant level was determined with intravenous saline in another six rats, using a run-time cutoff of 60 s. Runway dimensions and experimental details have been published previously (Wakonigg et al., 2003a,b, 2004).
In vivo microdialysis was performed during the behavioral experiments. The analytical probes were perfused with a 1 mm sodium phosphate-buffered solution containing (in mm) 147 NaCl, 3 KCl, 1 MgCl2, and 1.3 CaCl2 at 2 μl/min. The dialysate was immediately mixed with a stabilizing solution (10 mm HCl), collected in 10 min samples (void volume of the probe outlet, <0.2 μl), and stored at −70°C for off-line quantification. In some experiments, 10 μm atropine or 100 μm mecamylamine was added to the phosphate-buffered solution to administer the compound into the NACore by reverse microdialysis. These concentrations, 10-fold higher than those generally found in brain slice superfusion experiments, were chosen to compensate for the spatial and temporal limitations associated with drug administration by reverse microdialysis. DA, ACh, and drug were simultaneously quantified by tandem mass spectrometry (MS/MS). To that end, 85 μl stabilized dialysate and internal standard (d3-dopamine, 1913–5000 fmol on column) were directly injected into the HPLC/MS/MS instrumentation. Chromatographic separation was performed on a Waters Atlantis dC18 column (3 μm particle size; 2.1 × 150 mm) with a mobile phase of 5 mm formic acid in 16% acetonitrile at a flow rate of 0.35 ml/min or on a Waters Acquity BEH C18 column (1.7 μm particle size; 2.1 × 50 mm) with a mobile phase of 5 mm formic acid in 50% acetonitrile at a flow rate of 0.25 ml/min into a Micromass Quattro Ultima triple quadrupole spectrometer (Waters, Vienna, Austria). Chromatographic peaks were identified using the following mass transitions: DA, 154 > 137; d3-dopamine, 157 > 139; ACh, 146 > 87; COC, 304 > 182; RMF, both 377 > 317 (quantifier) and 377 > 113 (additional qualifier). For the simultaneous determination of DA and ACh, no acetylcholinesterase inhibitor was added to the perfusion Ringer's medium. The identity of ACh was further confirmed by the addition of 1 μm neostigmine to the perfusion Ringer's solution after the experiments proper, by spiking the dialysate samples with freshly dissolved external ACh, and by the addition of acetylcholinesterase to neostigmine-free dialysate (data not shown).
Analysis of data.
Unless indicated otherwise, values are given as means ± SEM of n determinations. Because of an experimenter-imposed cutoff of 60 s, group data for run times were first compared by the nonparametric repeated-measures-corrected Friedman test, followed by Dunn's multiple comparison test. All other group data were subjected to repeated-measures-corrected two-factor (time, treatment) ANOVA, followed by Tukey's multiple comparison test. The correlation of run times with ACh or DA peaks was investigated using Pearson's rank correlation test. Statistical tests were performed with Prism version 4 (GraphPad Software, San Diego, CA) or SPSS version 12 (SPSS, Chicago, IL).
Results
Figures 1 and 2 summarize the behavioral and neurochemical effects of contingent versus noncontingent intravenous RMF (0.032 mg/kg) (Fig. 1) or intravenous COC (0.33 mg/kg) (Fig. 2) and of contingent intravenous saline (operant level) (Fig. 1) in a rat runway procedure. Run times of the first run (i.e., before the first response–stimulus pairing) were not different between any of the treatment groups. Run times, however, continually decreased over the five consecutive runs of the experiment if intravenous RMF or intravenous COC was made available at the end of the runway (first vs fifth run: RMF, 26 ± 4 s vs 9 ± 1 s, p < 0.001; COC, 26 ± 3 vs 7 ± 2, p < 0.05), whereas run times for saline increased. At the level of grossly observable behavior, when drug infusions were made available in the goal area, the rats immediately left the start area (i.e., committed to approaching the drug-infusion-associated goal area immediately) and approached the goal area without engaging in alternative behavior (e.g., rearing or exploring the runway alley). In contrast, both a latency to leave the start area and an increase in alternative behavior in the runway alley were consistently observed if saline infusions were made available in the goal area of the runway. Thus, RMF or COC acquired a distinctive positive reinforcing effect within the five runs of the experiment [p < 0.001 for RMF vs saline in run 5 (Fig. 1); p < 0.001 for contingent vs noncontingent COC (Fig. 2)]. DA, ACh, and RMF or COC in the NACore peaked with each intravenous RMF or COC administration and decreased to pre-run baseline with half-lives of ∼10 min. As expected, RMF or COC peaks did not change continuously over the five runs, both after contingent and noncontingent administration. Accordingly, total RMF recovered from the NACore (i.e., cumulative RMF for the 10 min samples 4–23) was 401 ± 30 fmol after contingent and 347 ± 46 fmol after noncontingent RMF administration (p = 0.17). Run times were inversely correlated with ACh peak height after contingent RMF administration (r = −0.90; one-tailed, p = 0.019). ACh peaks for COC (samples 4–33) were 22,252 ± 1496 fmol for contingent and 19,140 ± 1156 fmol for noncontingent drug administration (p = 0.13). As was the case for RMF, run times were inversely correlated with ACh peak height after contingent COC administration (r = −0.80; one-tailed, p = 0.051).
Figure 1.

Operant responding for intravenous 0.032 mg/kg RMF and associated changes in levels of ACh, DA, and RMF in the NACore. x-axis, Consecutive number of 10 min samples (i.e., time); y-axis, femtomoles per 10 min sample (means ± 1 SEM). Rats either actively ran for intravenous RMF (i.e., contingent RMF; •; n = 6) or intravenous saline (i.e., operant level; ▿; n = 6) during samples 4 (run 1), 8 (run 2), 12 (run 3), 16 (run 4), and 20 (run 5) or passively received RMF within the confines of the start area of the runway and were then allowed to traverse the runway (○; n = 5). Run times for noncontingent RMF are not shown because rats that passively received RMF did not leave the start area because of the acute sedative and locomotor impairing effect of RMF. Baseline overflow (samples 1–3) of ACh and DA did not differ significantly between groups. Repeated-measures-corrected group comparisons were statistically significant for run times (saline vs contingent RMF; Friedman test, p = 0.0001; run times for run 1 vs runs 3–5, p < 0.001 each), ACh peaks (saline vs noncontingent RMF vs contingent RMF; 2-factor ANOVA each; p < 0.0001 each for time, treatment, and time-by-treatment interaction; ACh peak comparison for run 1 vs run 4 or 5, p < 0.05 each), and DA peaks (saline vs noncontingent RMF vs contingent RMF; p = 0.0073 each for time, treatment, and time-by-treatment interaction; DA peak comparison: run 1 vs run 2, p < 0.05; run 1 vs runs 3–5, p > 0.05 each).
Figure 2.
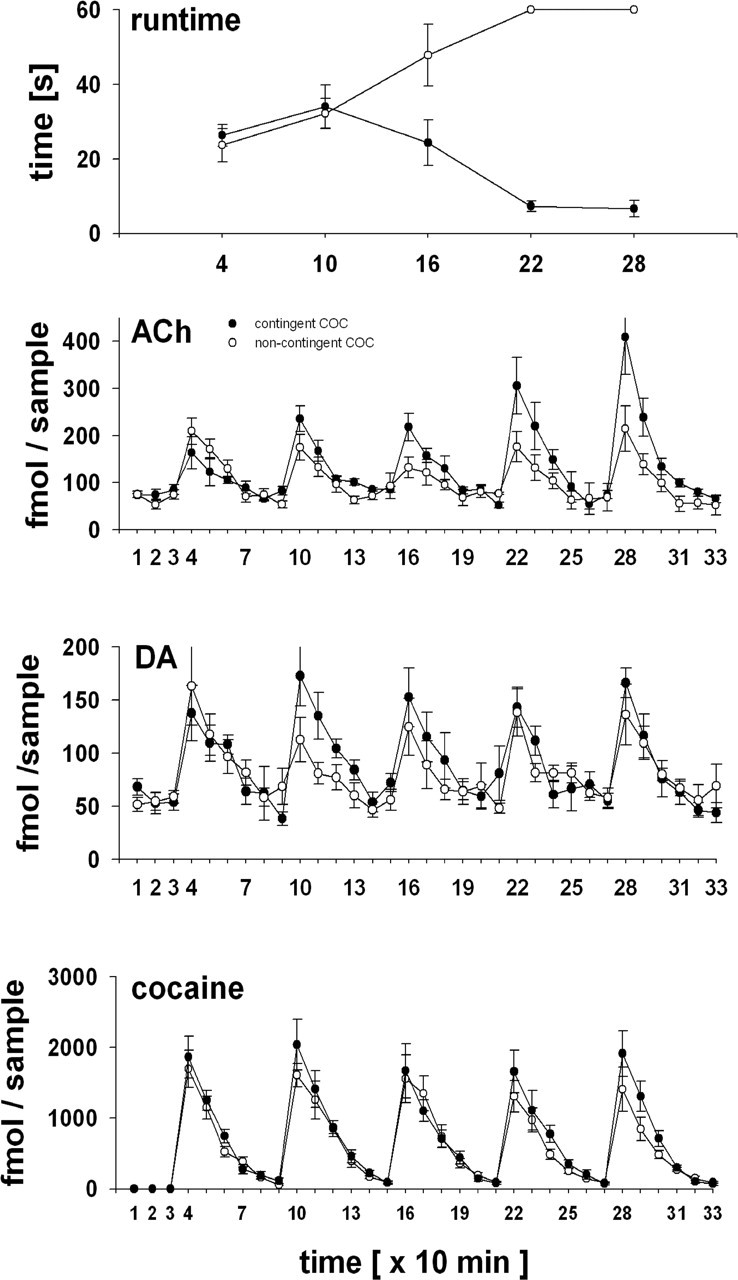
Operant responding for intravenous 0.33 mg/kg COC and associated changes in levels of ACh, DA, and COC in the NACore. x-axis, Consecutive number of 10 min samples (i.e., time); y-axis, femtomoles per 10 min sample (means ± 1 SEM). Rats either actively ran for intravenous COC (i.e., contingent COC; •; n = 6) during samples 4 (run 1), 10 (run 2), 16 (run 3), 22 (run 4), and 28 (run 5) or passively received intravenous COC within the confines of the start area of the runway and were then allowed to traverse the runway alley (i.e., noncontingent COC; ○; n = 6). Baseline overflow (samples 1–3) of ACh and DA did not differ significantly between the contingent and noncontingent groups. Repeated-measures-corrected group comparisons were significantly different for run times (contingent vs noncontingent COC; Friedman test, p = 0.0071; run times for run 1 vs runs 4 and 5, p < 0.05 each) and ACh (2-factor ANOVA; p = 0.012 for treatment; p < 0.0001 for time; p = 0.0003 for time-by-treatment interaction; ACh peak comparison for run 1 vs run 5, p < 0.01) but not for NACore DA (treatment, p = 0.3) or NACore COC (treatment, p = 0.13).
Contrary to expectations, however, NACore DA peaks did not continuously increase during the acquisition of RMF or COC reinforcement (run 1 vs run 5: RMF, 257 ± 35 vs 232 ± 38 fmol, p > 0.05; COC, 137 ± 26 vs 166 ± 14 fmol, p > 0.05), whereas ACh peaks increased 5-fold for RMF (run 1 vs run 5: 252 ± 1243 vs 1,243 ± 489 fmol/10 min sample; p < 0.001) (Fig. 1) and 2.5-fold for COC (run 1 vs run 5: 163 ± 34 vs 408 ± 79 fmol; p < 0.001) (Fig. 2). The total cumulative NACore DA overflow was 3192 ± 359 versus 2668 ± 154 fmol for contingent versus noncontingent RMF (one-tailed, p = 0.065) and 2728 ± 134 versus 2500 ± 162 (one-tailed, p = 0.15) for contingent versus noncontingent COC. The respective total cumulative NACore ACh overflow for RMF was 8081 ± 1750 versus 4311 ± 520 fmol (one-tailed, p = 0.045) and for COC was 4064 ± 259 versus 3148 ± 157 (one-tailed, p = 0.0064). In contrast to the ACh peaks associated with contingent RMF or COC administration, DA peaks did not correlate with run times (RMF: r = 0.15, one-tailed, p = 0.41; COC: r = 0.18, one-tailed, p = 0.39). Contingent administration of intravenous saline (operant level) did not affect DA or ACh levels (Fig. 1).
In another set of experiments, the acquisition of RMF reinforcement was tested in the presence of 10 μm of the muscarinic ACh receptor (mAChR) antagonist atropine or 100 μm of the nicotinic ACh receptor (nAChR) antagonist mecamylamine, applied locally into the NACore by reverse microdialysis. Figure 3 shows that the intra-accumbens administration of mecamylamine or atropine blocked the acquisition of RMF reinforcement while leaving RMF transients in the NACore unaffected. Mecamylamine abolished the continuous run-by-run increase in the NACore ACh peaks induced by run-contingent RMF (Fig. 1). Intra-accumbens atropine lead to an overall massive increase in RMF-induced ACh, with ACh levels never returning to baseline in the 40 min inter-run interval.
Figure 3.
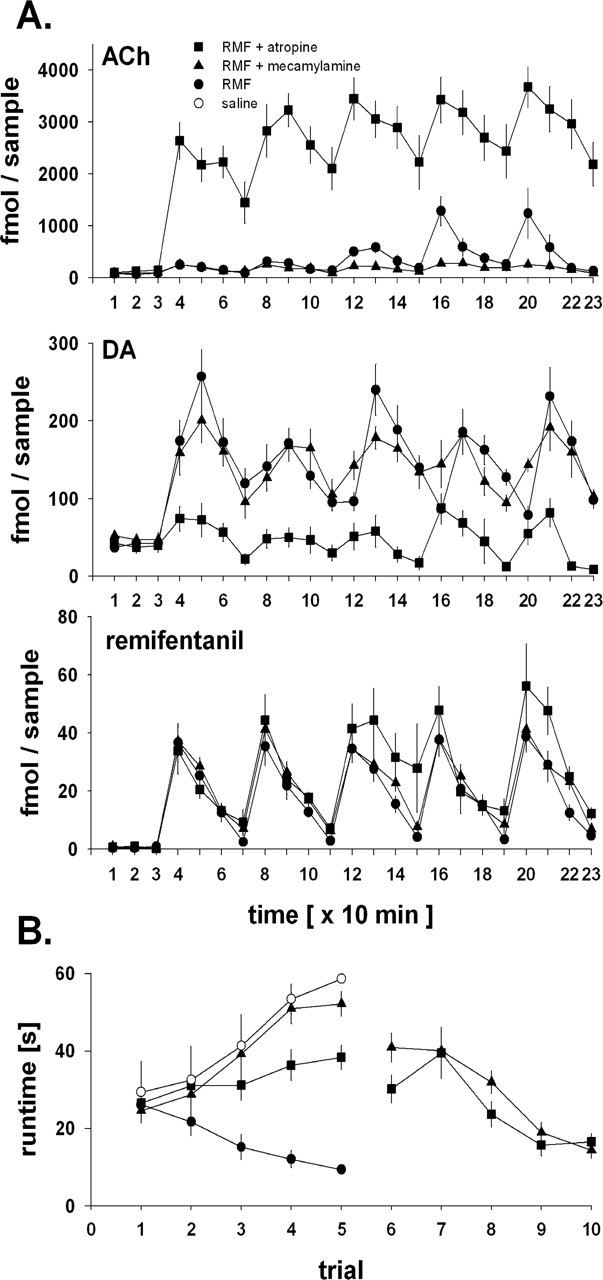
A, Effects of intra-accumbens core atropine and mecamylamine on the acquisition of RMF reinforcement. Operant responding for intravenous 0.032 mg/kg RMF and associated changes in levels of ACh, DA, and RMF in the NACore was determined in the absence (•; n = 6) or presence of atropine (10 μm; ▪; n = 6) or mecamylamine (100 μm; ▴; n = 10), which was locally administered into the NACore by reverse microdialysis starting 5 min into sample 3 (i.e., 5 min before the rat had the opportunity to run for the first RMF injection). x-axis, Consecutive number of 10 min samples (i.e., time); y-axis, femtomoles per 10 min sample (means ± 1 SEM). Rats ran for contingent intravenous RMF during samples 4 (run 1), 8 (run 2), 12 (run 3), 16 (run 4), and 20 (run 5). B, Reversibility of the effect of intra-accumbens atropine or mecamylamine on the acquisition of RMF reinforcement. On day 1 (left; runs 1–5), rats ran for intravenous saline (○; n = 6; see also Fig. 1) or intravenous 0.032 mg/kg RMF in the absence (•; n = 6; see also Fig. 1) or presence of intra-accumbens atropine (▪; n = 6; see also Fig. 2, top) or mecamylamine (▴; n = 10; see also Fig. 2, top). On day 4 (right; runs 6–10), the same experiment was repeated for those animals that had run for RMF in the presence of atropine (▪) or mecamylamine (▴) on day 1.
The complete block of the acquisition of RMF reinforcement by local accumbal atropine or mecamylamine was fully reversible: when the same animals were tested again 3 d after the atropine or mecamylamine experiments (Fig. 3B, right), the decrease in run times over the five runs of the experiment was essentially identical to the decrease in run times engendered by contingent administration of RMF in animals that had not been exposed to intra-accumbens atropine or mecamylamine (Fig. 3B, left).
Discussion
Acquisiton of the reinforcing effect of RMF or COC reinforcing in a rat runway procedure was paralleled by a continuous, run-by-run increase in ACh overflow in the NACore, whereas the overflow of DA, the expected prime neurotransmitter candidate for conditioning in drug reinforcement, did not increase after consecutive runs. Local intra-NACore antagonism of mAchRs or nAChRs completely and reversibly blocked the acquisition of RMF reinforcement, indicating (1) that activation of both mAchR and nAChR in the NACore is necessary for the conditioning of the positive reinforcing effect of RMF (i.e., for the animal's learning to associate the drug stimulus with the appropriate operant response) and (2) that the ACh release by cholinergic large aspiny interneurons, the second most prevalent neuron population in the striatum including the NACore (Squire et al., 2003), is instrumental for this learning process. Thus, the general rule that striatal cholinergic interneurons play an eminent role in the formation of stimulus–response associations (i.e., learning) (Aosaki et al., 1994; Calabresi et al., 2000; Suzuki et al., 2001; Kitabatake et al., 2003; Mansvelder et al., 2005) also seems to apply to the NACore, a striatal structure dedicated to processing stimuli of high emotional and motivational valence (Haber et al., 1985; Jongen-Relo et al., 1994). Striatal cholinergic interneurons are known to affect GABAergic medium spiny neurons, which constitute the major (95%) neuron population of the striatum and also serve as the major motivational/locomotor output of the striatum (Haber et al., 1985; Heimer et al., 1991; Zahm and Brog, 1992; Meredith and Chang, 1994; Squire et al., 2003; Voorn et al., 2004). In accordance with our findings, both mAChR and nAChR in the NAC have been shown to modulate the activity of the GABAergic medium spiny output neurons (de Rover et al., 2002). The fact that the learning-associated systematic increase in ACh peak amplitude could be completely blocked by the nAChR antagonist mecamylamine (Fig. 3) is in good agreement with the finding that activation of nAChR, albeit in the ventral tegmental area (VTA), has been shown to favor synchronized activation of presynaptic glutamatergic and postsynaptic dopaminergic neurons and that this nicotine-induced sychronization leads to long-term potentiation of the glutamatergic input (Mansvelder and McGehee, 2000; Mansvelder et al., 2005). The mAchR system itself seems to be able to modulate striatal output in opposite directions (for review, see Calabresi et al., 2000). Accordingly, in the present study, blocking mAChRs was sufficient to completely impair the learning of the operant response. Clearly, additional work will be necessary to define the relative contribution and mode of cooperation of the mAChRs and nAChRs in the acquisition of drug reinforcement.
Our findings on the instrumental role of the intra-NACore cholinergic system in the acquisition of drug reinforcement corroborate previous results by others showing that accumbal ACh was increased during multiple-injection lever press-based COC self-administration (Mark et al., 1999) with a time course that was very similar to that of accumbal DA (Hemby et al., 1997). Also in corroboration of these findings, in the present study, both DA and ACh levels were consistently higher during contingent RMF administration than during noncontingent administration, albeit to a much larger degree for ACh (87% increase) than for DA (20% increase (see also Zapata et al., 2003). The same held true for COC-induced ACh versus DA overflow (29 vs 9% increase). Our findings also corroborate the results of Smith et al. (2004), who demonstrated that neurotoxic ablation of cholinergic neurons in the posterior NAC and ventral pallidum by 129-IgG-saporin produced a flattening of the COC dose–response curve in rats self-administering intravenous COC.
Data on the cholinergic modulation of drug reinforcement, however, are equivocal. Since the pioneering study by Wilson and Schuster (1973), it has also been shown that inhibition rather than activation of the cholinergic system enhances drug reinforcement (Hikida et al., 2001, 2003). In the same vein, drug self-administration was paralleled by a decrease in ACh turnover in the NAC (Smith et al., 1984, 2004). Profound differences in experimental conditions, not least in the nature of the investigated behavior (i.e., running vs lever pressing or place preference), could be used to explain the differences between our findings and those cited above.
At first sight, the absence of an increase in NACore DA during the conditioning of the reinforcing effects of RMF or COC seems at odds with previous findings from two independent laboratories that showed increases of NACore DA during conditioning to a drug-associated visual stimulus (Ito et al., 2000) or a food-associated olfactory stimulus (Bassareo and DiChiara, 1999). However, in the present study, only the first five consecutive response–stimulus pairings were observed, whereas 2-h-long multiple-injection sessions were administered over a minimum of 10 d before a FR10(FR10:s) second-order schedule was established, totaling a minimum of 23+ days of conditioning of the drug-associated visual stimulus (Ito et al., 2000). It is conceivable that during the initial response–reinforcer pairings (present study), volume transmission (Garris et al., 1994) of NACore DA plays much less of a role than during a firmly established response–reinforcer association (Ito et al., 2000). This suggestion is supported by another experiment on conditioning-induced NACore DA overflow (DiChiara et al., 2004), in which a drug (i.e., subcutaneous morphine)-associated olfactory stimulus was presented for three trials before NACore DA release was assessed during a fourth trial. In accordance with the present study, no conditioned NACore DA release was found in this experiment (DiChiara et al., 2004). Thus, only three to five response–drug pairings do not seem to lead to a conditioned increase in NACore DA volume transmission. In the experiment involving food reinforcement, however, Bassareo and DiChiara (1999) showed that a single trial was sufficient to produce stimulation of NACore DA by the cheese-like smell of a snack food in a subsequent trial 5 d later. Maybe food reinforcers, being of much higher reinforcing strength for rats than drug reinforcers, at least in the runway procedure (Wakonigg et al., 2003b), are able to induce conditioned increases in NACore DA much faster than drug reinforcers.
When generalizing the present experimental data to RMF distribution in the brain, one must consider that RMF was determined in the NAC [as opposed to the VTA, i.e., the brain area thought to be the main site of μ-opioid agonist reinforcement (DeVries and Shippenberg, 2002; Chefer et al., 2003)]. However, when directly compared, striatal levels of the μ-opioid agonist morphine did not differ significantly from its midbrain levels (Bhargava et al., 1993), suggesting that NACore RMF levels provide a reasonable measure of RMF brain levels. The NACore COC data of the present study, on the other hand, are in direct agreement with a study on intra-NAC levels of COC itself and of COC-induced DA overflow both for contingent and noncontingent COC administration (Hemby et al., 1997).
One could argue that traversing a rat runway alley to obtain a reinforcer at its end constitutes less a bona fide operant response than a pavlovian approach (Cardinal et al., 2002, 2003). Thus, our behavioral paradigm might assess more the pavlovian incentive value or incentive salience (Cardinal et al., 2003; Everitt et al., 2003) or drug “wanting” (Berridge and Robinson, 2003) than the “goal status” or incentive value of the drugs (Cardinal et al., 2002). Despite this shortcoming, speed of approach does have excellent face value when assessing the attractiveness of a reinforcer.
Continuous increases in approach speed after repeated drug availability might not reflect conditioning of drug reinforcement but only locomotor sensitization. However, microanalysis of runway behavior clearly showed that drugs of abuse of different chemical classes and food dose-dependently increased the probability of the rat's decision to commit to a fast run across the runway to obtain the reinforcer, whereas running speed itself remained unchanged (Wakonigg et al., 2003b). This argues against simple locomotor sensitization. Accordingly, when the rats of the present study passively received COC within the confines of the start area of the runway and were then allowed to traverse the alley of the runway in the noncontingent COC condition, run times continuously increased, also indicating that simple locomotor sensitization was not causing the continuous decrease in run times engendered by run-contingent COC.
Finally, the lack of a change in NACore DA overflow in the present study as determined by in vivo microdialysis cannot address the important contribution of fast DA transients to the conditioning of drug reinforcement that could be demonstrated with the help of experimental approaches that offer much higher temporal and spatial resolution (Wightman and Robinson, 2002; Phillips et al., 2003) than in vivo microdialysis. Our experimental approach is also unable to capture any fast DA-mediated changes in its signal transduction cascade. The present findings, however, clearly indicate that local NACore volume transmission of ACh (Zhou et al., 2002) rather than volume transmission of DA (Garris et al., 1994) is instrumental for the acquisition of drug reinforcement.
Footnotes
This work was supported by Austrian Science Fund Grants P16394-B05 and P18787-B05 and by the Verein für Experimentelle Psychiatrie, Psychotherapie, und Pharmakologie. The animals used in this study were cared for in accordance with the guidelines of the National Institutes of Health Animal Care and Use Program and the National Institute on Drug Abuse–Intramural Research Program Animal Program, which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
References
- Acquas E, Marrocu P, Pisanu A, Cadoni C, Zernig G, Saria A, DiChiara G (2001). Intravenous administration of 3,4-methylenedioxymethamphetamine (“Ecstasy”) enhances cortical and striatal acetylcholine in vivo. Eur J Pharmacol 418:207–211. [DOI] [PubMed] [Google Scholar]
- Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel AM, Kimura M (1994). Responses of tonically active neurons in the primate's striatum undergo systematic changes during behavioral sensorimotor conditioning. J Neurosci 14:3969–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassareo V, DiChiara G (1999). Differential responsiveness of dopamine transmission to food-stimuli in nucleus accumbens shell/core compartments. Neuroscience 89:637–641. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE (2003). Parsing reward. Trends Neurosci 26:507–513. [DOI] [PubMed] [Google Scholar]
- Bhargava HN, Larsen AK, Rahmani NH, Villar VM (1993). Naltrexone-induced alterations of the distribution of morphine in brain regions and spinal cord of the rat. Brain Res 607:1–8. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G (2000). Acetylcholine-mediated modulation of striatal function. Trends Neurosci 23:120–126. [DOI] [PubMed] [Google Scholar]
- Cardinal RN, Parkinson JA, Hall J, Everitt BJ (2002). Emotion and motivation: the role of the amygdala, ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev 26:321–352. [DOI] [PubMed] [Google Scholar]
- Cardinal RN, Parkinson JA, Marbini HD, Toner AJ, Bussey TJ, Robbins TW, Everitt BJ (2003). Role of the anterior cingulate cortex in the control over behavior by Pavlovian conditioned stimuli in rats. Behav Neurosci 117:566–587. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Kieffer BL, Shippenberg TS (2003). Basal and morphine-evoked dopaminergic neurotransmission in the nucleus accumbens of MOR- and DOR-knockout mice. Eur J Neurosci 18:1915–1922. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Sanna PP, Weiss F (2001). Cocaine-predictive stimulus induces drug-seeking behavior and neural activation in limbic brain regions after multiple months of abstinence: reversal by D(1) antagonists. Proc Natl Acad Sci USA 98:1976–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox EH, Langemeijer MW, Gubbens-Stibbe JM, Muir KT, Danhof M (1999). The comparative pharmacodynamics of remifentanil and its metabolite, GR90291, in a rat electroencephalographic model. Anesthesiology 90:535–544. [DOI] [PubMed] [Google Scholar]
- de Rover M, Lodder JC, Kits KS, Schoffelmeer AN, Brussaard AB (2002). Cholinergic modulation of nucleus accumbens medium spiny neurons. Eur J Neurosci 16:2279–2290. [DOI] [PubMed] [Google Scholar]
- DeVries TJ, Shippenberg TS (2002). Neural systems underlying opiate addiction. J Neurosci 22:3321–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiChiara G, Bassareo V, Fenu S, DeLuca MA, Spina L, Cadoni C, Acquas E, Carboni E, Valentini V, Lecca D (2004). Dopamine and drug addiction:the nucleus accumbens shell connection. Neuropharmacology 47:227–241. [DOI] [PubMed] [Google Scholar]
- Ettenberg A (2004). Opponent process properties of self-administered cocaine. Neurosci Biobehav Rev 27:721–728. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Cardinal RN, Parkinson JA, Robbins TW (2003). Appetitive behavior: impact of amygdala-dependent mechanisms of emotional learning. Ann NY Acad Sci 985:233–250. [PubMed] [Google Scholar]
- Garris PA, Ciolkowski EL, Pastore P, Wightman RM (1994). Efflux of dopamine from the synaptic cleft in the nucleus accumbens. J Neurosci 14:6084–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geist TD, Ettenberg A (1990). A simple method for studying intravenous drug reinforcement in a runway. Pharmacol Biochem Behav 36:703–706. [DOI] [PubMed] [Google Scholar]
- Ghitza UE, Fabbricatore AT, Prokopenko V, Pawlak AP, West MO (2003). Persistent cue-evoked activity of accumbens neurons after prolonged abstinence from self-administered cocaine. J Neurosci 23:7239–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Groenewegen HJ, Grove EA, Nauta WJ (1985). Efferent connections of the ventral pallidum: evidence of a dual striato pallidofugal pathway. J Comp Neurol 235:322–335. [DOI] [PubMed] [Google Scholar]
- Haber SN, Kunishio K, Mizobuchi M, Lynd-Balta E (1995). The orbital and medial prefrontal circuit through the primate basal ganglia. J Neurosci 15:4851–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidar SH, Moreton JE, Liang Z, Hoke JF, Muir KT, Eddington ND (1997). Evaluating a possible pharmacokinetic interaction between remifentanil and esmolol in the rat. J Pharm Sci 86:1278–1282. [DOI] [PubMed] [Google Scholar]
- Heimer L, Zahm DS, Churchill L, Kalivas PW, Wohltmann C (1991). Specificity in the projection patterns of accumbal core and shell in the rat. Neuroscience 41:89–125. [DOI] [PubMed] [Google Scholar]
- Hemby SE, Co C, Koves TR, Smith JE, Dworkin SI (1997). Differences in extracellular dopamine concentrations in the nucleus accumbens during response-dependent and response-independent cocaine administration in the rat. Psychopharmacology (Berl) 133:7–16. [DOI] [PubMed] [Google Scholar]
- Hikida T, Kaneko S, Isobe T, Kitabatake Y, Watanabe D, Pastan I, Nakanishi S (2001). Increased sensitivity to cocaine by cholinergic cell ablation in nucleus accumbens. Proc Natl Acad Sci USA 98:13351–13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikida T, Kitabatake Y, Pastan I, Nakanishi S (2003). Acetylcholine enhancement in the nucleus accumbens prevents addictive behaviors of cocaine and morphine. Proc Natl Acad Sci USA 100:6169–6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito R, Dalley JW, Howes SR, Robbins TW, Everitt BJ (2000). Dissociation in conditioned dopamine release in the nucleus accumbens core and shell in response to cocaine cues and during cocaine-seeking behavior in rats. J Neurosci 20:7489–7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongen-Relo AL, Voorn P, Groenewegen HJ (1994). Immunohistochemical characterization of the shell and core territories of the nucleus accumbens in the rat. Eur J Neurosci 6:1255–1264. [DOI] [PubMed] [Google Scholar]
- Kitabatake Y, Hikida T, Watanabe D, Pastan I, Nakanishi S (2003). Impairment of reward-related learning by cholinergic cell ablation in the striatum. Proc Natl Acad Sci USA 100:7965–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Ahmed SH, Boutrel B, Chen SA, Kenny PJ, Markou A, O'Dell LE, Parsons LH, Sanna PP (2004). Neurobiological mechanisms in the transition from drug use to drug dependence. Neurosci Biobehav Rev 27:739–749. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS (2000). Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27:349–357. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, van Aerde KI, Couey JJ, Brussaard AB (2006). Nicotinic modulation of neuronal networks: from receptors to cognition. Psychopharmacology (Berl) 184:292–305. [DOI] [PubMed] [Google Scholar]
- Mark GP, Hajnal A, Kinney AE, Keys AS (1999). Self-administration of cocaine increases the release of acetylcholine to a greater extent than response-independent cocaine in the nucleus accumbens of rats. Psychopharmacology (Berl) 143:47–53. [DOI] [PubMed] [Google Scholar]
- McFarland K, Ettenberg A (1998). Haloperidol does not affect motivational processes in an operant runway model of food-seeking behavior. Behav Neurosci 112:630–635. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Chang HT (1994). Synaptic relationships of enkephalinergic and cholinergic neurons in the nucleus accumbens of the rat. Brain Res 667:67–76. [DOI] [PubMed] [Google Scholar]
- Neisewander JL, Baker DA, Fuchs RA, Tran-Nguyen LT, Palmer A, Marshall JF (2000). Fos protein expression and cocaine-seeking behavior in rats after exposure to a cocaine self-administration environment. J Neurosci 20:798–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan HT, Menacherry S, Justice J Jr (1991). Differences in the pharmacokinetics of cocaine in naive and cocaine- experienced rats. J Neurochem 56:1299–1306. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998). In: The rat brain in stereotaxic coordinates New York: Academic.
- Peoples LL, Lynch KG, Lesnock J, Gangadhar N (2004). Accumbal neural responses during the initiation and maintenance of intravenous cocaine self-administration. J Neurophysiol 91:314–323. [DOI] [PubMed] [Google Scholar]
- Phillips PE, Stuber GD, Heien MLAV, Wightman RM, Carelli RM (2003). Subsecond dopamine release promotes cocaine seeking. Nature 422:614–618. [DOI] [PubMed] [Google Scholar]
- Pontieri FE, Tanda G, DiChiara G (1995). Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the “shell” as compared with the “core” of the rat nucleus accumbens. Proc Natl Acad Sci USA 92:12304–12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shippenberg TS, Herz A, Spanagel R, Bals-Kubik R, Stein C (1992). Conditioning of opioid reinforcement: neuroanatomical and neurochemical substrates. Ann NY Acad Sci 654:347–356. [DOI] [PubMed] [Google Scholar]
- Smith JE, Co C, Lane JD (1984). Limbic acetylcholine turnover rates correlated with rat morphine-seeking behaviors. Pharmacol Biochem Behav 20:429–442. [DOI] [PubMed] [Google Scholar]
- Smith JE, Co C, Yin X, Sizemore GM, Liguori A, Johnson III WE, Martin TJ (2004). Involvement of cholinergic neuronal systems in intravenous cocaine self-administration. Neurosci Biobehav Rev 27:841–850. [DOI] [PubMed] [Google Scholar]
- Squire LR, Bloom FE, McConnell SK, Roberts JL, Spitzer NC, Zigmond MJ (2003). In: Fundamental neuroscience London: Academic.
- Suzuki T, Miura M, Nishimura K, Aosaki T (2001). Dopamine-dependent synaptic plasticity in the striatal cholinergic interneurons. J Neurosci 21:6492–6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taber MT, Zernig G, Fibiger HC (1998). Opioid receptor modulation of feeding-evoked dopamine release in the nucleus accumbens. Brain Res 785:24–30. [DOI] [PubMed] [Google Scholar]
- Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, Pennartz CM (2004). Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci 27:468–474. [DOI] [PubMed] [Google Scholar]
- Wakonigg G, Sturm K, Saria A, Zernig G (2003a). Methylenedioxymethamphetamine (MDMA, “Ecstasy”) serves as a robust positive reinforcer in a rat runway procedure. Pharmacology 69:180–182. [DOI] [PubMed] [Google Scholar]
- Wakonigg G, Sturm K, Saria A, Zernig G (2003b). Opioids, cocaine, and food change runtime distribution in a rat runway procedure. Psychopharmacology (Berl) 169:52–59. [DOI] [PubMed] [Google Scholar]
- Wakonigg G, Sturm K, Saria A, Zernig G (2004). Drug history overrides opioid reinforcement in a rat runway procedure. Pharmacology 72:225–230. [DOI] [PubMed] [Google Scholar]
- Wightman RM, Robinson DL (2002). Transient changes in mesolimbic dopamine and their association with “reward.”. J Neurochem 82:721–735. [DOI] [PubMed] [Google Scholar]
- Wilson MC, Schuster CR (1973). Cholinergic influence on intravenous cocaine self-administration by rhesus monkeys. Pharmacol Biochem Behav 1:643–649. [DOI] [PubMed] [Google Scholar]
- Zahm DS, Brog JS (1992). On the significance of suberritories in the “accumbens” part of the ventral striatum. Neuroscience 50:751–767. [DOI] [PubMed] [Google Scholar]
- Zapata A, Chefer VI, Ator R, Shippenberg TS, Rocha BA (2003). Behavioural sensitization and enhanced dopamine response in the nucleus accumbens after intravenous cocaine self-administration in mice. Eur J Neurosci 17:590–596. [DOI] [PubMed] [Google Scholar]
- Zernig G, Fibiger HC (1998). Acute and prolonged effects of clocinnamox and methoclocinnamox on nucleus accumbens dopamine overflow. Psychopharmacology (Berl) 135:17–21. [DOI] [PubMed] [Google Scholar]
- Zernig G, O'Laughlin I, Fibiger HC (1997). Nicotine and heroin augment cocaine-induced dopamine overflow in the nucleus accumbens. Eur J of Pharmacology 337:1–10. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Wilson CJ, Dani JA (2002). Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol 53:590–605. [DOI] [PubMed] [Google Scholar]