

Abstract
Chronic exposure to Δ9-tetrahydrocannabinol (THC) induces tolerance to cannabinoid-induced locomotor effects, which are mediated by cannabinoid receptors (CB1Rs) located in motor control regions, including the cerebellum. There is substantial evidence of cerebellar CB1R molecular adaptation and modifications in receptor signaling after prolonged cannabinoid exposure. However, very little is known about the effects of chronic cannabinoid administration on cerebellar synaptic plasticity, which may contribute to the development of cannabinoid behavioral tolerance.
In the cerebellar cortex, activation of CB1R inhibits excitatory synaptic transmission at parallel fiber (PF)–Purkinje cell (PC) synapses by decreasing neurotransmitter release. Our study aimed to investigate the neurophysiological adaptive responses occurring at cerebellar PF-PC cell synapses after repeated THC exposure. In THC-tolerant mice, an increase of the basal release probability was found at PF-PC synapses, in parallel with a facilitation of slow mGluR1 (metabotropic glutamate receptor type 1)-mediated excitatory postsynaptic currents and a reduced sensitivity to the inhibitory effects of the CB1R agonist CP55,940 [(−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol]. Additionally, after repeated THC exposures, presynaptic PF-PC long-term potentiation was blocked by A1R (adenosine receptor-1) activation. Inhibition of the extracellular signal regulated kinase (ERK) pathway prevented these alterations of cerebellar synaptic transmission and plasticity.
In summary, we provide evidence for ERK-dependent modulatory mechanisms at PF-PC synapses after chronic THC administration. This contributes to generation of forms of pathological synaptic plasticity that might play a role in cannabinoid dependence.
Keywords: cerebellum, synaptic plasticity, cannabinoid, THC, tolerance, ERK, adenosine
Introduction
In laboratory animals, cannabinoid dependence develops after prolonged administration (Abood et al., 1993; Maldonado and Rodriguez, 2002). This is also true for humans, in the case of heavy abusers or after prolonged therapeutic uses, where pharmacological tolerance has been reported for most of the effects of cannabis (Haney et al., 1999; Hart et al., 2002).
In the brain, cannabinoids interact with the CB1 cannabinoid receptor (CB1R), a seven transmembrane domain receptor coupled to G-proteins that inhibits adenylate cyclase activity (Mukhopadhyay et al., 2002). Δ9-Tetrahydrocannabinol (THC), the main psychoactive component of marijuana, impairs motor function in humans and laboratory animals (Adams and Martin, 1996) through the activation CB1R located in control motor regions, such as the basal ganglia and cerebellum (Chaperon and Thiebot, 1999; Patel and Hillard, 2001). Behavioral tolerance to the cannabinoid-induced locomotor effects (hypolocomotion, ataxia, and catalepsy) develops after chronic THC exposure, in parallel with CB1R downregulation and desensitization occurring in several brain areas, including the cerebellum (Martin et al., 2004; Rubino et al., 2004, 2005). Additionally, the THC withdrawal syndrome is associated with compensatory changes in the cAMP pathway occurring selectively in the cerebellum, indicating this area as a major neurobiological substrate for the chronic effects of THC (Tzavara et al., 2000).
Administration of THC in rodents activates the monomeric GTP-binding protein Ras/extracellular signal regulated kinase (ERK) cascade (Valjent et al., 2001, 2004; Derkinderen et al., 2003; Rubino et al., 2004). In vivo, the pharmacological block of the ERK pathway suppresses behavioral responses and CB1R-mediated plasticity in the cerebellum and basal ganglia, thereby suggesting a role for the Ras/ERK pathway in cannabinoid tolerance to the THC-induced hypolocomotor effects (Rubino et al., 2005). Similar results have been reported in RasGRF1 null mice (Rubino et al., 2004, 2005). RasGRF1 is a neuron-specific Ras guanine nucleotide exchange factor (Zippel et al., 1997) that acts on the Ras/ERK pathway and is activated in response to G-protein-coupled and ionotropic receptors (Feig, 1994; Zippel et al., 1996; Tian et al., 2004). Mice lacking RasGRF1 (−/−) do not develop behavioral tolerance after chronic treatment with THC (Rubino et al., 2004), thereby representing a useful genetic model to investigate the role of the Ras/ERK pathway in cannabinoid dependence.
In the cerebellar cortex, activation of CB1R inhibits excitatory synaptic transmission at parallel fiber (PF)–Purkinje cell (PC) synapses by modulation of Ca2+ and K + ion channels (Levenes et al., 1998; Daniel et al., 2004). Because the activity pattern of granule cells is crucial for the precise control of PC firing (Marr, 1969), chronic cannabinoid action on PF-PC synaptic transmission could affect cerebellar function. However, alterations of cerebellar synaptic plasticity induced by chronic cannabinoid exposure have not been explored so far. The aim of our study is to investigate the neurophysiological adaptive responses occurring at cerebellar PF-PC synapses after chronic THC exposure. The role of the Ras/ERK pathway in chronic THC-induced synaptic plasticity has been addressed by means of the genetically engineered mouse model RasGRF1 and by in vivo pharmacological block of ERK1,2 activation.
Materials and Methods
Drugs.
Δ9-Tetrahydrocannabinol (THC) was a generous gift from the National Institute on Drug Abuse (Foreigner Investigator Supply Program, Bethesda, MD). N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-pyrazole-3-carboxamide (SR141716A) was kindly provided by Sanofi Synthelabo Recherche (Longjumeau, France).
α [ Amino [ (aminophenyl) thio ] methylene ] 2 (trifluoromethyl) benezeneacetonitrile (SL327), 6,7-dinitroquinoxaline-2,3-dione (DNQX), dl-2-amino-5-phosphonopentanoic acid (dl-AP5), picrotoxin, forskolin, (−) - cis - 3 - [ 2 - hydroxy - 4 - (1,1 - dimethylheptyl) phenyl ] - trans - 4 - (3 - hydroxypropyl)cyclohexanol (CP55,940), and 6,7-dinitroquinoxaline-3-dione (DPCPX) were purchased from Tocris Bioscience (Avonmouth, UK).
THC was dissolved in ethanol, cremophor, and saline (1:1:18); SR141716A was dissolved in Tween 80, DMSO, and distilled water (1:2:7); SL327 was dissolved in DMSO.
Animals and drug treatment.
RasGRF +/+ and −/− mice originating from the leading strain described by Brambilla et al. (1997) on a C57BL/6 background were housed in a controlled environment at constant temperature and humidity, on a 12-h light/dark cycle, with free access to food and water. They were chronically treated with THC or its vehicle, subcutaneously administered at the dose of 10 mg/kg twice per day for 4.5 d. This is a well known chronic treatment protocol inducing tolerance in mice (Bass and Martin, 2000; Rubino et al., 2004). Bradykinesia and/or catalepsy were routinely observed during the initial THC exposure. For chronic studies with SR141716A (3 mg/kg), mice were intraperitoneally injected 20 min before THC treatment. In chronic SL327 experiments, mice were administered with 50 mg/kg intraperitoneally of the compound 1 h before every THC injection.
The total number of animals treated with chronic administration of THC was n = 30. The total number of animals injected with vehicle was n = 29. In the experiments using RasGRF1 −/− mice, the total number of chronically THC- and vehicle-treated mice was n = 16 for each group.
All animal protocols were performed in accordance with the Italian Ministry of Health guidelines (D.L. 116/92; D.L. 111/94-B) and with the European Community directives regulating animal research (86/609/EEC). All efforts were made to minimize the number of animals used and their suffering.
Slice preparation.
Parasagittal cerebellar slices (250–300 μm) were prepared from 40- to 50-d-old mice following an established technique (Levenes et al., 1998). Mice were decapitated 1 d after the final injection, and the cerebellar vermis was rapidly removed and chilled in ice-cold artificial CSF (ACSF) consisting of the following (in mm): 130 NaCl, 3.5 KCl, 1.2 NaH2PO4, 1.3 MgCl2, 2 CaCl2, 25 NaHCO3, and 11 glucose, saturated with 95% O2 and 5% CO2. Slices were incubated in ACSF at room temperature (RT) for ≥60 min before recordings. During recordings, slices were continuously superfused with ACSF (2 ml/min) at room temperature.
Electrophysiology.
Whole-cell recordings were obtained from the soma of Purkinje cells (pipette resistance, 3–4.5 MΩ), using an Axopatch 200B patch-clamp amplifier (Molecular Devices, Union City, CA) in voltage-clamp mode. The internal solution contained the following (in mm): 125 CsMetSO3, 10 CsCl, 10 HEPES, 0.2 EGTA, 2 MgATP, 0.2 NaGTP, pH 7.2, with CsOH. In the experiments measuring slow metabotropic glutamate receptor type 1 (mGluR1)-mediated EPSCs, CsMetSO3 was replaced by K-gluconate, pH 7.2, with KOH. Stock solutions of CP55,940, forskolin, SR141716A, and DPCPX were dissolved in DMSO. Dilutions to final concentrations were made just before the start of experiment in oxygenated ACSF. When the effect of CP55,940, forskolin and SR141716A was assessed, control solutions always contained the corresponding DMSO concentration (up to 0.2%). For electrical stimulation of PFs, current pulses (100 μs; 2–7 V) were delivered through a bipolar electrode positioned in the molecular layer. Purkinje cells were voltage clamped at −70 mV, and control currents were recorded at 0.5 or 0.1 Hz in the presence of 100 μm picrotoxin, to block GABAergic inputs. Current were filtered at 2 kHz and sampled at 20 kHz using a 1300A Digidata (Molecular Devices). Series resistance of the recorded cell (10–20 MΩ) was monitored throughout the experiment by a 5 mV hyperpolarizing voltage-step. Paired-pulse facilitation (PPF) was elicited by twin stimuli at different time intervals (50, 100, 150, 200, and 250 ms), and it was expressed as the ratio between the amplitude of the second and the first EPSC. To avoid series resistance errors and an underestimation of PPF, we compared EPSCs with similar amplitudes, ranging from 150 to 350 pA in response to the first stimulus, both in vehicle- and THC-treated mice. Slow mGluR1-mediated EPSCs were activated by high-frequency trains of stimuli to PFs (1–10 stimuli at 100 Hz) at fixed stimulus intensity (7 V), in the presence of ionotropic glutamate and GABA antagonists (100 μm picrotoxin, 40 μm dl-APV, 20 μm DNQX).
During the long-term depression (LTD) experiments, control traces were recorded at 0.5 Hz for at least 10 min. To induce LTD, PF stimulation at 1 Hz was paired with PC depolarization (to 0 mV for 100 ms) 120 times, over 2 min. For the LTP experiments, the external Ca2+ concentration was raised to 2.5 mm. Control traces were recorded at 0.05 Hz. LTP was induced by a brief repetitive stimulation of PF (8Hz, 15 s). In LTP experiments, 20 mm BAPTA was added to the pipette solution. The tetanus was given at least 15 min after the beginning of the recording to reach an adequate BAPTA diffusion in the dendritic tree. The baseline of the PF EPSC amplitude was kept at <300 pA to avoid sodium spikes that escaped voltage-clamp, during and after tetanization.
Statistical analysis.
Data are presented as mean ± SEM. Values obtained from different experimental groups were tested for statistical differences using an independent two-population t test (Origin software; OriginLab, Northampton, MA). Concentration–response curves were obtained using a sigmoidal concentration–response equation: Y = maximum − minimum/(1 − EC50/X), where X is the concentration, Y is the response amplitude, and EC50 is the concentration at which the half-maximal effect was observed.
Protein kinase A activity.
Protein kinase A (PKA) activity was assayed ex vivo according to the instructions of the SignaTECH PKA assay system (Promega, Madison WI). Briefly, 24 h after the last injection, cerebella from chronic THC- and vehicle-treated mice were homogenized in ice-cold buffer containing 0.25 m sucrose, 50 mm Tris-HCl, pH 7.5, 5 mm EGTA, 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 0.1 mm dithiothreitol, 0.1% Triton X-100, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Lysates were centrifuged at 13,000 × g at 4°C, and the supernatant was used as cellular extract. PKA activity was measured in the absence or in the presence of 100 μm exogenous cAMP in the reaction mixture. PKA activity measured in the absence of exogenous cAMP represents the free active catalytic subunit (active PKA) that is present in the extract. PKA activity measured in the presence of cAMP represents the total enzyme (total PKA) present in the cellular preparation. Kinase reaction was initiated by adding 25 μg of proteins and 100 μm biotinylated Kemptide (LRRASLG) to 25 μl of reaction mixture. After 5 min of incubation at 30°C, 12.5 μl of termination buffer was added to the reaction mixture containing γP32-ATP. An aliquot of the reaction mixture was spotted onto a phosphocellulose filter, and PKA activity was measured by scintillation counting.
Results
Altered parallel fiber-Purkinje cell synaptic transmission after chronic THC exposure
Cerebellar PCs receive excitatory inputs from the PFs, the axons of granule cells. Because acute activation of CB1R located presynaptically on PFs modulates granule cell synaptic release (Takahashi and Linden, 2000; Brown et al., 2004; Daniel et al., 2004), we investigated whether chronic cannabinoid treatment would alter PF-PC synaptic properties. The relationship between excitatory postsynaptic current (EPSCs) amplitudes (output) as a function of PF stimulus intensity (input) for each neuron was compared in cerebellar slices prepared from mice chronically exposed to THC (10 mg/kg) or to its vehicle (Fig. 1A). In THC-treated mice, the average values were significantly higher relative to the vehicle group (Fig. 1B). Because acute activation of CB1 receptors suppresses PF-PC synaptic transmission through a presynaptic mechanism, we tested the hypothesis that a compensatory synaptic potentiation occurred at a presynaptic level after prolonged CB1R stimulation. We compared PPF of EPSCs, an index of presynaptic short-term plasticity. The degree of PPF observed in THC-treated mice was reduced compared with animals injected with the vehicle alone, indicating an increased release probability after chronic cannabinoid exposure (Fig. 2A). This effect was prevented by pretreatment with SR141716A (3 mg/kg), a CB1R antagonist (Fig. 2B).
Figure 1.

Chronic THC exposure induced a long-lasting potentiation of basal PF-PC synaptic transmission. A, Representative traces of PF-PC EPSCs recorded at different stimulus intensities, in vehicle (Veh)- and THC-treated mice. Stimulus artifacts were suppressed for clarity. B, Input–output curves for PF-PC EPSCs recorded in cerebellar slices originating from mice chronically exposed to THC or to its vehicle. Asterisks indicate significant differences (p < 0.01). Values are expressed as mean ± SEM.
Figure 2.

Short-term synaptic plasticity was altered after chronic THC exposure (A). Pretreatment with SR141716A (SR; 3 mg/kg, i.p., 20 min before THC injection) prevented PPF reduction (B). Top, Purkinje cell EPSC PPF at intervals of 50, 100, 150, 200, and 250 ms after chronic vehicle (Veh) and chronic THC. Stimulus artifacts were suppressed for clarity. Bottom, PPF ratio values (mean ± SEM) plotted against paired-pulse intervals. Asterisks indicate significant differences (p < 0.01) from chronic vehicle.
Facilitated induction of slow metabotropic glutamate receptor type 1-mediated EPSCs in THC-treated mice
The increased release probability observed at PF-PC synapses after chronic THC exposure could result in an increased accumulation of perisynaptic and extrasynaptic glutamate during trains of PFs activity. Accumulation of glutamate is required for the activation of mGluR1-mediated EPSCs in PCs (Batchelor and Garthwaite, 1993; Tempia et al., 1998; Brown et al., 2004). We evaluated whether the induction of slow mGluR1-mediated EPSCs was facilitated in tolerant mice by delivering a high-frequency train of stimuli to PFs (e.g., 1–10 stimuli at 100 Hz) in the presence of ionotropic GABA and glutamate antagonists (100 μm picrotoxin, 40 μm dl-APV, 20 μm DNQX). This paradigm activated a slow EPSC (Fig. 3) that was blocked by the selective mGluR1 antagonist 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylates (CpCCOet; 300 nm) (Fig. 3A). As expected, the analysis of the input–output relationship revealed a facilitation of mGluR1-mediated EPSCs after chronic THC treatment (Fig. 3B,C).
Figure 3.
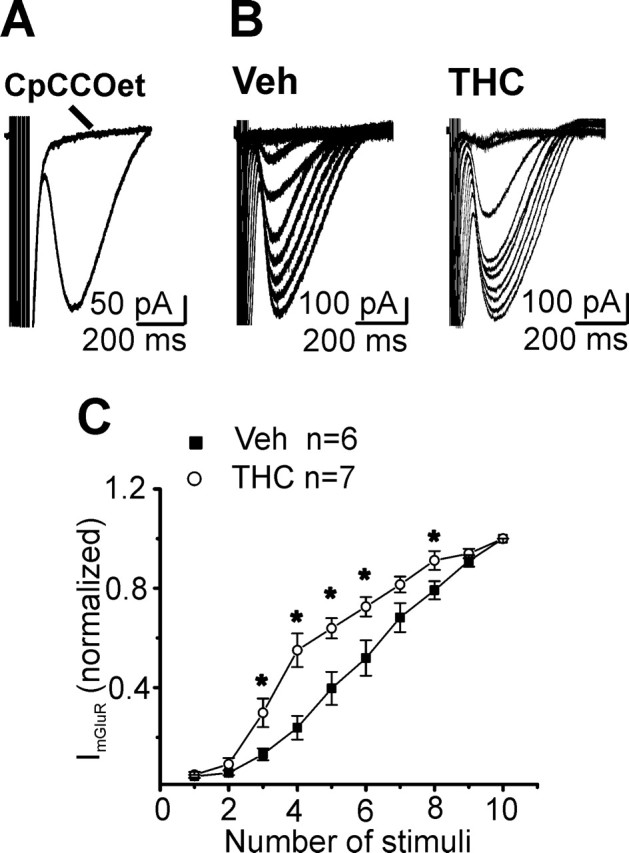
Facilitated induction of slow mGluR1-mediated EPSCs in THC-treated mice. A, High-frequency trains of stimuli (1–10 stimuli at 100 Hz) activated a slow EPSC that was blocked by the selective mGluR1 antagonist CpCCOet (300 nm). B, Sample traces of slow mGluR1-mediated EPSCs in vehicle (Veh)- and THC-exposed mice. C, Plot of the amplitudes of mGluR1-mediated EPSCs recorded at different stimulus intensities in vehicle- and THC-treated animals. Each data point represents the mean ± SEM of currents normalized to the response evoked by a train of 10 stimuli. Asterisks indicate significant differences (p < 0.05) from vehicle.
Tolerance to the synaptic inhibition induced by the CB1R-selective agonist CP55,940
CB1R activation suppresses PF synaptic activity by modulation of presynaptic Ca2+ and K + channels (Takahashi and Linden, 2000). As reported previously for other brain areas (Hoffman et al., 2003; Mato et al., 2004), chronic THC altered the response of PF-PC synapses to the selective CB1R agonist CP55,940 (Fig. 4). The EPSC depression induced by CP55,940 was reduced in mice exposed to THC compared with controls (Fig. 4A). Tolerance to the CP55,940 inhibitory effects could be attributable to residual THC accumulation in the slices, leading to persistent CB1R activation. This possibility was excluded, because bath application of the CBR1 antagonist SR141716A (1 μm) did not increase baseline synaptic transmission in THC-injected mice (98 ± 2% of control, n = 3). The specificity of the CP55,940 effect at high concentrations (CP55,940 10 μm) was confirmed in slices from vehicle-treated mice by its reversibility after SR141716A application (SR141716A 10 μm) (EPSC amplitude: 96 ± 6% of control, n = 3) (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 4.

Functional tolerance to the CP55,940-mediated inhibition of glutamate release at PF-PC synapse. A, Traces of EPSCs and inhibition by 20 μm CP55,940 in slices from a vehicle (Veh)- and THC-treated mice. B, Concentration–response curve for PF-PC EPSCs inhibition in cerebellar slices from mice after chronic vehicle (▪; EC50 = 3.7 ± 1.5 μm) or chronic THC (○; EC50 = 2 ± 1 μm). Each point represents the mean ± SEM of three to five cells.
Slices from THC-exposed mice showed a reduction in CP55,940 inhibition relative to the controls, not accompanied by a rightward shift of the concentration–response curve (Fig. 4B). A decreased number of CB1Rs or a reduced efficacy in CB1R-mediated signaling, rather than a modification of CB1R agonist affinity, may explain this effect. Consistent with these data, chronic THC administration (10 mg/kg, s.c., twice per day for 4.5 d) reduced the [3H]CP55,940 receptor binding and attenuated the cannabinoid-induced G-protein activation in the cerebellum originating from the same mouse strain (Rubino et al., 2005).
Chronic THC exposure did not affect PF-PC LTD but inhibited a presynaptic form of PF-PC LTP
The effects of cannabinoid tolerance include an impaired motor coordination (Chaperon and Thiebot, 1999). Long-term depression (LTD) of the PF-PC synapse is a cellular phenomenon that has been suggested to underlie forms of motor learning and coordination (Hansel et al., 2001). Additionally, PF-PC LTD is partially occluded by acute CB1R activation (Levenes et al., 1998). However, when we examined the properties of PF-PC LTD after chronic THC treatment we could not detect any significant difference relative to the chronic vehicle (Fig. 5A,B).
Figure 5.

Chronic THC exposure did not affect PF-PC LTD. A, Superimposed averaged records (5 traces) of PF responses from a PC, before (solid line) and 30 min after (dotted line) the LTD induction. B, Averaged time course (mean ± SEM) of normalized EPSC amplitudes. At t = 10 min, LTD was induced by pairing 1 Hz stimulation of PF for 2 min (horizontal bar; 120 stimuli) with PC depolarization (to 0 mV for 100 ms). Traces were acquired at 0.5 Hz and averaged every 60 s. Veh, Vehicle.
In addition to LTD, PFs express a form of long-term potentiation (LTP) that is triggered by presynaptic Ca2+ influx and is dependent on the activation of presynaptic adenylyl cyclase and protein kinase A (Salin et al., 1996; Chen and Regehr, 1997). The altered PPF measured in THC-exposed mice (Fig. 2A) and the involvement of the cAMP/PKA pathway in the development of cannabinoid tolerance (Rubino et al., 2000; Tzavara et al., 2000) suggest that cerebellar PKA-mediated LTP might be affected by chronic cannabinoid treatment. To test this hypothesis, we first analyzed changes in PF-PC synaptic strength induced by bath application of forskolin, an adenylyl cyclase activator. It is well known that activation of the cAMP/PKA pathway by forskolin causes a PKA-mediated phosphorylation of synaptic active zone proteins (Chen and Regehr, 1997; Lonart et al., 2003), leading to an increase of glutamate release probability. As expected, in vehicle-injected mice we observed a forskolin-induced enhancement of EPSCs (Fig. 6A). However, after chronic THC exposure, forskolin caused a depression of PF-PC transmission (Fig. 6A). On the same extent, brief tetanic PF stimulation (8 Hz, 15 s), known to induce a PKA-dependent LTP (Salin et al., 1996), elicited an enhancement of synaptic responses in slices from vehicle-treated mice, whereas it triggered a clear EPSC depression in THC-exposed mice (Fig. 6B). The EPSC depression, which persisted for many minutes after the end of the tetanic stimulation, was associated with an increase of PPF ratio (measured in a subset of experiments at 30 min after induction: 126 ± 2.1% of control, n = 3). This strongly suggests that the depression was caused, at least in part, by a decrease in transmitter release. Previous studies have demonstrated that both CB1 and adenosine A1 receptors (A1Rs) exhibit convergent regulation of adenylyl cyclase at PF-PC synapses (DeSanty and Dar, 2001; Selley et al., 2004). Like CB1R, A1R activation inhibits synaptic transmission (Fredholm et al., 1990; Mitchell et al., 1993; DeSanty and Dar, 2001). It has been reported that increases in cAMP level can lead to the formation of extracellular adenosine and the activation of adenosine receptors. This can be observed with both forskolin-induced stimulation of adenylyl cyclase (Brundege and Dunwiddie, 1998) and after synaptic stimulation (Mitchell et al., 1993; Manzoni et al., 1994). Because chronic THC treatment induces an upregulation of cerebellar basal cAMP level (Rubino et al., 2000), the activation of the adenosine receptors might contribute to the EPSC depression measured in tolerant mice. To address this hypothesis, we performed experiments on slices obtained from THC-treated mice in which PF tetanic stimulation was delivered in the presence of the selective A1R antagonist DPCPX (500 nm). DPCPX application not only prevented the stimulus-induced depression of EPSCs, but it fully rescued PF-PC LTP (Fig. 6C). To characterize the timing of A1R signaling relevant for the induction and expression of LTD, and to understand whether continued activation of A1R is necessary to maintain this form of synaptic depression, DPCPX was applied at increasing intervals after the stimulus. Application of DPCPX at 30 s and 5 min after tetanus, preceding the onset of LTD (see Fig. 6B), did not prevent LTD induction (see supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Additionally, continuous activation of A1R was not required for LTD maintenance, because application of DPCPX at 30 min after tetanus, when LTD was clearly expressed, did not reverse it (see supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). These findings suggest that the A1R activation necessary for LTD induction occurred during the tetanus, because LTD could only be prevented when DPCPX was applied before stimulation. Furthermore, they indicate that maintenance of LTD, 30 min after induction, did not require continuous A1R activation.
Figure 6.
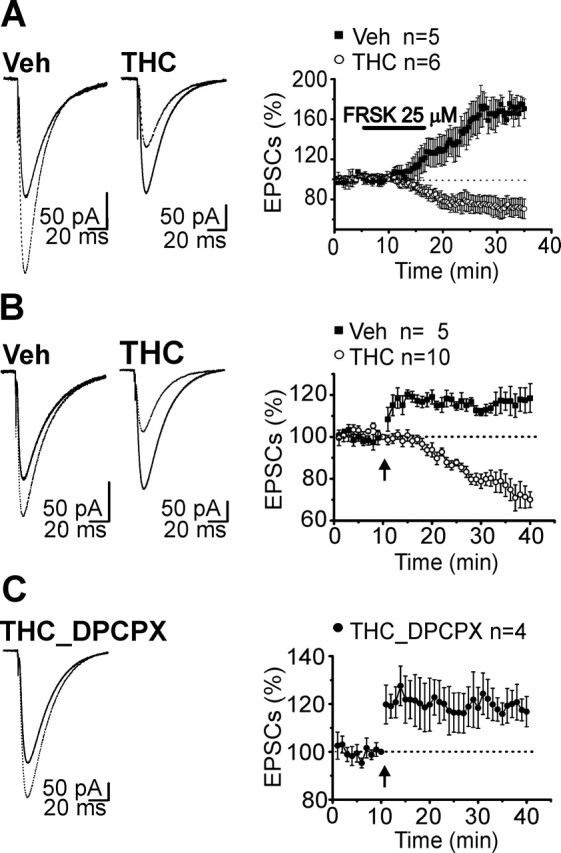
In THC tolerant mice, presynaptic PF-PC LTP was blocked by adenosine receptor activation. A, Activation of the cAMP/PKA pathway by forskolin application induced an enhancement of EPSCs in vehicle-injected mice. However, after chronic THC exposure, forskolin caused a depression of PF-PC synaptic transmission. B, Brief tetanic stimulation (8 Hz, 15 s) caused a long-lasting increase of the PF responses in slices from vehicle (Veh)-treated mice, whereas it triggered a clear EPSC depression in THC tolerant mice. C, Inhibition of A1R by application of DPCPX (500 nm) prevented the tetanus-induced depression and fully rescued PF-PC in THC-tolerant slices. DPCPX application started 30 min before stimulation and continued until the end of the experiment. A–C, Left, Superimposed averaged records (5 traces) of PF response from a PC before (solid line) and 30 min after forskolin (A) or tetanus application (B, C) (dotted line) in cerebellar slices from vehicle- and THC-treated mice. A–C, Right, Averaged time course (mean ± SEM) of forskolin- (A) or tetanus-induced (B, C) changes in normalized PF EPSC amplitudes. A, Traces were acquired every 0.033 Hz. B, C, Traces were acquired every 0.033 Hz and averaged every 10 s. Tetanus was given at t = 10 min (indicated by the arrow).
Application of DPCPX alone did not affect basal PF-PC synaptic transmission (at 25 min after DPCPX application: vehicle, 100.9 ± 6.5% of control; THC, 97.8 ± 2.1% of control, n = 3; see also supplemental Fig. 2C, available at www.jneurosci.org as supplemental material) in ether vehicle- or chronic THC-treated mice. Thus, after chronic THC administration, the basal extracellular adenosine tone was not elevated and adenosine was released most likely in response to the tetanic stimulation.
RasGRF1 knock-out mice did not show changes in PF-PC synaptic properties after repeated THC exposure
RasGRF1 −/− mice do not develop behavioral tolerance to cannabinoid-induced locomotor effects after chronic cannabinoid exposure (Rubino et al., 2004). Recently, it has been reported that acute THC exposure induces activation of the cerebellar ERK cascade (Rubino et al., 2004) and triggers cerebellar CB1R downregulation and uncoupling from Gi/o-protein in wild-type, but not in RasGRF1 null mice (Rubino et al., 2005). This suggests that the Ras/ERK pathway may be relevant for the establishment of cannabinoid behavioral tolerance in the cerebellum. We therefore investigated the involvement of the Ras/ERK cascade in the development of the synaptic alterations observed at PF-PC synapses after repeated THC administration.
Unlike what we observed in wild-type animals (Fig. 2), chronic THC treatment did not affect PF-PC short-term synaptic plasticity (Fig. 7A) or the induction of slow mGluR1-mediated EPSCs (Fig. 7A) in RasGRF1 −/− mice. It is worth noting the decreased PPF measured in RasGRF1 −/− compared with +/+ cerebellar slices both in vehicle-treated (PPF at 50 ms stimulus intervals: +/+, 2.2 ± 0.04, n = 17; −/−, 2.04 ± 0.06, n = 10; p < 0.05) (see also Figs. 2, 7) and in naive mice (PPF at 50 ms stimulus intervals: +/+, 2.25 ± 0.05, n = 15; −/−, 1.99 ± 0.1, n = 8; p < 0.05), indicating an increase in the basal release probability in RasGRF1 knock-out animals. In addition, slices obtained from knock-out, THC-treated mice did not show any tolerance to the inhibitory effect of CP55,940 (10 μm) (% of EPSC inhibition: vehicle, 58 ± 1.7%, n = 5 vs THC, 55 ± 4.4%, n = 7). In agreement with the lower coupling efficiency between CB1R and Gi/o transduction proteins measured in RasGRF1 −/− cerebellum relative to wild type (Rubino et al., 2005), efficacy, but not potency, of CP55,940 inhibition of −/− PF-PC EPSCs was reduced compared with controls (% of EPSCs inhibition: vehicle −/−, 58 ± 1.7%, n = 5 vs vehicle +/+, 71 ± 2.5%, n = 5, p < 0.01; vehicle −/−, EC50 = 3.5 ± 1.2 μm; vehicle +/+, EC50 = 3.7 ± 1.5 μm; see also supplemental Fig. 3, available at www.jneurosci.org as supplemental material). We then assessed the effect of chronic THC treatment on −/− PF-PC LTP. Tetanic stimulation elicited only a short-term enhancement of the EPSCs that returned to baseline levels at 10 min after the tetanus, both in THC- and vehicle-treated −/− mice (Fig. 7C). These results indicate that the PKA-dependent LTP as well as the A1R-induced EPSCs depression were abolished in RasGRF1 −/− mice, independently of the chronic THC exposure.
Figure 7.
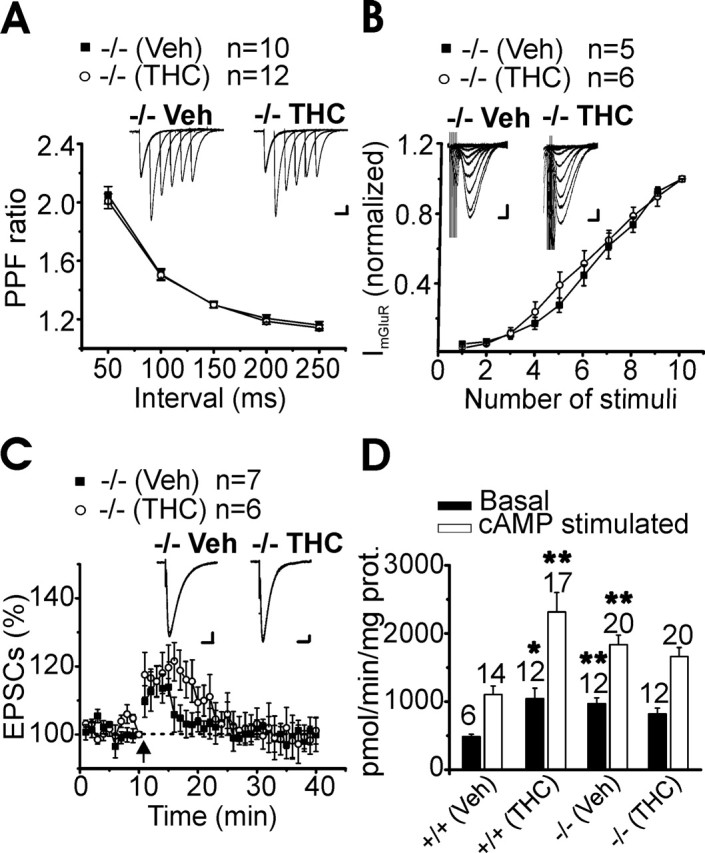
Mice knock-out for the RasGRF1 gene, a Ras/ERK pathway activator, did not show changes in PF-PC synaptic properties after repeated THC exposure. A, Summary of PF-PC PPF ratio values (mean ± SEM) measured at five interstimulus intervals (50, 100, 150, 200, and 250 ms) in cerebellar slices from RasGRF1 −/− vehicle (Veh)- and THC-injected animals. Inset, Representative traces of Purkinje cell EPSC PPF at intervals of 50, 100, 150, 200, and 250 ms after chronic vehicle and chronic THC. Stimulus artifacts were suppressed for clarity. Calibration: 50 pA, 50 ms. B, Amplitude of mGluR1-mediated EPSCs versus number of pulses. Each data point represent the mean ± SEM (▪, −/− chronic vehicle; ○, −/− chronic THC) normalized to the response evoked by a train of 10 stimuli. Inset, mGluR1-mediated EPSCs evoked in PCs by 1–10 train pulses (at 100 Hz) in a vehicle- and THC-treated cerebellar slice. Calibration: 50 pA, 100 ms. C, Averaged time course (mean ± SEM) of tetanus-induced changes in normalized RasGRF1 −/− PF EPSC amplitudes. Tetanus was given at t = 10 min, as indicated by the arrow. Traces were acquired every 0.033 Hz and averaged every 10 s. Inset, Superimposed traces of PF response from a PC before (solid line) and 30 min after tetanus application (dotted line) in cerebellar slices from RasGRF1 −/− vehicle- and THC-treated mice. Calibration: 25 pA, 25 ms. D, Effect of chronic in vivo treatment with THC on ex vivo basal- and cAMP-stimulated PKA activity in RasGRF1 −/− and +/+ cerebellum. Data are expressed as mean ± SEM of at least six mice in each group. n, number of samples for each group. *p < 0.05 vs vehicle; **p < 0.01 vs vehicle.
Repeated cannabinoid exposure enhances the cAMP cascade in several brain areas, leading to an upregulated PKA activity (Rubino et al., 2000). In agreement with these data, chronic THC exposure caused a significant increase of both active and total PKA activity (see Materials and Methods for details) in the cerebellum of wild-type mice, which was not detectable in RasGRF1 −/− mice (Fig. 7D). It is worth noting that both active and total PKA activities were higher in RasGRF1 −/− relative to +/+ vehicle-treated animals (active PKA: vehicle −/−, 970 ± 85 vs vehicle +/+, 485 ± 35, p < 0.01; total PKA: vehicle −/−, 2190 ± 140 vs vehicle +/+, 1135 ± 166, p < 0.01; PKA activity is expressed as picomoles of ATP per minute per milligram of protein).
Altogether, this evidence points to alterations of the basal CB1R- and cAMP-mediated signaling in RasGRF1−/− mice.
Pharmacological inhibition of the ERK pathway prevented chronic THC-induced alterations of cerebellar synaptic plasticity
The results obtained in RasGRF1 −/− mice suggest a role of the ERK pathway in the alterations of cerebellar synaptic plasticity caused by repeated THC exposure. However, the basal differences in release probability and the lack of the PKA-mediated LTP described in this mouse model, together with possible developmental effects of gene knock-out, prompted us to confirm the involvement of the ERK cascade in the observed phenomena by a pharmacological approach.
In wild-type mice, the in vivo pretreatment with SL327 (50 mg/Kg), a drug that crosses the blood–brain barrier and prevents the activation of ERK by inhibiting MEK (Atkins et al., 1998; Derkinderen et al., 2003), prevented the enhancement of PF-PC short-term synaptic plasticity (Fig. 8A) and the facilitation of mGluR1-mediated EPSCs, induced by chronic THC treatment (Fig. 8B). Additionally, inhibition of the ERK cascade during the chronic THC treatment blocked the A1R-dependent synaptic depression seen in tolerant mice (Fig. 8C). After pretreatment with SL327, tetanic stimulation of PF (8Hz, 15 s) elicited a clear EPSCs LTP in cerebellar slices from THC-exposed mice (Fig. 8C).
Figure 8.

In vivo SL327 pretreatment prevented chronic THC-induced alteration of PF-PC cerebellar synaptic plasticity. A, Summarized magnitude of paired-pulse responses (mean ± SEM) measured at five interstimulus intervals (50, 100, 150, 200, and 250 ms) in slices from SL323 pretreated mice exposed to chronic vehicle (Veh) or chronic THC. The number of cells analyzed are as indicated. B, Averaged Input–output curves (mean ± SEM) for slow mGluR1-mediated EPSCs (▪, SL327+veh; ○, SL327+THC). Current amplitudes were normalized to the response evoked by a train of 10 stimuli. C, Averaged time courses of tetanus induced changes in normalized EPSCs (mean ± SEM) for four and five Purkinje neurons recorded in cerebellar slices originating from SL323 pretreated mice exposed chronically to vehicle (▪) or to THC (○). □, The average time course of normalized EPSCs already shown in Figure 6 in non-SL327-pretreated mice exposed only to chronic THC and reported here for comparison. Tetanus (8 Hz, 15 s) was given at t = 10 min (indicated by the arrow). Traces were acquired every 0.033 Hz and averaged every 10 s.
Discussion
Cerebellar circuits are major neurobiological substrates for the chronic effects of THC and for the expression of THC withdrawal in vivo (Rubino et al., 2000; Tzavara et al., 2000; Ghozland et al., 2002; Maldonado and Rodriguez, 2002).
There is substantial evidence of changes in cerebellar CB1R molecular adaptations and modifications in receptor signaling taking place after prolonged THC exposure (Martin et al., 2004; Rubino et al., 2004). However, little is known about the effects of chronic THC treatment on cerebellar synaptic plasticity, which may contribute to the development of behavioral tolerance. Here, we demonstrated that neurophysiological adaptive mechanisms occur at cerebellar PF-PC synapses after repeated THC administration and that such neuroadaptations are dependent on the ERK pathway activation. These alterations deregulate the physiological long-term synaptic plasticity between PF and PC, potentially affecting cerebellar functions.
Tolerance results from compensatory mechanisms that maintain equilibrium by reducing drug effects (Berke and Hyman, 2000). In rat cerebellar slices, acute CB1R activation presynaptically suppresses PF synaptic inputs to PC (Levenes et al., 1998; Daniel et al., 2004). We have shown that chronic THC administration induced a potentiation of the synaptic transmission between PF and PC, persisting for at least 24 h after drug injection. This phenomenon was expressed at the presynaptic site, as inferred from the decreased PPF ratio measured in tolerant mice. The reduction in PPF, indicating an increased release probability (pr) (Stuart and Redman, 1991), was likely to be dependent on CB1R activation, because it was fully blocked by in vivo pretreatment with the CB1R antagonist SR141716A. Although there is evidence for multiple types of cannabinoid receptors in the CNS (Begg et al., 2005; Hoffman et al., 2005), CB1R is present at high levels in the presynaptic boutons of cerebellar granule cells (Pettit et al., 1998; Tsou et al., 1998; Kawamura et al., 2006). Additionally, in the cerebellum, CB1R is responsible for cannabinoid-dependent suppression of excitatory transmission (Kawamura et al., 2006) and for several forms of endocannabinoid (eCB)-mediated synaptic plasticity (Kreitzer and Regehr, 2001; Safo and Regehr, 2005).
pr is a function of modulation of synaptic vesicle exocytosis by second-messenger activated protein kinases (Salin et al., 1996; Zucker and Regehr, 2002; Leenders and Sheng, 2005). The higher pr observed in THC-treated mice may be caused by the upregulation of the cAMP/PKA pathway occurring in the cerebellum during cannabinoid tolerance (Rubino et al., 2000). The present results are consistent with reports of a PKA-dependent increase of transmitter release after prolonged exposure to other drugs of abuse (Bonci and Williams, 1997; Melis et al., 2002). However, we cannot exclude that the increase in pr may be dependent on the THC-induced activation of the Ras/ERK pathway (Valjent et al., 2000, 2001, 2004; Derkinderen et al., 2003). Recent reports demonstrate that activation of Ras/ERK signaling enhances docking of synaptic vesicle to active zones, thereby leading to an increase in the size of the readily releasable pool of vesicles (Kushner et al., 2005; Seeger et al., 2005).
A repetitive stimulation of PF-PC synapses is required for glutamate spillover into perisynaptic and extrasynaptic space, where it activates postsynaptic mGluR1 (Reichelt and Knopfel, 2002). The facilitation of the mGluR1-mediated EPSCs that we observed in tolerant mice may therefore be explained by the increase in pr.
mGluR1 activation in PC is essential for long-term depression and motor coordination (Aiba et al., 1994; Conquet et al., 1994). Although THC-treated mice did not exhibit an altered LTD, facilitation of mGluR1 may interfere with the postsynaptic mGluR1-dependent signaling. Synaptic activation of mGluR1 and the elevation of postsynaptic Ca2+ induce eCB biosynthesis and release (Brenowitz and Regehr, 2005). Depolarization-induced suppression of excitation (DSE) (Kreitzer and Regehr, 2001) relies instead on eCB release that is triggered by membrane depolarization, differing from eCB release induced by synaptic activation (Safo and Regehr, 2005). In THC tolerant mice, the facilitation of mGluR1-mediated EPSCs, together with the functional tolerance to the effects of the CB1R agonist CP55,940, may have different implications for these forms of synaptic plasticity that involve eCB signaling. The effects of chronic THC treatment on the eCB-mediated synaptic plasticity have not been analyzed here and are matter of a future investigation.
The PF-PC synapse displays a PKA-dependent type of LTP (Salin et al., 1996). In THC-tolerant mice, activation of the cAMP/PKA pathway by forskolin or by brief tetanic stimulation induces an adenosine A1R-mediated inhibition of synaptic release, instead of triggering LTP. Adenylyl cyclase activation by forskolin causes intracellular cAMP accumulation, transport, and conversion into adenosine by extracellular phosphodiesterase activity (Rosenberg and Dichter, 1989; Rosenberg et al., 1994; Rosenberg and Li, 1995). Adenosine, originating from the metabolism of intracellular cAMP, may also be released after synaptic stimulation (Mitchell et al., 1993). An increase of extracellular adenosine after the activation of adenylyl cyclase has been reported in several brain areas as a consequence of the upregulation of the cAMP cascade after chronic administration of drugs of abuse (Bonci and Williams, 1997; Chieng and Williams, 1998). In our experiments, the selective A1R antagonist DPCPX did not influence basal PF-PC synaptic transmission, indicating that the extracellular adenosine tone was not elevated in THC tolerant mice. However, the tetanus-induced depression of EPSCs was fully prevented by DPCPX application. After chronic THC treatment, the increase of cAMP might lead to an accumulation of intracellular adenosine that can be released in response to tetanic stimulation.
The contribution of the A1R to PF-PC long-term synaptic plasticity is of particular relevance. First, it suggests a functional interaction between cerebellar A1R- and CB1R-mediated signaling that may play a role in the behavioral tolerance to the THC-induced locomotor effects. Additionally, this interaction generates a form of pathological synaptic plasticity in THC tolerant mice, where brief tetanic stimulation induced LTD instead of LTP at the PF-PC synapse. Considering the possible function of presynaptic parallel fiber LTP during motor learning (Du et al., 1995; Perrett and Mauk, 1995; Kim and Thompson, 1997; Hansel et al., 2001), the conversion of LTP into a form of A1R-dependent LTD may have profound consequences for cerebellar information processing.
ERK regulates cerebellar and striatal CB1R receptor plasticity (desensitization and downregulation) underlying the tolerance to the THC-induced hypomotility (Rubino et al., 2004, 2005). As inferred from behavioral and biochemical work performed on RasGRF1 knock-out mice (Rubino et al., 2004) and on SL327 pretreated wild-type mice (Rubino et al., 2005), ERK activation is not necessary for triggering the acute effects of THC but is required for the chronic ones (Rubino et al., 2004, 2005). It has been recently proposed that THC-induced ERK activation would lead to phosphorylation of target proteins relevant for CB1R desensitization and internalization (Rubino et al., 2006). As suggested for other G-protein-coupled receptors (GPCRs) (Schmidt et al., 2000; Trincavelli et al., 2002), ERK activation may represent an initial signal for CB1R desensitization. Our results indicate that ERK activation is also relevant for the emergence of the PF-PC synaptic alterations induced by repeated THC exposure. Mice lacking RasGRF1, a Ras/ERK pathway activator, do not develop tolerance to the THC hypolocomotor effect or show cerebellar CB1R modifications after chronic treatment with THC (Rubino et al., 2004, 2005). Similarly, in RasGRF1 −/− animals, repeated THC treatment did not affect either PF-PC synaptic properties or the PF-PC synaptic response to the inhibitory action of CP55,940. RasGRF1 −/− mice are basally different from wild type in some biochemical and functional features. They show a lower coupling efficacy between cerebellar CB1Rs and the Gi/o proteins (Rubino et al., 2005) compared with wild type, which may account for the basal reduced sensitivity of PF-PC synapses to CP55,940. In RasGRF1 −/− mice, basal pr values measured at PF-PC synapses was higher relative to control mice. PKA-dependent LTP was also impaired in RasGRF1 −/− mice, both in vehicle- and THC-treated animals, suggesting an altered cAMP/PKA-mediated signaling in this mouse model. This hypothesis is supported by our results showing that RasGRF1 −/− mice displayed higher levels of PKA activity relative to wild-type animals.
The ERK-dependent modulation of PF-PC synaptic plasticity after chronic THC exposure is further confirmed by the results obtained in SL327-treated mice. The pharmacological inhibition of ERK during chronic THC exposure prevented the alterations in PF-PC synaptic plasticity described in THC tolerant mice, as summarized in the model proposed in the supplemental Figure 4 (available at www.jneurosci.org as supplemental material). These neurophysiological alterations of synaptic plasticity might be the consequence of CB1R desensitization and/or originate from independent presynaptic mechanisms triggered by THC exposure, including changes in gene expression.
In summary, this work reveals for the first time that prolonged THC exposure induces ERK-dependent neuronal adaptations at the PF-PC synapes that contribute to generate forms of pathological plasticity and might play a role in cannabinoid dependence.
Footnotes
This work was supported by the Italian Ministry of Education, University, and Research (Programmi di Ricerca di Rilevante Interesse Nazionale 2003 to R.Z. and R.T.). We thank the National Institute on Drug Abuse (Foreigner Investigator Supply Program) for the THC supply. We are grateful to Sanofi Synthelabo Recherche for the SR141716A supply. We also thank Dr. Paola Pedarzani for critical feedback during the preparation of this manuscript.
References
- Abood ME, Sauss C, Fan F, Tilton CL, Martin BR (1993). Development of behavioral tolerance to delta 9-THC without alteration of cannabinoid receptor binding or mRNA levels in whole brain. Pharmacol Biochem Behav 46:575–579. [DOI] [PubMed] [Google Scholar]
- Adams IB, Martin BR (1996). Cannabis: pharmacology and toxicology in animals and humans. Addiction 91:1585–1614. [PubMed] [Google Scholar]
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S (1994). Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell 79:377–388. [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD (1998). The MAPK cascade is required for mammalian associative learning. Nat Neurosci 1:602–609. [DOI] [PubMed] [Google Scholar]
- Bass CE, Martin BR (2000). Time course for the induction and maintenance of tolerance to delta(9)-tetrahydrocannabinol in mice. Drug Alcohol Depend 60:113–119. [DOI] [PubMed] [Google Scholar]
- Batchelor AM, Garthwaite J (1993). Novel synaptic potentials in cerebellar Purkinje cells: probable mediation by metabotropic glutamate receptors. Neuropharmacology 32:11–20. [DOI] [PubMed] [Google Scholar]
- Begg M, Pacher P, Batkai S, Osei-Hyiaman D, Offertaler L, Mo FM, Liu J, Kunos G (2005). Evidence for novel cannabinoid receptors. Pharmacol Ther 106:133–145. [DOI] [PubMed] [Google Scholar]
- Berke JD, Hyman SE (2000). Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25:515–532. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT (1997). Increased probability of GABA release during withdrawal from morphine. J Neurosci 17:796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Gnesutta N, Minichiello L, White G, Roylance AJ, Herron CE, Ramsey M, Wolfer DP, Cestari V, Rossi-Arnaud C, Grant SG, Chapman PF, Lipp HP, Sturani E, Klein R (1997). A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature 390:281–286. [DOI] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2005). Associative short-term synaptic plasticity mediated by endocannabinoids. Neuron 45:419–431. [DOI] [PubMed] [Google Scholar]
- Brown SP, Safo PK, Regehr WG (2004). Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci 24:5623–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundege JM, Dunwiddie TV (1998). Metabolic regulation of endogenous adenosine release from single neurons. NeuroReport 9:3007–3011. [DOI] [PubMed] [Google Scholar]
- Chaperon F, Thiebot MH (1999). Behavioral effects of cannabinoid agents in animals. Crit Rev Neurobiol 13:243–281. [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG (1997). The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci 17:8687–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng B, Williams JT (1998). Increased opioid inhibition of GABA release in nucleus accumbens during morphine withdrawal. J Neurosci 18:7033–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Conde F (1994). Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature 372:237–243. [DOI] [PubMed] [Google Scholar]
- Daniel H, Rancillac A, Crepel F (2004). Mechanisms underlying cannabinoid inhibition of presynaptic Ca2+ influx at parallel fibre synapses of the rat cerebellum. J Physiol (Lond) 557:159–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H, Ledent C, Trzaskos J, Caboche J, Girault JA (2003). Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci 23:2371–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSanty KP, Dar MS (2001). Cannabinoid-induced motor incoordination through the cerebellar CB(1) receptor in mice. Pharmacol Biochem Behav 69:251–259. [DOI] [PubMed] [Google Scholar]
- Du LS, Raymond JL, Sejnowski TJ, Lisberger SG (1995). Learning and memory in the vestibulo-ocular reflex. Annu Rev Neurosci 18:409–441. [DOI] [PubMed] [Google Scholar]
- Feig LA (1994). Guanine-nucleotide exchange factors: a family of positive regulators of Ras and related GTPases. Curr Opin Cell Biol 6:204–211. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Duner-Engstrom M, Fastbom J, Hu PS, van der Ploeg I (1990). Role of G proteins, cyclic AMP, and ion channels in the inhibition of transmitter release by adenosine. Ann NY Acad Sci 604:276–288. [DOI] [PubMed] [Google Scholar]
- Ghozland S, Aguado F, Espinosa-Parrilla JF, Soriano E, Maldonado R (2002). Spontaneous network activity of cerebellar granule neurons: impairment by in vivo chronic cannabinoid administration. Eur J Neurosci 16:641–651. [DOI] [PubMed] [Google Scholar]
- Haney M, Ward AS, Comer SD, Foltin RW, Fischman MW (1999). Abstinence symptoms following smoked marijuana in humans. Psychopharmacology (Berl) 141:395–404. [DOI] [PubMed] [Google Scholar]
- Hansel C, Linden DJ, D'Angelo E (2001). Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci 4:467–475. [DOI] [PubMed] [Google Scholar]
- Hart CL, Ward AS, Haney M, Comer SD, Foltin RW, Fischman MW (2002). Comparison of smoked marijuana and oral delta(9)-tetrahydrocannabinol in humans. Psychopharmacology (Berl) 164:407–415. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Oz M, Caulder T, Lupica CR (2003). Functional tolerance and blockade of long-term depression at synapses in the nucleus accumbens after chronic cannabinoid exposure. J Neurosci 23:4815–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AF, Macgill AM, Smith D, Oz M, Lupica CR (2005). Species and strain differences in the expression of a novel glutamate-modulating cannabinoid receptor in the rodent hippocampus. Eur J Neurosci 22:2387–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M, Ohno-Shosaku T, Kano M (2006). The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J Neurosci 26:2991–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Thompson RF (1997). Cerebellar circuits and synaptic mechanisms involved in classical eyeblink conditioning. Trends Neurosci 20:177–181. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001). Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727. [DOI] [PubMed] [Google Scholar]
- Kushner SA, Elgersma Y, Murphy GG, Jaarsma D, van Woerden GM, Hojjati MR, Cui Y, LeBoutillier JC, Marrone DF, Choi ES, De Zeeuw CI, Petit TL, Pozzo-Miller L, Silva AJ (2005). Modulation of presynaptic plasticity and learning by the H-ras/extracellular signal-regulated kinase/synapsin I signaling pathway. J Neurosci 25:9721–9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenders AG, Sheng ZH (2005). Modulation of neurotransmitter release by the second messenger-activated protein kinases: implications for presynaptic plasticity. Pharmacol Ther 105:69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenes C, Daniel H, Soubrie P, Crepel F (1998). Cannabinoids decrease excitatory synaptic transmission and impair long-term depression in rat cerebellar Purkinje cells. J Physiol 510:867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudhof TC, Linden DJ (2003). Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell 115:49–60. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Rodriguez dF (2002). Cannabinoid addiction: behavioral models and neural correlates. J Neurosci 22:3326–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni OJ, Manabe T, Nicoll RA (1994). Release of adenosine by activation of NMDA receptors in the hippocampus. Science 265:2098–2101. [DOI] [PubMed] [Google Scholar]
- Marr D (1969). A theory of cerebellar cortex. J Physiol (Lond) 202:437–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Sim-Selley LJ, Selley DE (2004). Signaling pathways involved in the development of cannabinoid tolerance. Trends Pharmacol Sci 25:325–330. [DOI] [PubMed] [Google Scholar]
- Mato S, Chevaleyre V, Robbe D, Pazos A, Castillo PE, Manzoni OJ (2004). A single in-vivo exposure to delta 9THC blocks endocannabinoid-mediated synaptic plasticity. Nat Neurosci 7:585–586. [DOI] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci (2002). A long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci 22:2074–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JB, Lupica CR, Dunwiddie TV (1993). Activity-dependent release of endogenous adenosine modulates synaptic responses in the rat hippocampus. J Neurosci 13:3439–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Shim JY, Assi AA, Norford D, Howlett AC (2002). CB(1) cannabinoid receptor-G protein association: a possible mechanism for differential signaling. Chem Phys Lipids 121:91–109. [DOI] [PubMed] [Google Scholar]
- Patel S, Hillard CJ (2001). Cannabinoid CB(1) receptor agonists produce cerebellar dysfunction in mice. J Pharmacol Exp Ther 297:629–637. [PubMed] [Google Scholar]
- Perrett SP, Mauk MD (1995). Extinction of conditioned eyelid responses requires the anterior lobe of cerebellar cortex. J Neurosci 15:2074–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit DA, Harrison MP, Olson JM, Spencer RF, Cabral GA (1998). Immunohistochemical localization of the neural cannabinoid receptor in rat brain. J Neurosci Res 51:391–402. [DOI] [PubMed] [Google Scholar]
- Reichelt W, Knopfel T (2002). Glutamate uptake controls expression of a slow postsynaptic current mediated by mGluRs in cerebellar Purkinje cells. J Neurophysiol 87:1974–1980. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Dichter MA (1989). Extracellular cAMP accumulation and degradation in rat cerebral cortex in dissociated cell culture. J Neurosci 9:2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg PA, Li Y (1995). Adenylyl cyclase activation underlies intracellular cyclic AMP accumulation, cyclic AMP transport, and extracellular adenosine accumulation evoked by beta-adrenergic receptor stimulation in mixed cultures of neurons and astrocytes derived from rat cerebral cortex. Brain Res 692:227–232. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Knowles R, Knowles KP, Li Y (1994). Beta-adrenergic receptor-mediated regulation of extracellular adenosine in cerebral cortex in culture. J Neurosci 14:2953–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino T, Vigano' D, Massi P, Spinello M, Zagato E, Giagnoni G, Parolaro D (2000). Chronic delta-9-tetrahydrocannabinol treatment increases cAMP levels and cAMP-dependent protein kinase activity in some rat brain regions. Neuropharmacology 39:1331–1336. [DOI] [PubMed] [Google Scholar]
- Rubino T, Forlani G, Vigano D, Zippel R, Parolaro D (2004). Modulation of extracellular signal-regulated kinases cascade by chronic delta 9-tetrahydrocannabinol treatment. Mol Cell Neurosci 25:355–362. [DOI] [PubMed] [Google Scholar]
- Rubino T, Forlani G, Vigano D, Zippel R, Parolaro D (2005). Ras/ERK signalling in cannabinoid tolerance: from behaviour to cellular aspects. J Neurochem 93:984–991. [DOI] [PubMed] [Google Scholar]
- Safo PK, Regehr WG (2005). Endocannabinoids control the induction of cerebellar LTD. Neuron 48:647–659. [DOI] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA (1996). Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron 16:797–803. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Schulz S, Klutzny M, Koch T, Handel M, Hollt V (2000). Involvement of mitogen-activated protein kinase in agonist-induced phosphorylation of the mu-opioid receptor in HEK 293 cells. J Neurochem 74:414–422. [DOI] [PubMed] [Google Scholar]
- Seeger G, Gartner U, Arendt T (2005). Transgenic activation of Ras in neurons increases synapse formation in mouse neocortex. J Neural Transm 112:751–761. [DOI] [PubMed] [Google Scholar]
- Selley DE, Cassidy MP, Martin BR, Sim-Selley LJ (2004). Long-term administration of delta9-tetrahydrocannabinol desensitizes CB1-, adenosine A1-, and GABAB-mediated inhibition of adenylyl cyclase in mouse cerebellum. Mol Pharmacol 66:1275–1284. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Redman SJ (1991). Mechanisms of presynaptic inhibition studied using paired-pulse facilitation. Neurosci Lett 126:179–183. [DOI] [PubMed] [Google Scholar]
- Takahashi KA, Linden DJ (2000). Cannabinoid receptor modulation of synapses received by cerebellar Purkinje cells. J Neurophysiol 83:1167–1180. [DOI] [PubMed] [Google Scholar]
- Tempia F, Miniaci MC, Anchisi D, Strata P (1998). Postsynaptic current mediated by metabotropic glutamate receptors in cerebellar Purkinje cells. J Neurophysiol 80:520–528. [DOI] [PubMed] [Google Scholar]
- Tian X, Gotoh T, Tsuji K, Lo EH, Huang S, Feig LA (2004). Developmentally regulated role for Ras-GRFs in coupling NMDA glutamate receptors to Ras, Erk and CREB. EMBO J 23:1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trincavelli ML, Tuscano D, Marroni M, Klotz KN, Lucacchini A, Martini C (2002). Involvement of mitogen protein kinase cascade in agonist-mediated human A(3) adenosine receptor regulation. Biochim Biophys Acta 1591:55–62. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM (1998). Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83:393–411. [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Valjent E, Firmo C, Mas M, Beslot F, Defer N, Roques BP, Hanoune J, Maldonado R (2000). Cannabinoid withdrawal is dependent upon PKA activation in the cerebellum. Eur J Neurosci 12:1038–1046. [DOI] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J (2000). Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci 20:8701–8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pages C, Rogard M, Besson MJ, Maldonado R, Caboche J (2001). Delta 9-tetrahydrocannabinol-induced MAPK/ERK and Elk-1 activation in vivo depends on dopaminergic transmission. Eur J Neurosci 14:342–352. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pages C, Herve D, Girault JA, Caboche J (2004). Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci 19:1826–1836. [DOI] [PubMed] [Google Scholar]
- Zippel R, Orecchia S, Sturani E, Martegani E (1996). The brain specific Ras exchange factor CDC25 Mm: modulation of its activity through Gi-protein-mediated signals. Oncogene 12:2697–2703. [PubMed] [Google Scholar]
- Zippel R, Gnesutta N, Matus-Leibovitch N, Mancinelli E, Saya D, Vogel Z, Sturani E (1997). Ras-GRF, the activator of Ras, is expressed preferentially in mature neurons of the central nervous system [Erratum (1997) 52:170]. Brain Res Mol Brain Res 48:140–144. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002). Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. [DOI] [PubMed] [Google Scholar]