

Abstract
The protein HPC-1/syntaxin 1A is abundantly expressed in neurons and localized in the neuronal plasma membrane. It forms a complex with SNAP-25 (25 kDa synaptosomal-associated protein) and VAMP-2 (vesicle-associated membrane protein)/synaptobrevin called SNARE (a soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor) complex, which is considered essential for synaptic vesicle exocytosis; thus, HPC-1/syntaxin 1A is considered crucial for synaptic transmission. To examine the physiological function of HPC-1/syntaxin 1A in vivo, we produced knock-out (KO) mice by targeted gene disruption. Although HPC-1/syntaxin 1A expression was completely depleted without any effect on the expression of other SNARE proteins, the KO mice were viable. They grew normally, were fertile, and displayed no difference in appearance compared with control littermate. In cultured hippocampal neurons derived from the KO mice, the basic synaptic transmission in vitro was normal. However, the mutant mice had impaired long-term potentiation in the hippocampal slice. Also, although KO mice exhibited normal spatial memory in the hidden platform test, consolidation of conditioned fear memory was impaired. Interestingly, the KO mice had impaired conditioned fear memory extinction. These observations suggest that HPC-1/syntaxin 1A may be closely related to synaptic plasticity.
Keywords: extinction, fear memory, HPC-1/syntaxin 1A, LTP, SNARE, synaptic plasticity
Introduction
Neurons communicate with other neurons via neurotransmitters. The modulation of neurotransmission by both postsynaptic and/or presynaptic mechanisms may cause neuronal plasticity (Malenka and Nicoll, 1999; Sheng and Kim, 2002; Krupa and Liu, 2004). In presynapse, plastic change may be mediated by neurotransmitter release, which is initiated by Ca2+ influx through the presynaptic Ca2+ channel, followed by fusion of the synaptic vesicle with the presynaptic plasma membrane. Elucidating the mechanism of neurotransmitter release is therefore important in understanding neuronal plasticity as well as synaptic function.
Neurotransmitter release is mediated by the soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor (SNARE) protein complex (Lin and Scheller, 2000; Rizo and Sudhof, 2002), and HPC-1/syntaxin 1A has postulated as an abundantly expressed neuronal target-membrane-associated SNARE (t-SNARE) (Bennett et al., 1992; Inoue et al., 1992). HPC-1/syntaxin 1A forms a ternary SNARE protein complex by interacting with the synaptosomal-associated protein of 25 kDa (SNAP-25) and vesicle-associated membrane protein-2 (VAMP-2)/synaptobrevin. The inhibition of complex formation modulates neurotransmitter release in vitro (Sollner et al., 1993; Yamaguchi et al., 1997; Xu et al., 1999; Fujiwara et al., 2001; Mishima et al., 2002), which suggests that HPC-1/syntaxin 1A is necessary for neurotransmitter release. As well as HPC-1/syntaxin 1A, syntaxin 1B, which is one of the syntaxin isoforms, is also localized to the plasma membrane in neurons. These two proteins are highly homologous and have similar distribution within individual neurons. It is supposed that these proteins might perform similar function as neuronal t-SNARE.
Interestingly, HPC-1/syntaxin 1A interacts with N- and P/Q-type Ca2+ channels and regulates their function (Bezprozvanny et al., 1995; Mochida et al., 1996; Degtiar et al., 2000), suggesting its involvement in the regulation of presynaptic Ca2+ influx. Furthermore, HPC-1/syntaxin 1A modulates the activity of the plasma membrane neurotransmitter transporters GAT-1 (GABA transporter), NET (norepinephrine transporter), and SERT (serotonin transporter) (Quick et al., 1997; Quick, 2003; Sung et al., 2003). This implies that HPC-1/syntaxin 1A plays an important role in the release and uptake of neurotransmitters and in regulating plastic change by modulating the concentration of neurotransmitters in the synaptic cleft during neurotransmission.
These results strongly suggest that HPC-1/syntaxin 1A is closely associated with synaptic plasticity. Nakayama et al. (1997, 1998) stated that the HPC-1/syntaxin 1A gene is included in the deleted region of Williams–Beuren syndrome (WS), which is caused by hemizygous microdeletion of chromosome 7q11.23. Patients with WS exhibit highly specific cognitive deficit profiles (Bellugi et al., 1999). Although several other genes contribute to the unique cognitive profile of WS patients, the deletion of the HPC-1/syntaxin 1A gene is likely associated with the neuropsychological phenotype of WS through the regulation of synaptic transmission and/or neuronal plasticity.
We generated knock-out (KO) mice to study the role of HPC-1/syntaxin 1A in synaptic transmission and plasticity. The mice showed normal synaptic transmission but, unexpectedly, impaired synaptic plasticity.
Materials and Methods
Generation of HPC-1/syntaxin 1A KO mice.
A bacterial artificial chromosome clone containing a mouse HPC-1/syntaxin 1A gene fragment was purchased from Incyte Genomics (St. Louis, MO). A 16 kbp fragment containing exons 4–10 was subcloned into pBlueskript. The region from exon 9 to exon 10 that encoded the H3 and transmembrane domains was replaced with a neomycin-resistant gene (see Fig. 1A). The diphtheria toxin gene (DT-A) was attached to the 3′ end of the construct for selection against random insertion. The linearized targeting vector was transfected into embryonic stem (ES) cells, which were selected from a culture medium containing G418 (Murata et al., 1999). G418-resistant ES cell colonies were screened by PCR and Southern blot analysis. Nine of 376 colonies were confirmed to be clones with homologous recombination. KO ES cell clones were injected into blastocysts and implanted into pseudopregnant ICR females. The resultant male chimeric mice were bred with C57BL/6J to generate heterozygous mutant (HT) mice, and genotypes of the offspring were determined by PCR and Southern blot analysis. Finally, the heterozygote was backcrossed with C57BL/6J for four to five generations.
Figure 1.
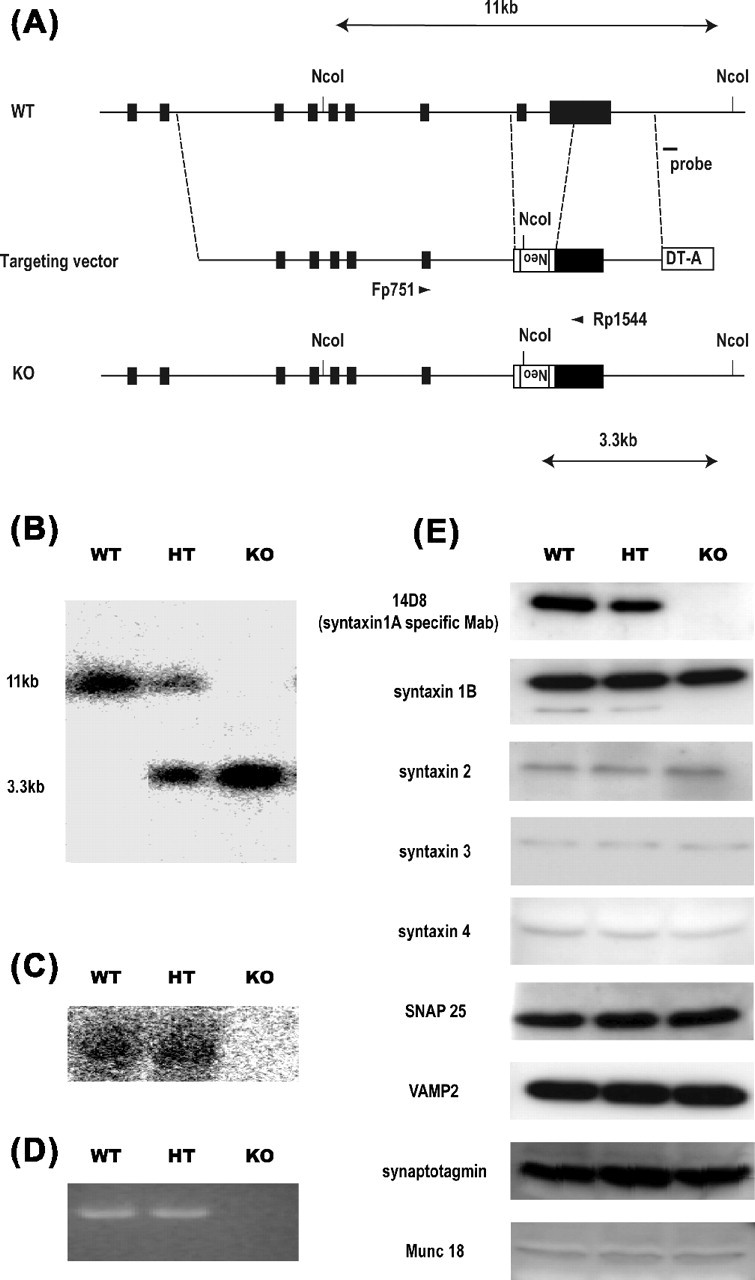
Generation of HPC-1/syntaxin 1A KO mice. A, Schematic showing the WT mouse HPC-1/syntaxin 1A locus, targeting vector construct, and targeted allele. The solid bar indicates HPC-1/syntaxin 1A exons 2–10. The targeting vector contains exons 4–10 of the gene. The region from exon 9 to exon 10 was replaced with a neomycin expression cassette (Neo). A diphtheria toxin expression cassette (DT-A) was attached to the 3′ end of the construct for negative selection. A probe for Southern blot analysis was indicated. B, Southern blot analysis of genomic DNA from a litter obtained by HT intercrossing. Each genomic DNA was digested by NcoI and hybridized with an external probe indicated in A. The 11 kbp WT and 3.3 kbp mutated DNA band were determined. C, Northern blot analysis of HPC-1/syntaxin 1A mRNA expression. Total RNA was prepared from postnatal day 7 mouse brain and hybridized with HPC-1/syntaxin 1A cDNA. The expression of HPC-1/syntaxin 1A mRNA was not detectable in KO mice. D, RT-PCR analysis of HPC-1/syntaxin 1A mRNA expression. A pair of primers, which corresponded to exon 8 and exon 10, was used. The expression of HPC-1/syntaxin 1A mRNA was completely lacking in KO mice. E, Western blot analysis of HPC-1/syntaxin 1A and other SNARE proteins. Whole-brain homogenate was prepared from postnatal day 7 mice. The expression of HPC-1/syntaxin 1A protein was lacking in KO, but other SNARE proteins were not affected.
Western blot analysis.
Whole-brain homogenate was obtained from newborn mice and applied to Western blotting as described previously (Fujiwara et al., 2001). Briefly, 5 mg of protein was loaded into a SDS-PAGE and transferred to the polyvinylidene difluoride membrane. Immunoreactive bands were visualized using ECL (Amersham Biosciences, Arlington Heights, IL). The HPC-1/syntaxin 1A-specific monoclonal antibody 14D8 (Kushima et al., 1997) and the polyclonal antibody against HPC-1/syntaxin 1A, syntaxin 1B, were obtained as described previously (Iwahashi et al., 2003). Polyclonal antibodies against syntaxin 3 and syntaxin 4 were raised against recombinant protein in rabbit as reported previously (Inoue et al., 1992). Antibodies against SNAP-25, VAMP-2/synaptobrevin, and synaptotagmine were purchased from Wako Pure Chemical Industries (Osaka, Japan). The polyclonal antibody against syntaxin 2 was purchased from Chemicon (Temecula, CA), and the monoclonal antibody against Munc 18 was purchased from Transduction Laboratories (Lexington, KY).
Northern blot and reverse transcription-PCR analysis.
Total RNA was prepared from newborn mouse brains using Trisol reagent (Life Technologies, Gaithersburg, MD). Ten micrograms of total RNA were loaded onto agarose gels and transferred to Hybond-N+ membrane (Amersham Biosciences). This membrane was hybridized with [α-32P]dCTP-labeled mouse HPC-1/syntaxin 1A cDNA probe, washed, and visualized by autoradiography.
For reverse transcription-PCR (RT-PCR) analysis, 1 mg of total RNA was used in the reaction with Moloney murine leukemia virus reverse transcriptase (Amersham Biosciences, Piscataway, NJ). First-strand cDNA was analyzed by PCR using a pair of primers corresponding to the sequence of exon 8 (Fp751, CATGTTCATGGACATGGCCATGCTGGTGGA) and exon 10 (Rp1544, CTCAAAGCAAGGCTGGAGATGACACCAC) (see Fig. 1A) as described by Iwahashi et al. (2003). We used a PCR amplification cycle of 20, and the products were electrophoresed on 1.5% agarose gels and visualized by staining with Syber Green.
Hippocampal primary culture.
Primary cultured neurons were obtained from postnatal day 0 mouse hippocampus as reported by Mishima et al. (2002). The dissected hippocampal tissue fragments were digested with papain and dissociated by pipetting through plastic tips. The dissociated neurons were then plated on a glial feeder layer at low density (3–4 × 103/cm2). The neurons were cultured at 37°C in a humidified incubator with 95% air and 5% CO2 in DMEM containing 2% B-27 supplement and used on days 7–22 in vitro.
Electrophysiology.
Synaptic transmission in vitro was monitored with dual whole-cell patch-clamp recordings from presynaptic and postsynaptic neurons with soma within 100 mm. The currents were collected through two amplifiers, EPC-9 (HEKA Elektronik, Lambrecht/Pfalz, Germany) for the postsynaptic neuron and CEZ-2300 (Nihon Koden, Tokyo, Japan) for presynaptic neurons, and low-pass filtered at 2 kHz. The data were stored on DAT (RD-120; Teac, Tokyo, Japan), later digitized at 10 kHz, and analyzed using Power Lab (AD Instruments, Colorado City, CO). The series resistance was monitored continuously throughout the experiment by measuring the capacitative current response to a 5 mV voltage step and was compensated 60%. If the resistance changed by >10%, the experiment was discarded. Recordings were made in modified Tyrode’s solution containing (in mm) 135 NaCl, 3.5 KCl, 1.8 CaCl2, 1.5 MgCl2, 10 HEPES, and 10 glucose. The internal solution for presynaptic neurons contained (in mm) 130 K-gluconate, 10 KCl, 5 HEPES, 1 EGTA, 0.1 CaCl2, 2 MgCl2, 2 MgATP, and 0.2 NaGTP; that for postsynaptic neurons contained (in mm) 115 Cs-gluconate, 10 CsCl, 10 tetraethylammonium, 5 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide], 5 HEPES, 1 EGTA, 0.1 CaCl2, 2 MgCl2, 2 MgATP, and 0.2 NaGTP. Spontaneous release was recorded with 1 mm tetrodotoxin in the medium.
Long-term potentiation in hippocampal slices.
Adult mice (8–20 weeks old) were deeply anesthetized, and their brains were dissected. Horizontal hippocampal slices (400 mm thick) were obtained using a vibratome in an ice-cold cutting solution containing (in mm) 125 choline Cl, 20 sucrose, 25 NaHCO3, 10 glucose, 2.5 KCl, 1.25 NaH2PO4, 8 MgCl2, and 0.5 CaCl2. The slices were maintained for at least 2 h at room temperature in 95% O2 and 5% CO2-bubbled artificial CSF (ACSF) containing (in mm) 123 NaCl, 25 NaHCO3, 10 glucose, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, and 1 MgCl2. In the recording chamber, the slices were superfused continuously with 95% O2 and 5% CO2-bubbled ACSF (1–2 ml/min). Schaffer collaterals were stimulated with a bipolar tungsten electrode, and field EPSPs (fEPSPs) were recorded from the stratum radiatum of the CA1 using glass pipettes containing 500 mm NaCl. Basal synaptic transmission was monitored at 0.05 Hz. Long-term potentiation (LTP) was induced by theta-burst stimulation (TBS), following Pang et al. (2004) (i.e., 12 bursts at 5 Hz, each consisting of four pulses delivered at 100 Hz). The duration of the pulses was 0.1 ms, and the stimulation strength was set to obtain a fEPSP with 35–40% maximal amplitude.
Histology.
Twelve-week-old mouse brains were dissected under deep anesthesia and fixed with 4% paraformaldehyde/PBS. The brains were cryoprotected with 20% sucrose/PBS. Cryostat sections (18 mm) were stained with hematoxylin and eosin and examined under a light microscope.
For electron microscopic analysis of the hippocampal synaptic structure, brains obtained from 12-week-old male mice were fixed with 2.5% glutaraldehyde. These were further fixed with OsO4, and uranyl acetate staining was performed as described previously (Koh et al., 1993). The quantitative analysis was performed using NIH Image J as described by Leenders et al. (2001). Asymmetric synapse with clearly visible active zone from wild-type (WT) and KO mice were selected randomly for analysis. The distance between the center of each synaptic vesicle and the active zone was measured. Docked vesicles were defined as located within 50 nm distance from the active zone.
Open-field test.
All of the tests were conducted during the light phase (8:00 A.M. to 8:00 P.M.). Each mouse was housed individually, and food and water were available ad libitum in the home cage. Ten- to 13-week-old male mice were tested blind to the genotype.
The apparatus for the open-field test was a circular arena (75 cm diameter) with a white floor. The mice were placed on the floor, a novel environment for the mice, and locomotor activity was monitored with a CCD camera and analyzed using a video tracking system (Muromachi, Tokyo, Japan). The open field was divided into center and peripheral zones, and the time spent in the center zone was calculated to study anxiety behavior, as reported previously (Prut and Belzung, 2003).
Morris water maze test.
The apparatus for the Morris water maze test consisted of a circular pool (120 cm diameter). A transparent circular platform (10 cm diameter) was placed in the pool and used as an escape platform. The water maze was located in a room with distal visual cues at several locations. At the beginning of each training day, the pool was filled with 22°C water containing skim milk. Each mouse was examined by a hidden platform test in the milk pool (Meng et al., 2002). Each mouse was trained to find the platform that was submerged 1 cm below the surface of the milk–water at a fixed location in the pool. The training was repeated four times daily, at 20 min intertrial intervals, for 5 d. The mice were allowed to swim until they reached the platform. The mice that failed to reach the platform within 60 s were guided to it and were allowed to remain there for 30 s. The behavior was monitored by a CCD camera and analyzed with a video tracking system (Muromachi). The average swim path distance to reach the platform and swim speed were calculated each training day. A probe test was conducted 24 h after 5 d of training, in which the platform was removed and the mice were placed in the pool for 60 s. The movement of each mouse was monitored and analyzed for the time spent in each of the four quadrants in the pool.
Conditioned fear memory.
Two conditioning chambers of different shape (context A, square; context B, triangle) were used for the conditioned fear memory test. Each chamber consisted of a plastic box with a shock grid floor made of stainless rod (Muromachi). The floor in context B was covered with an acrylic insert. Each mouse was exposed to an audible cue [conditioned stimulus (CS), 20 dB] coterminating with an electrical footshock [unconditioned stimulus (UCS): 0.6 mA, 2 s] in a conditioning chamber (see Fig. 7B) as described by Powell et al. (2004). After pairing, the mice were allowed to remain in the conditioning chamber for 30 s before being returned to their home cage. Approximately 24 h after pairing, each mouse was placed in context A to study contextual fear memory and was scored for freezing response for 3 min. To study cued fear memory, the mice were returned to context B and scored for freezing response to the CS for 57 s without the UCS. In the overtraining protocol, the CS–UCS pairing was repeated three times at 90 s intertrial intervals (see Fig. 7D). One day or 2 weeks after CS–UCS pairing, each mouse underwent the contextual fear memory or cued fear memory test. Their behaviors were recorded with a video camera and scored for freezing behavior, which was defined as a motionless posture without respiratory movements.
Figure 7.
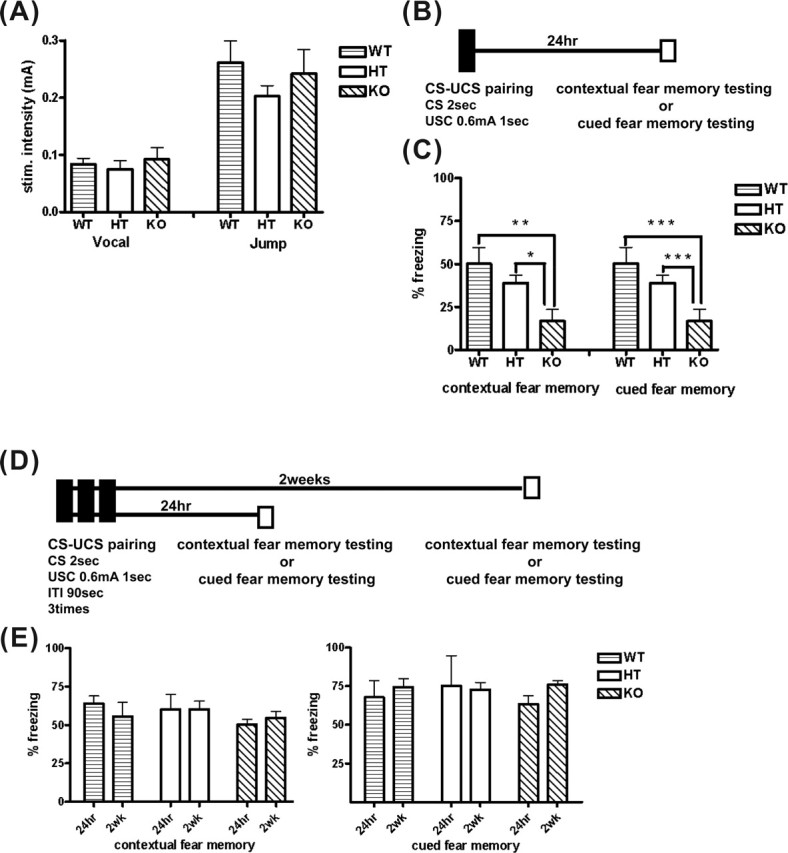
Consolidation of contextual and cued fear memory was impaired in the HPC-1/syntaxin 1A KO mice. A, Pain threshold to electrical footshock. Mice were exposed to a 2 s electrical footshock. The minimal intensity of a shock generating vocalizing or jumping response was determined. There were no significant differences among genotypes (WT, n = 5; HT, n = 5; KO, n = 5). B, Schematic showing a single CS–UCS pairing protocol. Each mouse received only a CS–UCS pairing in a conditioning chamber. Approximately 24 h after pairing, contextual fear memory testing and cued fear memory testing was conducted. C, Consolidation of contextual and cued fear memory by this training protocol was significantly impaired in KO mice compared with WT and HT mice (WT, n = 6; HT, n = 6; KO, n = 6). *p < 0.01; **p < 0.05. D, Schematic showing the overtraining protocol. Each mouse received CS–UCS pairings three times in a conditioning chamber. One day or 2 weeks after pairing, contextual fear memory testing or cued fear memory testing was conducted. E, Contextual (left) and cued fear (right) memory 24 h or 2 weeks after a CS–UCS pairing in KO mice was not significantly different compared with WT and HT mice (WT, n = 8; HT, n = 8; KO, n = 10). Data represent the mean ± SD.
For the cued fear memory extinction test, each mouse received five CS–UCS pairings in context A. Approximately 24 h after pairings, each mouse received an extinction trial, which consisted of the CS for 57 s five times, at 90 s intertrial intervals, without the UCS in context B (see supplemental Fig. S4, available at www.jneurosci.org as supplemental material). This extinction trial was repeated for 4 d. To study memory retention without extinction trials, each mouse was maintained for 4 d in the home cage after the CS–UCS pairings before the cued fear memory was examined.
Results
HPC-1/syntaxin 1A KO mice displayed normal growth
We constructed an HPC-1/syntaxin 1A targeting vector that contained the replaced exon 9 and a portion of exon 10 with a MC-Neor gene (Fig. 1A). These exons encoded an α-helical coiled-coil domain (H3 domain) of HPC-1/syntaxin 1A that is important for SNARE complex formation (Sollner et al., 1993; Xu et al., 1999; Fujiwara et al., 2001). Using the targeting vector, we obtained nine KO ES clones, three of which were used to generate chimeric founder mice. HT mice obtained from these chimera mice were bred with C57BL/6J. The HT mice were indistinguishable from the wild type in appearance and were viable and fertile.
The loss of syntaxin 1 expression in Drosophila and Caenorhabditis elegans has been reported to be lethal (Schulze et al., 1995; Saifee et al., 1998), because it led to a failure in synaptic transmission. To examine whether HPC-1/syntaxin 1A manipulation in homozygous mutant mice would be lethal, HT mice were intercrossed, and we genotyped the newborn mice (Fig. 1B). Unexpectedly, the homozygous mutant mice (KO) were viable, unlike in Drosophila and C. elegans, and no apparent difference was observed among the WT, HT, and KO mice at birth, although the ratio of KO in all the pups was slightly lower than that expected (19.1% of 365 mice).
To confirm that KO mice completely lacked the expression of HPC-1/syntaxin 1A, we conducted a Western blot analysis using a specific monoclonal antibody (Kushima et al., 1997). As shown in Figure 1E (top panel), KO mice showed a total lack of HPC-1/syntaxin 1A expression. In HT mice, the content of the protein was reduced by ∼50% compared with the WT mice. The same results were observed using HPC-1/syntaxin 1A polyclonal antibody (data not shown). Northern blot analysis did not detect any mRNA expression in KO mice (Fig. 1C). To investigate whether the mutant mice completely lacked mRNA expression, we performed RT-PCR. We used a pair of primers, located on exon 8 and the 3′ region on exon 10 (Fig. 1A). The expression of HPC-1/syntaxin 1A mRNA was not detected in KO mice by RT-PCR (Fig. 1D). Furthermore, no signal was detected using a pair of primers located on exon 2 and exon 5, indicating that no shorter mRNA was expressed in KO mice (data not shown).
To examine the growth of the mutant mice, we measured body weight gain. KO mice showed no marked difference compared with control littermates (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Also, KO mice were viable for at least 12 months and fertile. These results show that HPC-1/syntaxin 1A is not essential for growth and weight gain.
Lack of HPC-1/syntaxin 1A expression did not affect the expression of other SNARE proteins
We examined whether the loss of HPC-1/syntaxin 1A gene expression would affect the expression of other SNARE proteins. Several HPC-1/syntaxin 1A homologs are expressed in the brain, including syntaxin 1B, which is said to have a function similar to HPC-1/syntaxin 1A in the exocytotic process. However, there was no apparent change in the protein content of syntaxin 1B in the brains of KO mice (Fig. 1E); none of the content of syntaxin 2, syntaxin 3, or syntaxin 4, all of which are plasma membrane localizing syntaxins, changed in the brains of KO mice. These results revealed that the loss of HPC-1/syntaxin 1A gene expression does not affect the expression of other plasma membrane localizing syntaxins in the brain. We also examined the expression of SNAP-25, VAMP-2/synaptobrevin, synaptotagmine, and Munc-18/nSec-1 in the mutant mice because these interact with HPC-1/syntaxin 1A and play an important role in exocytosis. The content of these proteins also showed no marked difference among the genotypes (Fig. 1E), indicating that the lack of HPC-1/syntaxin 1A expression did not result in major compensatory changes of SNARE or other related gene expression.
HPC-1/syntaxin 1A KO mice exhibited a normal brain structure
HPC-1/syntaxin 1A was shown to be involved in axonal growth in vitro (Igarashi et al., 1996; Yamaguchi et al., 1996). Therefore, we examined the brain structures to determine whether they were morphologically normal in the KO mice. Light microscopy showed no apparent differences in the hippocampus and cerebral cortex among the genotypes (Fig. 2A). We used electron microscopy to examine the synaptic structure in detail (Fig. 2B). The appearance of presynaptic structure and postsynaptic densities on the hippocampus in KO mice did not differ from WT mice. Quantitative analysis revealed that the total amount of synaptic vesicles per square micrometer of the presynaptic terminal was not significantly different between WT and KO mice (WT, 200.4 ± 19.97; KO, 188.8 ± 9.65; p > 0.1). The distribution of synaptic vesicles in presynaptic terminals was not significantly different between WT and KO mice (Fig. 2C). The number of docked vesicles that were defined as located within 50 nm distance from the active zone was also not significantly different between WT and KO mice (WT, 21.33 ± 2.13; KO, 22.53 ± 9.65; p > 0.1). These results suggested that deletion of HPC-1/syntaxin 1A may not affect the morphological development of brain synapses.
Figure 2.
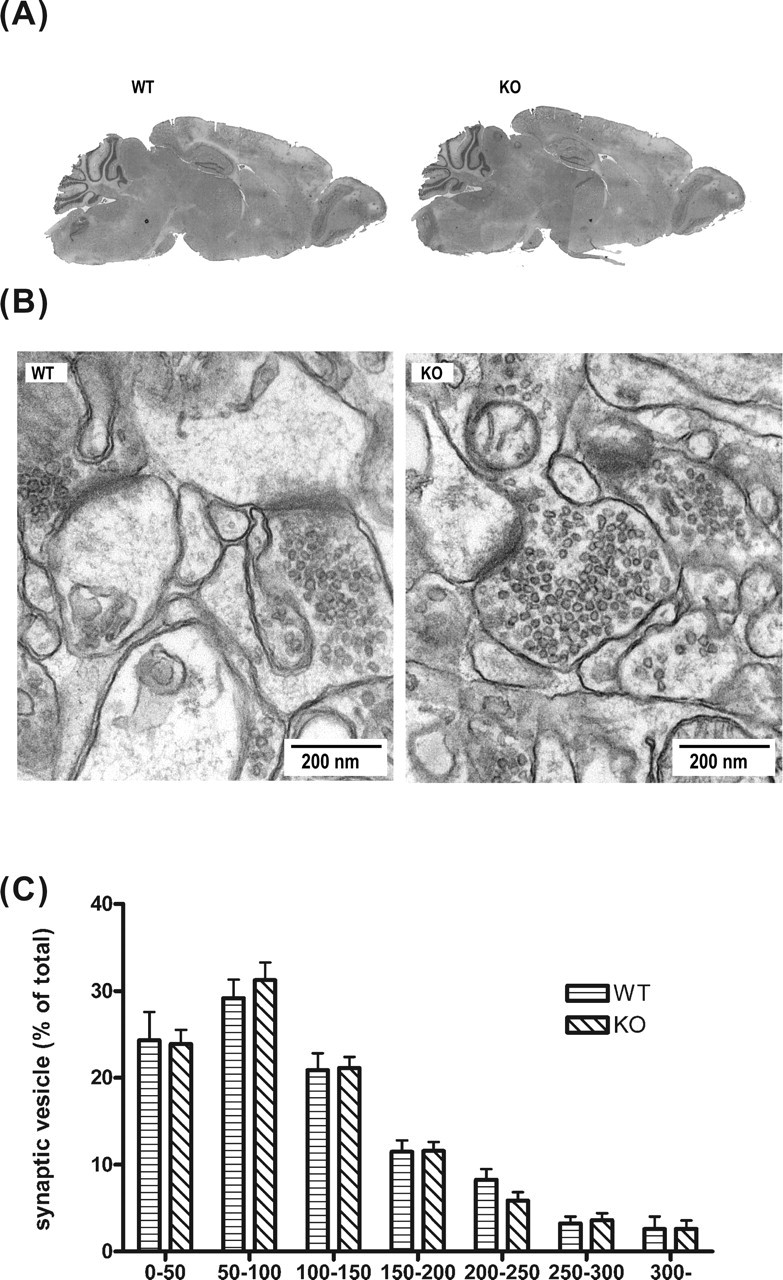
HPC-1/syntaxin 1A KO mice exhibit normal brain morphology and synaptic structure. A, Sagittal sections of an adult brain from WT and KO mice were stained with hematoxylin and eosin. There was no difference between genotypes. B, Electron micrograms of the adult hippocampus. Scale bar, 200 nm. C, Distribution of synaptic vesicles. All synaptic vesicles in presynaptic terminals were counted, and the distance between the center of vesicles and active zone was measured. Vesicles were collected in 50 nm bins, and the percentage of vesicles per bin was plotted. There were no significant differences between WT and KO (WT synapse, n = 35; KO synapse, n = 35). Data represent the mean ± SD.
Analysis of basal neurotransmitter release in cultured hippocampal neurons
In the dissociated hippocampal culture condition without using AMPA receptor blockers, the neurons show spontaneous epileptiform discharges with high frequency, and stimulation of the presynaptic neuron causes recurrent EPSCs, which overlaid on the evoked EPSC (data not shown). Almost similar results were obtained from KO mice. Therefore, we thought that basic glutamatergic transmission seems to be normal in KO. Although it was necessary to analyze the property of the evoked excitatory synaptic transmission in vitro, it is hard to perform because of recurrent EPSCs mentioned above. So, we tried to analyze miniature excitatory currents (mEPSCs) of AMPA synapse and evoked GABAergic inhibitory synaptic transmission instead of evoked EPSCs, because basic machinery of synaptic transmission is thought to be same between excitatory and inhibitory synapse. AMPA receptor-mediated spontaneous mEPSC in cultured hippocampal neurons were recorded at a holding potential of −70 mV in the presence of d,l-APV (100 μm) and bicuculine (50 μm) to block NMDA and GABAA receptors, respectively. The waveform of the mEPSCs did not differ in the neurons of either WT or KO mice (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). The mean frequency of mEPSC was not significantly different between these genotypes in immature [7–8 d in vitro (DIV); WT: 0.98 ± 0.17 Hz, n = 25; KO: 0.45 ± 0.12 Hz, n = 11) and mature (14–15 DIV; WT: 2.68 ± 0.69 Hz, n = 13; KO: 5.41 ± 1.15 Hz, n = 20) neurons in vitro (Fig. 3A). Although the mean amplitude of mEPSCs in immature neurons varied between WT and KO mice (7–8 DIV; WT, 28.73 ± 0.53 pA; KO, 24.88 ± 0.46 pA; p < 0.001), the difference was not significant in mature neurons (14–15 DIV; WT, 25.21 ± 0.34 pA; KO, 23.71 ± 0.17 pA; p > 0.1). We next studied evoked IPSCs. There was no significant difference in the mean amplitude of evoked IPSCs between the two genotypes (WT, 1494.16 ± 272.948 pA; KO, 1679.75 ± 429.878 pA; p > 0.1). We also examined the paired-pulse ratio of IPSCs by varying the interstimulus interval. At GABAergic synapses, the response to the second stimulus was always smaller than to the first. There was no difference in paired-pulse ratios between WT and KO mice (Fig. 3B). We reported previously that suppressing HPC-1/syntaxin 1A affects neurotransmitter release by retarding the refilling of readily releasable vesicles. Therefore, we examined the recovery process after depletion of synaptic vesicles by tetanus stimulation (20 Hz, 10 s). Readily releasable synaptic vesicles recovered fully within 15 s of tetanus stimulation in these neuron genotypes, and no difference was observed (Fig. 3C). These data indicate normal basic synaptic transmission in the cultured neurons obtained from HPC-1/syntaxin 1A KO mice.
Figure 3.

Basal synaptic transmission in cultured hippocampal neurons from WT and KO mice. A, Cumulative histograms of mEPSC amplitude from individual events from KO or WT cultures at 7–8 and 14–15 DIV. At 7–8 DIV, the mean mEPSC amplitude of control cultures were larger than that of KO cultures (WT vs KO, 28.73 ± 0.53 vs 24.88 ± 0.46 pA), but at 14–15 DIV, no difference was observed (WT vs KO, 25.21 ± 0.34 vs 23.71 ± 0.17 pA; mean ± SE). B, Paired-pulse ratio of IPSC. There was no significant difference between cultured neurons from KO (n = 6) and WT (n = 12) mice. C, Recovery from depletion of synaptic vesicle by tetanic stimulation (20 Hz, 10 s). There was no significant difference between cultured neurons from KO (n = 3) and WT (n = 6) mice. Data represent the mean ± SE.
LTP in the hippocampal CA1 region
To examine synaptic plasticity in KO mice, we analyzed LTP in the hippocampal slice (Fig. 4). The TBS of Schaffer collaterals produced post-tetanic potentiation (PTP) in all of the slices. The maximal value of PTP within 2 min of TBS was 249.3 ± 16.0% for WT (n = 10), 269.8 ± 17.1% for HT (n = 13), and 256.3 ± 13.4% for KO (n = 12) mice. There was no significant difference in the levels of PTP (WT, n = 3; KO, n = 3; p > 0.1). The levels of LTP at 60 min after TBS were 187.7 ± 10.5%, 168.1 ± 4.1%, and 150.1 ± 6.2%, for WT, HT, and KO mice, respectively. TBS evoked a lesser increase in the slope of fEPSP recorded from KO mice compared with WT and HT mice (p < 0.05). These results indicate that HPC-1/syntaxin 1A may be involved in LTP in the hippocampal CA1 region.
Figure 4.
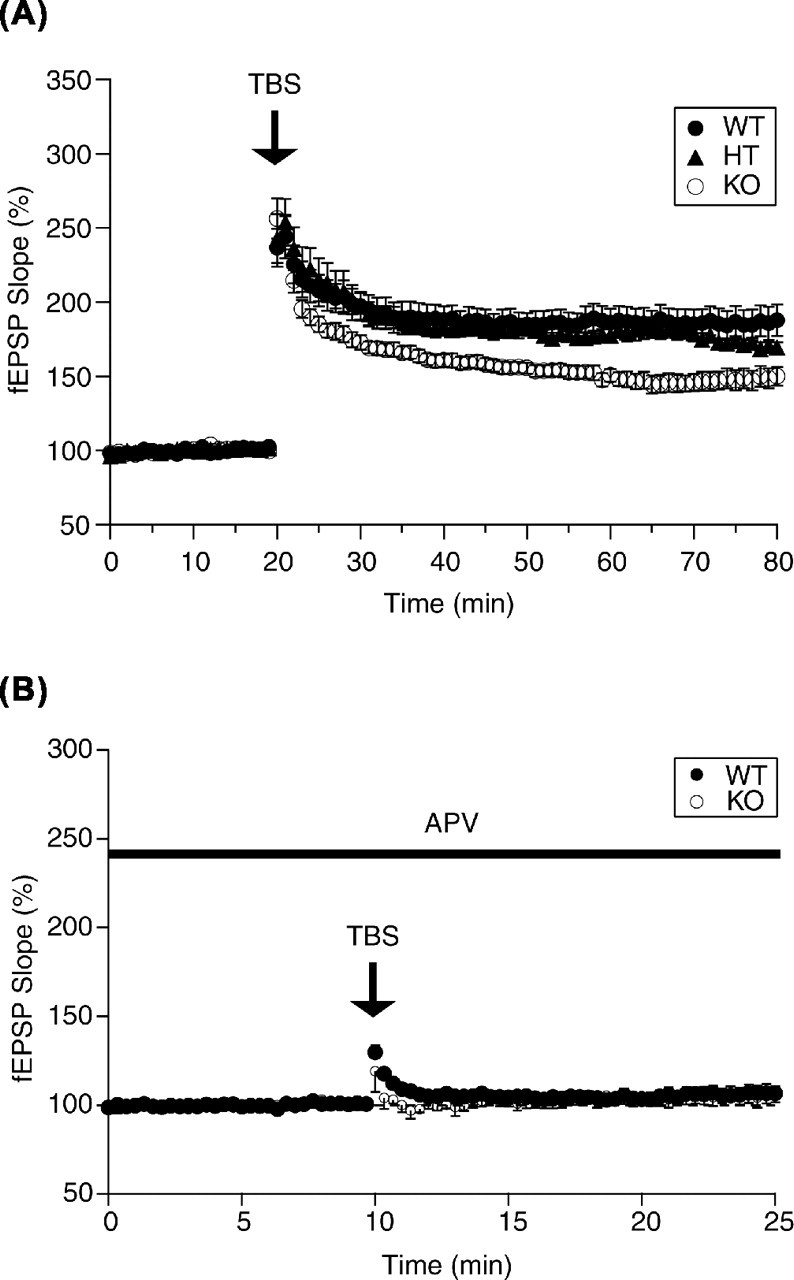
LTP in the CA1 region was impaired in KO mice. A, LTP recorded from slices from WT, HT, and KO. TBS of Schaffer collaterals evoked a high increase in the slope of fEPSPs recorded from slices of WT and HT than KO (WT, n = 6; HT, n = 6; KO, n = 6; p < 0.05). The mean slope of fEPSPs recorded 0–20 min before TBS was taken as 100%. Data represent the mean ± SE. B, The application of 50 mm APV abolished LTP in slices from each genotype.
HPC-1/syntaxin 1A KO exhibited normal spatial memory in the Morris water maze test
Impairment of hippocampal LTP may affect hippocampus-dependent memory (D’Hooge and De Deyn, 2001). To determine whether HPC-1/syntaxin 1A KO mice displayed normal hippocampal-dependent memory, we conducted the hidden platform test in a Morris water maze. KO mice learned their platform position in the milk pool normally (Fig. 5). The path length to reach the platform during 5 d of training was not significantly different among genotypes (Fig. 5A). The time required to reach the platform was also not significantly different (data not shown). The swimming speed of each mouse on each training day was not significantly different, although KO mice tended to swim slightly faster than WT mice (Fig. 5B). A probe test after 5 d of training revealed no significant difference among genotypes (Fig. 5C). These results indicate that the HPC-1/syntaxin 1A KO mice displayed normal spatial memory acquisition, although LTP in the hippocampal slice was reduced.
Figure 5.
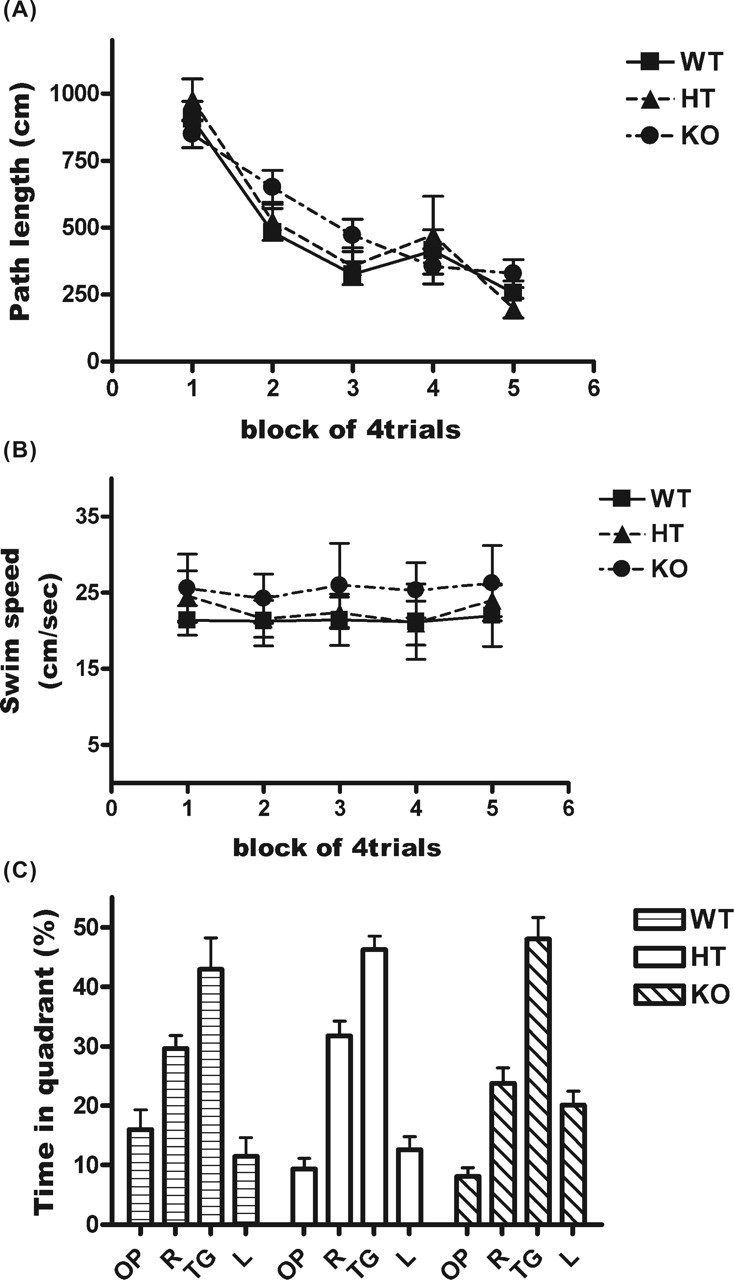
HPC-1/syntaxin 1A KO mice exhibited normal spatial memory in the hidden platform test. Each mouse was handled for 3–5 d before training and subjected to the hidden platform test in a Morris water maze (WT, n = 9; HT, n = 7; KO, n = 19). Data represent mean ± SD. A, The average swim path length to reach the escape platform on each training day is shown. A similar decrease in swim path length to reach the platform during 5 d of training was observed for each genotype. There was no significant difference among genotypes. B, The average swim speed on each training day is shown. There were no significant differences among genotypes. C, After 5 d of training, each mouse underwent a probe test. The average percentage of time spent in each of the four quadrants of the pool is shown. There were no significant differences among genotypes. TG, Quadrant where the platform was located during training for the hidden platform test; OP, opposite; R, right; L, left.
HPC-1/syntaxin 1A KO displayed normal locomotor activity in a novel environment
Mutant coloboma mice in which SNAP-25, a binding partner of HPC-1/syntaxin 1A, was deleted display spontaneous hyperactivity and are considered a model of attention-deficit hyperactivity disorder (Hess et al., 1992). Therefore, we studied the locomotor activity of KO mice via an open-field test. However, we found that the locomotor activity of KO mice was similar to that of WT and HT mice during the initial session (Fig. 6A). The locomotor activities of these mice gradually decreased, possibly because of habituation to the environment. The activities of the last session were not noticeably different among genotypes. These results showed that KO mice exhibited neither hyperactivity nor impairment of habituation to a novel environment. The time spent in the center zone of the open field was also calculated to study anxiety-like behavior (Prut and Belzung, 2003), but there were no significant differences among genotypes (Fig. 6B). These results suggest that KO mice did not show a significant impairment in the open-field test.
Figure 6.

HPC-1/syntaxin 1A KO mice exhibit normal behavior in the open-field test. A, The behavior in a novel environment was monitored for 30 min, in 3 min sessions, and analyzed using a video tracking system. Locomotor activity in the environment was not significantly different among genotypes (WT, n = 6; HT, n = 8; KO, n = 11). B, The average time spent in the center zone of the open field during these 30 min was calculated. There were no significant differences among genotypes (WT, n = 6; HT, n = 8; KO, n = 11). Data represent the mean ± SD.
Consolidation of conditioned fear memory was impaired in HPC-1/syntaxin 1A KO mice
In addition to the hidden platform test, we conducted the conditioned fear memory test to study a type of memory ability in HPC-1/syntaxin 1A KO mice. Before this test, sensitivity to electrical footshock was examined for each genotype because it affects fear-conditioned memory. However, there was no noticeable difference among genotypes (Fig. 7A). Each mouse acquired conditioned fear memory by single CS–UCS pairings, followed by the contextual fear memory or cued fear memory test (Fig. 7B). Contextual fear memory was considered hippocampus- and amygdala-dependent memory, whereas cued fear memory was considered amygdala-dependent memory (Rogan and LeDoux, 1996). Interestingly, 24 h after the CS–UCS pairing, the freezing response to training context and to an audible cue in KO mice significantly decreased compared with WT and HT mice (Fig. 7C), indicating that consolidation of both contextual and cued fear memory was impaired in KO mice. We further examined fear memory by repeated training protocol. Each mouse received CS–UCS pairings three times during training (Fig. 7D). Both contextual and cued fear memory at 24 h after pairing were not significantly different among genotypes (Fig. 7E). Similar results were observed 2 weeks after pairing.
Extinction of conditioned fear memory was impaired in HPC-1/syntaxin 1A KO mice
We examined the extinction of conditioned fear memory in HPC-1/syntaxin 1A KO mice. Each mouse received CS–UCS pairings five times to acquire a cued fear memory. The freezing response to CS at 24 h after pairing was not significantly different among genotypes. Each mouse then received extinction training five times per day. The freezing response to CS decreased gradually in WT and HT mice (Fig. 8). Interestingly, the KO mice displayed more frequent freezing responses compared with WT and HT mice during each extinction trial, suggesting that the memories of the KO mice were resistant to them. It is possible that the impairment of memory extinction was because the cued fear memory was more stable in KO mice. Therefore, we examined whether the retention of cued fear memory by this CS–UCS pairing condition was normal in the KO mice. However, the freezing response at 4 d after pairing without extinction trials was not significantly different among genotypes (Fig. 8, open symbols). These results indicate that the extinction process of cued fear memory was impaired in HPC-1/syntaxin 1A KO mice.
Figure 8.

Cued fear memory extinction was impaired in HPC-1/syntaxin 1A KO mice. Each mouse received CS–UCS pairings five times in a conditioning chamber. Approximately 24 h after pairing, each mouse received cued fear memory extinction trials in a novel test chamber. The extinction trials consisted of the CS for 57 s five times per day without the USC in a novel test chamber. The extinction trials were repeated for 4 d. The freezing response to the CS in each extinction trial was scored. Cued fear memory extinction in KO mice was impaired compared with WT and HT mice, although consolidation and retention of the memory was indistinguishable among all genotypes (WT, n = 8; HT, n = 8; KO, n = 8). *p < 0.01; **p < 0.005. Data represent mean ± SD.
Discussion
HPC-1/syntaxin 1A null mutant mice grew normally
HPC-1/syntaxin 1A acts as a t-SNARE and forms a SNARE complex by interacting with SNAP-25 and VAMP-2/synpto-brevin. Recently, it was reported that SNAP-25 or VAMP-2/synptobrevin gene KO mice died immediately after birth because of the loss of evoked synaptic transmission (Schoch et al., 2001; Washbourne et al., 2002). The loss of syntaxin 1 expression in Drosophila and C. elegans was also reported to be lethal because of the failure of evoked synaptic transmission (Schulze et al., 1995; Saifee et al., 1998). Therefore, we expected HPC-1/syntaxin 1A KO mice to follow the same course. However, we found that the null mutant mice were viable. They showed normal body growth (supplemental Fig. 1S, available at www.jneurosci.org as supplemental material) and exhibited normal basic synaptic transmission in cultured hippocampal neurons (Fig. 3). These results suggest that HPC-1/syntaxin 1A may not be essential for development as a whole and may be dispensable for basic synaptic transmission. There are several possible explanations, the most likely being compensation by other syntaxin protein for the loss of HPC-1/syntaxin 1A expression, because several syntaxin isoforms are expressed in the brain (Bennett et al., 1993). Syntaxin 1B, which is generated by a distinct gene from HPC-1/syntaxin 1A, is one such isoform and is also abundantly expressed as well as HPC-1/syntaxin 1A in the brain (Fig. 1E). Both HPC-1/syntaxin 1A and syntaxin 1B are thought to be similar to neuronal t-SNARE in synaptic transmission. Therefore, HPC-1/syntaxin 1A KO mice may not show severe impairment in basal synaptic transmission possibly because of compensation by syntaxin 1B. However, the content of syntaxin 1B did not change in the KO mice (Fig. 1E), and other plasma membrane localized syntaxin proteins were also unchanged. These results suggest that the presence of syntaxin 1B is enough for maintaining the basic synaptic transmission. However, it was reported previously that distribution of HPC-1/syntaxin 1A and syntaxin 1B was slightly different in some area of CNS (Ruiz-Montasell et al., 1996). More detailed analysis may be needed to determine whether neuronal transmission is normal in the area in which syntaxin 1B is low.
HPC-1/syntaxin 1A involvement in synaptic plasticity
Although basic synaptic transmission in cultured hippocampal neurons was almost normal, LTP of the hippocampal CA1 region was reduced in the KO mice (Fig. 4). In the hippocampus, the content of HPC-1/syntaxin 1A and syntaxin 1B is almost equivalent (Kushima et al., 1997) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Therefore, the sum of syntaxin 1 (HPC-1/syntaxin 1A and syntaxin 1B) in the hippocampus of KO mice is reduced ∼50% in WT mice. It is possible that the syntaxin 1B in neurons lacking HPC-1/syntaxin 1A may be sufficient for basic synaptic transmission, but not for synaptic plasticity. Interestingly, it was reported that the function of HPC-1/syntaxin 1A and syntaxin 1B might be different (Nagamatsu et al., 1996; Perez-Branguli et al., 2002). This difference may produce the distinctive roles of these proteins in LTP of the hippocampal CA1 region. Currently, however, there is no evidence to validate any of these possibilities.
Although LTP of the CA1 region is mainly mediated by a postsynaptic mechanism (Malenka and Nicoll, 1999), presynaptic mechanisms may also be involved (Krupa and Liu, 2004; Huang et al., 2005). It is important to consider how the presynaptic membrane protein HPC-1/syntaxin 1A contributes to synaptic plasticity in the CA1 region. Recent reports have indicated that LTP of the CA1 region can be reduced by inhibition of PKA-mediated synaptic modulation (Castillo et al., 1997; Huang et al., 2005). In HPC-1/syntaxin 1A KO mice, the activation of PKA, which is mediated by Ca2+/calmodulin-sensitive adenylyl cyclase, may be affected because HPC-1/syntaxin 1A is involved in the regulation of presynaptic Ca2+ channels (Bezprozvanny et al., 1995; Mochida et al., 1996; Yang et al., 1999; Degtiar et al., 2000). According to this hypothesis, Ca2+-dependent short-term plasticity may also be affected in KO mice. However, we did not observe any change in the paired-pulse ratio, at least in the cultured hippocampal neurons (Fig. 3A). Therefore, additional analysis is required to answer whether the function of presynaptic Ca2+ channels and PKA-dependent synaptic plasticity are affected in KO mice.
Although the LTP of the hippocampal slice was reduced in KO mice, the spatial memory as revealed by the hidden platform test was normal (Fig. 5). It is important to note that there is still disagreement concerning the relationship between the LTP of hippocampal and spatial memory using the Morris water maze (Cain, 1998). To determine whether HPC-1/syntaxin 1A KO mice exhibit normal hippocampus-dependent memory, additional analysis on both spatial and reference memory is necessary.
Impairment of conditioned fear memory in HPC-1/syntaxin 1A KO mice
Our observations revealed that HPC-1/syntaxin 1A KO mice have impaired consolidation of contextual and cued fear memory (Fig. 7) and impaired extinction of cued fear memory (Fig. 8). These results may not be attributable to increased vulnerability to anxiety in KO mice because anxiety-like behavior in the open-field test was normal (Fig. 6B). Impairment of the conditioned fear memory in HPC-1/syntaxin 1A KO mice may not be directly related to the function of neurotransmitter release at presynapse. For example, Powell et al. (2004) reported that a decrease in release probability or loss of LTP alone did not cause impairment of learning and memory ability. HPC-1/syntaxin 1A KO mice may exhibit an impaired conditioned fear memory without dysfunction of neurotransmitter release for several reasons. 5-HT, norepinephrine, or GABA neurotransmission may be related to synaptic plasticity, and HPC-1/syntaxin 1A modulates the transporters of the neurotransmitters SERT (Quick, 2003), NET (Sung et al., 2003), and GAT-1 (Quick et al., 1997). The modulation of the function of these transporters could regulate the neurotransmitter concentration in the synaptic cleft during neurotransmission, resulting in plastic change. Furthermore, mice with mutated transporters exhibit impaired synaptic plasticity and abnormal behavior (Chiu et al., 2005; Haller et al., 2002; Jones et al., 1998; Murphy et al., 2003). Therefore, it is possible that impairment of conditioned fear memory in HPC-1/syntaxin 1A KO mice may be attributable to a disturbance in the modulation of the transporter functions.
Moreover, the expression of HPC-1/syntaxin 1A mRNA is likely regulated by neuronal activity (Fujino et al., 1997; Hu et al., 2003), suggesting that upregulation of HPC-1/syntaxin 1A expression may occur during memory and/or learning processes. It was reported that HPC-1/syntaxin 1A expression increases during inhibitory avoidance training (Igaz et al., 2004). Interestingly, the expression of SNAP-25 also transiently increases after fear conditioning training (Hou et al., 2004). These observations suggest that an increase in some presynaptic SNARE proteins may be closely related to memory and/or learning mechanisms.
Finally, HPC-1/syntaxin 1A is involved in axonal growth (Igarashi et al., 1996) and/or sprouting (Yamaguchi et al., 1996). Because neurite growth and/or sprouting may occur during plastic change in the CNS (Marrone and Petit, 2002), perturbation of axonal growth and/or the sprouting process may cause turbulence of synaptic plasticity. Although we observed neither abnormal brain structure nor abnormal synaptic structure in the resting condition (Fig. 2), axonal growth and/or sprouting may be impaired in HPC-1/syntaxin 1A KO mice during plastic change. In supporting this possibility, we observed abnormal plastic change in the spinal cord after ligation of the sciatic nerve in HPC-1/syntaxin 1A KO mice (our unpublished data). However, more detailed analysis is needed to determine whether HPC-1/syntaxin 1A is involved in axonal growth and/or sprouting during memory and/or learning processes.
It was reported previously that gene KO of LIMK1 or CYLN2, which are involved in the WS deleted region, resulted in behavioral abnormalities (Hoogenraad et al., 2002; Meng et al., 2002). Therefore, these genes may be involved in some of the abnormalities observed in WS patients. Whether the impairment of synaptic plasticity and/or conditioned fear memory in HPC-1/syntaxin 1A KO mice is linked to the neuropsychological profile (i.e., specific cognitive function, memory profile, sociability and spatial deficits) of a WS patient is an interesting question. A recent report suggested that the abnormality of amygdala activation (Meyer-Lindenberg et al., 2005) is related to hypersociality and marked fear in WS patients, and that impairment of fear memory in HPC-1/syntaxin 1A KO subjects may cause these peculiar phenotypic profiles in WS. However, to determine whether the behavioral abnormalities in HPC-1/syntaxin 1A mutant mice were related to the phenotypes in the WS patient, additional behavioral analyses of heterozygous and homozygous mutant mice are needed.
Conclusions
Our data show that HPC-1/syntaxin 1A KO mice do not have defective basic synaptic transmission. The function of synaptic vesicle exocytosis might be compensated by syntaxin 1B or other syntaxin proteins. However, we observed that HPC-1/syntaxin 1A KO mice exhibit impaired LTP in the hippocampal slice and impaired memory consolidation and extinction in the conditioned fear memory test. Because these phenomena rely on synaptic plasticity, this indicates that HPC-1/syntaxin 1A is closely associated with neuronal plasticity.
Footnotes
This work was supported in part by a Grant in Aid for Encouragement of Young Scientists to T.F. (Grant 15700292) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and by a Grant in Aid from the Promotion and Mutual Aid Cooperation for Private Schools of Japan to K.A.
References
- Bellugi U, Lichtenberger L, Mills D, Galaburda A, Korenberg JR (1999). Bridging cognition, the brain and molecular genetics: evidence from Williams syndrome. Trends Neurosci 22:197–207. [DOI] [PubMed] [Google Scholar]
- Bennett MK, Calakos N, Scheller RH (1992). Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 257:255–259. [DOI] [PubMed] [Google Scholar]
- Bennett MK, Garcia-Arraras JE, Elferink LA, Peterson K, Fleming AM, Hazuka CD, Scheller RH (1993). The syntaxin family of vesicular transport receptors. Cell 74:863–873. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Scheller RH, Tsien RW (1995). Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature 378:623–626. [DOI] [PubMed] [Google Scholar]
- Cain DP (1998). Testing the NMDA, long-term potentiation, and cholinergic hypotheses of spatial learning. Neurosci Biobehav Rev 22:181–193. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Janz R, Sudhof TC, Tzounopoulos T, Malenka RC, Nicoll RA (1997). Rab3A is essential for mossy fiber long-term potentiation in the hippocampus. Nature 388:590–593. [DOI] [PubMed] [Google Scholar]
- Chiu CS, Brickley S, Jensen K, Southwell A, Mckinney S, Cull-Candy S, Mody I, Lester HA (2005). GABA transporter deficiency causes tremor, ataxia, nervousness, and increased GABA-induced tonic conductance in cerebellum. J Neurosci 25:3234–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degtiar VE, Scheller RH, Tsien RW (2000). Syntaxin modulation of slow inactivation of N-type calcium channels. J Neurosci 20:4355–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Hooge R, De Deyn PP (2001). Applications of the Morris water maze in the study of learning and memory. Brain Res Brain Res Rev 36:60–90. [DOI] [PubMed] [Google Scholar]
- Fujino I, Fujiwara T, Akagawa K (1997). Transient decrease of HPC-1/syntaxin-1A mRNA in the rat hippocampus by kainic acid. Neurosci Res 28:243–247. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Yamamori T, Akagawa K (2001). Suppression of transmitter release by Tat HPC-1/syntaxin 1A fusion protein. Biochim Biophys Acta 1539:225–232. [DOI] [PubMed] [Google Scholar]
- Haller J, Bakos N, Rodriguiz RM, Caron MG, Wetsel WC, Liposits Z (2002). Behavioral responses to social stress in noradrenaline transporter knockout mice: effects on social behavior and depression. Brain Res Bull 58:279–284. [DOI] [PubMed] [Google Scholar]
- Hess EJ, Jinnah HA, Kozak CA, Wilson MC (1992). Spontaneous locomotor hyperactivity in a mouse mutant with a deletion including the Snap gene on chromosome 2. J Neurosci 12:2865–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad CC, Koekkoek B, Akhmanova A, Krugers H, Dortland B, Miedema M, van Alphen A, Kistler WM, Jaegle M, Koutsourakis M, Van Camp N, Verhoye M, van der Linden A, Kaverina I, Grosveld F, De Zeeuw CI, Galjart N (2002). Targeted mutation of Cyln2 in the Williams syndrome critical region links CLIP-115 haploinsufficiency to neurodevelopmental abnormalities in mice. Nat Genet 32:116–127. [DOI] [PubMed] [Google Scholar]
- Hou Q, Gao X, Zhang X, Kong L, Wang X, Bian W, Tu Y, Jin M, Zhao G, Li B, Jing N, Yu L (2004). SNAP-25 in hippocampal CA1 region is involved in memory consolidation. Eur J Neurosci 20:1593–1603. [DOI] [PubMed] [Google Scholar]
- Hu JY, Meng X, Schacher S (2003). Redistribution of syntaxin mRNA in neuronal cell bodies regulates protein expression and transport during synapse formation and long-term synaptic plasticity. J Neurosci 23:1804–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Zakharenko SS, Schoch S, Kaeser PS, Janz R, Sudhof TC, Siegelbaum SA, Kandel ER (2005). Genetic evidence for a protein-kinase-A-mediated presynaptic component in NMDA-receptor-dependent forms of long-term synaptic potentiation. Proc Natl Acad Sci USA 102:9365–9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi M, Kozaki S, Terakawa S, Kawano S, Ide C, Komiya Y (1996). Growth cone collapse and inhibition of neurite growth by botulinum neurotoxin C1: a t-SNARE is involved in axonal growth. J Cell Biol 134:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz LM, Bekinschtein P, Izquierdo I, Medina JH (2004). One-trial aversive learning induces late changes in hippocampal CaMKIIalpha, Homer 1a, Syntaxin 1a and ERK2 protein levels. Brain Res Mol Brain Res 132:1–12. [DOI] [PubMed] [Google Scholar]
- Inoue A, Obata K, Akagawa K (1992). Cloning and sequence analysis of cDNA for a neuronal cell membrane antigen, HPC-1. J Biol Chem 267:10613–10619. [PubMed] [Google Scholar]
- Iwahashi K, Kuji N, Fujiwara T, Tanaka H, Takahashi J, Inagaki N, Komatsu S, Yamamoto A, Yoshimura Y, Akagawa K (2003). Expression of the exocytotic protein syntaxin in mouse oocytes. Reproduction 126:73–81. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG (1998). Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci USA 95:4029–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S, Yamamoto A, Inoue A, Inoue Y, Akagawa K, Kawamura Y, Kawamoto K, Tashiro Y (1993). Immunoelectron microscopic localization of the HPC-1 antigen in rat cerebellum. J Neurocytol 22:995–1005. [DOI] [PubMed] [Google Scholar]
- Krupa B, Liu G (2004). Does the fusion pore contribute to synaptic plasticity? Trends Neurosci 27:62–66. [DOI] [PubMed] [Google Scholar]
- Kushima Y, Fujiwara T, Sanada M, Akagawa K (1997). Characterization of HPC-1 antigen, an isoform of syntaxin-1, with the isoform-specific monoclonal antibody, 14D8. J Mol Neurosci 8:19–27. [DOI] [PubMed] [Google Scholar]
- Leenders AG, Lopes da Silva FH, Ghijsen WE, Verhage M (2001). Rab3a is involved in transport of synaptic vesicles to the active zone in mouse brain nerve terminals. Mol Biol Cell 12:3095–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RC, Scheller RH (2000). Mechanisms of synaptic vesicle exocytosis. Annu Rev Cell Dev Biol 16:19–49. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1999). Long-term potentiation–a decade of progress? Science 285:1870–1874. [DOI] [PubMed] [Google Scholar]
- Marrone DF, Petit TL (2002). The role of synaptic morphology in neural plasticity: structural interactions underlying synaptic power. Brain Res Brain Res Rev 38:291–308. [DOI] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z (2002). Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35:121–133. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Hariri AR, Munoz KE, Mervis CB, Mattay VS, Morris CA, Berman KF (2005). Neural correlates of genetically abnormal social cognition in Williams syndrome. Nat Neurosci 8:991–993. [DOI] [PubMed] [Google Scholar]
- Mishima T, Fujiwara T, Akagawa K (2002). Reduction of neurotransmitter release by the exogenous H3 domain peptide of HPC-1/syntaxin 1A in cultured rat hippocampal neurons. Neurosci Lett 329:273–276. [DOI] [PubMed] [Google Scholar]
- Mochida S, Sheng ZH, Baker C, Kobayashi H, Catterall WA (1996). Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron 17:781–788. [DOI] [PubMed] [Google Scholar]
- Murata S, Kawahara H, Tohma S, Yamamoto K, Kasahara M, Nabeshima Y, Tanaka K, Chiba T (1999). Growth retardation in mice lacking the proteasome activator PA28gamma. J Biol Chem 274:38211–38215. [DOI] [PubMed] [Google Scholar]
- Murphy DL, Uhl GR, Holmes A, Ren-Patterson R, Hall FS, Sora I, Detera-Wadleigh S, Lesch KP (2003). Experimental gene interaction studies with SERT mutant mice as models for human polygenic and epistatic traits and disorders. Genes Brain Behav 2:350–364. [DOI] [PubMed] [Google Scholar]
- Nagamatsu S, Fujiwara T, Nakamichi Y, Watanabe T, Katahira H, Sawa H, Aagawa K (1996). Expression and functional role of syntaxin 1/HPC-1 in pancreatic beta cells. Syntaxin 1A, but not 1B, plays a negative role in regulatory insulin release pathway. J Biol Chem 271:1160–1165. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Fujiwara T, Miyazawa A, Asakawa S, Shimizu N, Shimizu Y, Mikoshiba K, Akagawa K (1997). Mapping of the human HPC-1/syntaxin 1A gene (STX1A) to chromosome 7 band q11.2. Genomics 42:173–176. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Matsuoka R, Kimura M, Hirota H, Mikoshiba K, Shimizu Y, Shimizu N, Akagawa K (1998). Hemizygous deletion of the HPC-1/syntaxin 1A gene (STX1A) in patients with Williams syndrome. Cytogenet Cell Genet 82:49–51. [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B (2004). Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306:487–491. [DOI] [PubMed] [Google Scholar]
- Perez-Branguli F, Muhasisen A, Blasi J (2002). Munc 18a binding to syntaxin 1A and 1B isoforms defines its localization at the plasma membrane and blocks SNARE assembly in a three-hybrid system assay. Mol Cell Neurosci 20:169–180. [DOI] [PubMed] [Google Scholar]
- Powell CM, Schoch S, Monteggia L, Barrot M, Matos MF, Feldmann N, Sudhof TC, Nestler EJ (2004). The presynaptic active zone protein RIM1alpha is critical for normal learning and memory. Neuron 42:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prut L, Belzung C (2003). The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol 463:3–33. [DOI] [PubMed] [Google Scholar]
- Quick MW (2003). Regulating the conducting states of a mammalian serotonin transporter. Neuron 40:537–549. [DOI] [PubMed] [Google Scholar]
- Quick MW, Corey JL, Davidson N, Lester HA (1997). Second messengers, trafficking-related proteins, and amino acid residues that contribute to the functional regulation of the rat brain GABA transporter GAT1. J Neurosci 17:2967–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Sudhof TC (2002). Snares and Munc18 in synaptic vesicle fusion. Nat Rev Neurosci 3:641–653. [DOI] [PubMed] [Google Scholar]
- Rogan MT, LeDoux JE (1996). Emotion: systems, cells, synaptic plasticity. Cell 85:469–475. [DOI] [PubMed] [Google Scholar]
- Ruiz-Montasell B, Aguado F, Majo G, Chapman ER, Canals JM, Marsal J, Blasi J (1996). Differential distribution of syntaxin isoforms 1A and 1B in the rat central nervous system. Eur J Neurosci 8:2544–2552. [DOI] [PubMed] [Google Scholar]
- Saifee O, Wei L, Nonet ML (1998). The Caenorhabditis elegans unc-64 locus encodes a syntaxin that interacts genetically with synaptobrevin. Mol Biol Cell 9:1235–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET (2001). SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294:1117–1122. [DOI] [PubMed] [Google Scholar]
- Schulze KL, Broadie K, Perin MS, Bellen HJ (1995). Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell 80:311–320. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim MJ (2002). Postsynaptic signaling and plasticity mechanisms. Science 298:776–780. [DOI] [PubMed] [Google Scholar]
- Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE (1993). SNAP receptors implicated in vesicle targeting and fusion. Nature 362:318–324. [DOI] [PubMed] [Google Scholar]
- Sung U, Apparsundaram S, Galli A, Kahlig KM, Savchenko V, Schroeter S, Quick MW, Blakely RD (2003). A regulated interaction of syntaxin 1A with the antidepressant-sensitive norepinephrine transporter establishes catecholamine clearance capacity. J Neurosci 23:1697–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002). Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5:19–26. [DOI] [PubMed] [Google Scholar]
- Xu T, Rammner B, Margittai M, Artalejo AR, Neher E, Jahn R (1999). Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell 99:713–722. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Nakayama T, Fujiwara T, Akagawa K (1996). Enhancement of neurite-sprouting by suppression of HPC-1/syntaxin 1A activity in cultured vertebrate nerve cells. Brain Res 740:185–192. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Takada M, Fujimori K, Tsuchimoto Y, Kushima Y, Sanada M, Fujiwara T, Akagawa K (1997). Enhancement of synaptic transmission by HPC-1 antibody in the cultured hippocampal neuron. NeuroReport 8:3641–3644. [DOI] [PubMed] [Google Scholar]
- Yang S-N, Larsson O, Branstrom R, Bertorello AM, Leibiger B, Leibiger IB, Moede T, Kohler T, Meister B, Berggren P-O (1999). Syntaxin interacts with the LD subtype of voltage-gated Ca2+ channels in pancreatic β cells. Proc Natl Acad Sci USA 96:10164–10169. [DOI] [PMC free article] [PubMed] [Google Scholar]