

Abstract
Inflammation could be involved in some neurodegenerative disorders that accompany signs of inflammation. However, because sensitivity to inflammation is not equal in all brain structures, a direct relationship is not clear. Our aim was to test whether some physiological circumstances, such as stress, could enhance susceptibility to inflammation in the prefrontal cortex (PFC), which shows a relative resistance to inflammation. PFC is important in many brain functions and is a target for some neurodegenerative diseases. We induced an inflammatory process by a single intracortical injection of 2 μg of lipopolysaccharide (LPS), a potent proinflammogen, in nonstressed and stressed rats. We evaluated the effect of our treatment on inflammatory markers, neuronal populations, BDNF expression, and behavior of several mitogen-activated protein (MAP) kinases and the transcription factor cAMP response element-binding protein. Stress strengthens the changes induced by LPS injection: microglial activation and proliferation with an increase in the levels of the proinflammatory cytokine tumor necrosis factor-α; loss of cells such as astroglia, seen as loss of glial fibrillary acidic protein immunoreactivity, and neurons, studied by neuronal-specific nuclear protein immunohistochemistry and GAD67 and NMDA receptor 1A mRNAs expression by in situ hybridization. A significant increase in the BDNF mRNA expression and modifications in the levels of MAP kinase phosphorylation were also found. In addition, we observed a protective effect from RU486 [mifepristone (11β-[p-(dimethylamino)phenyl]-17β-hydroxy-17-(1-propynyl)estra-4,9-dien-3-one)], a potent inhibitor of the glucocorticoid receptor activation. All of these data show a synergistic effect between inflammation and stress, which could explain the relationship described between stress and some neurodegenerative pathologies.
Keywords: prefrontal cortex, stress, inflammation, neuronal death, BDNF, MAP kinases
Introduction
Pathological processes of neurodegeneration such as those observed in Alzheimer’s (AD) and Parkinson’s diseases are accompanied by an inflammatory reaction that is believed to contribute to pathogenesis (Akiyama et al., 2000). We previously studied the involvement of inflammatory reactions in the degeneration of the nigrostriatal dopaminergic system (Tomás-Camardiel et al., 2004), which is preceded by extensive microglial activation in substantia nigra (SN), suggesting the possible relationship between the two processes. However, despite the fact that evidence for a chronic inflammatory reaction in the brain is particularly strong in AD (McGeer and McGeer, 2004), no direct relationship has been reported between the degenerative processes and inflammation in brain structures.
Lipopolysaccharide (LPS) is a potent inductor of inflammation and produces the activation of microglia and astrocytes (Lieberman et al., 1989; Benveniste et al., 1990; Chung and Benveniste, 1990) and the induction of diverse cytokines (Lee et al., 1993; Chao et al., 1995; Jeohn et al., 1998). Injection of LPS in the prefrontal cortex (PFC) does not have a similar effect as in SN (Kim et al., 2000). This suggests a lower response to LPS challenge in PFC than in other structures such as SN. Thus, we sought for conditions that could enhance the inflammatory response to LPS injection. Regarding this, some conditions have been suggested as circumstances to increase the risk of developing AD. Stress is a condition of human experience and includes not only major life events but also the hassles of daily life that elevate activities of physiological systems so as to cause disruption of ongoing homeostasis. It reflects individual experience and genetic background; therefore, reactions to a stressful incident are highly variable (McEwen, 2000, 2002). Stress is widely acknowledged as a predisposing and precipitating factor in psychiatric illnesses (Kendler and Karkowski-Shuman, 1997; Caspi et al., 2003). Repeated stress causes changes in the PFC and amygdala (Mazure et al., 1995; Yokoyama and Sasaki, 1999; Wellman, 2001; Trentani et al., 2002; Vyas et al., 2002; Gerrits et al., 2003; Radley et al., 2004). Evidence illustrates atrophy and cell death of stress-vulnerable neurons in PFC; stress has been recognized as strongly influencing cognitive and emotional processes subserved by PFC (Arnsten, 1998).
The aim of the present work was to test whether a chronic variate stress could enhance the LPS-induced damage in PFC. Immunohistochemical and molecular biology techniques were combined in an effort to elucidate the effects of chronic stress, intracortical LPS injection, and a combination of both on different cell types of the PFC. We found that stress significantly increased the inflammatory damage induced by LPS. Thus, we also studied the effect produced by RU486 [mifepristone (11β-[p-(dimethylamino)phenyl]-17β-hydroxy-17-(1-propynyl)estra-4,9-dien-3-one)], a potent inhibitor of the glucocorticoid receptor (GR) activation, on the different parameters assayed.
Materials and Methods
Animals and surgery.
Male albino Wistar rats (200–300 g) were used for these studies. The rats were kept at constant room temperature of 22 ± 1°C and relative humidity (60%) with a 12 h light/dark cycle with access to food and water ad libitum. Rats were anesthetized with chloral hydrate (400 mg/kg) and positioned in a stereotaxic apparatus (Kopf Instruments, Tujunga, CA) to conform to the brain atlas of Paxinos and Watson (1986). Injections into the PFC were made 3.2 mm anterior, 1.3 mm lateral, and 3.5 mm ventral to the bregma.
Experiments were performed in accordance with the Guidelines of the European Union Council (86/609/EU), following the Spanish regulations (BOE 67/8509-12, 1988) for the use of laboratory animals and approved by the Scientific Committee of the University of Seville.
Five groups of animals were established according to the different treatments. (1) The vehicle-injected group received a single dose of 2 μl of Monastral Blue inert tracer (Sigma, St. Louis, MO) (1% in PBS) in the left PFC. (2) The vehicle/stressed group received a single dose of 2 μl of Monastral Blue inert tracer in the left PFC and was stressed for 9 d. (3) The LPS-injected group received a single dose of LPS (2 μg; from Escherichia coli, serotype 026:B6; Sigma) dissolved in 2 μl of vehicle in the left PFC. (4) The LPS/stressed group received a single intracortical injection of 2 μg of LPS and was stressed for 9 d. (5) The LPS/stressed/RU486 group received a single intracortical injection of 2 μg of LPS, was stressed for 9 d, and received a daily dose of RU486 (Sigma) (20 mg/kg in saline 20% DMSO) 1 h after the 9 d stress. All animals were killed by decapitation 10 d after surgery. At least five animals were used for each group. Body weight was measured at the beginning and at the end of the treatment. After killing the animals, the adrenal gland weight was evaluated as an indirect parameter of hypothalamic–pituitary–adrenal (HPA) axis activation.
Stress model.
Chronic variate stress was adapted from other models of variate stress (Willner et al., 1987; Konarska et al., 1990; Papp et al., 1991; Murua and Molina, 1992; Muscat et al., 1992; Gamaro et al., 2003) with modifications. Animals were divided in stressed and nonstressed groups. Nonstressed animals were kept undisturbed in their home cages during the 10 d of treatment. A 9 d variate-stressor paradigm was used for animals in the stressed groups. Individual stressors and length of time applied each day are listed in Table 1. The following stressors were used: (1) 24 h of food deprivation; (2) 24 h of water deprivation; (3) 1–3 h of restraint, as described below; (4) 1.5–2 h of restraint at 4°C; and (5) forced swimming during 10 min, as described below. Application of stress started at a different time everyday to minimize predictability. Restraint was performed by placing the animal in a 21 × 6 cm plastic tube and adjusting it with plaster tape on the outside so that the animal was unable to move. There was a 6 cm hole at the far end for breathing. Forced swimming was performed by placing the animal in a glass tank measuring 44 × 33 × 30 cm with 22 cm of water depth at 23 ± 2°C.
Table 1.
Schedule of stressing agents used during the chronic stress treatment
| Day of treatment | Agent used | Duration |
|---|---|---|
| 1 | Forced swimming | 10 min |
| 2 | Restraint | 3 h |
| 3 | Water deprivation | 24 h |
| 4 | Restrain at 4°C | 1.5 h |
| 5 | Restraint | 3 h |
| 6 | Food deprivation | 24 h |
| 7 | Water deprivation | 24 h |
| 8 | Restrain at 4°C | 2 h |
| 9 | Food deprivation | 24 h |
Immunohistological evaluation: glial fibrillary acidic protein, OX-6, and neuronal-specific nuclear protein.
Rats were perfused through the heart under deep anesthesia (chloral hydrate) with 150–200 ml of 4% paraformaldehyde in phosphate buffer, pH 7.4. The brains were removed and then cryoprotected serially in sucrose in PBS, pH 7.4, first in 10% sucrose for 24 h and then in 30% sucrose until sunk (2–5 d). Brains were then frozen in isopentane at −15°C, and 25 μm sections were cut on a cryostat and mounted in gelatin-coated slides. Primary antibodies used were mouse-derived anti-glial fibrillary acidic protein (GFAP) (1:300; Chemicon, Temecula, CA), mouse-derived OX-6 (1:200; Serotec, Oxford, UK) and mouse-derived anti-neuronal nuclei [neuronal-specific nuclear protein (NeuN), 1:2000; Chemicon]. Sections were washed and then treated with 0.3% hydrogen peroxide in methanol for 30 min, washed again, and incubated in a solution containing TBS and 1% horse serum for 60 min in a humid chamber. Slides were drained and further incubated with the primary antibody in TBS containing 1% horse serum and 0.25% Triton X-100 for 24 h. Sections were then incubated for 2 h with biotinylated horse anti-mouse IgG (1:200; Vector Laboratories, Burlingame, CA), followed by a second 1 h incubation with ExtrAvidin peroxidase solution (1:100; Sigma). The antibody was diluted in TBS containing 0.25% Triton X-100 (TBST), and its addition was preceded by three 10 min rinses in TBS. The peroxidase was visualized with a standard diaminobenzidine/hydrogen reaction for 5 min.
Histochemistry data analysis: quantification of tissue areas.
An AnalySIS image software (Soft Imaging System, Münster, Germany) coupled to a Polaroid (Bedfordshire, UK) DMC camera attached to a Leica (Nussloch, Germany) light microscope was used for all measurements. Areas lacking GFAP and NeuN immunoreactivity were measured. Cells showing OX-6 immunoreactivity were counted using five fields per section and five sections per animal.
Measurement of dopamine and 3,4-dihydroxyphenylacetic.
HPLC with electrochemical detection was used for the determination of dopamine (DA) and its metabolite 3,4-dihydroxyphenylacetic (DOPAC). Analyses were performed as described previously (Herrera et al., 1993). Forty-five animals were used for these experiments.
Preparation of riboprobes.
pBluescript SK plasmids, containing the cDNA sequence for glutamic acid decarboxylase 67 (GAD67) as a 3.2 kb EcoRI insert (clones 14 and 18), were kindly provided by Dr. A. Tobin (University of California, Los Angeles, Los Angeles, CA). The GAD67 cDNA was isolated from a 1 gt-11 cDNA library made from poly(A) RNA from adult rat brain (Erlander et al., 1991). To prepare the GAD67 antisense transcript, clone 14 was digested with SalI and used as a template with the T3 RNA polymerase. To prepare the GAD67 sense transcript, clone 18 was digested with SalI and used as a template with the T3 RNA polymerase.
pBluescript SK plasmids, containing the cDNA sequence for NMDA receptor 1A (NMDAR1A) were kindly provided by Dr. S. Heinemann (Salk Institute, San Diego, CA). To prepare the NMDAR1A antisense riboprobe, plasmid pBluescript SK was linearized with EcoRI and used as a template with the T7 RNA polymerase. NMDAR1A sense riboprobe was generated after XholI digestion by using the T3 RNA polymerase.
BDNF-specific riboprobes were transcribed from a 460 bp template of rat BDNF codon subcloned into a PGEM-4Z vector (Promega, Madison, WI) kindly provided by Genentech (South San Francisco, CA). Sense and antisense transcripts were generated using SP6 and T7 polymerases, respectively (Research Biochemicals, Natick, MA) from HindIII and EcoRI fragments of the vector.
Sense and antisense riboprobes used for in situ hybridization were transcribed in the presence of [35S]UTP (1300 Ci/mmol; Amersham Biosciences, Little Chalfont, UK).
Isotopic in situ hybridization histochemistry.
In situ hybridization on brain frozen sections was performed following a modification of a procedure described in detail previously (Pasinetti et al., 1989). Thaw-mounted 12 μm sections were postfixed for 30 min in 4% paraformaldehyde, followed by three 10 min washes in PBS, pH 7.4. Sections were treated for 1 min in 0.1 m triethanolamine, followed by 10 min in acetic anhydride/0.1 m triethanolamine to decrease nonspecific binding. After a 1 min wash in 2× SSC, sections were dehydrated in a series of increasing concentrations of ethanol and then air dried. The sections were hybridized for 3 h at 50°C with the [35S] cRNA, rinsed in 4× SSC/20 mm DTT, and then 4× SSC alone. Sections were subjected to 30 min of RNase digestion at 37°C (20 μg/ml RNase A in 0.5 m NaCl, 0.01 m Tris-HCl, and 0.001 m EDTA, pH 8.0) and washed for 2 h in 2× SSC at 25°C, followed by 0.1× SSC at 60°C for 1 h, dehydrated in a series of ethanols, air dried, and processed for emulsion autoradiography. Autoradiograms were generated by apposing the labeled tissue to βmax Hyperfilm (Amersham Biosciences) for 2 weeks.
In situ hybridization data analysis.
For quantification of GAD and NMDAR1A mRNA expression, films from dry-film autoradiography were scanned at high resolution. For measurement of the areas lacking GAD and NMDAR1A mRNA, we used an AnalySIS image software (Soft Imaging System).
SDS-PAGE and Western blotting of mitogen-activated protein kinases.
PFC was lysed in the following (in mm): 15 Tris-HCl, pH 7.5, 150 NaCl, 1 EDTA, 1 EGTA, and 1 PMSF (all from Sigma). The homogenized was centrifuged at 12,000 × g for 20 min at 4°C. Protein content of the samples was estimated by the method of microLowry using standard of BSA (Fryer et al., 1986); 25–50 μg of protein was loaded for each lane. Protein samples were separated by SDS-PAGE (10%) and transferred to a nitrocellulose membrane (Hybond-C extra; Amersham Biosciences). Membranes were blocked with blocking buffer (5% milk in TBS: 20 mm Tris-HCl, pH 7.5, 500 mm NaCl, and 0.05% Tween 20) for 1 h at room temperature. Membranes were then incubated using the following antibodies: anti-stress-activated protein kinase (SAPK)/c-Jun N-terminal protein kinase (JNK), anti-p38, anti-extracellular signal-regulated kinase 1/2 (ERK1/2), anti-Akt1, and anti-cAMP response element-binding protein 1 (CREB-1) (1:1000) for 1 h at room temperature in blocking buffer (5% milk in TBS); and anti-phosphorylated (P)-SAPK/JNK (Thr183/Tyr185), anti-P-p38 (Tyr 182), anti-P-Akt (Thr 308), anti-P-CREB-1 (Ser 133), and anti-P-ERK1/2 (1:500) overnight at 5°C in blocking buffer (5% milk in TBS). All antibodies used were from Santa Cruz Biotechnology (Santa Cruz, CA), and anti-SAPK/JNK and anti-P-SAPK/JNK were from Cell Signaling Technology (Lake Placid, NY). After incubation with the primary antibodies, all membranes were washed in TBST, incubated with peroxidase-conjugated anti-Ig secondary antibodies (DakoCytomation, Produktionsvej, Denmark) at a dilution of 1:3000 for 1 h at room temperature in TBST. Proteins were visualized using Western blotting chemiluminescence luminal reagent (Santa Cruz Biotechnology). Nonphosphorylated antibodies were used as loading controls. All experiments were repeated in triplicate. The products were analyzed by densitometry using the Quantity One 1-D Analysis software (Bio-Rad, Hercules, CA).
Reverse transcriptase-PCR.
The left PFC was dissected from each rat 6 h after treatment, snap frozen in liquid nitrogen, and stored at −80°C. Tissue samples were homogenized in TRIzol reagent (Invitrogen, Carlsbad, CA), using 1 ml of TRIzol per 50–100 mg of tissue. Total RNA was extracted from the tissue according to the suggested protocol of the manufacturer. The RNA was resuspended in PCR-grade water, and the concentration was calculated by A260 measurement. RNA quality was assessed by electrophoresis on a 1% agarose gel.
First-strand cDNA was synthesized from 3 μg of each RNA using 200 ng of random hexamer primers and 0.5 mm dNTPs. The samples were incubated at 25°C for 10 min and heated at 65°C for 5 min. Afterward, 10 mm DTT, 100 U RNAseOUT Recombinant Ribonuclease Inhibitor (Invitrogen), and 1× first-strand buffer (in mm: 250 Tris-HCl, pH 8.3, 375 KCl, and 15 MgCl2) were added. Samples were heated at 42°C for 2 min and then 200 U of Moloney murine leukemia virus reverse transcriptase (RT) (SuperScript II; Invitrogen) were added to each tube. After incubation at 42°C for 50 min, the reaction was inactivated by heating at 70°C for 15 min.
Triplicate reverse transcription reactions were performed along with an additional reaction in which the reverse transcriptase enzyme was omitted to allow for assessment of genomic DNA contamination in each sample.
PCR was performed with 5 μl of cDNA in 20 μl of PCR reaction mix containing 40 pmol of each primer (Table 2), 0.4 mm dNTPs, 2.5 U of TaqDNA polymerase, 1.5 mm MgCl2, and 1× PCR buffer (200 mm Tris-HCl, pH 8.4, and 500 mm KCl). To determine optimal experimental conditions for relative quantification of transcripts, RT-PCR was preliminary tested at several cycle numbers (20–40 cycles) with variable cDNA concentrations. Samples were placed in a PTC-100 PCR system (MJ Research, Watertown, MA) at 94°C for 5 min, followed sequentially by a cyclic phase of 26 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 45 s, and extension at 72°C for 45 s, followed by a final elongation step at 72°C for 7 min. Concurrently, RT-PCR amplification of β-actin was performed as an internal control for variations in the efficiencies of RNA isolation and RT.
Table 2.
Primers for RT-PCR
| Target mRNA | Forward (F) and reverse (R) primers | Expected product size (bp) | Reference |
|---|---|---|---|
| IL-1β | F, 5′-CAGGATGAGGACATGAGCACC-3′ | 447 | Rostworowski et al., 1997 |
| R, 5′-CTCTGCAGACTCAAACTCCAC-3′ | |||
| TNF-α | F, 5′-TACTGAACTTCGGGGTGATTGGTCC-3′ | 297 | Meltzer et al., 1998 |
| R, 5′-CAGCCTTGTCCCTTGAAGAGAACC-3′ | |||
| IL-6 | F, 5′-AAAATCTGCTCTGGTCTTCTGG-3′ | 290 | Lee et al., 2005 |
| R, 5′-GGTTTGCCGAGTAGACCTCA-3′ | |||
| β-Actin | F,5′-TGTGATGGTGGGAATGGGTCAG-3′ | 514 | Rostworowski et al., 1997 |
| R,5′-TTTGATGTCACGCACGATTTCC-3′ |
Amplified products were separated by electrophoresis on a 1% agarose gel, visualized over a UV transilluminator, and then photographed. The products were analyzed by densitometry using AnalySIS software, and quantities of each product were calculated relative to β-actin.
Statistical analysis.
Results are typically expressed as mean ± SD. Means were compared by one-way ANOVA. The least significance difference (LSD) test was used for post hoc multiple range comparisons.
Results
Changes in body and adrenal gland weights
Stress exerted a negative effect on the bodyweight gain after the injection of LPS: whereas nonstressed animals maintained their weight through the experiment, stressed rats lost 12.7% (p < 0.01) (Fig. 1A). Conversely, stress caused an increase of 26.5% in the adrenal gland weight (p < 0.05) (Fig. 1B) and 43.9% in the adrenal gland weight/body weight ratio (p < 0.01) (Fig. 1B). Bodyweight and adrenal weight alterations are typical effects of stress; these results agree with those of Gamaro et al. (2003), showing that the variate stress model applied to actually stressed animals.
Figure 1.

Effect of stress on the body and adrenal gland weights of LPS-injected animals. A, Body weight at the beginning and the end of the treatment of nonstressed and stressed animals. Results are expressed in grams and are the mean ± SD of five independent experiments. Statistical significance (Student’s t test) compared with the nonstressed rats: *p < 0.01. B, Adrenal weight (in milligrams; bars) and adrenal weight/body weight ratio (in milligrams per gram; circles) at the beginning and the end of the treatment of nonstressed and stressed animals. Results are mean ± SD of five independent experiments. Statistical significance (Student’s t test) compared with the nonstressed rats: #p < 0.05 for body weight; *p < 0.01 for the adrenal weight/body weight ratio.
Effect of stress on the cortical amounts of DA and DOPAC
Variate stress increased amounts of DA in the cortex, with peaks of nearly fourfold and twofold over the control values at days 2 and 8, respectively, after the beginning of the treatment (Table 3). The effect on DOPAC was similar, with peaks of approximately twofold and threefold over the control values at days 2 and 4, respectively (Table 3). These results are in agreement with others, showing alterations in dopamine content after stress (Gamaro et al., 2003) and a special sensitivity of PFC dopaminergic neurons to different types of stress (Feenstra et al., 1998; Abekawa et al., 2000) compared with subcortical regions (Matsumoto et al., 2005). The changes observed in the parameters mentioned above may be considered an index of the effectiveness of the chronic variate stress model.
Table 3.
Cortical amounts of DA and DOPAC in control and stressed rats
| Time (d) | DA |
DOPAC |
||
|---|---|---|---|---|
| Control | Stressed | Control | Stressed | |
| 0 | 45.1 ± 2.5 | 45.8 ± 2.2 | 57.0 ± 8.8 | 56.9 ± 6.7 |
| 2 | 45.2 ± 2.2 | 168.9 ± 22.3** | 56.2 ± 7.9 | 122.8 ± 22.0** |
| 4 | 45.3 ± 2.6 | 47.3 ± 11.4 | 55.8 ± 7.0 | 183.9 ± 31.8** |
| 8 | 45.5 ± 3.2 | 85.8 ± 15.2* | 59.8 ± 3.9 | 26.2 ± 4.5** |
| 10 | 45.7 ± 1.7 | 55.4 ± 14.4 | 54.8 ± 5.3 | 62.6 ± 12.8 |
At different time points after treatment (0, 2, 4, 8, and 10 d), animals were killed, and the prefrontal cortex was dissected out and processed for DA and DOPAC quantification by HPLC, as described in Material and Methods. Numbers are expressed as nanograms per gram of wet tissue and are mean ± SD of five independent experiments. *p < 0.05, **p < 0.01, statistical significance (Student’s t test) compared with control animals.
Effect of LPS and stress on microglia
Microglial cells become activated (change of morphology from resting resident ramified microglia with two or three fine processes to round cells resembling tissue macrophages) and proliferate when challenged. Injection of vehicle in nonstressed animals (Fig. 2B,G) produced a slight microglial reaction, and it had a mild effect on stressed animals (174.9% of control values; p < 0.01) (Fig. 2C,G), whereas LPS injection induces a stronger activation (259.0% compared with the vehicle-injected animals; p < 0.01) (Fig. 2D,G). Combination of stress and LPS produced an additive effect (415.9% compared with the vehicle-injected animals; p < 0.01) (Fig. 2E,G) that was highly reduced for RU486 (218.1% compared with the vehicle-injected animals) (Fig. 2F,G).
Figure 2.

Effect of stress and LPS in microglia. A, Schematic representation of the injections site in the prefrontal cortex. B, Coronal section showing a slight OX-6 immunoreactivity in a saline-injected nonstressed animal. C, OX-6 activation in a vehicle-injected stressed animal. D, Immunoreactivity in an LPS-injected nonstressed animal. The arrow points to the site of injection of 2 μg of LPS. Microglial reaction was mild. E, OX-6 immunoreactivity in an LPS-injected stressed animal. Microglial reaction is stronger and more widely distributed around the injection point (arrow). F, Treatment with RU486 reduces OX-6 activation in an LPS-injected stressed animal. Scale bar, 250 μm. G, Quantification of changes on the microglial population at the end of the treatments. Results are mean ± SD of four independent experiments and are expressed as positive cells per square millimeter. Statistical signification (one-way ANOVA, followed by the LSD post hoc test for multiple comparisons): a, compared with the control, vehicle-injected nonstressed animals; b, compared with the vehicle-injected stressed animals; c, compared with the LPS-injected nonstressed animals; d, compared with the LPS-injected stressed animals; p < 0.01. C, Control, vehicle injected in the PFC of nonstressed animals; S, vehicle injected in the PFC of stressed animals; L, LPS injected in the PFC of nonstressed animals; SL, LPS injected in the PFC of stressed animals; SLRU, stressed animals intracortically injected with LPS and treated with RU486.
Effect of LPS and stress on astroglia
We found a moderate astrogliosis around the injection site of vehicle in nonstressed animals without loss of GFAP immunostaining (Fig. 3B,G), although we observed damage in stressed animals (0.15 mm2; p < 0.01 compared with control) (Fig. 3C,G). Astroglia disappeared around the injection site of LPS, showing an area absent of GFAP-positive structures but surrounded by hyper-reactive astrocytes (0.34 mm2; p < 0.01 compared with the vehicle-injected animals) (Fig. 3D,G). Stress reinforces the effect of LPS in a synergistic manner, producing an increase in the area lacking astrocytes (0.83 mm2; p < 0.01 compared with the vehicle-injected animals) (Fig. 3E,G). This effect was reduced for RU486 (0.15 mm2) (Fig. 3F,G).
Figure 3.

Effect of stress and LPS in astroglia. A, Schematic representation of the injection sites in the prefrontal cortex. B, Coronal section showing GFAP immunoreactivity in a saline-injected nonstressed animal. Loss of astrocytes is restricted to the needle track. C, Loss of GFAP immunoreactivity in a vehicle-injected stressed animal. D, Coronal section showing GFAP immunoreactivity in an LPS-injected nonstressed animal. The arrow points to the site of injection of 2 μg of LPS. GFAP expression was more intense around the area lacking immunoreactivity. E, GFAP immunoreactivity in an LPS-injected stressed animal. The area lacking immunostaining is clearly larger. F, RU486 prevented the loss of astrocytes in an LPS-injected stressed animal. Scale bar, 250 μm. G, Quantification of the areas losing GFAP immunostaining. Results are mean ± SD of four independent experiments and are expressed as square millimeters. Statistical significance (one-way ANOVA, followed by the LSD post hoc test for multiple comparisons): a, compared with the control, vehicle-injected nonstressed animals; b, compared with the vehicle-injected stressed animals; c, compared with the LPS-injected nonstressed animals; d, compared with the LPS-injected stressed animals; p < 0.01. C, Control, vehicle injected in the PFC of nonstressed animals; S, vehicle injected in the PFC of stressed animals; L, LPS injected in the PFC of nonstressed animals; SL, LPS injected in the PFC of stressed animals; SLRU, stressed animals intracortically injected with LPS and treated with RU486.
Effect of LPS and stress on neuronal populations
Additional NeuN immunostaining was performed to detect neurons in general and in situ hybridization for GAD67 and NMDAR1A mRNA expression, which coexpressed in PFC, for detecting GABAergic neurons. Immunohistochemistry with NeuN resulted in the staining of both nucleus and cytoplasm (Fig. 4). This pattern was also seen in the PFC of vehicle-injected rats (Fig. 4B,G) and in the contralateral (untouched) PFC after LPS injection. Stress induces a loss of NeuN-positive neurons around the injection site of vehicle (0.52 mm2, p < 0.05 compared with the loss produced for the injection of vehicle in control animals, 0.02 mm2) (Fig. 4C,G) similar to that observed after LPS-injection in nonstressed animals (p < 0.05 compared with control) (Fig. 4D,G). When injected in stressed rats, an additive effect was observed for LPS (p < 0.05 compared with control) (Fig. 4E,G). This effect was reduced for RU486 (0.27 mm2) (Fig. 4F,G).
Figure 4.
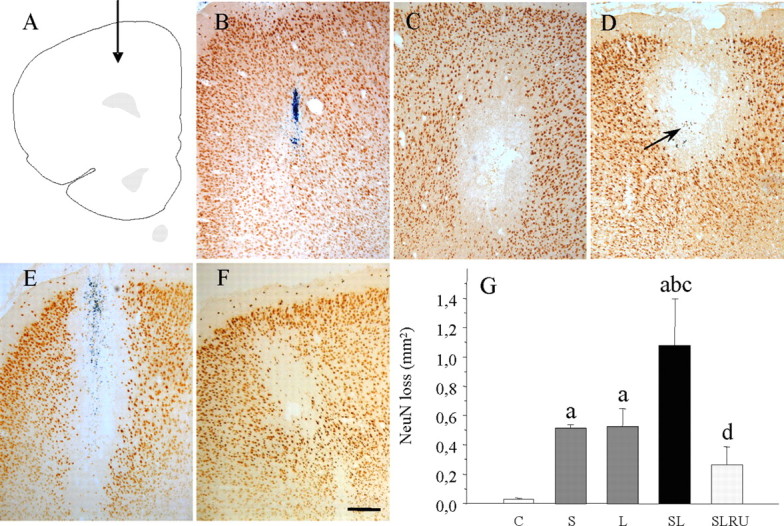
Effect of stress and LPS on cortical neurons. A, Schematic representation of the injection site into the prefrontal cortex. B, Coronal section showing NeuN immunoreactivity after the injection of vehicle in nonstressed animals. The loss was restricted to the injection track. C, NeuN immunoreactivity after the injection of vehicle in stressed animals. Stress causes an appreciable loss of cortical neurons. D, Coronal section showing NeuN immunoreactivity after the injection of 2 μg of LPS into the prefrontal cortex of nonstressed rats. Evenly distributed staining is disrupted around the injection site (arrow). E, NeuN immunoreactivity after the injection of 2 μg of LPS into the prefrontal cortex of stressed rats. The area lacking staining is larger. F, The treatment with RU486 highly diminished the loss of NeuN-positive neurons caused by the combined action of LPS and stress. Scale bar, 250 μm. G, Quantification of the areas losing NeuN immunostaining. Results are mean ± SD of four independent experiments and are expressed as square millimeters. Statistical significance (one-way ANOVA, followed by the LSD post hoc test for multiple comparisons): a, compared with the control, vehicle-injected nonstressed animals; b, compared with the vehicle-injected stressed animals; c, compared with the LPS-injected nonstressed animals; d, compared with the LPS-injected stressed animals; p < 0.01. C, Control, vehicle injected in the PFC of nonstressed animals; S, vehicle injected in the PFC of stressed animals; L, LPS injected in the PFC of nonstressed animals; SL, LPS injected in the PFC of stressed animals; SLRU, stressed animals intracortically injected with LPS and treated with RU486.
The GABAergic system in the PFC represents an abundant neuronal population, therefore showing a high level of expression of GAD67 mRNA (Fig. 5). We evaluated the effect of stress and LPS on the GABAergic population by injecting either vehicle or LPS in nonstressed and stressed animals. The specificity of the signals generated by the antisense-strand RNA probe was also confirmed using a sense probe (data not shown). Neuronal loss was not significant after injection of vehicle in nonstressed rats (Fig. 5B,G) and increased slightly in stressed animals (152.2% of control values) (Fig. 5C,G) but was more evident after the injection of LPS in nonstressed animals (280.0% compared with the vehicle-injected animals; p < 0.01) (Fig. 5D,G). Stress and LPS produced a synergistic effect (890.0% compared with the vehicle-injected animals; p < 0.01) (Fig. 5E,G). RU486 reduced the areas losing expression of GAD67 mRNA to 200.0% (compared with the vehicle-injected animals) (Fig. 5F,G).
Figure 5.

Effect of stress and LPS on cortical neurons expressing GAD67 mRNA. A, Schematic representation of the injection site into the prefrontal cortex. B, Coronal section showing GAD67 mRNA expression after the injection of vehicle in nonstressed animals. The loss can be seen closely around the injection track (dashed-lined area). C, GAD67 mRNA expression after the injection of vehicle in stressed animals. Stress causes a slight loss of cortical neurons (dashed-lined area). D, Coronal section showing GAD67 mRNA in situ hybridization after the injection of 2 μg of LPS into the prefrontal cortex of nonstressed rats. Normal expression is disrupted (dashed-lined area) around the injection site. Arrow shows the inert tracer. E, GAD67 mRNA expression after the injection of 2 μg of LPS into the prefrontal cortex of stressed rats. The area lacking staining (dashed-lined area) is larger. F, RU486 exerted a protective effect on the expression of GAD67 mRNA in stressed rats injected with LPS. Scale bar, 1 mm. G, Quantification of the areas losing GAD67 mRNA expression. Results are mean ± SD of five independent experiments and are expressed as square millimeters. Statistical significance (ANOVA, followed by the LSD post hoc test for multiple comparisons): a, compared with the control, vehicle-injected nonstressed animals; b, compared with the vehicle-injected stressed animals; c, compared with the LPS-injected nonstressed animals; d, compared with the LPS-injected stressed animals; p < 0.01. C, Control, vehicle injected in the PFC of nonstressed animals; S, vehicle injected in the PFC of stressed animals; L, LPS injected in the PFC of nonstressed animals; SL, LPS injected in the PFC of stressed animals; SLRU, stressed animals intracortically injected with LPS and treated with RU486.
NMDAR1A mRNA detected by in situ hybridization was sparsely distributed throughout the PFC (Fig. 6). Sections hybridized with the sense NMDAR1A probe were used as control for nonspecific labeling (data not shown). The loss of NMDAR1A mRNA expression observed after vehicle injection in nonstressed animals was reduced to the injection track (0.37 mm2) (Fig. 6B,G). Vehicle-injection in stressed animals (181.8% compared with the vehicle-injected animals; p < 0.01) (Fig. 6C,G) was similar in LPS-injected animals (207.3% compared with the vehicle-injected animals; p < 0.01) (Fig. 6D,G). NMDAR1A mRNA loss increased when LPS was injected into stressed rats (298.0% compared with the vehicle-injected animals; p < 0.01) (Fig. 6E,G). Again, this effect was reduced for RU486 near to control values (122.7% compared with the vehicle-injected animals) (Fig. 6F,G).
Figure 6.

Effect of stress and LPS on cortical neurons expressing NMDAR1A mRNA. A, Schematic representation of the injection site in the prefrontal cortex. B, Coronal section showing NMDAR1A mRNA expression after the injection of vehicle in nonstressed animals. The loss can be seen closely around the injection track (dashed-lined area). C, NMDAR1A mRNA expression after the injection of vehicle in stressed animals. Stress causes an appreciable loss of cortical neurons (dashed-lined area). D, NMDAR1A mRNA in situ hybridization after the injection of 2 μg of LPS into the prefrontal cortex of nonstressed rats. Evenly distributed expression is disrupted (dashed-lined area) around the injection site. Arrow shows the inert tracer. E, NMDAR1A mRNA expression after the injection of 2 μg of LPS into the prefrontal cortex of stressed rats. The area lacking expression (dashed-lined area) is larger. F, Loss of NMDAR1A mRNA expression induced by LPS and stress is reduced by the treatment with RU486. Scale bar, 1 mm. G, Quantification of the areas losing NMDAR1A mRNA expression. Results are mean ± SD of four independent experiments and are expressed as square millimeters. Statistical significance (one-way ANOVA, followed by the LSD post hoc test for multiple comparisons): a, compared with the control, vehicle-injected nonstressed animals; b, compared with the vehicle-injected stressed animals; c, compared with the LPS-injected nonstressed animals; d, compared with the LPS-injected stressed animals; p < 0.01. C, Control, vehicle injected in the PFC of nonstressed animals; S, vehicle injected in the PFC of stressed animals; L, LPS injected in the PFC of nonstressed animals; SL, LPS injected in the PFC of stressed animals; SLRU, stressed animals intracortically injected with LPS and treated with RU486.
Effect of LPS and stress on the expression of BDNF mRNA
Animals injected with vehicle displayed few BDNF mRNA-expressing cells in this structure. These cells were so scarce that we could not find more than one or two positive cells per section (Fig. 7A,F). Similar results were found when vehicle was injected into stressed animals (Fig. 7B,F). LPS injections increased this number by a factor of ∼12 (p < 0.01) (Fig. 7C,F), whereas the addition of stress synergistically increased this factor to nearly 43 (p < 0.01) (Fig. 7D,F). This strong upregulation of a subset of cortical neurons expressing BDNF mRNA was confined to around the injection site. RU486 reduced the number of BDNF mRNA-expressing cells to ninefold of the control (Fig. 7E,F).
Figure 7.
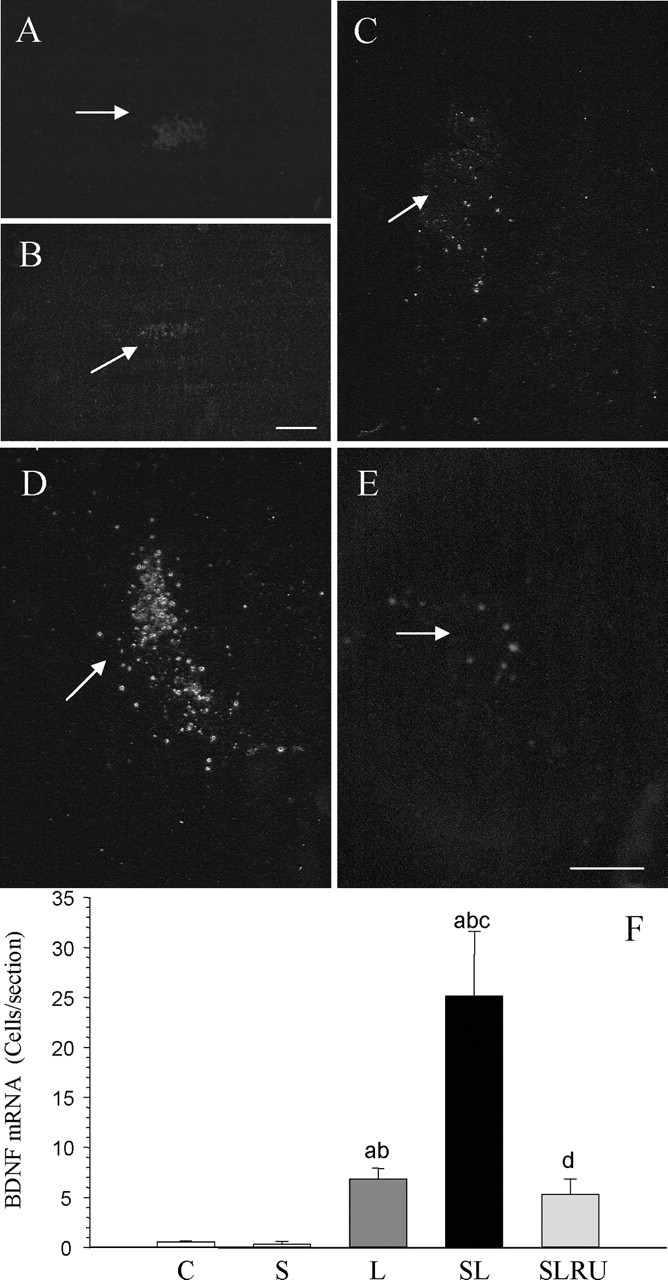
Effect of stress and LPS on BDNF mRNA expression in the prefrontal cortex. Dark-field photographs were taken from emulsion-dipped slide sections. A, BDNF mRNA expression after the injection of 2 μl of vehicle in the prefrontal cortex of nonstressed rats. Virtually no cells are detectable around the injection site (arrow). B, BDNF mRNA expression after the injection of 2 μl of vehicle into the prefrontal cortex of stressed rats. Cells are absent around the injection track. C, BDNF mRNA expression after the injection of 2 μg of LPS into the prefrontal cortex of nonstressed rats. A few cells can be seen around the injection site (arrow). D, BDNF mRNA expression after the injection of 2 μg of LPS into the prefrontal cortex of stressed rats. The number of cells around the injection site (arrow) has increased. E, The increase in BDNF mRNA expression induced by LPS and stress is diminished by the treatment with RU486. Scale bars, 250 μm. F, Quantification of the BDNF mRNA expression. Results are mean ± SD of four independent experiments and are expressed as cells per section. Statistical significance (one-way ANOVA, followed by the LSD post hoc test for multiple comparisons): a, compared with the control, vehicle-injected nonstressed animals; b, compared with the vehicle-injected stressed animals; c, compared with the LPS-injected nonstressed animals; d, compared with the LPS-injected stressed animals; p < 0.01. C, Control, vehicle injected in the PFC of nonstressed animals; S, vehicle injected in the PFC of stressed animals; L, LPS injected in the PFC of nonstressed animals; SL, LPS injected in the PFC of stressed animals; SLRU, stressed animals intracortically injected with LPS and treated with RU486.
Effect of LPS and stress at the tumor necrosis factor-α, interleukin-1β, and interleukin-6 levels
Given that stress exacerbated the LPS effect, we next sought to determine whether it was attributable to the increase of inflammatory mediators or to a direct effect of stress on microglia activation. RT-PCR analysis showed that the mRNAs for the proinflammatory cytokines tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and Il-6 were induced 6 h after LPS treatment. The expression levels of TNF-α mRNA in the PFC (Fig. 8A,B) were affected by stress and LPS. So, stress increased expression level to 128.6% of control values (p < 0.01). Similar results were found after LPS injection in nonstressed animals (136.9% compared with vehicle-injected animals; p < 0.01). When LPS was injected in stressed animals, expression levels of TNF-α reached 153.6% of control vehicle-injected nonstressed animals (p < 0.01). RU486 treatment reduced expression levels to 120.2% of controls (p < 0.01).
Figure 8.

Effect of stress and LPS on the expression levels of TNF-α, IL-1β, and IL-6 mRNAs. Expression of TNF-α, IL-1β, and IL-6 mRNAs were measured by RT-PCR in the prefrontal cortex of rats from the different treatments assayed: C, control, vehicle injected in the PFC of nonstressed animals; S, vehicle injected in the PFC of stressed animals; L, LPS injected in the PFC of nonstressed animals; SL, LPS injected in the PFC of stressed animals; SLRU, stressed animals intracortically injected with LPS and treated with RU486. A, Photographs from agarose gels showing expression of mRNAs 6 h after injection of either vehicle or LPS into the left PFC. β-Actin mRNA expression served as control. B, Quantification of TNF-α mRNA expression. Stress and LPS increased expression levels in a similar way. TNF-α mRNA induction was higher when LPS and stress were combined. RU486 diminished expression levels. C, Quantification of IL-1β mRNA expression. Stress had no effect, whereas induction of IL-1β levels attributable to LPS injection was similar in nonstressed and stressed animals. RU486 reduced IL-1β levels to control values. D, Quantification of IL-6 mRNA expression. Similar to IL-1β, stress had no effect on IL-6 mRNA expression levels, and the increase induced by LPS was reduced by RU486. Results are mean ± SD of four independent experiments and are expressed as percentage of β-actin mRNA expression. Statistical significance (one-way ANOVA, followed by the LSD post hoc test for multiple comparisons): a, compared with the control, vehicle-injected nonstressed animals; b, compared with the vehicle-injected stressed animals; c, compared with the LPS-injected nonstressed animals; d, compared with the LPS-injected stressed animals; p < 0.01.
The expression levels of IL-1β mRNA (Fig. 8A,C) did not change in vehicle-injected stressed animals (100.9% compared with control) but increased in LPS-injected nonstressed animals (156.4% of vehicle-injected nonstressed animals; p < 0.01). Similar levels were found when LPS was injected in stressed animals (153.8%; p < 0.01). RU486 reduced IL-1β expression levels to control values (108.6% of controls).
Similar to IL-1β, the expression levels of IL-6 in PFC (Fig. 8A,D) did not change in vehicle-injected stressed animals (113.5% compared with control) but increased in LPS-injected nonstressed animals (158.7% of vehicle-injected nonstressed animals; p < 0.01). Expression levels in LPS-injected stressed animals reached 165.9% (p < 0.01) of controls. RU486 reduced expression levels to 130.0% of controls (p < 0.01).
Effect of LPS and stress on the levels of total and phosphorylated JNK, p38, ERK, Akt, and CREB
Activation of mitogen-activated protein kinases (MAPKs) was determined by Western blot analysis using specific antibodies of the phosphorylated and nonphosphorylated forms of JNK, p38, ERK, Akt, and the transcription factor CREB. Neither stress nor LPS produced significant changes in the total amounts of these proteins (Fig. 9). On the contrary, phosphorylated levels were altered. LPS induced an increase in P-JNK (from 31.8% in control animals to 40.6% in LPS-injected animals; p < 0.05; percentage values are relative to the total amount of the studied protein in control animals). This increase was reinforced for stress (49.9%; p < 0.05) but decreased to 23.9% of control animals after the treatment with RU486. P-p38 MAPK levels were also altered: LPS induced a strong increase (from 17.0% in control to 33.9% in LPS-injected animals; p < 0.01), whereas LPS injected in stressed animals raised the levels to 43.8% of controls (p < 0.01). Again, RU486 reduced the levels to 24.9% of controls. P-ERK levels changed after the injection of vehicle in stressed animals, increasing from 78.1% in control to 102.8% in stressed animals (p < 0.01). The effect was similar in nonstressed LPS-injected animals (105.6% of controls; p < 0.01) and reinforced when LPS was injected in stressed animals (130.3% of controls; p < 0.01). RU486 treatment prevents these changes, keeping P-ERK levels close to control values. The term Akt generally refers to Akt1, one of the three known members of the Akt gene family (Brazil and Hemmings, 2001). Although it is likely that the three Akt proteins have at least some nonoverlapping functions, they have yet to be fully understood. We found that Akt phosphorylation levels were similar in all treatments and not significantly different from controls. Phosphorylation of CREB was also examined by Western blot analysis (Fig. 9). P-CREB was reduced after the injection of vehicle (19.6% of controls; p < 0.01) and LPS (22.2% of control; p < 0.01) in nonstressed animals and after the injection of LPS into stressed animals (21.9%; p < 0.01) compared with the control values (66.8% of control). Again, RU486 prevented these changes, keeping P-CREB levels close to control values (83.5% of control).
Figure 9.
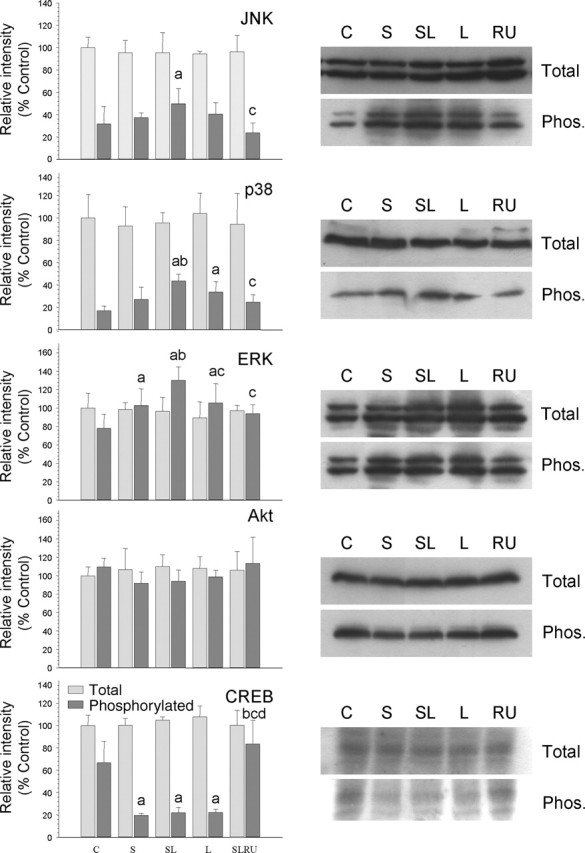
Effect of LPS and stress on several kinases and the transcription factor CREB in the prefrontal cortex. Proteins from the cortex of rats from the different treatments assayed (C, control, vehicle injected in the PFC of nonstressed animals; S, vehicle injected in the PFC of stressed animals; SL, LPS injected in the PFC of stressed animals; L, LPS injected in the PFC of nonstressed animals; RU, stressed animals intracortically injected with LPS and treated with RU486) were separated by electrophoresis, transferred to nitrocellulose membranes, and stained using anti-JNK, anti P-JNK, anti-p38, anti-P-p38, anti-ERK, anti-P-ERK, anti-Akt, anti-P-Akt, anti-CREB, and anti-P-CREB antibodies. Total optical density of each band was calculated. Results are mean ± SD of four independent experiments and are expressed as relative intensity of control bands. Statistical significance (one-way ANOVA, followed by the LSD post hoc test for multiple comparisons): a, compared with the control, vehicle-injected nonstressed animals; b, compared with the vehicle-injected stressed animals; c, compared with the LPS-injected stressed animal; d, compared with the LPS-injected nonstressed animals; p < 0.01.
Discussion
It has been shown previously that sensitivity of neurons to inflammation depends on both its duration and the region affected (Willard et al., 1999; Herrera et al., 2000; Kim et al., 2000). For instance, neurons from the dorsal raphe nuclei, hippocampus, or cortex were relatively insensitive to a single LPS injection at doses as high as 10 μg. Interestingly, our results show that the response to LPS in PFC was significantly stronger in stressed than in nonstressed animals: it was shown as a stronger OX-6 reaction, along with the induction of the proinflammatory cytokines TNF-α but not IL-1β and Il-6. Proinflammatory cytokines are known to increase in the inflammatory process induced by LPS. We also found a greater loss of GFAP immunoreactivity in the stressed animals. This pattern is maintained in the neuronal populations, showing a greater loss of NeuN-positive neurons in the stressed animals accompanied by the loss of cells expressing GAD67 and NMDAR1A mRNAs, which coexpressed in PFC GABAergic neurons. The effect of vehicle injection in stressed animals is generally lower than that found after the injection of LPS in nonstressed animals. The effects produced by LPS alone can be caused exclusively by the inflammatory response because it is not believed to exert a major direct influence on the HPA axis (Seo and Rivier, 2001). However, the increased neuronal damage produced by LPS in stressed animals could indicate a cooperative effect of chronic stress and inflammation.
Stress is able to enhance neuronal damage in some conditions; previous exposure to social stress increases infarction volume and exacerbates cognitive deficits associated with transient cerebral ischemia (Sugo et al., 2002). The mechanism underlying the effects of stress on stroke outcome likely involves corticosterone acting through GR to increase subsequent ischemia-induced neuronal death (Craft et al., 2005).
Deleterious consequences of prolonged exposure to an excess of glucocorticoids have been mainly described for the hippocampus; atrophy of dendrites and neuronal loss has been reported after corticosterone treatment (Sapolsky et al., 1985; Woolley et al., 1990). In the presence of glucocorticoids, both in vitro and in vivo, hippocampal neurons become more vulnerable to ischemia (Sapolsky and Pulsinelli, 1985; Morse and Davis, 1990), hypoglycemia (Tombaugh and Sapolsky, 1992; Tombaugh et al., 1992), and exposure to antimetabolites (Packan and Sapolsky, 1990; McIntosh et al., 1998) and various neurotoxins (Hortnagl et al., 1993).
Damages found in PFC could be caused by the increase of radical oxygen species (ROS), extracellular glutamate levels, excessive Ca2+ influx, and BDNF induced by LPS in stressed animals. Involvement of ROS in the degenerative effects induced by LPS intranigral injection has been described previously (Tomás-Camardiel et al., 2004). Moreover, it is known that stress induces the formation of ROS (McIntosh and Sapolsky, 1996) and leads to oxidative injury in various tissues, including blood (Oishi et al., 1999) and brain (Liu et al., 1996). Overexcitation of glutamate receptors seems to contribute to neurodegeneration at early stages of LPS infusion (Glezer et al., 2003). Stress activates the glutamatergic system in the PFC more intensely than in other regions (Gilad et al., 1990; Moghaddam, 1993; Karreman and Moghaddam, 1996). The stress-induced increase in dopamine release found by us in the PFC at the beginning of the stressing treatment could be a consequence of an increased glutamatergic neurotransmission in this region (Jedema and Moghaddam, 1996); this glutamate may excite GABA neurons and induce their death (Li et al., 2002), presumably with the increase of intracellular Ca2+ levels (Elliott and Sapolsky, 1993). At the same time, Harvey et al. (2004) reported that stress–restress-mediated glucocorticoids release produced a downregulation of hippocampal NMDA receptors and a decrease in GABA. Li et al. (2002) demonstrated in acute slices of rat limbic cortex that NMDA receptors on the GABAergic neurons were disproportionately more sensitive to the antagonistic effects of MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] than to those on pyramidal neurons. Thus, downregulation of presynaptic markers for GABAergic interneurons may reflect a chronic loss of the trophic influences of NMDA receptor activation (Paulson et al., 2003).
Another interesting result is the increase in BDNF mRNA expression in PFC during the injection of LPS in stressed rats. Expression of BDNF changes in response to different stressors and depends on the brain structure studied (Pizarro et al., 2004; Xu et al., 2004; Rosenbrock et al., 2005). The increase in BDNF expression found in our experiment could be a synergistic effect produced by LPS toxicity and stress through the loss of GABAergic neurons rather than being produced by stress directly. GABA represses BDNF expression (Zafra et al., 1991; Heese et al., 2000; Marmigere et al., 2003), whereas LPS and stress activate glutamatergic (Karreman and Moghaddam, 1996; Glezer et al., 2003) and dopaminergic (Abekawa et al., 2000; Gamaro et al., 2003; Matsumoto et al., 2005) systems, which induces BDNF expression (Zafra et al., 1990, 1991; Kuppers and Beyer, 2001). Although BDNF increases in pathological situations (Felderhoff-Mueser et al., 2002) and seems to exert a protective activity (Tapia-Arancibia et al., 2004), some evidence suggests that neurotrophins strengthen or may even cause neuronal damage under certain pathological conditions through the induction of NADPH-oxidase (Koh et al., 1995; Samdani et al., 1997; Kim et al., 2002). Thus, BDNF could be involved in the increase of the damage induced by inflammation in stressed animals.
All of these data allow us to suggest that inflammation along with chronic stress induces a special situation that leads to increased damage and loss of neurons. However, damage produced by stress can be prevented. Inhibition of the rise of corticosteroids levels during ischemia preserves cell survival after ischemia and prevents seizures (Krugers et al., 1995, 1998, 1999; Smith-Swintoski et al., 1996); also, GR blockade with RU486 (a potent inhibitor of GR activation) prevents CA1 neuron loss 24 h after constriction nerve injury (McCullers et al., 2002). It is also known that RU486 reverses glucocorticoid-mediated vulnerability to glutamate in a cultured murine hippocampal cell line (Behl et al., 1997a). We found that treatment with RU486 produced a significant protection, reducing OX-6 reaction and loss of GFAP immunoreactivity. RU486 also prevented the increase of the three cytokines studied.
This pattern is maintained in the neuronal populations, showing a significant reduction of the loss of NeuN-positive neurons and cells expressing GAD67 and NMDAR1A mRNAs. Moreover, RU486 treatment also inhibits the increase in BDNF mRNA expression in PFC during the injection of LPS in stressed rats. The protective effect of RU486 could be caused by its antiglucocorticoid effects combined with its antioxidant properties (Behl et al., 1997b). The antiglucocorticoid effects could prevent excessive intracellular Ca2+ accumulation in neurons (Elliott and Sapolsky, 1993; McCullers et al., 2002). RU486 antioxidant effects may mediate neuroprotection in CA1 pyramidal cells. It also prevents oxidation of the low-density lipoprotein (Parthasarathy et al., 1994) and the oxidative modification of proteins (Carpenter et al., 1996).
This is also in agreement with the results found for MAPK cascades, responsible for the transduction of neurotrophic signals. We found that stress, LPS (through cytokines such as TNF-α and IL-1β), and the combination of both produced an increase in the phosphorylation levels of JNK, p38, and ERK; this was prevented by RU486 treatment. Activation of JNK/SAPK and p38 is associated with promotion of apoptosis and plays an important role in the pathogenesis of inflammatory responses (Walton et al., 1998). Our results also show a marked reduction of CREB phosphorylation that is prevented by RU486 treatment. This is in agreement with previous work reporting that chronic stress increased P-ERK but significantly reduced P-CREB expression in PFC (Kuipers et al., 2003). We found that TNF-α release induced by LPS is reinforced by stress and is prevented by RU486.
All of these results suggest a relationship between the inflammatory process and the increased basal activity of the HPA axis that have been repeatedly demonstrated in AD, including increase in basal concentration of cortisol in plasma (Belanoff et al., 2001) along with a higher incidence of chronic stress adaptation failure in patients with AD (Pascualy et al., 2000). It is well known that amyloid deposits are not exclusive of AD patients but appear in most nondemented people after the age of 65 years (Sparks et al., 1993). PFC and probably other CNS structures could be less sensitive to inflammation than to stress; a cooperative effect between the two processes could explain the suggested relationship between stress and AD, because a high susceptibility to distress leads to a double risk of developing this disease (Wilson et al., 2003). This is also in agreement with the first evidence of a genetic association between the glucocorticoid system and AD reported by de Quervain et al. (2004). Set-association analysis revealed a rare haplotype in the 5′ regulatory region of the gene encoding 11β-hydroxysteroid dehydrogenase type 1 (HSD11B1) associated with a sixfold increased risk for sporadic AD. HSD11B1 controls tissue levels of biologically active glucocorticoids and thereby influences neuronal vulnerability. These results also point to the possibility of using RU486 in AD, because different assays show that RU486 slows the progression of cognitive decline in this disease (Belanoff et al., 2002; DeBattista and Belanoff, 2005).
Footnotes
This work was supported by Spanish Ministerio de Educacíon y Ciencia Grants SAF2002-01952 and SAF2004-06601. R.M.d.P and R.F.V. thank the Spanish Ministerio de Educación y Ciencia for Formación del Profesorado Universitario and Formación de Personal Investigador fellowships, respectively. A.J.H. thanks the Junta de Andalucía for a Contrato de Retorno. We thank J. P. Calero for his skilful technical assistance and Diane Haun for language and style revision of this manuscript.
References
- Abekawa T, Ohmori T, Ito K, Koyama T (2000). D1 dopamine receptor activation reduces extracellular glutamate and GABA concentrations in the medial prefrontal cortex. Brain Res 867:250–254. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL (2000). Inflammation and Alzheimer disease. Neurobiol Aging 21:383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF (1998). The biology of being frazzled. Science 280:1711–1712. [DOI] [PubMed] [Google Scholar]
- Behl C, Trapp T, Skutella T, Holsboer F (1997a). Protection against oxidative stress-induced neuronal cell death—a novel role for RU486. Eur J Neurosci 9:912–920. [DOI] [PubMed] [Google Scholar]
- Behl C, Lezoualc’h F, Trapp T, Widmann M, Skutella T, Holsboer F (1997b). Glucocorticoids enhance oxidative stress-induced cell death in hippocampal neurons in vitro. Endocrinology 138:101–106. [DOI] [PubMed] [Google Scholar]
- Belanoff JK, Gross K, Yager A, Schatzberg AF (2001). Corticosteroids and cognition. J Psychiatr Res 35:127–145. [DOI] [PubMed] [Google Scholar]
- Belanoff JK, Jurik J, Schatzberg LD, DeBattista C, Schatzberg AF (2002). Slowing the progression of cognitive decline in Alzheimer’s disease using RU486. J Mol Neurosci 19:201–206. [DOI] [PubMed] [Google Scholar]
- Benveniste EN, Sparacio SM, Norris JG, Grenett HE, Fuller GM (1990). Induction and regulation of interleukin-6 gene expression in rat astrocytes. J Neuroimmunol 30:201–212. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Hemmings BA (2001). Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci 26:657–664. [DOI] [PubMed] [Google Scholar]
- Carpenter SE, Santanam N, Murphy AA, Rock JA, Parthasarathy S (1996). Inhibition of oxidative modification of proteins by RU486. Fertil Steril 66:90–94. [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301:386–389. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Peterson PK (1995). Glia, cytokines and neurotoxicity. Crit Rev Neurobiol 9:189–205. [PubMed] [Google Scholar]
- Chung IY, Benveniste EN (1990). Tumor necrosis factor-alpha production by astrocytes. Induction by lipopolysaccharide, IFN-gamma, and IL-1 beta. J Immunol 144:2999–3007. [PubMed] [Google Scholar]
- Craft TK, Glasper ER, McCullough L, Zhang N, Sugo N, Otsuka T, Hurn PD, DeVries AC (2005). Social interaction improves experimental stroke outcome. Stroke 36:2006–2011. [DOI] [PubMed] [Google Scholar]
- DeBattista C, Belanoff J (2005). C-1073 (RU486) in the adjunctive treatment of Alzheimer’s disease. Curr Alzheimer Res 2:125–129. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Poirier R, Wollmer MA, Grimaldi LM, Tsolaki M, Streffer JR, Hock C, Nitsch RM, Mohajeri MH, Papassotiropoulos A (2004). Glucocorticoid-related genetic susceptibility for Alzheimer’s disease. Hum Mol Genet 13:47–52. [DOI] [PubMed] [Google Scholar]
- Elliott EM, Sapolsky RM (1993). Corticosterone impairs hippocampal neuronal calcium regulation: possible mediating mechanisms. Brain Res 602:84–90. [DOI] [PubMed] [Google Scholar]
- Erlander MG, Tillakaratne NJ, Feldblum S, Patel N, Tobin AJ (1991). Two genes encode distinct glutamate decarboxylases. Neuron 7:91–100. [DOI] [PubMed] [Google Scholar]
- Feenstra MG, Botterblom MH, van Uum JF (1998). Local activation of metabotropic glutamate receptors inhibits the handling-induced increased release of dopamine in the nucleus accumbens but not that of dopamine or noradrenaline in the prefrontal cortex: comparison with inhibition of ionotropic receptors. J Neurochem 70:1104–1113. [DOI] [PubMed] [Google Scholar]
- Felderhoff-Mueser U, Sifringer M, Pesditschek S, Kuckuck H, Moysich A, Bittigau P, Ikonomidou C (2002). Pathways leading to apoptotic neurodegeneration following trauma to the developing rat brain. Neurobiol Dis 11:231–245. [DOI] [PubMed] [Google Scholar]
- Fryer HJ, Davis GE, Manthorpe M, Varon S (1986). Lowry protein assay using an automatic microtiter plate spectrophotometer. Anal Biochem 153:262–266. [DOI] [PubMed] [Google Scholar]
- Gamaro GD, Manoli LP, Torres IL, Silveira R, Dalmaz C (2003). Effects of chronic variate stress on feeding behavior and on monoamine levels in different rat brain structures. Neurochem Int 42:107–114. [DOI] [PubMed] [Google Scholar]
- Gerrits M, Westenbroek C, Fokkema DS, Jongsma ME, Den Boer JA, Ter Horst GJ (2003). Increased stress vulnerability after a prefrontal cortex lesion in female rats. Brain Res Bull 61:627–635. [DOI] [PubMed] [Google Scholar]
- Gilad GM, Gilad VH, Wyatt RJ, Tizabi Y (1990). Region-selective stress-induced increase of glutamate uptake and release in rat forebrain. Brain Res 525:335–338. [DOI] [PubMed] [Google Scholar]
- Glezer I, Munhoz CD, Kawamoto EM, Marcourakis T, Avellar MC, Scavone C (2003). MK-801 and 7-Ni attenuate the activation of brain NF-kappa B induced by LPS. Neuropharmacology 45:1120–1129. [DOI] [PubMed] [Google Scholar]
- Harvey BH, Oosthuizen F, Brand L, Wegener G, Stein DJ (2004). Stress-restress evokes sustained iNOS activity and altered GABA levels and NMDA receptors in rat hippocampus. Psychopharmacology (Berl) 175:494–502. [DOI] [PubMed] [Google Scholar]
- Heese K, Otten U, Mathivet P, Raiteri M, Marescaux C, Bernasconi R (2000). GABA(B) receptor antagonists elevate both mRNA and protein levels of the neurotrophins nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) but not neurotrophin-3 (NT-3) in brain and spinal cord of rats. Neuropharmacology 39:449–462. [DOI] [PubMed] [Google Scholar]
- Herrera AJ, Machado A, Cano J (1993). Ageing and monoamine turnover in the lateral geniculate nucleus and visual cortex of the rat. Neurochem Int 22:531–539. [DOI] [PubMed] [Google Scholar]
- Herrera AJ, Castaño A, Venero JL, Cano J, Machado A (2000). The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis 7:429–447. [DOI] [PubMed] [Google Scholar]
- Hortnagl H, Berger ML, Havelec L, Hornykiewicz O (1993). Role of glucocorticoids in the cholinergic degeneration in rat hippocampus induced by Ethylcholine Aziridinium (AF64A). J Neurosci 13:2939–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedema HP, Moghaddam B (1996). Characterization of excitatory amino acid modulation of dopamine release in the prefrontal cortex of conscious rats. J Neurochem 66:1448–1453. [DOI] [PubMed] [Google Scholar]
- Jeohn GH, Kong LY, Wilson B, Hudson P, Hong JS (1998). Synergistic neurotoxic effects of combined treatments with cytokines in murine primary mixed neuron/glia cultures. J Neuroimmunol 85:1–10. [DOI] [PubMed] [Google Scholar]
- Karreman M, Moghaddam B (1996). Effect of a pharmacological stressor on glutamate efflux in the prefrontal cortex. Brain Res 716:180–182. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Karkowski-Shuman L (1997). Stressful life events and genetic liability to major depression: genetic control of exposure to the environment? Psychol Med 27:539–547. [DOI] [PubMed] [Google Scholar]
- Kim SH, Won SJ, Sohn S, Kwon HJ, Lee JY, Park JH, Gwag BJ (2002). Brain-derived neurotrophic factor can act as a pronecrotic factor through transcriptional and translational activation of NADPH oxidase. J Cell Biol 159:821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS (2000). Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci 20:6309–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JY, Gwag BJ, Lobner D, Choi DW (1995). Potentiated necrosis of cultured cortical neurons by neurotrophins. Science 268:573–575. [DOI] [PubMed] [Google Scholar]
- Konarska M, Stewart RE, McCarty R (1990). Predictability of chronic intermittent stress: effects on sympathetic-adrenal medullary responses of laboratory rats. Behav Neural Biol 53:231–243. [DOI] [PubMed] [Google Scholar]
- Krugers HJ, Knollema S, Kemper RH, Ter Horst GJ, Korf J (1995). Down-regulation of the hypothalamo-pituitary-adrenal axis reduces brain damage and number of seizures following hypoxia/ischaemia in rats. Brain Res 690:41–47. [DOI] [PubMed] [Google Scholar]
- Krugers HJ, Kemper RH, Korf J, Ter Horst GJ, Knollema S (1998). Metyrapone reduces rat brain damage and seizures after hypoxia-ischemia: an effect independent of modulation of plasma corticosterone levels? J Cereb Blood Flow Metab 18:386–390. [DOI] [PubMed] [Google Scholar]
- Krugers HJ, Maslam S, Van Vuuren SM, Korf J, Joels M (1999). Postischemic steroid modulation: effects on hippocampal neuronal integrity and synaptic plasticity. J Cereb Blood Flow Metab 19:1072–1082. [DOI] [PubMed] [Google Scholar]
- Kuipers SD, Trentani A, Den Boer JA, Ter Horst GJ (2003). Molecular correlates of impaired prefrontal plasticity in response to chronic stress. J Neurochem 85:1312–1323. [DOI] [PubMed] [Google Scholar]
- Kuppers E, Beyer C (2001). Dopamine regulates brain-derived neurotrophic factor (BDNF) expression in cultured embryonic mouse striatal cells. NeuroReport 12:1175–1179. [DOI] [PubMed] [Google Scholar]
- Lee da Y, Oh YJ, Jin BK (2005). Thrombin-activated microglia contribute to death of dopaminergic neurons in rat mesencephalic cultures: dual roles of mitogen-activated protein kinase signaling pathways. Glia 51:98–110. [DOI] [PubMed] [Google Scholar]
- Lee SC, Dickson DW, Liu W, Brosnan CF (1993). Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccgaride and IL-1β. J Immunol 150:2659–2667. [PubMed] [Google Scholar]
- Li Q, Clark S, Lewis DV, Wilson WA (2002). NMDA receptor antagonists disinhibit rat posterior cingulated and retrosplenial cortices: a potential mechanism of neurotoxicity. J Neurosci 22:3070–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman AP, Pitha PM, Shin HS, Shin ML (1989). Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc Natl Acad Sci USA 86:6348–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang X, Shigenaga MK, Yeo HC, Mori A, Ames BN (1996). Immobilization stress causes oxidative damage to lipid, protein, and DNA in the brain of rats. FASEB J 10:1532–1538. [PubMed] [Google Scholar]
- Marmigere F, Rage F, Tapia-Arancibia L (2003). GABA-glutamate interaction in the control of BDNF expression in hypothalamic neurons. Neurochem Int 42:353–358. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Togashi H, Kaku A, Kanno M, Tahara K, Yoshioka M (2005). Cortical GABAergic regulation of dopaminergic responses to psychological stress in the rat dorsolateral striatum. Synapse 56:117–121. [DOI] [PubMed] [Google Scholar]
- Mazure CM, Kincare P, Schaffer CE (1995). DSM-III-R Axis IV: clinician reliability and comparability to patients’ reports of stressor severity. Psychiatry 58:56–64. [DOI] [PubMed] [Google Scholar]
- McCullers DL, Sullivan PG, Sheff SW, Hermen JP (2002). RU486 protects CA1 hippocampal neurons following traumatic brain injury in rat. Neuroscience 109:219–230. [DOI] [PubMed] [Google Scholar]
- McEwen BS (2000). The neurobiology of stress: from serendipity to clinical relevance. Brain Res 886:172–189. [DOI] [PubMed] [Google Scholar]
- McEwen BS (2002). Protective and damaging effects of stress mediators: the good and bad sides of the response to stress. Metabolism 51:2–4. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG (2004). Inflammation and the degenerative diseases of aging. Ann NY Acad Sci 1035:104–116. [DOI] [PubMed] [Google Scholar]
- McIntosh LJ, Sapolsky RM (1996). Glucocorticoids may enhance oxygen radical-mediated neurotoxicity. Neurotoxicology 17:873–882. [PubMed] [Google Scholar]
- McIntosh LJ, Cortopassi KM, Sapolsky RM (1998). Glucocorticoids may alter antioxidant enzyme capacity in the brain: kainic acid studies. Brain Res 791:215–222. [DOI] [PubMed] [Google Scholar]
- Meltzer JC, Sanders V, Grimm PC, Stern E, Rivier C, Lee S, Rennie SL, Gietz RD, Hole AK, Watson PH, Greenberg AH, Nance DMProduction of digoxigenin-labelled RNA probes and the detection of cytokina mRNA in rat spleen and brain by in situ hybridization. Brain Res Brain Res Protoc 2:339–351. [DOI] [PubMed] [Google Scholar]
- Moghaddam B (1993). Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J Neurochem 60:1650–1657. [DOI] [PubMed] [Google Scholar]
- Morse JK, Davis JN (1990). Regulation of ischemic hippocampal damage in the gerbil: adrenalectomy alters the rate of CA1 cell disappearance. Exp Neurol 110:86–92. [DOI] [PubMed] [Google Scholar]
- Murua VS, Molina VA (1992). Effects of chronic variable stress and antidepressant drugs on behavioral inactivity during an uncontrollable stress: interaction between both treatments. Behav Neural Biol 57:87–89. [DOI] [PubMed] [Google Scholar]
- Muscat R, Papp M, Willner P (1992). Reversal of stress-induced anhedonia by the atypiacal antidepressants, fluoxetine and maprotiline. Psychopharmacology 106:821–826. [DOI] [PubMed] [Google Scholar]
- Oishi K, Yokoi M, Maekawa S, Sodeyama C, Shiraishi T, Kondo R, Kuriyama T, Machida K (1999). Oxidative stress and haematological changes in immobilized rats. Acta Physiol Scand 165:65–69. [DOI] [PubMed] [Google Scholar]
- Packan DR, Sapolsky RM (1990). Glucocorticoid endangerment of the hippocampus: tissue, steroid and receptor specificity. Neuroendocrinology 51:613–618. [DOI] [PubMed] [Google Scholar]
- Papp M, Willner P, Muscat R (1991). An animal model of anhedonia: attenuation of sucrose consumption and place preference conditioning by chronic unpredictable mild stress. Psychopharmacology (Berl) 104:255–259. [DOI] [PubMed] [Google Scholar]
- Parthasarathy S, Morales AJ, Murphy AA (1994). Antioxidant: a new role for RU-486 and related compounds. J Clin Invest 94:1990–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascualy M, Petrie EC, Brodkin K, Peskind ER, Wilkinson CW, Raskind MA (2000). Hypothalamic pituitary adrenocortical and sympathetic nervous system responses to the cold pressor test in Alzheimer’s disease. Biol Psychiatry 48:247–254. [DOI] [PubMed] [Google Scholar]
- Pasinetti GM, Lerner SP, Johnson SA, Morgan DG, Telford NA, Finch CE (1989). Chronic lesions differentially decrease tyrosine hydroxylase messenger RNA in dopaminergic neurons of the substantia nigra. Brain Res Mol Brain Res 5:203–209. [DOI] [PubMed] [Google Scholar]
- Paulson L, Martin P, Persson A, Nilsson CL, Ljung E, Westman-Brinkmalm A (2003). Comparative genome and proteome analysis of cerebral cortex from MK-801-treated rats. J Neurosci Res 7 1:526–533. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1986). In: The rat brain in stereotaxic coordinates San Diego: Academic.
- Pizarro JM, Lumley LA, Medina W, Robison CL, Chang WE, Alagappan A, Bah MJ, Dawood MY, Shah JD, Mark B, Kendall N, Smith MA, Saviolakis GA, Meyerhoff JL (2004). Acute social defeat reduces neurotrophin expression in brain cortical and subcortical areas in mice. Brain Res 1025:10–20. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, Hof PR, McEwen BS, Morrison JH (2004). Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience 125:1–6. [DOI] [PubMed] [Google Scholar]
- Rosenbrock H, Koros E, Bloching A, Podhorna J, Borsini F (2005). Effect of chronic intermittent restraint stress on hippocampal expression of marker proteins for synaptic plasticity and progenitor cell proliferation in rats. Brain Res 1040:55–63. [DOI] [PubMed] [Google Scholar]
- Rostworowski M, Balasingam V, Chabot S, Owens T, Yong VW (1997). Astrogliosis i the neonatal and adult murine brain post-trauma: elevation of inflammatory cytokines and the lack of requirement for endogenous interferon-gamma. J Neurosci 17:3664–3674. Samdani AF, Newcamp C, Resink A, Facchinetti F, Hoffman BE, Dawson VL, Dawson TM(1997)Differential susceptibility to neurotoxicity mediated by neurotrophins and neuronal nitric oxide synthase. J Neurosci 17:4633–4641.9169524 [Google Scholar]
- Sapolsky RM, Pulsinelli WA (1985). Glucocorticoids potentiate ischemic injury to neurons: therapeutic implications. Science 229:1397–1400. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS (1985). Prolonged glucocorticoid exposure reduces hippocampal neuron number: implications for aging. J Neurosci 5:1222–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo DO, Rivier C (2001). Microinfusion of a nitric oxide donor in discrete brain regions activates the hypothalamic-pituitary-adrenal axis. J Neuroendocrinol 13:925–933. [DOI] [PubMed] [Google Scholar]
- Smith-Swintoski VL, Pettigrew LC, Sapolsky RM, Phares C, Craddock SD, Brooke SM, Mattson MP (1996). Metyrapone, an inhibitor of glucocorticoid production, reduces brain injury induced by focal and global ischemia and seizures. J Cereb Blood Flow Metab 16:585–598. [DOI] [PubMed] [Google Scholar]
- Sparks DL, Liu H, Scheff SW, Coyne CM, Hunsaker JC 3rd (1993). Temporal sequence of plaque formation in the cerebral cortex of non-demented individuals. J Neuropathol Exp Neurol 52:135–142. [DOI] [PubMed] [Google Scholar]
- Sugo N, Hurn PD, Morahan MB, Hattori K, Traystman RJ, DeVries AC (2002). Social stress exacerbates focal cerebral ischemia in mice. Stroke 33:1660–1664. [DOI] [PubMed] [Google Scholar]
- Tapia-Arancibia L, Rage F, Givalois L, Arancibia S (2004). Physiology of BDNF: focus on hypothalamic function. Front Neuroendocrinol 25:77–107. [DOI] [PubMed] [Google Scholar]
- Tomás-Camardiel M, Herrera AJ, Venero JL, Cano J, Machado A (2004). Inflammatory process as a determinant factor for the degeneration of substantia nigra dopaminergic neurons: possible relevance to the etiology of Parkinson’s disease. Curr Med Chem 4:223–233. [DOI] [PubMed] [Google Scholar]
- Tombaugh GC, Sapolsky RM (1992). Corticosterone accelerates hypoxia-and cyanide-induced ATP loss in cultured hippocampal astrocytes. Brain Res 588:154–158. [DOI] [PubMed] [Google Scholar]
- Tombaugh GC, Yang SH, Swanson RA, Sapolsky RM (1992). Glucocorticoids cxacerbate hypoxic and hypoglycemic hippocampal injury in vitro: biochemical correlates and a role for astrocytes. J Neurochem 59:137–146. [DOI] [PubMed] [Google Scholar]
- Trentani A, Kuipers SD, Ter Horst GJ, Den Boer JA (2002). Selective chronic stress-induced in vivo ERK1/2 hyperphosphorylation in medial prefrontocortical dendrites: implications for stress-related cortical pathology? Eur J Neurosci 15:1681–1691. [DOI] [PubMed] [Google Scholar]
- Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S (2002). Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci 22:6810–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton KM, DiRocco R, Bartlett BA, Koury E, Marcy VR, Jarvis B, Schaefer EM, Bhat RV (1998). Activation of p38MAPK in microglia after ischemia. J Neurochem 70:1764–1767. [DOI] [PubMed] [Google Scholar]
- Wellman CL (2001). Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol 49:245–253. [DOI] [PubMed] [Google Scholar]
- Willard LB, Hauss-Wegrzyniak B, Wenk GL (1999). Pathological and biochemical consequences of acute and chronic neuroinflammation within the basal forebrain cholinergic system of rats. Neuroscience 88:193–200. [DOI] [PubMed] [Google Scholar]
- Willner P, Towell A, Sampson D, Sophokleous S, Muscat R (1987). Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology (Berl) 93:358–364. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Barnes LL, Bennett DA, Li Y, Bienias JL, Mendes de Leon CF, Evans DA (2003). Proneness to psychological distress and risk of Alzheimer disease in a biracial community. Neurology 64:380–382. [DOI] [PubMed] [Google Scholar]
- Woolley CS, Gould E, McEwen BS (1990). Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res 531:225–231. [DOI] [PubMed] [Google Scholar]
- Xu H, Luo C, Richardson JS, Li XM (2004). Recovery of hippocampal cell proliferation and BDNF levels, both of which are reduced by repeated restraint stress, is accelerated by chronic venlafaxine. Pharmacogenomics J 4:322–331. [DOI] [PubMed] [Google Scholar]
- Yokoyama C, Sasaki K (1999). Regional expressions of Fos-like immunoreactivity in rat cerebral cortex after stress; restraint and intraperitoneal lipopolysaccharide. Brain Res 816:267–275. [DOI] [PubMed] [Google Scholar]
- Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D (1990). Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J 9:3545–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Castren E, Thoenen H, Lindholm D (1991). Interplay between glutamate and gamma-aminobutyric acid transmitter systems in the physiological regulation of brain-derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proc Natl Acad Sci USA 88:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]