

Abstract
It is widely accepted that cannabinoids regulate GABA release by activation of cannabinoid receptor type 1 (CB1). Results obtained from a variety of brain regions consistently indicate that cannabinoid agonists can also reduce glutamatergic synaptic transmission. However, there are still conflicting data concerning the role of CB1 in cannabinoid-induced inhibition of glutamatergic transmission in cortical areas. Here, we provide direct evidence that activation of CB1 on terminals of principal neurons controls excitatory synaptic responses in the forebrain. In slices of the basolateral amygdala, the CA1 region of the hippocampus, and the primary somatosensory cortex of wild-type mice, application of the CB1 agonist (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone (WIN55,212-2; WIN) (5 μm) reduced evoked excitatory postsynaptic responses. In contrast, in slices obtained from conditional mouse mutants lacking CB1 in all principal forebrain neurons but not in GABAergic interneurons (CB1f/f;CaMKIIαCre), WIN no longer affected glutamatergic synaptic transmission in any of the brain regions tested. Compatible with a presynaptic mechanism, WIN did not change the sensitivity to focally uncaged l-glutamate. WIN reduced glutamatergic responses in slices obtained from mice lacking CB1 exclusively in GABAergic neurons (CB1f/f;Dlx5/6-Cre), thus excluding the involvement of CB1 expressed on GABAergic neurons in this effect of the drug. The present data strongly indicate that excitatory synaptic transmission in forebrain areas is directly modulated by CB1 expressed on presynaptic axon terminals originating from glutamatergic neurons.
Keywords: CB1, cannabinoids, glutamatergic, amygdala, hippocampus, cortex, principal neurons, caged glutamate, AMPA-EPSC
Introduction
Neuroanatomical, pharmacological, and electrophysiological data (Hofmann and Lupica, 2000; Katona et al., 2001; Wilson and Nicoll, 2001; Marsicano et al., 2002; Freund et al., 2003) have clearly shown that cannabinoids regulate GABAergic neurotransmission via the activation of cannabinoid receptor type 1 (CB1). Nevertheless, results from several brain regions such as the hippocampus (Misner and Sullivan, 1999), the prefrontal cortex (Auclair et al., 2000), the nucleus accumbens (Robbe et al., 2001), the amygdala (Azad et al., 2003), and other cortical and noncortical areas (Riegel and Lupica, 2004; Freiman and Szabo, 2005; Köfalvi et al., 2005) consistently indicate that CB1 agonists can also reduce glutamatergic synaptic transmission. Furthermore, endocannabinoids released by brief depolarization or tetanic stimulation induce either short-term or long-term depression of excitatory synaptic transmission in different brain areas (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Gerdeman et al., 2002; Robbe et al., 2002).
Evidence for the presence of CB1 on glutamatergic terminals has been provided only for noncortical areas, such as the striatum (Robbe et al., 2001; Rodriguez et al., 2001; Köfalvi et al., 2005). The role of CB1 in cannabinoid-induced inhibition of glutamatergic transmission in cortical areas is still being debated, because detailed immunohistochemical investigations were unable to detect CB1 immunostaining in axon terminals forming asymmetric, glutamatergic synapses (e.g., in the hippocampus) (Katona et al., 1999; Hajos et al., 2000). Also, experiments on neurotransmitter release in the CA1 region seem to support the view that cannabinoid receptors are located exclusively on inhibitory axon terminals (Hoffman et al., 2003). Furthermore, in the hippocampus of CB1-deficient mice (CB1−/−), the synthetic CB1 agonist (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone (WIN55,212-2; WIN) no longer reduced GABAergic synaptic transmission, whereas it was still able to affect glutamatergic transmission (Hajos et al., 2001) and to decrease glutamate release (Köfalvi et al., 2003). These observations led to the hypothesis that cannabinoid actions on glutamatergic transmission in the hippocampus could be mediated through a novel cannabinoid receptor, distinct from the CB1, which, however, has not yet been molecularly characterized.
In the amygdala, the hippocampus, and the neocortex, immunohistochemical data revealed that CB1 is expressed only in a subpopulation of GABAergic interneurons (Katona et al., 2001; Hoffman et al., 2003). However, in these brain regions, CB1 mRNA is present also in non-GABAergic cells (Marsicano and Lutz, 1999; Hermann et al., 2002), suggesting that these receptors may also control other transmitter systems. To further clarify the role of CB1 in the regulation of excitatory neurotransmission, the present electrophysiological study investigated glutamatergic synaptic transmission in slices of the amygdala, hippocampus, and neocortex of conditional mouse mutants lacking CB1 either in all principal forebrain neurons, but not in GABAergic interneurons (CB1f/f;CaMKIIαCre) (Marsicano et al., 2003), or in all GABAergic neurons (CB1f/f;Dlx5/6-Cre).
Materials and Methods
Animals.
Conditional mutant CB1f/f;CaMKIIα-Cre mice, lacking CB1 expression in all forebrain principal neurons and, consequently, expressing CB1 in the forebrain only in cortical GABAergic interneurons, were generated as described previously (Marsicano et al., 2003). More recently, obtained conditional mutant CB1f/f;Dlx5/6-Cre mice lack CB1 expression in all GABAergic neurons. Their generation is described elsewhere (Monory et al., unpublished results). All wild-type and mutant mice were littermates and were genotyped before and regenotyped after the experiment by PCR. Electrophysiological experiments were performed in accordance with the guidelines of the Ethical Committee on the Use and Care of Animals (Government of Bavaria, Germany). For slice preparation, the animals were anesthetized with isoflurane and decapitated. The brains were rapidly removed and placed in ice-cold artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 25 d-glucose, and 1.25 NaH2PO4, pH 7.4, bubbled with a 95% O2/5% CO2 mixture. Slices of each brain region (400 μm thick) were prepared using a vibroslicer (FTB, Weinheim, Germany). After incubation in a holding chamber with ACSF (22–25°C) for at least 60 min, the slices were placed in a submerged recording chamber and superfused with ACSF at a flow rate of 1.5 ml/min (for additional details, see Simon et al., 2001; Rammes et al., 2003; Schierloh et al., 2004).
Electrophysiology.
Stimulation, recording, and analysis of EPSCs, field potentials (FPs), and EPSPs in the CA1 region of the hippocampus and the basolateral amygdala (BLA) were performed as described previously (Simon et al., 2001; Rammes et al., 2003). EPSCs in layer II neurons of the primary somatosensory cortex were evoked by stimulation in layer II. To avoid polysynaptic responses, we used an ultra-fine bipolar electrode (12 μm; FHC, Bowdoinham, ME), which was positioned in layer II close to the apical dendrite of the recorded neuron. Isolated AMPA receptor-mediated EPSCs were measured in the presence of 50 μm d-(−)-2-amino-5-phosphonopentanoic acid (d-AP-5), 50 μm picrotoxin (neocortex, 30 μm), and 200 μm 3-aminopropyl(diethoxymethyl)phosphonic acid (CGP 35348).
Statistical analysis was performed using the Student's paired t test to compare the values of 10 min of a stable baseline and the 10 min period after stable effects had been established. p < 0.05 was considered as a significant difference. Data are presented as the percentage (mean ± SEM) of the average baseline response. Only the last 10 min of the baseline are depicted in all graphs.
Photostimulation.
For photostimulation, caged l-glutamate was released by focal photolysis, as described previously (Dodt et al. 1999). Briefly, the beam of a UV laser (355 nm wavelength, frequency-tripled Nd:YVO4, 100 kHz pulse repetition rate; DPSS Lasers, San Jose, CA) was focused by the objective (60 W, 0.9 numerical aperture; Olympus, Tokyo, Japan) on a small spot (5 μm in diameter) positioned on a dendrite ∼10–20 μm from the soma. Laser stimulation was delivered alternating with synaptic stimulation in intervals at 15 s. Once a stable whole-cell recording had been obtained, “caged-glutamate” γ-CNB (α-carboxy-2- nitrobenzyl)-caged glutamate (Sigma, St. Louis, MO) was added to the perfusate (0.125 mm), which was recirculated (11 ml volume). Caged-glutamate had no discernible effect on neurons per se (Dodt et al., 1999). Glutamate was released by Q-switching brief laser pulses (3–5 ms; intensity, 1–2 mW), applied at regular intervals of 30 s throughout the experiment.
Drugs.
The following pharmacological compounds were used: WIN, picrotoxin, d-AP-5, and CGP 35348 (Sigma/RBI, Natick, MA). Stock solutions of WIN (10 mm) were prepared either in DMSO or in HCl (0.1N) and stored at −20°C. Final DMSO concentrations were ≤0.05%. Before all experiments, fatty acid-free bovine serum albumin (1 mg/ml) was rinsed through the system to avoid binding of WIN to the walls of the tubing.
Results
In the limbic system, activation of CB1 does not affect EPSC amplitude in mice lacking CB1 expression in principal neurons
To study the pharmacological effect of CB1 activation on excitatory synaptic transmission, we performed extracellular recordings of FPs in BLA slices of mice lacking the expression of CB1 in principal neurons. In the BLA of CB1f/f [which express CB1 in a wild-type manner (Marsicano et al., 2003)], WIN reduced the FP amplitude to 69.5 ± 5.2% of baseline (n = 9), whereas in the BLA of CB1f/f;CaMKIIαCre littermates, FPs were significantly increased to 107.4 ± 5.0% (n = 9; p < 0.05 compared with baseline; CB1f/f vs CB1f/f;CaMKIIαCre, p < 0.05) (Fig. 1A). FPs reflect the concerted action of both glutamatergic and GABAergic synaptic transmission. To gauge the contribution of the glutamatergic system, isolated glutamatergic FPs were recorded in the presence of the GABAA receptor antagonist picrotoxin (50 μm) and the GABAB receptor antagonist CGP 35348 (200 μm). FPs were decreased by WIN in CB1f/f but not in CB1f/f;CaMKIIαCre littermates (FPs of CB1f/f: 73.1 ± 8.4%, n = 9, p < 0.05; FPs of CB1f/f;CaMKIIaCre: 105.3 ± 5.0%, n = 9, p > 0.05) (Fig. 1B). Furthermore, in whole-cell experiments, we studied the influence of WIN (5 μm) on isolated AMPA receptor-mediated glutamatergic currents (AMPA-EPSCs) by the additional application of the NMDA receptor antagonist AP-5 (50 μm). Although the CB1 agonist significantly reduced the AMPA-EPSC amplitude to 55.3 ± 6.4% (n = 8; p < 0.05) in slices of CB1f/f, it did not have any effect on the AMPA-EPSC amplitude in CB1f/f;CaMKIIαCre (Fig. 1C).
Figure 1.
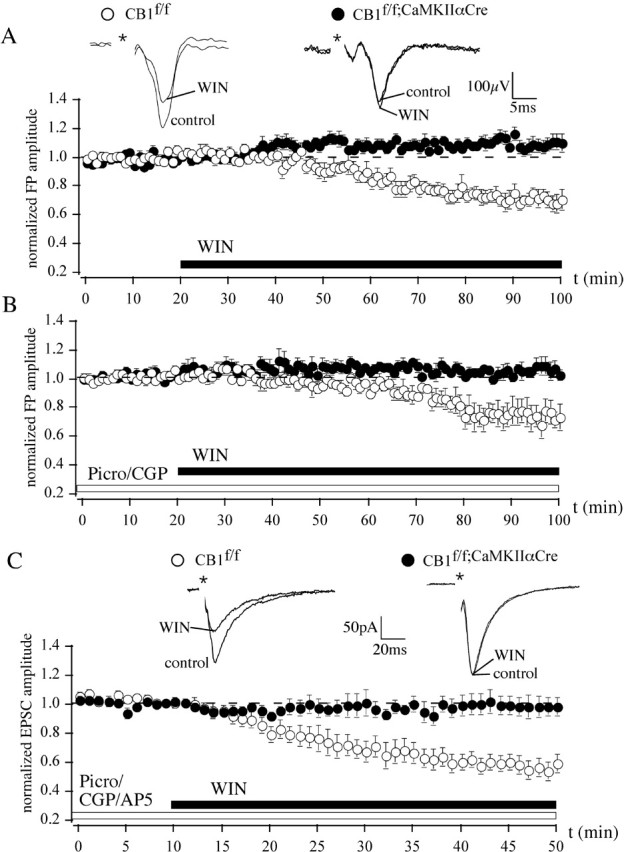
In the BLA, activation of CB1 does not affect FP and EPSC amplitudes of mice lacking CB1 expression in principal neurons. A, WIN (5 μm) reduces FP amplitudes in wild-type CB1f/f mice to 69.5 ± 5.2% of baseline (n = 9). This effect is completely abolished in CB1f/f;CaMKIIαCre mice (5 μm WIN; 107.4 ± 5.0%; n = 9). B, C, WIN significantly reduces isolated glutamatergic FPs (B) and AMPA-EPSCs (C) in CB1f/f mice to 73.1 ± 8.4% (n = 9) and to 55.3 ± 6.4% (n = 8) of baseline, respectively, but not in the CB1f/f;CaMKIIαCre mice. Representative traces are shown. Data are normalized to the respective baseline values (10 min of baseline). Asterisks represent stimulation artifacts. Picro, Picrotoxin; CGP, CGP 35348.
To exclude possible interactions between CB1 expressed in either principal neurons or inhibitory interneurons in the BLA, we tested the WIN-mediated inhibition of FPs and AMPA-EPSCs in BLA slices of mice lacking the expression of CB1 on GABAergic interneurons. In both (CB1f/f and CB1f/f;Dlx5/6-Cre) genotypes, WIN significantly depressed FP amplitudes (CB1f/f: 60.2 ± 10.3%, n = 7; CB1f/f;Dlx5/6-Cre: 44.0 ± 10.4%, n = 7; CB1f/f vs CB1f/f;Dlx5/6-Cre, p > 0.05) (Fig. 2A). Activation of CB1 by WIN depressed isolated AMPA-EPSCs in CB1f/f to 47.1 ± 10.0% (n = 8) and in CB1f/f;Dlx5/6-Cre to 44.2 ± 6.5% (n = 9; CB1f/f vs CB1f/f;Dlx5/6-Cre, p > 0.05) of baseline (Fig. 2B). These data indicate that CB1 receptors expressed on GABAergic interneurons do not participate in the WIN-mediated regulation of glutamatergic transmission in the BLA and thereby confirm that this effect is solely mediated by CB1 present in principal neurons.
Figure 2.
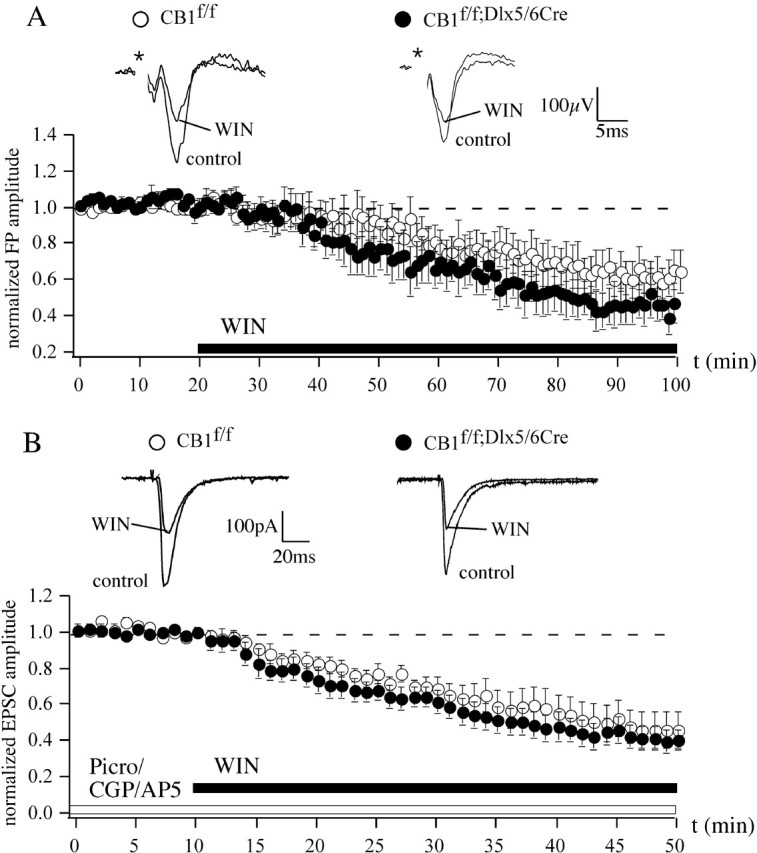
In the BLA, CB1 activation reduces FP and EPSC amplitudes of mice lacking CB1 expression in GABAergic interneurons. A, In CB1f/f and CB1f/f;Dlx5/6-Cre, WIN (5 μm) depresses FP amplitudes significantly (CB1f/f: 60.2 ± 10.3%, n = 7; CB1f/f;Dlx5/6-Cre: 44.0 ± 10.4%, n = 7). B, WIN reduces the amplitude of isolated AMPA-EPSCs to 47.1 ± 10.0% (n = 8) and to 44.2 ± 6.5% of baseline in CB1f/f and in CB1f/f;Dlx5/6-Cre, respectively (n = 9). Picro, Picrotoxin; CGP, CGP 35348.
In the CA1 region of the hippocampus, WIN reduced evoked field EPSPs (fEPSPs) to 49.6 ± 4.3% (n = 12) (Fig. 3A) in CB1f/f mice. However, in slices of CB1f/f;CaMKIIαCre mice, WIN produced a slight but significant increase in the fEPSP slope (109.9 ± 4.2%; n = 12; p < 0.05) (Fig. 3A). When testing isolated AMPA-EPSCs, we obtained similar results as in the BLA. In CB1f/f mice, WIN inhibited EPSC amplitude, whereas in the CB1f/f;CaMKIIαCre mice, EPSCs were not affected (EPSCs of CB1f/f: 52.7 ± 11.1%, n = 6; EPSCs of CB1f/f;CaMKIIαCre: 96.9 ± 8.2%, n = 6; CB1f/f vs CB1f/f;CaMKIIαCre, p < 0.05) (Fig. 3B). Because the effectiveness of WIN primarily depends on the solvent used, we also applied WIN dissolved in 0.1N HCl. In the CA1 of CB1f/f mice, WIN reduced fEPSPs to 64.1 ± 5.3% (n = 5; p < 0.05). In slices of CB1f/f;CaMKIIαCre mice, fEPSPs were not affected (99.1 ± 5.4%; n = 5; data not shown).
Figure 3.
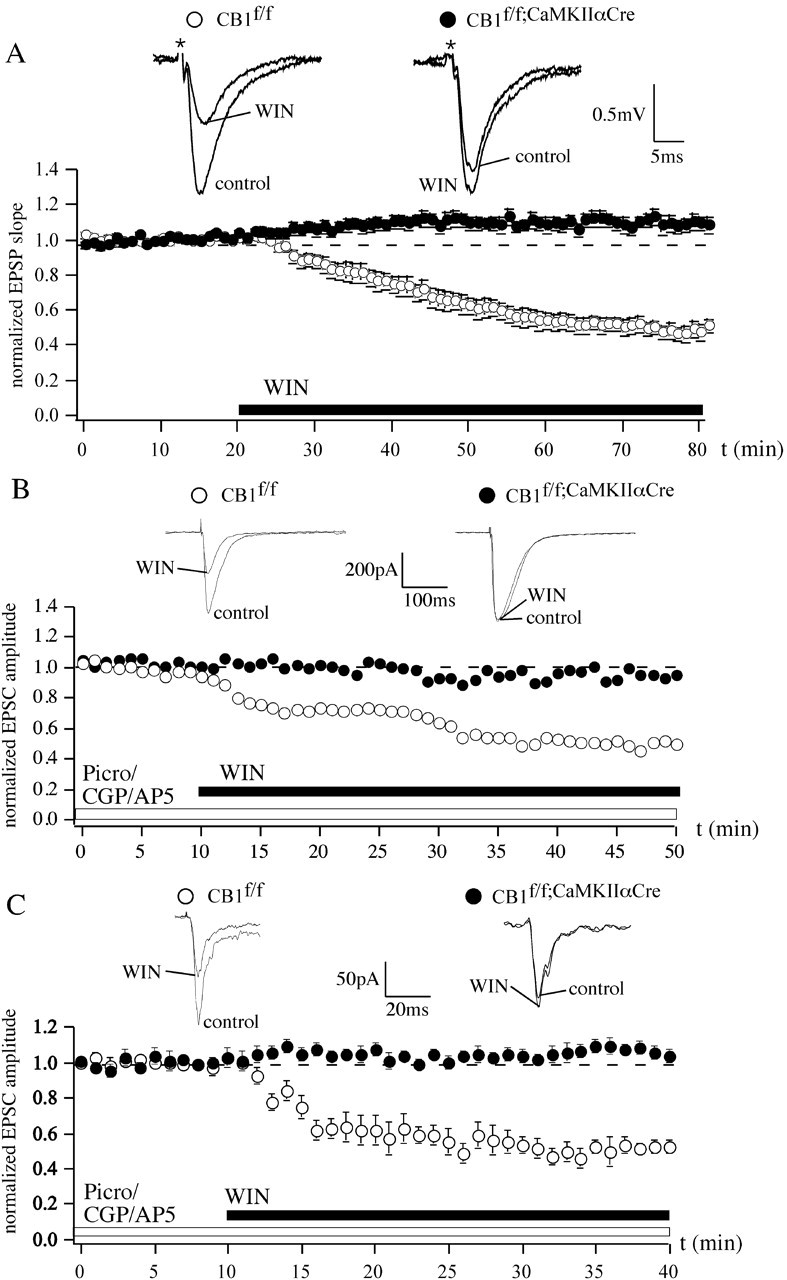
In the hippocampus and somatosensory cortex, CB1 on glutamatergic neurons is necessary for the WIN-induced inhibition of glutamatergic transmission. A, In the CA1 region of the hippocampus, EPSPs are reduced to 49.6 ± 4.3% (n = 12) after application of WIN (5 μm) in CB1f/f mice but not in slices of CB1f/f;CaMKIIαCre mice (109.9 ± 4.2%; n = 12). B, AMPA-EPSC amplitudes are decreased by WIN only in CB1f/f mice (EPSCs of CB1f/f: 52.7 ± 11.1%, n = 6; EPSCs of CB1f/f;CaMKIIαCre: 96.9 ± 8.2%, n = 6). C, In the somatosensory cortex of layer II neurons, WIN reduces the AMPA-EPSC amplitude in CB1f/f mice to 49.7 ± 5.0% (n = 8), whereas in CB1f/f;CaMKIIαCre mice, WIN has no effect (105.4 ± 4.3%; n = 8). Representative traces are shown. Picro, Picrotoxin; CGP, CGP 35348.
In the neocortex of mice lacking CB1 expression in principal neurons, WIN does not reduce glutamatergic transmission
We also tested the effect of CB1 activation on glutamatergic synaptic transmission in layer II neurons of the somatosensory cortex. In this region, WIN reduced the AMPA-EPSC amplitude in CB1f/f mice to 49.7 ± 5.0% (n = 8), whereas in CB1f/f;CaMKIIαCre mice, WIN had no effect (105.4 ± 4.3%; n = 8; p < 0.05) (Fig. 3C).
WIN does not modify postsynaptic effects of photolysis-uncaged l-glutamate
Results from a variety of cortical tissue preparations consistently indicate that cannabinoid-mediated modulation of neurotransmission is exerted at a presynaptic level (Schlicker and Kathmann, 2001). This mechanism has also been proposed for the reduction in excitatory synaptic neurotransmission (Misner and Sullivan, 1999; Auclair et al., 2000; Azad et al., 2003). To further strengthen these findings, we used photolysis to rapidly uncage l-glutamate locally at dendrites of cortical neurons (Dodt et al., 1999). This method has been shown to be sensitive enough to detect alterations of AMPA receptors and allows to closely mimic the synaptic activation of AMPA receptors in slices (Rammes et al., 2003). Thus, photostimulation is suitable for elucidating postsynaptic mechanisms. AMPA-EPSCs and AMPA receptor responses to focal glutamate were obtained alternately. WIN (5 μm) induced a clear reduction in evoked AMPA-EPSCs in slices of the BLA (69.9 ± 4.4%; n = 7) (Fig. 4A, filled circles) and the hippocampus (67.8 ± 6.0%; n = 7) (Fig. 4B, filled circles). In contrast, responses to focally uncaged l-glutamate were never reduced by WIN in either region. Thirty minutes after washin, responses were 105.1 ± 4.6 and 102.2 ± 2.6% of baseline in the BLA and hippocampus, respectively (n = 7) (Fig. 4). These results demonstrate that the CB1-dependent reduction in glutamatergic synaptic responses is not exerted by the modulation of the activity of postsynaptic glutamate receptors, thereby indicating that presynaptic CB1 receptors are responsible for this effect of WIN.
Figure 4.
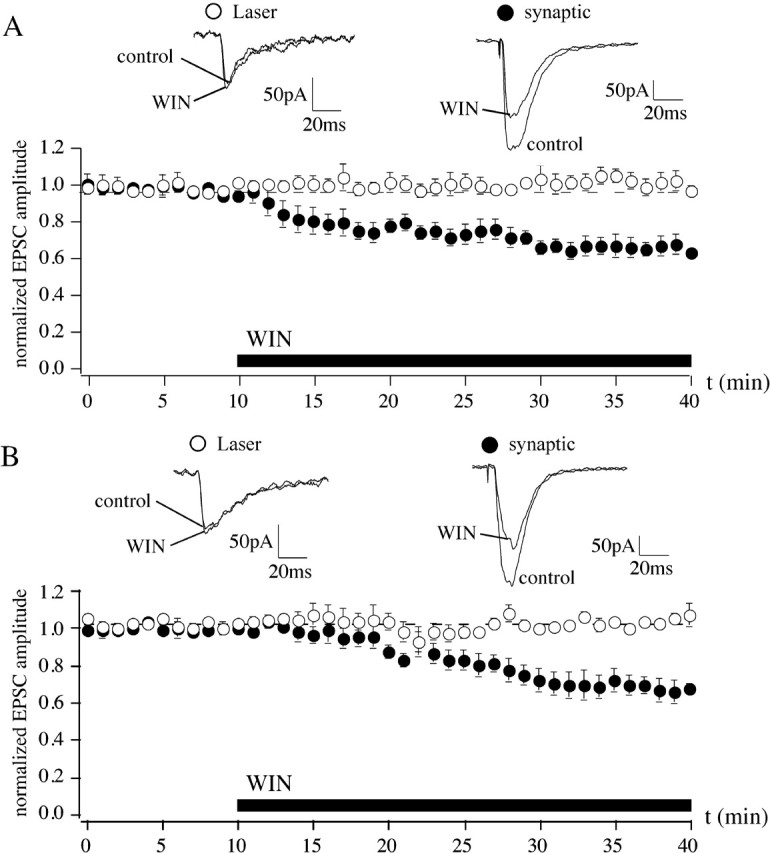
In neurons of limbic structures, cannabinoids reduce glutamatergic synaptic transmission exclusively through presynaptic mechanisms. A, Although the CB1 agonist WIN (5 μm) reduces evoked AMPA-EPSCs in the BLA (to 69.9 ± 6.0%; n = 7; filled circles), inward currents induced by the uncaging of glutamate (3–5 ms, 1 mW, 1 μm laser spot diameter; size of uncaging area, ∼10 μm) are not affected by WIN (105.1 ± 4.6%; n = 7; open circles). B, In the CA1 region of the hippocampus, only synaptic responses are decreased by WIN (AMPA-EPSC: 67.8 ± 6.0, n = 7, filled circles; laser-induced responses: 105.1 ± 4.6%, n = 7, open circles).
Discussion
The present findings provide for the first time clear evidence that WIN reduces excitatory responses directly via CB1 located on glutamatergic synapses in forebrain areas. In the hippocampus, the BLA, and the somatosensory cortex, we found that WIN was unable to reduce glutamatergic synaptic transmission in the absence of CB1 on principal neurons, thus demonstrating that these receptors are responsible for WIN-induced reduction in glutamatergic neurotransmission in the cortex. In cortical areas, CB1 is highly expressed in interneurons that contain GABA (Katona et al., 1999; Marsicano and Lutz, 1999; Freund et al., 2003), but evidence exists for its presence also in principal neurons (e.g., of the hippocampus) (Marsicano and Lutz, 1999). Thus, we generated a mouse line [CB1f/f;CaMKIIαCre (Marsicano et al., 2003)] in which CB1 is deleted in all principal neurons of the forebrain but maintains its expression in cortical GABAergic interneurons and in cerebellar neurons. In these animals, we were able to investigate the role of CB1 expressed on principal neurons in modulating excitatory synaptic transmission.
Previous studies demonstrating that low micromolar concentrations of WIN and anandamide inhibit glutamate release in the hippocampus of CB1−/− mice (Hajos et al., 2001; Hajos and Freund, 2002; Köfalvi et al., 2003) and stimulate [35S]GTPγS binding in the entire brain and in cerebellar membranes (Monory et al., 2002; Breivogel et al., 2004) of CB1−/− mice prompted the idea that the effect of cannabinoids on glutamatergic synaptic transmission could be mediated by a novel non-CB1 cannabinoid receptor. However, in the present study, we could not detect any effect of WIN on the amplitude of excitatory responses recorded in the hippocampus, the BLA, and the somatosensory cortex of CB1f/f;CaMKIIαCre mice. This discrepancy might be explained by the differences between the conventional deletion of the receptor in CB1−/− mice and the conditional mutation of CB1f/f;CαMKIIαCre mice. Conditional mutagenesis, using the Cre/LoxP or similar recombination systems, has been developed to overcome major drawbacks of conventional gene inactivation strategies, such as the lack of spatial and temporal selectivity. The aim is to limit the possibility of compensatory alterations caused by the long-lasting lack of the gene of interest throughout the entire prenatal and postnatal life of the animals (Le and Sauer, 2001; Morozov et al., 2003). In contrast, because the CaMKIIα gene is expressed by postmitotic neurons, Cre-mediated recombination under the control of its regulatory sequences in CB1f/f;CaMKIIαCre occurs only at late embryonic or even early postnatal stages (Casanova et al., 2001). It is therefore conceivable that a putative compensatory overexpression of the proposed novel non-CB1 cannabinoid receptor, able to mediate cannabinoid-dependent control of glutamatergic transmission (Hajos et al., 2001; Freund et al., 2003), might occur in CB1−/− mice but not in CB1f/f;CaMKIIαCre mice, in which the genetic manipulation is less invasive and much more limited in time and space. In this frame, a scenario emerges in which the expression of the putative non-CB1 cannabinoid receptor in glutamatergic neurons would be an exceptional event occurring as compensation of the long-lasting deletion of CB1 in conventional CB1−/− mice. Conversely, in CB1f/f;CaMKIIαCre mice, this compensatory overexpression does not appear to occur. Moreover, strain differences might be present in the compensatory expression of the putative non-CB1 cannabinoid receptor on cortical glutamatergic neurons. Indeed, Hajos et al. (2001) observed an effect of WIN on glutamatergic transmission of CB1−/− mice backcrossed on the CD1 background, whereas our previous results showed that, at least in the BLA, this effect of the cannabinoid drug is abolished in CB1−/− mice backcrossed into the C57BL/6N strain (Azad et al., 2003). This observation is in agreement with a differential expression of the putative non-CB1 cannabinoid receptor in different species and strains (Hoffman et al., 2005).
Furthermore, based on our results, we can also exclude a developmental or functional interaction between CB1 expressed on glutamatergic synapses with that expressed on GABAergic synapses, because in slices of CB1f/f;Dlx5/6-Cre mice, which are devoid of CB1 in GABAergic neurons, WIN decreases excitatory and AMPA receptor-mediated responses to the same extent as seen in the wild-type mice. In general, our results do not exclude the existence of a novel non-CB1 receptor per se, but rather provide strong evidence for the control of glutamatergic synaptic transmission by CB1 under physiological conditions.
Interestingly, in both BLA and CA1 slices of CB1f/f;CaMKIIαCre, WIN was able to slightly but significantly increase FPs, which reflect the concerted action of excitatory and inhibitory neurotransmission. These observations suggest that pharmacologically applied cannabinoids decrease both excitatory and inhibitory neurotransmission through CB1 receptors expressed in glutamatergic and GABAergic neurons, respectively. When CB1 is selectively deleted from principal neurons, the residual effect of the CB1 agonist is exerted on GABAergic neurotransmission, thereby leading to the increase in FPs. However, additional experiments are necessary to analyze the details of such dualistic effects of CB1 agonists.
The release of caged l-glutamate by focal photolysis clearly demonstrated that WIN acts on presynaptically localized CB1 to decrease glutamatergic synaptic transmission. We found a pronounced synaptic depression of AMPA-EPSCs that is uncorrelated with any reduction in glutamate sensitivity, even when the temporal and spatial specific photolytic release of glutamate induced responses that closely mimic synaptic currents (Rammes et al., 2003). This conclusion is consistent with anatomical and electrophysiological observations that CB1 presynaptically regulates the release of certain types of neurotransmitters from axon terminals (Freund et al., 2003).
In conclusion, using advanced genetic and electrophysiological tools, our study provides clear evidence that CB1 receptors, expressed on glutamatergic terminals of the BLA, the CA1 region of hippocampus, and the somatosensory cortex of mice, directly mediate the effects of WIN on glutamatergic transmission. These results reveal a novel function of CB1 receptors in the brain and open new possibilities to explain the effects of pharmacological cannabinoid administration on animal behavior.
Footnotes
*M.R.D. and S.C.A. contributed equally to this work.
This work was supported in part by Deutsche Forschungsgemeinschaft Grant BL 755/1-4 to B.L. We thank Christine Hilf for excellent technical assistance, Martina Reents and Tanja Orschmann for mouse genotyping, and Carola Hetzel for secretarial work. We also thank Günther Schütz and Emilio Casanova for providing the CB1f/f;CaMKIIαCre mice and John Rubenstein and Marc Ekker for providing the CB1f/f;Dlx5/6-Cre mice.
References
- Auclair N, Otani S, Soubrie P, Crepel F (2000). Cannabinoids modulate synaptic strength and plasticity at glutamatergic synapses of rat prefrontal cortex pyramidal neurons. J Neurophysiol 83:3287–3293. [DOI] [PubMed] [Google Scholar]
- Azad SC, Eder M, Marsicano G, Lutz B, Zieglgänsberger W, Rammes G (2003). Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learn Mem 10:116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breivogel CS, Walker JM, Huang SM, Roy MB, Childers SR (2004). Cannabinoid signaling in rat cerebellar granule cells: G-protein activation, inhibition of glutamate release and endogenous cannabinoids. Neuropharmacology 47:81–91. [DOI] [PubMed] [Google Scholar]
- Casanova E, Fehsenfeld S, Mantamadiotis T, Lemberger T, Greiner E, Stewart AF, Schutz G (2001). A CamKIIalpha iCre BAC allows brain-specific gene inactivation. Genesis 31:37–42. [DOI] [PubMed] [Google Scholar]
- Dodt H, Eder M, Frick A, Zieglgänsberger W (1999). Precisely localized LTD in the neocortex revealed by infrared-guided laser stimulation. Science 286:110–113. [DOI] [PubMed] [Google Scholar]
- Freiman I, Szabo B (2005). Cannabinoids depress excitatory neurotransmission between the subthalamic nucleus and the globus pallidus. Neuroscience 133:305–313. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83:1017–1066. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM (2002). Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5:446–451. [DOI] [PubMed] [Google Scholar]
- Hajos N, Freund TF (2002). Distinct cannabinoid sensitive receptors regulate hippocampal excitation and inhibition. Chem Phys Lipids 121:73–82. [DOI] [PubMed] [Google Scholar]
- Hajos N, Katona I, Naiem SS, MacKie K, Ledent C, Mody I, Freund TF (2000). Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci 12:3239–3249. [DOI] [PubMed] [Google Scholar]
- Hajos N, Ledent C, Freund TF (2001). Novel cannabinoid-sensitive receptor mediates inhibition of glutamatergic synaptic transmission in the hippocampus. Neuroscience 106:1–4. [DOI] [PubMed] [Google Scholar]
- Hermann H, Marsicano G, Lutz B (2002). Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience 109:451–460. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR (2000). Mechanisms of cannabinoid inhibition of GABAA synaptic transmission in the hippocampus. J Neurosci 20:2470–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AF, Riegel AC, Lupica CR (2003). Functional localization of cannabinoid receptors and endogenous cannabinoid production in distinct neuron populations of the hippocampus. Eur J Neurosci 18:524–534. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Macgill AM, Smith D, Oz M, Lupica CR (2005). Species and strain differences in the expression of a novel glutamate-modulating cannabinoid receptor in the rodent hippocampus. Eur J Neurosci 22:2387–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF (1999). Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19:4544–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, Freund TF (2001). Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci 21:9506–9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köfalvi A, Vizi ES, Ledent C, Sperlagh B (2003). Cannabinoids inhibit the release of [3H]glutamate from rodent hippocampal synaptosomes via a novel CB1 receptor-independent action. Eur J Neurosci 18:1973–1978. [DOI] [PubMed] [Google Scholar]
- Köfalvi A, Rodrigues RJ, Ledent C, Mackie K, Vizi ES, Cunha RA, Sperlagh B (2005). Involvement of cannabinoid receptors in the regulation of neurotransmitter release in the rodent striatum: a combined immunochemical and pharmacological analysis. J Neurosci 16:2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001). Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727. [DOI] [PubMed] [Google Scholar]
- Le Y, Sauer B (2001). Conditional gene knockout using Cre recombinase. Mol Biotechnol 17:269–275. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Lutz B (1999). Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse brain. Eur J Neurosci 11:4213–4225. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B (2002). The endogenous cannabinoid system controls extinction of aversive memories. Nature 418:530–534. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, van der Stelt M, Lopez-Rodriguez ML, Casanova E, Schutz G, Zieglgänsberger W, Di Marzo V, Behl C, Lutz B (2003). CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302:84–88. [DOI] [PubMed] [Google Scholar]
- Misner DL, Sullivan JM (1999). Mechanism of cannabinoid effects on long-term potentiation and depression in hippocampal CA1 neurons. J Neurosci 19:6795–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monory K, Tzavara ET, Lexime J, Ledent C, Parmentier M, Borsodi A, Hanoune J (2002). Novel, not adenylyl cyclase-coupled cannabinoid binding site in cerebellum of mice. Biochem Biophys Res Commun 292:231–235. [DOI] [PubMed] [Google Scholar]
- Morozov A, Kellendonk C, Simpson E, Tronche F (2003). Using conditional mutagenesis to study the brain. Biol Psychiatry 54:1125–1133. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M (2001). Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29:729–738. [DOI] [PubMed] [Google Scholar]
- Rammes G, Palmer M, Eder M, Dodt HU, Zieglgansberger W, Collingridge GL (2003). Activation of mGlu receptors induces LTD without affecting postsynaptic sensitivity of CA1 neurons in rat hippocampal slices. J Physiol (Lond) 546:455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegel AC, Lupica CR (2004). Independent presynaptic and postsynaptic mechanisms regulate endocannabinoid signaling at multiple synapses in the ventral tegmental area. J Neurosci 24:11070–11078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ (2001). Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci 21:109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ (2002). Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA 99:8384–8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JJ, Mackie K, Pickel VM (2001). Ultrastructural localization of the CB1 cannabinoid receptor in μ-opioid receptor patches of the rat caudate putamen nucleus. J Neurosci 21:823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierloh A, Eder M, Zieglgänsberger W, Dodt HU (2004). Effects of sensory deprivation on columnar organization of neuronal circuits in the rat barrel cortex. Eur J Neurosci 20:1118–1124. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Kathmann M (2001). Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci 22:565–572. [DOI] [PubMed] [Google Scholar]
- Simon W, Hapfelmeier G, Kochs E, Zieglgänsberger W, Rammes G (2001). Isoflurane blocks synaptic plasticity in the mouse hippocampus. Anesthesiology 94:1058–1065. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001). Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410:588–592. [DOI] [PubMed] [Google Scholar]