

Abstract
The activation of ligand-gated ion channels is thought to depend solely on the binding of chemical neurotransmitters. In this study, we demonstrate that kainate (KA) ionotropic glutamate receptors (iGluRs) require not only the neurotransmitter l-glutamate (l-Glu) but also external sodium and chloride ions for activation. Removal of external ions traps KA receptors (KARs) in a novel inactive state that binds l-Glu with picomolar affinity. Moreover, occupancy of KARs by l-Glu precludes external ion binding, demonstrating crosstalk between ligand- and ion-binding sites. AMPA iGluRs function normally in the absence of external ions, revealing that even closely related iGluR subfamilies operate by distinct gating mechanisms. This behavior is interchangeable via a single amino acid residue that operates as a molecular switch to confer AMPA receptor behavior onto KARs. Our findings identify a novel allosteric site that singles out KARs from all other ligand-gated ion channels.
Keywords: agonist, glutamate receptor, desensitization, gating, epilepsy, activation
Introduction
In the vertebrate brain, ligand-gated ion channels are an important class of signaling protein designed to respond to a specific chemical neurotransmitter such as acetylcholine (ACh) (Colquhoun and Sakmann, 1998), l-glutamate (l-Glu) (Erreger et al., 2004), glycine (Lynch, 2004), or GABA (Maconochie et al., 1994). Although neurotransmitter substances are numerous in number (Krnjevic, 1974), all ligand-gated ion channels are thought to undergo conformations into the activated state by harnessing the energy from neurotransmitter binding (Colquhoun, 1998). There are no exceptions to this rule, although it has been recognized for some time that basal ion-channel activity is regulated by other factors such as phosphorylation and, more recently, by interactions with scaffolding proteins. Previously, we have shown that external anions and cations regulate both the response amplitude and channel kinetics of kainate (KA) ionotropic glutamate receptors (iGluRs) (Bowie, 2002; Bowie and Lange, 2002). Specifically, the rate of channel closure (i.e., deactivation) is ion dependent, suggesting that the stability of the activated/open state of the receptor is regulated by external ions as well as neurotransmitter binding. As yet, the molecular basis of this effect is not understood, although closely related AMPA receptors (AMPARs) are insensitive to external anions and cations (Bowie, 2002; Bowie and Lange, 2002; Paternain et al., 2003).
Here, we tested two opposing mechanisms to account for the effect of external ions on KA receptors (KARs). Experiments in ion-free solutions reveal that external anions and cations do not simply modulate basal receptor activity but instead are an absolute requirement for activation. This observation demonstrates unequivocally that external ions are coactivators of KARs. Furthermore, we identify allosteric cooperativity between ligand- and ion-binding sites and show that KARs enter into a novel inactive state when external sodium and chloride ions are absent.
Materials and Methods
Cell culture.
tsA201 cells were transiently cotransfected with cDNA encoding wild-type (wt) or mutant GluR6 or GluR1 subunits and enhanced green fluorescent protein (eGFPS65T) as described previously (Bowie, 2002; Bowie and Lange, 2002). After transfection for 8–10 h (GluR6) or 12 h (GluR1), cells were washed and maintained in fresh medium. Electrophysiological recordings were performed 24–48 h later.
Mutagenesis.
Mutation of GluR6(Q) was performed using the Stratagene (La Jolla, CA) Quickchange II XL site-directed mutagenesis kit. Mutant cDNAs were amplified, purified, and initially identified by restriction digest and confirmed by automated DNA sequencing of the entire GluR6 coding region (McGill University and Genome Quebec Innovation Center, Montreal, Québec, Canada).
Electrophysiology.
Experiments were performed on outside-out patches, and agonist solutions were applied using a piezo-stack-driven perfusion system (Bowie, 2002; Bowie and Lange, 2002; Bowie et al., 2003). Solution exchange (10–90% rise time, 25–50 μs) was determined at the end of each experiment by measuring the liquid junction current. Recordings were performed with an Axopatch 200B amplifier (Molecular Devices, Palo Alto, CA) using borosilicate glass pipettes (4–6 MΩ) coated with dental wax. Current records were filtered at 10 kHz and digitized at 50–100 kHz, and series resistances (7–12 MΩ) were compensated by 95%. The reference electrode was connected to the bath via a 3 m KCl agar bridge. Data acquisition was performed using pClamp9 (Molecular Devices) and illustrated using Origin 7 (Microcal, Northampton, MA). All experiments were performed at room temperature.
Solutions.
External solutions contained the following: 5 mm HEPES, 0.1 mm CaCl2, 0.1 mm MgCl2, and 2% phenol red, to which 1–405 mm NaCl was added as required. For solutions containing 150 mm external NaCl or less, the osmotic pressure was adjusted to 290 mOsm using sucrose. For solutions with higher NaCl (>150 mm), the osmotic pressure was adjusted to 760 mOsm. pH was adjusted to 7.3 using 5N NaOH, with the exception of experiments presented in Figure 1 in which different cations were compared. In this case, pH was adjusted with the corresponding hydroxide solution (e.g., LiOH for LiCl). The internal solution contained the following (in mm): 115 NaCl, 10 NaF, 5 HEPES, 5 Na4BAPTA, 0.5 CaCl2, 1 MgCl2, and 10 Na2ATP, pH was adjusted to 7.3 with 5N NaOH, and the osmotic pressure was adjusted with sucrose to correspond with external solutions.
Figure 1.
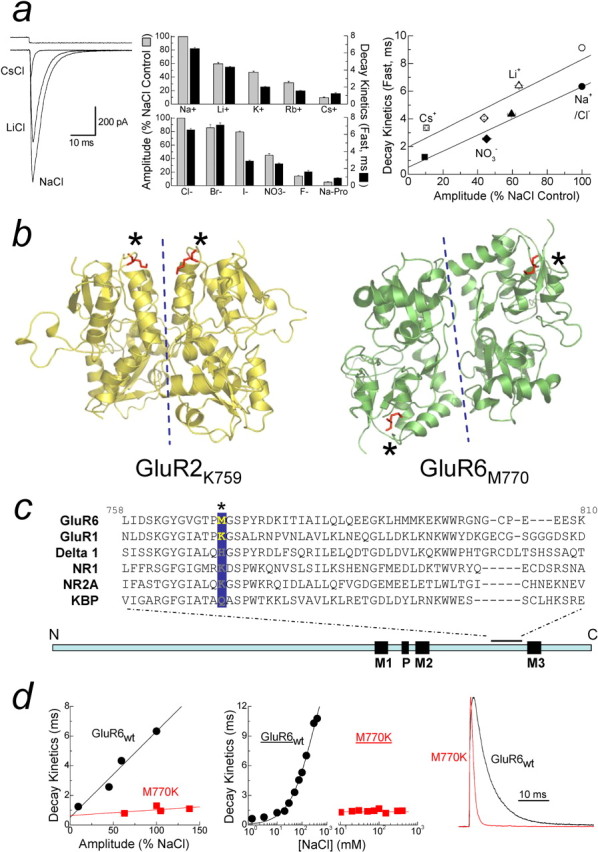
External anions and cations regulate GluR6 KARs. a, Left, Membrane currents elicited by 10 mm l-Glu in 150 mm Na+, Li+, or Cs+, with top trace showing solution exchange (patch 040419p2). Middle, Summary bar graph of external ion effects on GluR6 amplitude (gray bars) and decay kinetics (black bars). Right, Comparison between the amplitude and decay kinetics of GluR6 in different ion concentrations (150 mm, filled symbols; 405 mm, open symbols). Solid lines represent linear regression fits of the data at each ion concentration. b, Crystal dimer structures drawn using Pymol show GluR2 (Protein Data Bank number 1FTJ) and GluR6 (Protein Data Bank number 1S7Y), with M770 and K759 in red and marked by asterisks. Dotted lines show dimer interface. c, Sequence alignment of several iGluRs at the extracellular M2–M3 linker region, with the GluR6 M770 position highlighted in yellow. d, GluR6M770K is unaffected by ion type [left, 150 mm Na+ (Cl−), Li+, NO3−, and Cs+] or concentration (middle, 1–405 mm NaCl) and has faster decay kinetics than GluR6wt (right, 150 mm NaCl).
In experiments shown in Figures 2 and 3, solutions lacking external NaCl contained 100 μm each of CaCl2 and MgCl2 to improve patch stability, sucrose to maintain the osmotic pressure at 290 mOsm, and 5 mm Tris or 5 mm ammonium bicarbonate (NH5CO3) as pH buffers. With Tris, the pH was adjusted to 7.4 using 5N HCl, whereas the pH of NH5CO3-containing solutions was maintained by a gas mixture of 95%O2/5%CO2. For agonist solutions, the free acid of l-glutamate was dissolved in NaCl-free solution, and the pH was adjusted using 2.5 m Tris (for Tris buffer) or 2 m NH5CO3 (for NH5CO3-buffered solution).
Figure 2.

GluR6 KARs have an absolute requirement for external ions. a, Superimposed family of membrane currents evoked by 1 mm l-Glu acting on GluR6wt (patch 050311p1), GluR1wt (patch 050321p2), and GluR6M770K (patch 050405p2) receptors in solutions lacking external NaCl (range, −100 to +110 mV, 15 mV increments). b, Averaged current–voltage plots in 0 mm (filled circles), 10 mm (open squares), and 150 mm (open triangles) NaCl for each iGluR tested.
Figure 3.

External ions are not a prerequisite for agonist binding to KARs. a, Typical experimental traces (Vh of +50 mV) in which the effect of preincubating GluR6 KARs in NaCl-lacking solutions but containing 1 mm l-Glu was tested (patch 050711p1). Left, GluR6 receptors elicited robust responses to 1 mm l-Glu when preincubated in 150 mm external NaCl. Middle, In contrast, GluR6 receptors were unresponsive to NaCl when pretreated in 0 mm NaCl and 1 mm l-Glu. Right, The response was fully recovered during the addition of 150 mm NaCl to external solutions. b, Same experiment as in a using 40 mm AA instead of l-Glu (patch 050818p1). c, Experimental traces from b superimposed for comparison.
Analysis.
Concentration–response curve to external NaCl (see Fig. 1d, middle) was fit with the following equation: τNaCl = τmax/1 + (EC50/[NaCl])N, where τNaCl represents the observed decay kinetics at any concentration of NaCl, τmax is the slowest time constant for the fast decay component, which assumes that NaCl has a saturable effect, EC50 is the concentration of NaCl that elicits half-maximal decay kinetics, and N is the slope. Inhibition curves shown in Figure 4 were fit with a single- and double-binding site isotherm of the following forms: (for single) IGlu = Imax/1 + ([Glu]/IC50)N and (for double) IGlu = Imax(High)/1 + ([Glu]/IC50(High))N + (1 − Imax(High))/1 + ([Glu]/IC50(Low))N, where Imax is the response to 10 mm l-Glu in the absence of preapplied l-Glu, IC50 is the concentration of l-Glu that elicits half-maximal inhibition, N is the slope, and the low-affinity component (ILow) of inhibition by l-Glu is 1 − Imax(High). Data in all experiments are expressed as mean ± SEM from at least five patches.
Figure 4.

External ions regulate occupancy of a novel, high-affinity inactive state. a, Typical experiment showing the onset of and recovery from inhibition of peak GluR6 responses by l-Glu. b, Individual traces shown in a superimposed to show response profile in detail (patch 030613p1). c, Family of inhibition curves to l-Glu in 5 mm (filled triangles), 10 mm (open triangles), 150 mm (filled circles), and 405 mm (open circles) NaCl. Solid lines are fits to single- or double-binding site isotherms. The IC50 of the low-affinity state was concentration independent (1.5 μm), whereas the Hill coefficient, nh, was 0.8 in 5 mm NaCl and 1.1 in 10–405 mm NaCl. The IC50 for the high-affinity inactive state was concentration dependent, being 70 nm (nh = 0.5) in 10 mm NaCl and 10 pm (nh = 1.0) in 5 mm NaCl. d, Inhibition curve observed in 5 mm external NaCl in more detail showing contribution of high- and low-affinity states (dotted curves). e, Effect of NaCl on the fractional occupancy of high- and low-affinity states. Dotted lines represent fit extrapolations.
Results
External anions and cations modulate KAR amplitude and channel kinetics
Figure 1a shows typical effects of external monovalent ions on electrophysiological responses mediated by recombinant GluR6 KARs. Replacement of external Na+ with an equimolar equivalent of either Li+ or Cs+ elicits a reduction in peak response amplitude as well as acceleration in decay (i.e., desensitization) kinetics (Fig. 1a, left). Similar findings were observed with an extended series of monovalent cations in which the degree of modulation was dependent on ion species (Fig. 1a, middle). Interestingly, substitution of external Cl− with equimolar concentrations of other anions had a comparable effect to cation replacement (Fig. 1a, middle). Together, these data suggest that the chemical nature of the external solution and not its ionic strength regulates KAR gating behavior (Bowie, 2002).
The conventional explanation for these observations as described for other voltage- and ligand-gated ion channels (Yellen, 1997) is that external ions regulate the basal gating behavior of GluR6 KARs but are not an absolute requirement. In this case, the effect of external Cs+ or propionate ions is to reduce channel activity, whereas it is increased by external Na+ or Cl−. An alternative is that GluR6 KARs exhibit an absolute requirement for external anions and cations; that is, external ions act as coactivators of KARs. In this case, external Na+ or Cl− ions are more effective in stabilizing KARs in the open state compared with Cs+ or propionate ions. We will use the term “coactivator” throughout to include two possible mechanisms: (1) that ions affect KARs simply by binding or (2) ion binding causes conformational changes in the receptor, which affects function. To test whether external ions are coactivators of KARs, we compared GluR6 responses in 150 and 405 mm external ion solutions (Fig. 1a, right). Increasing external ion concentration to 405 mm prolonged GluR6 decay kinetics with each external anion (Cl−, NO3−) or cation (Na+, Li+, Cs+) tested (Fig. 1a, right). Interestingly, a parallel shift in the relationship between response amplitude and decay kinetics was observed with the rank order of potency for each ion unchanged (Fig. 1a, right). This observation is inconsistent with a mechanism whereby external ions modulate the basal gating properties of KARs. In this case, Cs+ ions would be expected to further accelerate decay kinetics at higher concentrations (i.e., 405 mm). However, the slowing of decay kinetics with all ion species tested supports the hypothesis that external ions are coactivators of KARs.
A single amino acid residue delineates between channel kinetics and response amplitude
It is interesting that KARs are the only iGluR whose response amplitude and decay kinetics is regulated by external anions and cations (Bowie, 2002; Bowie and Lange, 2002) despite structural (Mayer, 2005b) and functional (Dingledine et al., 1999) similarities with other family members, particularly AMPARs. In view of this, we further hypothesized that the gating mechanism of other iGluRs, such as AMPARs, do not have an absolute requirement for external ions; that is, agonist-induced conformational changes into the open state can still occur in the absence of external ions. Furthermore, given their considerable homology, we reasoned that it should be possible to interconvert the gating behavior of KA and AMPARs. In support of this, it has been shown that replacement of methionine-770 (M770) in GluR6 with its equivalent lysine (K752 for GluR1) residue in AMPARs (Fig. 1b,c) blocks ion modulation of the KAR response amplitude (Paternain et al., 2003). Interestingly, recent x-ray diffraction studies of GluR6 KAR (Mayer, 2005a) and GluR2 AMPAR (Armstrong and Gouaux, 2000) dimers places these residues in different locales of the quaternary structure (Fig. 1b, red labels and asterisks). An important caveat, however, is that another group has suggested a different dimer organization closer to that of GluR2 AMPARs (Nanao et al., 2005). Although the precise nature of the dimer interface awaits additional study, the structure reported by Mayer (2005) is consistent with KARs possessing a unique ion-binding site(s) that regulates the peak response amplitude. Given the concomitant effect of external ions on amplitude and decay kinetics (Bowie, 2002), we were therefore interested in testing whether M770K also affects GluR6 decay kinetics.
Figure 1d summarizes a series of experiments in which desensitization kinetics of GluR6M770K and GluR6wt were compared. In contrast to GluR6wt, the decay kinetics of GluR6M770K were almost identical for all external anions (i.e., Cl− or NO3−) and cations (i.e., Na+, Li+, and Cs+) tested (Fig. 1d, left), suggesting that inclusion of a positively charged lysine at the 770 site is sufficient in abolishing ion-sensitive effects on channel kinetics. Unexpectedly, external anions and cations continued to regulate the peak response amplitude of GluR6M770K, contrary to Paternain et al. (2003). In this case, the rank order of potency was different between GluR6M770K [NO3− > Na+ (or Cl−) ∼ Li+ > Cs+] and GluR6wt [Na+ (or Cl−) > Li+ > NO3− > Cs+] (Fig. 1d, left). GluR6M770K mutant also blocked the effect of changing the external Na+ and Cl− ion concentration on desensitization kinetics (Bowie, 2002; Bowie and Lange, 2002) (Fig. 1d, middle), further supporting the pivotal role of M770 in controlling KAR gating behavior. Interestingly, in 150 mm external NaCl, the decay kinetics of GluR6M770K were several-fold faster than GluR6wt (Fig. 1d, right) or GluR1 AMPARs (data not shown), suggesting that amino acid residues other than the M/K site may be involved in endowing KARs with ion-dependent gating. As reported by others (Paternain et al., 2003), mutant AMPA receptors containing a Met residue instead of Lys express poorly, and, therefore, we were unable to examine the ion sensitivity of GluR1K752M.
External anions and cations are an absolute requirement for KAR activation
To directly test whether KAR activation has an absolute requirement for external ions, we recorded GluR6 responses in the absence of external NaCl at a range of membrane potentials (−100 to +110 mV, 15 mV increments) (Fig. 2). To do this, experiments were performed using the free acid of l-glutamate and either Tris or bicarbonate buffers to maintain an external pH of 7.3 (see Materials and Methods). For comparison, we repeated experiments on GluR1 AMPARs and the KAR mutant GluR6M770K. GluR6wt were entirely unresponsive in the absence of external NaCl at all membrane potentials tested (Fig. 2, left), consistent with our hypothesis that external ions are coactivators of KARs. Figure 2 (bottom, left) shows the current–voltage (I–V) relationships observed in 0 mm NaCl (filled circles) compared with I–V plots in 10 mm (open squares) and 150 mm (open triangles) external NaCl. In contrast, GluR1 AMPARs were fully responsive in the absence of external NaCl (Fig. 2, middle), suggesting that external ions are not an absolute requirement for the gating behavior of this iGluR subfamily. The membrane current observed represents the outward movement of permeating ions (i.e., Na+) from the internal solution of the patch pipette. Interestingly, GluR6M770K was also responsive in solutions lacking external NaCl (Fig. 2, right), supporting the pivotal role of the M/K site in determining KAR gating behavior. Moreover, this observation eliminates the possibility that the functional effects observed in low ionic strength solutions are not attributable to denaturation of the quaternary structure of the intact KARs.
Crosstalk between agonist- and ion-binding sites
Our observations cannot be explained by the failure of agonist binding to KARs in NaCl-free solutions. In support of this, GluR6 KARs failed to respond to both NaCl and l-Glu when preincubated in solutions lacking external ions but containing l-Glu (Fig. 3a). This demonstrates that agonist binding (and subsequent receptor desensitization) can occur in the absence of external ions. If agonist binding had not occurred, GluR6 receptors would be expected to respond to the application of 150 mm NaCl (Fig. 3a, middle). Unexpectedly, however, we did not observe an equilibrium current typically associated with desensitized GluR6 receptors (Bowie and Lange, 2002; Bowie et al., 2003). We therefore hypothesized that agonist binding precludes the ability of external ions to bind. Because it was difficult to resolve equilibrium responses with l-Glu, we repeated the experiments using l-aminoadipate (AA) (40 mm), which elicits larger equilibrium responses (Fig. 3b, left). As with l-Glu, application of NaCl failed to elicit an equilibrium response after pretreatment with AA (Fig. 3b, right, c). Together, we have shown that, although external ions are required for KAR functionality, GluR6 receptors are able to bind agonists in solutions lacking external ions. Furthermore, conformational changes elicited by agonist binding prevent subsequent ion binding. To explain the failure of GluR6 receptors to respond in solutions lacking external ions, we show below that their removal accumulates KARs in a novel inactive state with high agonist affinity.
Identification of a novel inactive state with picomolar agonist affinity
Figure 4, a and b, shows a typical experiment in which the occupancy of the desensitized states was determined from inhibition of 10 mm l-Glu responses after incubation in l-Glu (0.1–50 μm). Similar experiments were also performed in 5, 10, 75, 150, and 405 mm external NaCl. A family of curves observed in different concentrations of NaCl were then fit with a single- or double-binding site model of inhibition as shown in Figure 4c. The inhibition of l-Glu responses in 150 and 405 mm NaCl (Fig. 4c, open and filled circles) were best fit with a single binding site isotherm estimating the IC50 to be 0.49 ± 0.04 and 0.56 ± 0.09 μm, respectively, in good agreement with previous studies of GluR6 (Wilding and Huettner, 1997; Paternain et al., 1998). At lower NaCl levels, inhibition plots were biphasic, revealing a high affinity, NaCl-dependent binding site with IC50 values of 50 ± 20 pm in 5 mm NaCl and 0.8 ± 0.6 nm in 10 mm NaCl (Fig. 4c,d). Extending our observations, <5 mm NaCl was not possible because membrane currents were small in amplitude, making measurement and analysis difficult. Moreover, patch stability was compromised in low ionic strength solutions. However, extrapolated fits of occupancy of the high- and low-affinity states (Fig. 4e) revealed that GluR6 receptors accumulate into this high-affinity inactive state as external ions are lowered. This finding explains the failure of KARs to gate in the absence of external NaCl (Fig. 2a, left).
Discussion
Classically, it has been thought that ligand-gated ion channels depend solely on chemical neurotransmitters for activation. This is exemplified by work on nicotinic ACh receptors in which channel lifetime is solely dependent on the nature of the ligand (Colquhoun and Sakmann, 1985). Here we show that external ions are coactivators of KARs, suggesting, unexpectedly, that channel lifetime is not only controlled by the ligand but also by ions. As yet, it is not clear whether external ions control KARs simply by binding or whether an additional conformational change is required in much the same way that glycine acts as a coagonist at NMDARs (Kleckner and Dingledine, 1988). It is also unclear whether M770 represents the ion-binding site(s) or a residue critical in the transduction process. Intuitively, it would be expected that anions and cations bind to discrete sites to satisfy electrostatic principles; however, other mechanisms, such as the establishment of a dipole, are possible (Bowie, 2002). Differentiating between these mechanisms can only be resolved through additional structure–function analysis of KARs.
How ligand and external ions determine the stability of the open state is not clear, but two possibilities may be considered. First, if external ions have a lower affinity (i.e., shorter residency time) than ligands, the time the KAR ion channel remains in the open state will be dependent on the rate of ion unbinding. This mechanism, however, fails to account for the slowing of channel kinetics at elevated ion levels (Bowie, 2002). Moreover, because agonist occupancy prevents ion rebinding (Fig. 3), KARs would also be expected to accumulate into the novel inactive state during prolonged agonist application. In this case, a decline in the equilibrium response would occur as ions unbind. However, we observe a well maintained equilibrium response, arguing against this mechanism (Fig. 3b). Alternatively, external ions may stabilize the ligand-binding cleft, which would account for the slowing of KAR deactivation and desensitization at high ion concentrations as well as the sustained equilibrium response. However, this mechanism seems inconsistent with the observation that l-Glu activation curves are weakly ion dependent (Bowie, 2002). Clearly, if KAR gating is to be elucidated, an important step will be to resolve the interplay between ligand and external ions.
Although the M770 residue is restricted to GluR6 and GluR7 subunits, equivalent residues in other KAR subunits also confer sensitivity to external Cs+ block (Paternain et al., 2003). In contrast, all AMPA and NMDA iGluRs have a conserved lysine residue in the M/K position (Fig. 1c), suggesting that only members of the KAR family are coactivated by external ions. Recent crystallographic work has indicated that full and partial agonists elicit different degrees of closure in the ligand-binding core of GluR6 KARs (Mayer, 2005a; Nanao et al., 2005). From work on AMPAR crystal structures, the degree of domain closure has been shown to be directly correlated to agonist efficacy (Jin et al., 2003). Our results, however, suggest that agonist behavior is not governed solely by conformations in the agonist-binding domain but that occupancy of a novel ion-binding site(s) must also be considered.
Finally, KARs are therapeutic targets in the treatment of several neurological diseases, including neuropathic pain (Palecek et al., 2004) and epilepsy (Smolders et al., 2002). Although some success has been achieved in developing selective KAR antagonists, a recurrent obstacle is that most, if not all, strategies rely on exploiting differences between the agonist-binding domain of iGluR subtypes (Bleakman et al., 2002). This is particularly problematic for AMPARs and KARs whose agonist-binding domains have overlapping pharmacology (Dingledine et al., 1999), which is expected given their significant structural homology (Mayer and Armstrong, 2004; Mayer, 2005a; Nanao et al., 2005) (Fig. 1). In principle, drug selectivity is best achieved when a unique pharmacological target can be identified. Our findings point to a mechanism that may be exploited to design drugs with a high selectivity for KARs that are useful in the treatment of neurological conditions.
Footnotes
This work was supported by operating grants from the National Institutes of Health and Canadian Institutes of Health Research (D.B.). A.Y.C.W. was supported by the David T. W. Lin fellowship, and A.-M.L.F. was supported by a predoctoral fellowship from the Fond de Recherche en Santé du Québec. D.B. is the recipient of a Canada Research Chair award. We thank Drs. J. Johnson, B. Collier, E. Cooper, and especially P. S. Swain for discussions and careful reading of this manuscript.
References
- Armstrong N, Gouaux E (2000). Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28:165–181. [DOI] [PubMed] [Google Scholar]
- Bleakman D, Gates MR, Ogden AM, Mackowiak M (2002). Kainate receptor agonists, antagonists and allosteric modulators. Curr Pharm Des 8:873–885. [DOI] [PubMed] [Google Scholar]
- Bowie D (2002). External anions and cations distinguish between AMPA and kainate receptor gating mechanisms. J Physiol (Lond) 539:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Lange GD (2002). Functional stoichiometry of glutamate receptor desensitization. J Neurosci 22:3392–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Garcia EP, Marshall J, Traynelis SF, Lange GD (2003). Allosteric regulation and spatial distribution of kainate receptors bound to ancillary proteins. J Physiol (Lond) 547:373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D (1998). Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol 125:923–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B (1985). Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol (Lond) 369:501–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B (1998). From muscle endplate to brain synapses: a short history of synapses and agonist-activated ion channels. Neuron 20:381–387. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999). The glutamate receptor ion channels. Pharmacol Rev 51:7–61. [PubMed] [Google Scholar]
- Erreger K, Chen PE, Wyllie DJ, Traynelis SF (2004). Glutamate receptor gating. Crit Rev Neurobiol 16:187–224. [DOI] [PubMed] [Google Scholar]
- Jin R, Banke TG, Mayer ML, Traynelis SF, Gouaux E (2003). Structural basis for partial agonist action at ionotropic glutamate receptors. Nat Neurosci 6:803–810. [DOI] [PubMed] [Google Scholar]
- Kleckner NW, Dingledine R (1988). Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 241:835–837. [DOI] [PubMed] [Google Scholar]
- Krnjevic K (1974). Chemical nature of synaptic transmission in vertebrates. Physiol Rev 54:418–540. [Google Scholar]
- Lynch JW (2004). Molecular structure and function of the glycine receptor chloride channel. Physiol Rev 84:1051–1095. [DOI] [PubMed] [Google Scholar]
- Maconochie DJ, Zempel JM, Steinbach JH (1994). How quickly can GABAA receptors open? Neuron 12:61–71. [DOI] [PubMed] [Google Scholar]
- Mayer ML (2005a). Crystal structures of the GluR5 and GluR6 ligand binding cores: molecular mechanisms underlying kainate receptor selectivity. Neuron 45:539–552. [DOI] [PubMed] [Google Scholar]
- Mayer ML (2005b). Glutamate receptor ion channels. Curr Opin Neurobiol 15:282–288. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Armstrong N (2004). Structure and function of glutamate receptor ion channels. Annu Rev Physiol 66:161–181. [DOI] [PubMed] [Google Scholar]
- Nanao MH, Green T, Stern-Bach Y, Heinemann SF, Choe S (2005). Structure of the kainate receptor subunit GluR6 agonist-binding domain complexed with domoic acid. Proc Natl Acad Sci USA 102:1708–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek J, Neugebauer V, Carlton SM, Iyengar S, Willis WD (2004). The effect of a kainate GluR5 receptor antagonist on responses of spinothalamic tract neurons in a model of peripheral neuropathy in primates. Pain 111:151–161. [DOI] [PubMed] [Google Scholar]
- Paternain AV, Rodriguez-Moreno A, Villarroel A, Lerma J (1998). Activation and desensitization properties of native and recombinant kainate receptors. Neuropharmacology 37:1249–1259. [DOI] [PubMed] [Google Scholar]
- Paternain AV, Cohen A, Stern-Bach Y, Lerma J (2003). A role for extracellular Na+ in the channel gating of native and recombinant kainate receptors. J Neurosci 23:8641–8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolders I, Bortolotto ZA, Clarke VR, Warre R, Khan GM, O’Neill MJ, Ornstein PL, Bleakman D, Ogden A, Weiss B, Stables JP, Ho KH, Ebinger G, Collingridge GL, Lodge D, Michotte Y (2002). Antagonists of GLU(K5)-containing kainate receptors prevent pilocarpine-induced limbic seizures. Nat Neurosci 5:796–804. [DOI] [PubMed] [Google Scholar]
- Wilding TJ, Huettner JE (1997). Activation and desensitization of hippocampal kainate receptors. J Neurosci 17:2713–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G (1997). Single channel seeks permeant ion for brief but intimate relationship. J Gen Physiol 110:83–85. [DOI] [PMC free article] [PubMed] [Google Scholar]