

Abstract
Genomic recombination requires cutting, processing, and rejoining of DNA by endonucleases, polymerases, and ligases, among other factors. We have proposed that DNA recombination mechanisms may contribute to long-term memory (LTM) formation in the brain. Our previous studies with the nucleoside analog 1-β-d-arabinofuranosylcytosine triphosphate (ara-CTP), a known inhibitor of DNA ligases and polymerases, showed that this agent blocked consolidation of conditioned taste aversion without interfering with short-term memory (STM). However, because polymerases and ligases are also essential for DNA replication, it remained unclear whether the effects of this drug on consolidation were attributable to interference with DNA recombination or neurogenesis. Here we show, using C57BL/6 mice, that ara-CTP specifically blocks consolidation but not STM of context fear conditioning, a task previously shown not to require neurogenesis. The effects of a single systemic dose of cytosine arabinoside (ara-C) on LTM were evident as early as 6 h after training. In addition, although ara-C impaired LTM, it did not impair general locomotor activity nor induce brain neurotoxicity. Importantly, hippocampal, but not insular cortex, infusions of ara-C also blocked consolidation of context fear conditioning. Separate studies revealed that context fear conditioning training significantly induced nonhomologous DNA end joining activity indicative of DNA ligase-dependent recombination in hippocampal, but not cortex, protein extracts. Finally, unlike inhibition of protein synthesis, systemic ara-C did not block reconsolidation of context fear conditioning. Our results support the idea that DNA recombination is a process specific to consolidation that is not involved in the postreactivation editing of memories.
Keywords: DNA recombination, DNA ligase, protein synthesis, fear conditioning, consolidation, reconsolidation, nonhomologous DNA end joining
Introduction
Memory consolidation requires the regulation of gene expression and function. As with protein synthesis inhibitors (Flexner et al., 1963; Schafe and LeDoux, 2000; Hernández et al., 2002; Scharf et al., 2002; Naghdi et al., 2003; Agnihotri et al., 2004; Quevedo et al., 2004; Santini et al., 2004), transcription inhibitors block long-term memory (LTM), while not interfering with short-term memory (STM) (Squire and Barondes, 1970; Ohi, 1977; Nguyen et al., 1994; Bailey et al., 1996; Frey et al., 1996; Pedreira et al., 1996; Wustenberg et al., 1998; Igaz et al., 2002; Calixto et al., 2003). Specific transcription factors (Alberini et al., 1994; Kida et al., 2002; Malkani et al., 2004) have been implicated in LTM. Additionally, modulation of chromatin structure by histone modifications and the regulation of translation may also have a role in consolidation (Korzus, 2003; Si et al., 2003; Levenson et al., 2004; Levenson and Sweatt, 2005).
We suggested that a mechanism of gene rearrangement, similar to variable/diversity/joining or V(D)J [V(D)J] recombination in the immune system, might also play a part in LTM (Peña de Ortiz and Arshavsky, 2001; Peña de Ortiz et al., 2004). V(D)J recombination involves the successive cutting, processing, and eventual religation, via DNA ligase IV-dependent nonhomologous DNA end joining (NHEJ), of gene segments encoding the variable portions of antigen receptors (Lieber et al., 2004; Schatz, 2004). Our previous studies showed that the expression of the gene encoding terminal deoxynucleotidyl transferase (TdT), a template-independent DNA polymerase involved in V(D)J recombination, is upregulated in brain neurons by learning and is required for the beneficial cognitive effects of enriched experiences (Peña de Ortiz et al., 2003). Finally, we demonstrated that the nucleoside analog 1-β-d-arabinofuranosylcytosine triphosphate (ara-CTP) inhibited brain NHEJ and impaired LTM, but not STM, of conditioned taste aversion (CTA) in rats when infused intracereventricularly before learning (Wang et al., 2003). The amnesic effects of ara-CTP were also observed with its precursor agent, cytosine arabinoside (ara-C; 1-β-d-arabinofuranosylcytosine).
By acting as an inhibitor of DNA ligases (Lamballe et al., 1988; Zittoun et al., 1989, 1991) and DNA polymerases (Gandhi et al., 1997; Han et al., 2000; Wills et al., 2000), ara-CTP is able to block the processes of DNA recombination and also of DNA replication. The fact that the effects of ara-CTP on CTA consolidation were observed as early as 4 h after training suggested that its effects were related to blockade of DNA recombination, rather than replication and neurogenesis (Wang et al., 2003). Here, we questioned whether recombination processes sensitive to ara-CTP are involved in the consolidation of context fear conditioning, a task that is independent of neurogenesis (Shors et al., 2002; Pham et al., 2005). The data show that ara-C specifically impaired consolidation, without interfering with reconsolidation of context fear memory. We also show that context fear conditioning elicits a rapid and transient activation of hippocampal NHEJ activity. The studies suggest that DNA recombination is one of the initial processes used by the brain for the storage of information and is not part of the molecular mechanisms activated after memory retrieval.
Materials and Methods
Subjects and drug treatments
The procedures were approved by the Institutional Animal Care and Use Committee of the Río Piedras Campus of the University of Puerto Rico in compliance with National Institutes of Health (NIH) guidelines for the care and use of laboratory animals (Department of Health and Human Services–NIH publication number 86-23). Experiments used male C57BL/6 mice [8 weeks of age (systemic studies) or 12 weeks of age (intracerebral studies)] obtained from Harlan Sprague Dawley (Indianapolis, IN). Food and water were available at all times, and the animals were kept on a 12 h light/dark cycle. The mice received intraperitoneal injections of 1000 mg/kg body weight ara-C (Sigma, St. Louis, MO), 75 mg/kg body weight anisomycin (ANI) (Sigma), or vehicle (PBS). The dose of ara-C was determined in a dose–response study. ANI was dissolved in PBS, and the pH was adjusted to 7.4 with 1N HCl. As in our previous studies (Wang et al., 2003), the dose of ara-C used in the intracerebral studies was 1 mm.
Context fear conditioning
Apparatus.
Our conditioning chamber (30 × 20 × 18 cm) was made of transparent Plexiglas on two sides and stainless steel on the other two sides. Each of the steel sides had a speaker and a 24 V light. The chamber had a 36-bar-insulated shock grid floor made of stainless steel rods (Coulbourn Instruments, Allentown, PA). The system included a white-noise generator to provide background noise (70 dB). The floor was removable and was cleaned with 75% ethanol after each subject was trained, re-exposed, or tested. Each bar (1.5 cm in diameter) was connected through a harness to a programmable Master Shocker (model 82404SS; Coulbourn Instruments) that delivered scrambled footshocks to each of the bars in the grid floor. A mini camera (Silent Witness Enterprises, Surrey, British Columbia, Canada) installed directly behind one of the two Plexiglas sides of the conditioning chamber was connected via a processor to a computer system for videotaping and scoring of freezing using the Xpress SDK software, which is a PCI bus mastering wavelet video compression/decompression and capture board (Integral Technologies, Indianapolis, IN).
Consolidation studies.
Context conditioning was done essentially as described previously (Bourtchouladze et al., 1994; Kida et al., 2002). Mice were given injections of ara-C or vehicle 1 h before training, 1 h after training, or both 1 h before training and 1 h before LTM testing. Animals were then taken from their home cages and placed into traveling cages in which they were transported into a dimly lit room and placed individually into the conditioning chamber. The mice were placed inside the conditioning chamber and allowed to explore for 2 min (habituation). Animals then received three consecutive footshocks (0.75 mA, 2 s duration each) that were each separated by a 1 min time period. After an additional 30 s, the animals were returned to their traveling cages and then to their home cages.
Reconsolidation studies.
The protocols were based on those described by Kida et al. (2002). On day 1, mice were subjected to fear conditioning training as above, but without receiving any injections. On day 2, mice were divided into two groups: re-exposure (reconsolidation) and no re-exposure. The re-exposure groups received ara-C or vehicle injections intraperitoneally 1 h before a 90 s re-exposure to the conditioning context. Animals in the no re-exposure groups received ara-C or vehicle (intraperitoneally) in their home cages. On day 3, all mice were tested for LTM in the conditioned context as detailed below. For the ANI experiments, both the drug and vehicle were injected 30 min before memory reactivation on day 2.
Testing.
Different animals were used to assess memory at each time point. We tested the animals by placing them back into the conditioning chamber for 5 min and measuring their freezing response. Memory was assessed by measuring freezing behavior. An animal was determined to be freezing when it adopted a motionless posture, refraining from all but respiratory movement. Freezing behavior, calculated as the percentage of freezing, was quantified in a blind manner by observing the videotape of mice behavior during the conditioning and memory tests. The images recorded during training and testing were divided in one frame per second. For an animal to be scored as freezing, it had to remain motionless for the entire 1 s observation. Finally, freezing behavior was expressed as a percentage of frames spent freezing.
Analysis of open-field behavior
Apparatus.
The activity chamber was made of transparent acrylic material and consisted of an arena of 10 × 10 × 16 inches and a base plate of 21 square inches. It had a sensor ring that sensed in two dimensions (X and Y), and the same ring was used for floor plane and rearing. All three measures can be performed simultaneously. Each link has a microprocessor on board to control scanning, calculate the coordinate sets for each of the rings, and report to the host computer.
Locomotor activity measurements.
Locomotor activity was monitored individually over a period of 60 min by a Tru Scan Photobeam Activity System (Coulbourn Instruments). Spontaneous ambulation and rearing behaviors were evaluated by continuous automated counting of photobeam interruptions. All counts were automatically totaled and recorded at 10 min intervals during 60 min. Several behavioral measures were determined, including the total number of movements, the number of vertical and horizontal movements, the number of center entries, and the time spent in the center of the arena, as well as measures relating to stereotypic behavior.
Histology
Cell density analysis.
Mice were killed after their last behavioral test. The brains were obtained and frozen on dry ice. Sagittal sections (14 μm) were stained with thionin as described by us previously (Peña de Ortiz et al., 2000). The sections were examined with a light microscope, and photomicrographs were obtained with a digital camera. Estimates of the number of neurons in hippocampal subfields, amygdala nuclei, and cerebellar Purkinje and molecular cell layers were obtained using Image J for stereological counting and determination of cell density (number of cells/area in square millimeters).
TdT-mediated dUTP end labeling.
Frozen brains were used to obtain sagittal sections with a thickness of 10 μm. TdT-mediated dUTP end labeling (TUNEL) assays were performed using the NeuroTACS II system (R & D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Sections were incubated with TdT and biotin-labeled dUTP. Sections used as reaction-positive controls were preincubated with TACS-Nuclease. DNA end-labeling was detected by incubation with streptavidin-conjugated horseradish peroxidase and diaminobenzidene. Counterstaining of cell nuclei was performed with hematoxylin, which generated a blue color, whereas TUNEL-positive nuclei appeared as brown. The sections were examined with a light microscope, and photomicrographs were obtained with a digital camera.
Surgery, cannulations, and intracerebral microinfusions
Mice were handled for several days before undergoing surgery. For surgery, the animals were anesthetized with sodium pentobarbital (60 mg/kg, i.p.) and placed in a stereotaxic apparatus, with the nose set to interaural zero. After a scalp incision was made, lambda and bregma were located and the skull was leveled. For the intrahippocampal infusions, bilateral guide cannulas (23 gauge) were implanted 1.0 mm above the target regions. The following coordinates were for CA3: anteroposterior (AP), −2.0 mm; mediolateral (ML), ±2.2 mm from bregma; dorsoventral (DV), −1.1 mm from the skull surface. For insular cortex infusions, bilateral guide cannulas (23 gauge) were implanted 1.0 mm above the target regions. The following coordinates were used: AP, 1.3 mm; ML, ± 3.5 mm from bregma; DV, −1.4 mm from the skull surface. The implanted cannulas were fixed on the skull with small screws and dental cement. A stainless steel wire stylet (33 gauge) was then inserted into the guide cannula to prevent leakage of the CSF, as well as to prevent obstruction of the cannula. After surgery, animals were allowed to rest for 3–5 d before training. To assess the effectiveness of the infusing pump system and to get the animals used to receiving intracerebral infusions, each animal was subjected to bilateral infusions (2 min at 0.25–0.5 μl/min) of 0.9% saline on the day before training. Infusions were done by inserting 33 gauge stainless steel internal injectors into the guide cannulas so that they extended 1.5 mm beyond the tip of the guide cannulas. After infusion, the injectors were removed, the stylets were replaced, and the animals were returned to their cages until the time of training 1 h later.
Brain dissection and protein extraction
C57BL/6 mice (8 weeks of age) were trained in the contextual fear conditioning chamber, as described above, but without receiving any drug or vehicle treatments. Animals were killed immediately, 10 min, 30 min, or 1 h after training. An untrained (naive) group was used as a control. The brains were obtained immediately after the animals were killed, chilled on dry ice, and used to dissect dorsal hippocampi and cortices. Dissected tissue was submerged in 1× PBS. Hippocampus and cortex samples were collected and separated in pools, each containing tissue from three mice per each time point. The experiment was conducted three times, ending with n = 3 pools, each representing nine animals per condition. Pooled tissue was stored at −80°C until used for protein extraction. Protein extracts were prepared as described by us previously (Ren and Peña de Ortiz, 2002; Wang et al., 2003). Tissues were homogenized in extraction buffer [30 mm HEPES/KOH, pH 7.9, 0.5 m KCl, 5 mm MgCl2,1 mm EDTA, 2 mm dithiothreitol (DTT), 20% glycerol, 1 mm phenylmethylsulfonyl fluoride (PMSF), and 1 μg/ml each of leupeptin and aprotinin] and incubated for 1 h in ice. The extract was centrifuged at 14,000 rpm for 1 h at 4°C. The supernatant was then dialyzed for 5 h in dialysis buffer (30 mm HEPES/KOH, pH 7.9, 50 mm KCl, 2 mm EDTA, 5 mm MgCl2, 1 mm DTT, 10% glycerol, 1 mm PMSF, and 1 μg/ml each of leupeptin and aprotinin). Dialyzed fractions were centrifuged at 14,000 rpm for 30 min at 4°C. Protein extracts were stored at −80°C until used. The protein concentration of the extract was determined by the Bradford method as detailed by us previously (Ren and Peña de Ortiz, 2002; Wang et al., 2003; Santini et al., 2004).
Substrate DNA and NHEJ assays
DNA substrates were also prepared as described by Ren and Peña de Ortiz (2002). To generate double-stranded DNA substrates with free 5′ and 3′ cohesive ends, pBluescript plasmid was linearized using the EcoRI restriction enzyme (Promega, Madison, WI). An NHEJ reaction mixture (total volume, 50 μl) contained a final concentration of 50 μg of protein extract, NHEJ buffer (30 mm HEPES/KOH, pH 7.9, 50 mm KCl, 2 mm EDTA, 5 mm MgCl2, 1 mm DTT, 10% glycerol), 1 mm ATP, 10 μg of BSA, and 20 ng of EcoRI-generated double-stranded DNA end substrates. Mixed reactants were incubated at room temperature for 1.25 h. The negative control consisted of heat-inactivating the protein extract at 100°C for 2 h before incubation with DNA substrates. Next, protein–DNA mixtures were heated at 70°C to terminate the reactions, followed by a 30 min incubation in 10% SDS and 5 μl of 1.0 mg/ml proteinase K (Stratagene, La Jolla, CA) at 37°C. DNA substrates and recombinant products were purified by phenol/chloroform extraction and ethanol precipitation. DNA from a single reaction was loaded onto a 1% agarose gel and electrophoresed for 85 min in 1× TAE buffer (40 mm Tris-acetate and 2 mm of EDTA, pH 8.5). Separated DNAs in the gels were transferred onto nylon membrane (GeneScreen; PerkinElmer Life Sciences, Wellesley, MA) following standard blotting procedures (Ren and Peña de Ortiz, 2002). The EcoRI-generated DNA substrate was labeled with (α-32P)dCTP by random priming (RediPrime TM II kit; Amersham Biosciences, Piscataway, NJ). Blotted membranes were subjected to Southern hybridization with the labeled DNA probes as described by us previously (Ren and Peña de Ortiz, 2002; Wang et al., 2003). Humid membranes were wrapped in plastic (Saran Wrap; SC Johnson, Sydney, Australia) to avoid drying and were then exposed to PhosphorImager screens for 24 h. Images were obtained in a PhosphorImager (Molecular Imager FX; Bio-Rad, Hercules, CA) and analyzed using the PhosphorImager Quantity-One Program (Bio-Rad). To normalize the phosphorimaging optical density (OD; counts per square millimeter) data obtained for different membranes exposed at different times, the following formula was used: (measured OD − minimum OD on membrane)/(maximum OD on membrane + minimum OD on membrane). Next, the generation of recombinant DNA products (dimers) was normalized against the OD measurements obtained for the substrate (monomer) to adjust for possible differences in the amount of substrate DNA used for each reaction. The normalized substrate value was obtained as follows: recombinant dimer OD/monomer OD. Each pool of hippocampal or cortex protein extracts (prepared with tissue from three animals; see above) was subjected to NHEJ assays three times. Averaged data were obtained from the triplicate NHEJ assays.
Statistical analysis
For all experiments, we assumed statistical significance at p < 0.05. For analysis of acquisition data, differences in mean and SEM between groups with respect to trials and treatment were analyzed using repeated-measures two-way ANOVA. For each memory time point, the differences between treatment and vehicle were analyzed using Student’s t tests. The data obtained from histological analyses were also subjected to Student’s t tests. Finally, the results of our NHEJ assays were analyzed using two-way ANOVA and Bonferroni’s post-testing.
Results
Systemic ara-C does not impair acquisition, STM, or general locomotor activity
The experimental design used initially in our studies is depicted in Figure 1(top). Male C57BL/6 mice were given intraperitoneal injections of ara-C or vehicle 1 h before training. The mice were then subjected to context fear conditioning, a STM test 1 h after training, and a behavioral toxicity test 24 h after training. For context fear conditioning, the mice were placed inside a conditioning chamber [conditioned stimulus (CS)] before receiving three consecutive footshocks (unconditioned stimuli). As seen in Figure 1 (bottom), ara-C had no effects on acquisition of fear conditioning, measured as the progressive enhancement of freezing behavior during a 30 s after-shock period. Repeated-measures two-way ANOVA showed no effect by the treatment (F(1,72) = 0.4402; p > 0.5), whereas a significant effect was found by the training (F(3,72) = 63.98; ***p < 0.0001). These results indicate that both groups (n = 12–14 each) were similarly capable of learning the task. Likewise, the STM test revealed no differences in the mean percentage of freezing of the animals from the vehicle (56.96 ± 3.597) or ara-C (54.07 ± 5.182) groups in terms of their ability to recall learned fear to the CS 1 h after training (Student’s t test; t(24) = 0.4441; p > 0.6).
Figure 1.
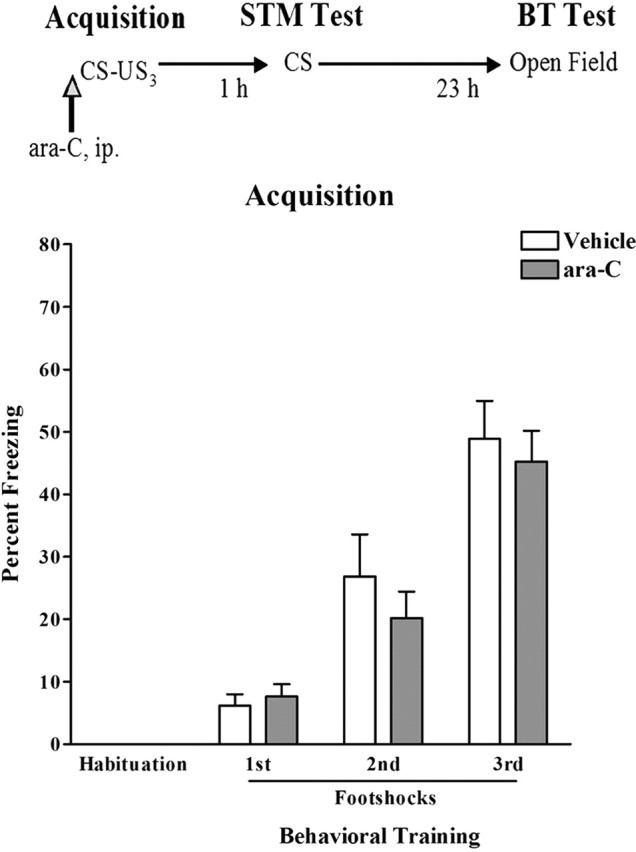
Pretraining ara-C does not affect acquisition or STM of context fear conditioning. Top, Schematic diagram depicting the experimental design. Mice received ara-C or vehicle intraperitoneally 1 h before training (vertical arrow). STM was assessed 1 h after training (STM test). The open-field behavior and general locomotor activity of the animals [behavioral toxicity (BT) test] was assessed 23 h later. US, Unconditioned stimulus. Bottom, Bar graph depicting the freezing responses (y-axis) during training observed for animals treated with vehicle (open bars) or ara-C (filled bars) (n = 12–14 in each group). Data are presented for the period of habituation to the conditioning chamber and for the three 30 s periods after each footshock. ara-C had no effects on acquisition of context fear conditioning, measured as the progressive enhancement of freezing behavior during a 30 s after-shock period (repeated-measures two-way ANOVA; treatment factor, p > 0.5; training factor, p < 0.0001). Error bars indicate SEM.
We then used the same set of animals to examine several measures of locomotor activity within an open field at 24 h after training (Fig. 1, top). We considered it important for clear interpretation of our next consolidation experiments to determine whether ara-C caused decreases in locomotor activity 24 h after training, because this time point would be used by us to examine LTM measured as freezing behavior within the conditioning cage. The obtained data were analyzed using Student’s t tests. ara-C-treated mice did not show differences, compared with vehicle-treated controls, in the number of total movements (vehicle, 284.5 ± 5.789 vs ara-C, 282.9 ± 7.050; t(24) = 0.74; p > 0.4), the number of vertical movements (rearing; vehicle, 194.8 ± 20.50 vs ara-C, 185.4 ± 13.48; t(24) = 0.86; p > 0.3), the amount of time (seconds) spent in the center of the open arena (vehicle, 246.9 ± 18.53 vs ara-C, 291.7 ± 35.51; t(24) = 0.71; p > 0.4), or the number of stereotypic movements (vehicle, 230.6 ± 7.29 vs ara-C, 234.8 ± 10.49; t(24) = 1.47; p > 0.1). The fact that ara-C had no effect on any of the behavioral parameters measured in an open field 24 h after training suggested to us that, at the dose used in our study, ara-C did not cause brain toxicity that could be reflected by altered general behavior in the animals. Specifically, the lack of effects on the time spent in the center and in the number of rearing movements suggests that the drug has no effect on exploratory behavior or the unconditioned fear to open spaces. In addition, because there were no differences in the number of total movements between the groups, we could conclude that ara-C had no effect on general locomotor activity 24 h after training and thus went on to test whether the drug would affect LTM of context fear conditioning as measured by the freezing responses of treated and trained animals.
Systemic ara-C is an effective blocker of both early and late memory consolidation
For our next set of experiments, we decided to test the effects of the drug on both the early (tested at 6 h after training) and late (tested at 24 h after training) stages of consolidation of context fear memory. Data from our experiments were analyzed using Student’s t tests. The results showed that ara-C-treated animals displayed significantly less freezing responses to the conditioning context than controls at both 6 h (vehicle, 62.05 ± 5.038 vs ara-C, 44.63 ± 3.373; t(24) = 2.87; **p < 0.01; n = 12) and 24 h (vehicle, 70.85 ± 5.066 vs ara-C, 37.69 ± 5.826; t(24) = 4.30; ***p < 0.0005; n = 12) after training. Thus, pretraining ara-C systemic injections interfered with LTM tested at 6 h as well as 24 h after conditioning.
The sensitivity of consolidation to ara-C is restricted to the time of conditioning
To determine whether ara-C-sensitive consolidation processes are restricted to the time of context fear conditioning, we trained mice in the task and gave them systemic injections of ara-C or vehicle (n = 10 mice per group) 1 h after training. Animals were then tested for LTM at the 24 h post-training time point. The data indicated that, unlike in the pretraining injection experiment, both the ara-C- and vehicle-treated mice displayed similar levels of conditioned freezing to the training context during the LTM test (vehicle, 73.13 ± 5.734 vs ara-C, 68.90 ± 4.325; t(18) = 0.5897; p > 0.5). Thus, for LTM of context fear conditioning to be established, DNA recombination processes inhibited by ara-C must be active at the time of learning.
The effects of ara-C are not related to state-dependent learning
One possible explanation for our findings, which we did not address in our previous studies on CTA (Wang et al., 2003), is whether the effects of ara-C on LTM are related to the phenomenon of state-dependent learning. State-dependent learning is evident when animals are only capable of retrieving previously acquired information if they are experiencing the same sensory, physiological, and chemical states that were present at the time of learning (Shulz et al., 2000). To elucidate whether the observed effects of ara-C on LTM were related to state-dependent learning, we used an additional set of mice that received intraperitoneal ara-C injections both 1 h before training and 1 h before LTM testing, which was done 24 h after conditioning. Like the results obtained when ara-C was administered only before conditioning, the results showed that treated animals displayed significantly less freezing responses to the conditioning context than controls at 24 h after training (vehicle, 65.44 ± 5.280 vs ara-C, 33.14 ± 5.559; t(24) = 4.214; ***p < 0.0005; n = 12). Because LTM was impaired in this experiment, we concluded that state dependency could not explain the amnesic effects of ara-C.
The effects of ara-C are not attributable to neurotoxicity
Together with the fact that acute exposure to ara-C does not affect open-field behavior at 24 h after training (see above), the lack of effect of post-training ara-C injections on conditioned freezing responses measured 24 h after training support the notion that the amnesic effects of the drug are unrelated to cellular toxicity. To further strengthen this conclusion, we performed stereological analysis of thionine-stained cells and examined cellular density (number of cells per square millimeter) within the hippocampus (dentate gyrus, CA3, CA1), amygdala, and cerebellum of the mice tested at 6 and 24 h after training. Representative stained dentate gyri from vehicle- and ara-C-treated mice are shown in Figure 2A (top and bottom, respectively). The results of our cellular density analysis of hippocampal pyramidal and granule cells indicated no significant differences between the groups (vehicle, 733.3 ± 104.4 vs ara-C, 729.7 ± 101.3; t(13) = 0.024; p > 0.9; n = 7–8). No group differences in cell density were observed within the amygdalar complex (vehicle, 1197 ± 49.97 vs ara-C, 1240 ± 63.43; t(13) = 0.522; p > 0.6; n = 7–8) or the cerebellum (vehicle, 898.7 ± 208.3 vs ara-C, 944.2 ± 174.7; t(13) = 0.169; p > 0.9; n = 7–8). In addition, TUNEL assays did not detect apoptotic cells in sagittal mouse brain sections of animals treated with ara-C (Fig. 2B) or vehicle. The nuclease-treated, TUNEL-positive control is shown in Figure 2C.
Figure 2.
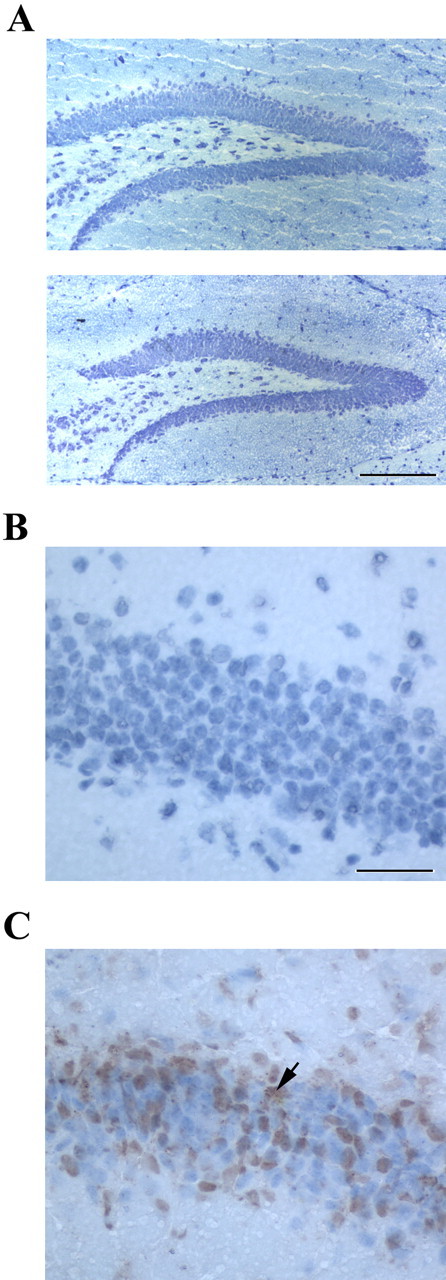
Acute systemic exposure to ara-C does not result in brain neurotoxicity. The brains of animals tested at 6 and 24 h after training were obtained and subjected to histopathological analysis focusing in the hippocampus, amygdala, and cerebellum. A, Representative photomicrographs (10×; scale bar, 0.10 mm) obtained from dentate gyrus Nissl staining examination are shown for vehicle-treated (top) and ara-C-treated (bottom) mice. B, No evidence of apoptosis was observed in brain sections, including the dentate gyrus (40×; scale bar, 0.4 mm), of ara-C-treated or of vehicle-treated (not shown) mice. C, Dentate gyrus granule cells (40×) showing TUNEL staining after pretreatment with TACS-nuclease (positive control). The arrow depicts a positively stained nucleus within the granule cell layer of the dentate gyrus.
Intrahippocampal infusions of ara-C specifically interfere with consolidation of context fear conditioning
Several studies have emphasized the role of the hippocampus (Phillips and LeDoux, 1992; Maren and Fanselow, 1995; Richmond et al., 1999; Yin et al., 2002; Lee and Kesner, 2004) in context fear conditioning. Thus, we decided to determine whether we could replicate the systemic effects of ara-C on LTM, by infusing the drug directly into this brain structure. Animals were subjected to surgical cannulations directed to the CA3 hippocampal subregion. Animals received ara-C [1 mm, as in the study by Wang et al. (2003)] or vehicle intracerebral infusions 1 h before conditioning. Our results showed that animals receiving CA3 ara-C infusions displayed significantly less freezing responses to the conditioning context than vehicle controls (vehicle, 47.31 ± 6.695 vs ara-C, 26.55 ± 5.312; t(26) = 2.46; *p < 0.05; n = 13–15). Next, to evaluate the brain regional specificity of the observed effects of ara-C, we examined the effects of the drug infused into the insular cortex on consolidation of context fear conditioning. We chose the insular cortex as a negative control site because evidence exists suggesting that this brain region is not involved in context fear conditioning (Brunzell and Kim, 2001). Accordingly, unlike the effects of CA3 ara-C infusions, infusion of the drug into the insular cortex had no effect on consolidation of context fear conditioning as measured by percentage of freezing (vehicle, 43.03 ± 8.356 vs ara-C, 47.12 ± 5.869; t(20) = 0.401; p > 0.6; n = 11). Together, the data from this set of experiments suggest that the role of DNA recombination in LTM of context fear conditioning is specific to brain regions shown to be important in this task.
Context fear conditioning induces hippocampal NHEJ activity
The results presented so far demonstrated that ara-C can inhibit consolidation of contextual fear conditioning. However, it remained unclear whether training in this task induces DNA ligase IV-dependent recombination processes, such as NHEJ. Previous studies demonstrated that the adult rat brain maintains an active NHEJ machinery (Ren and Peña de Ortiz, 2002). In addition, we showed that ara-CTP is a potent inhibitor of NHEJ (Wang et al., 2003). Based on these previous findings and those reported here, we set out to determine whether context fear conditioning training induces enhanced NHEJ activity in hippocampal protein extracts. Figure 3A shows representative results of NHEJ assays performed with hippocampal protein samples prepared from mice killed at different times after training, as well as from a control naive group. The results showed that hippocampal protein extracts prepared from mice killed at 10 and 60 min after training generated higher levels of recombinant dimer (rD) and trimer (rT) NHEJ products than protein extracts from naive controls or animals killed immediately after training. Figure 3B shows the results of the OD analysis for the generation of rD NHEJ products by protein extracts [normalized against the DNA substrate (S)] prepared from the dorsal hippocampus and cortex. Two-way ANOVA of the OD data confirmed a significant effect of training (F(4,20) = 6.765; **p < 0.005) and of brain region (F(1,20) = 47.76; ***p < 0.0001). Multiple-comparisons analysis indicated significantly higher generation of rD NHEJ products by hippocampal compared with cortex extracts at the 10 and 60 min post-training time points (***p < 0.001 each). The high hippocampal NHEJ activity levels observed 60 min after training do not contradict the absence of LTM effects when ara-C was injected 1 h after training (Fig. 2C) because this drug is a precursor that requires cellular conversion into its active form, ara-CTP. Previous studies have shown that the conversion rate of ara-C into ara-CTP is from 60 to 90 min (Jamieson et al., 1990; Hiddemann et al., 1992). Thus, we can conclude that hippocampal but not cortex NHEJ activity is enhanced shortly after fear conditioning. These results are the first to show that NHEJ is a learning-regulated biochemical process.
Figure 3.
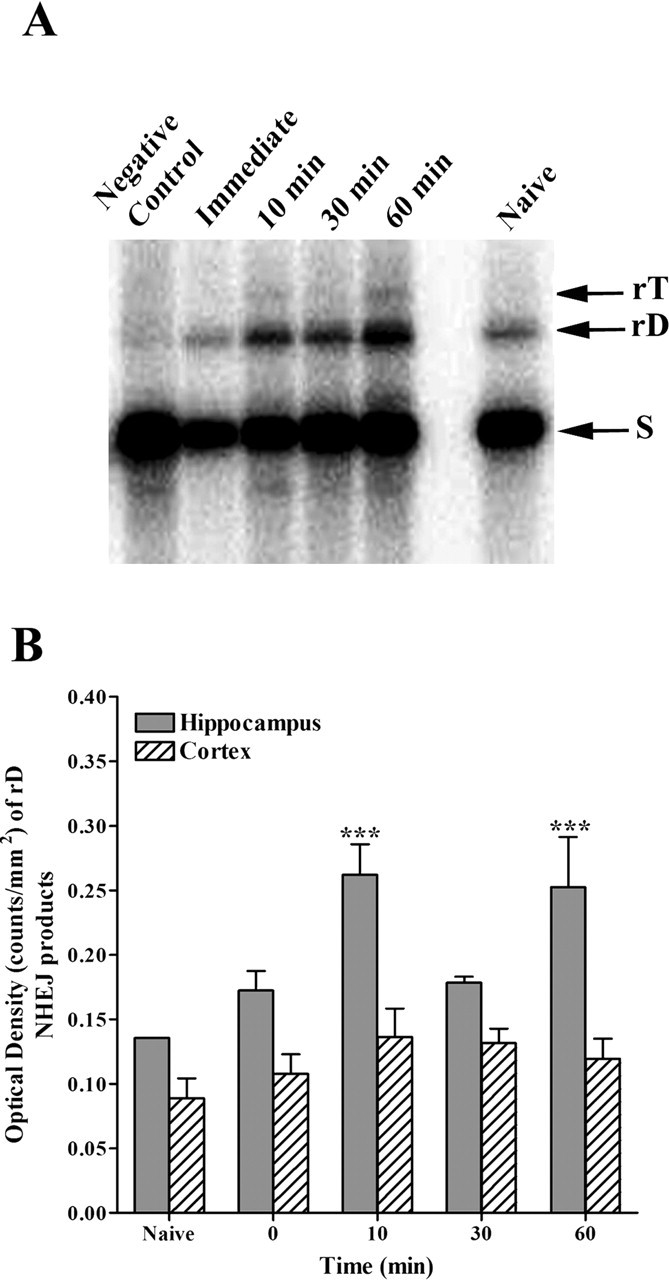
Hippocampal DNA recombination is related to consolidation of context fear conditioning. A, Representative Southern blot showing NHEJ rD and rT products generated by hippocampal protein extracts. S, DNA substrate. Lane 1, Negative control (DNA end-joining results when using heat-denatured protein); lanes 2–5, DNA end-joining products generated by hippocampal protein extracts prepared from mice killed immediately, 10 min, 30 min, or 60 min after training; lane 6, empty, nothing loaded; lane 7, DNA end-joining products generated by hippocampal protein extracts prepared from naive mice. B, Bar graph showing the relative generation of rD NHEJ products by hippocampal (filled bars) and cortical (hatched bars) protein extracts. The hippocampal peaks of NHEJ activity at 10 and 60 min were statistically significant (***p < 0.0001 each). Error bars indicate SEM.
Systemic ara-C has no effect on memory reconsolidation of context fear conditioning
Our next experiments were designed to assess whether ara-C-sensitive DNA recombination processes play a role in memory reconsolidation for context fear conditioning. The experiment design is depicted in Figure 4. Mice were trained for context fear conditioning on day 1 as before, except that they received no injections. To test the effect of ara-C on memory reconsolidation, a set of animals were injected with ara-C or vehicle (n = 14–16, each treatment) 1 h before exposing them to the conditioning context for only 90 s on day 2 (re-exposure groups). Other animals received the injections on day 2 but remained in their home cages and were not exposed to the conditioning chamber (no re-exposure groups; n = 13–14, each treatment). Importantly, we found that ara-C had no effect on the reactivation of fear memory, because the treated animals showed the same freezing responses as the controls (vehicle, 58.18 ± 6.863 vs ara-C, 54.44 ± 5.165; t(28) = 0.4422; p > 0.6). Finally, on day 3, all animals were placed in the conditioning context and were tested for LTM. Surprisingly, we found no difference in fear responses (percentage of freezing) between the vehicle- and ara-C-treated animals that had undergone memory reactivation (vehicle, 51.31 ± 4.950 vs ara-C, 52.66 ± 4.335; test, t(28) = 0.2273; p > 0.8), or those that remained in their home cages on day 2 (vehicle, 59.38 ± 5.764 vs ara-C, 60.74 ± 2.777; t(17) = 0.2714; p > 0.8).
Figure 4.

ara-C does not affect memory reconsolidation of context fear conditioning. Schematic diagram depicting the experiment design used in our studies. ara-C, ANI, or vehicle were injected intraperitoneally before memory reactivation on day 2 (open arrow). The effects of ara-C or ANI on memory reconsolidation were assessed on day 3. US, Unconditioned stimulus.
It has been suggested that the requirement of gene activation and protein synthesis for reconsolidation is subject to constraints related to the length of exposure to the conditioned stimulus or the conditioning strength among others (Biedenkapp and Rudy, 2004; Suzuki et al., 2004). To confirm that our behavioral protocol indeed elicited a protein synthesis-dependent reconsolidation process, we repeated the above experiment but used ANI instead of ara-C (Fig. 4). Mice were trained for context fear conditioning on day 1 without receiving any injections. On day 2, a set of animals were given injections of ANI or vehicle (n = 14–15, each treatment) 30 min before exposing them to the conditioning context for only 90 s. Other animals received the injections on day 2 but remained in their home cages and were not exposed to the conditioning chamber (no re-exposure groups; n = 13–14, each treatment). Similar to previous studies (Kida et al., 2002) and to the results obtained with ara-C, we found that ANI had no effect on the reactivation of fear memory, because the treated animals showed the same freezing responses as the controls (vehicle, 61.18 ± 3.052 vs ANI, 61.68 ± 3.798; t(27) = 0.1431; p > 0.8). Also similar to previous studies, but different to our results with ara-C, ANI-treated animals displayed significantly less freezing than vehicle controls (vehicle, 56.93 ± 3.437 vs ANI, 30.15 ± 4.170; t(26) = 4.955; ***p < 0.0001) when tested for LTM on day 3. As expected, we found no effect between ANI- and vehicle-treated mice that remained in their home cages on day 2 (vehicle, 63.07 ± 4.957 vs ANI, 67.12 ± 4.136; t(25) = 0.6999, p > 0.5). These results suggest that, unlike cAMP response element-binding protein (CREB) inactivation and general protein synthesis inhibition, blockade of DNA recombination processes during memory reactivation does not interfere with reconsolidation of fear conditioning.
Discussion
Our results demonstrate that ara-C impairs consolidation of context conditioning without interfering with reconsolidation nor affecting acquisition, STM, recall, or open-field behavior. ara-CTP, the active metabolite of ara-C, directly inhibits DNA ligase (Lamballe et al., 1988; Zittoun et al., 1989, 1991) and DNA ligase-dependent brain NHEJ (Wang et al., 2003). ara-CTP can also be incorporated into newly synthesized DNA and thereby interrupt replication (Gandhi et al., 1997; Ohno et al., 1998; Abdel-Aziz et al., 2000; Han et al., 2000; Wills et al., 2000). DNA-incorporated ara-CTP interferes with the religation activity of topoisomerases (Cline and Osheroff, 1999; Pourquier et al., 2000; Chrencik et al., 2003; Gmeiner et al., 2003) or with transcription factor binding (Zhang and Kiechle, 2004). Importantly, however, ara-CTP does not act as a direct inhibitor of transcription (Wang et al., 2003).
The amnesic effects of ara-C are associated with inhibition of DNA recombination rather than neurogenesis
The effects of protein synthesis inhibition on LTM of context fear conditioning can be seen as early as 3–6 h after training (Bourtchuladze et al., 1994). Similarly, the amnesic effects of ara-C on the same task can be detected as early as 6 h after conditioning. To block DNA replication, ara-C must first be continually incorporated into DNA for at least 6 h (Hamada et al., 2002). Thus, the rapid effects of ara-C on LTM are probably not attributable to blockade of DNA replication leading to neurogenesis. Importantly, dentate gyrus granule cells are still immature 48 h after DNA replication and require 4 additional weeks to become integrated into the neuronal circuitry (van Praag et al., 2002; Brandt et al., 2003). In addition, blocking hippocampal neurogenesis has no effect on consolidation of context conditioning (Shors et al., 2002). Accordingly, Pham et al. (2005) showed that context conditioning caused a moderate attenuation of neurogenesis in the dentate gyrus. Together, these results suggest that the amnesic effects of ara-C are related to its potential effects on gene expression regulation via blockade of DNA recombination processes and not to blockade of DNA replication and thereby neurogenesis.
The amnesic effects of ara-C are not related to neurotoxicity
Prolonged exposure to high doses of ara-C results in the death of postmitotic neurons (Wallace and Johnson, 1989; Martin et al., 1990; Deckwerth and Johnson, 1993; Tomkins et al., 1994; Dessi et al., 1995; Anderson and Tolkovsky, 1999). ara-C neurotoxicity in cancer patients has been documented within the cerebellum, brainstem, medulla, and spinal cord (Lazarus et al., 1981; Winkelman and Hines, 1983; Sylvester et al., 1987; Vogel and Horoupian, 1993). The neurotoxicity of ara-C is dose dependent, requires chronic exposure, and appears to be mediated by the DNA damage-activated, p53-dependent apoptosis pathway (Lazarus et al., 1981; Wallace and Johnson, 1989; Martin et al., 1990; Deckwerth and Johnson, 1993; Tomkins et al., 1994; Dessi et al., 1995; Enokido et al., 1996; Park et al., 1998). Similarly, DNA ligase IV mutations cause massive apoptosis of postmitotic neurons via the p53 pathway (Gao et al., 1998; Frank et al., 2000). Data from our experiments suggests that the observed effects of acute exposure to ara-C on LTM are not attributable to neurotoxicity. The effects of ara-C on LTM tested at 24 h after training were unrelated to general malaise, motor incoordination, sedation, or anxiety. Additionally, we found no evidence of neuronal loss or apoptosis in the brains of ara-C-treated mice. Similarly, ara-C injected 1 h after training did not affect freezing responses tested at 24 h after training. Finally, localized hippocampal infusion of ara-C had the same effect on LTM than systemic injections, indicating that the effects were related to the specific blockade of memory and not to systemic toxicity.
The specificity of ara-C-sensitive DNA recombination in consolidation
As in our previous studies with CTA (Wang et al., 2003), the data obtained from the post-training injections of ara-C indicates that there is a short time window for the requirement of DNA recombination mechanisms in consolidation of conditioned context fear. Our results with NHEJ assays suggest that DNA recombination activity is enhanced in the hippocampus as early as 10 min after context conditioning. The data also suggest that, similar to protein synthesis (Bourtchouladze et al., 1998), there might be more than one wave of NHEJ-mediated recombination in the hippocampus during consolidation of context fear conditioning. In addition, our NHEJ data suggest that DNA recombination is restricted initially to the hippocampus and does not involve the cortex, agreeing with the notion that biochemical memory consolidation first occurs within relevant subcortical structures. Accordingly, direct pretraining infusions of ara-C into the CA3 region of the hippocampus, but not into the insular cortex, blocked the consolidation of fear memory tested 24 h after conditioning. These results point to the existence of a functional DNA recombination mechanism in the hippocampus associated with consolidation of context fear conditioning. The fact that factors specifically related to V(D)J recombination, such as RAG-1 (recombination activating gene-1) (Chun et al., 1991) and TdT (Peña de Ortiz et al., 2003), have been shown to be expressed in this brain region and to be related to learning and memory processes, as is the case of TdT, supports this idea as well.
Consolidated memories can return to a labile state when they are reactivated and can be disrupted by administering ANI immediately after reactivation (Nader et al., 2000a; Taubenfeld et al., 2001; Debiec et al., 2002; Duvarci and Nader, 2004; Gruest et al., 2004). Thus, some have suggested that memory reactivation destabilizes consolidated information and elicits a cascade of molecular events that lead to reconsolidation or memory restabilization (Nader et al., 2000b). Importantly, however, there are contradicting studies reporting the absence of a protein synthesis-dependent reconsolidation process after memory reactivation (Cammarota et al., 2004; Hernández and Kelley, 2004). These contradictory findings could be explained in part by the fact that the requirement of protein synthesis for reconsolidation is subject to constraints related to the length of exposure to the CS or the conditioning strength (Biedenkapp and Rudy, 2004; Suzuki et al., 2004).
Together, these findings underscore the importance of answering the question of whether memory reconsolidation uses the same biochemical processes used during initial memory storage. The fact that in most studies so far protein synthesis inhibition, as well as inactivation of CREB (Kida et al., 2002), blocks both consolidation and reconsolidation processes supports this view. However, several studies indicate that there are important differences between the molecular players involved in consolidation versus reconsolidation processes. Taubenfeld et al. (2001) showed that consolidation, but not reconsolidation, of inhibitory avoidance memory requires the expression of the CAAT/enhancer binding protein β in the hippocampus. Lee et al. (2004) found opposite roles of brain-derived neurotrophic factor and Zif268 with respect to consolidation and reconsolidation of context fear conditioning. Others have shown that consolidation and reconsolidation of context fear memory differentially activate immediate-early gene expression (von Hertzen and Giese, 2005). Moreover, studies with CTA support the notion that consolidation and reconsolidation have different molecular and neuroanatomical substrates (Bahar et al., 2004). As suggested by Alberini (2005), reconsolidation might be a memory reactivation-dependent modulatory system that is part of the consolidation processes but that is not in itself a long-lasting memory storage mechanism. This is, in fact, supported by the findings obtained by Lattal and Abel (2004), who showed that when ANI injections followed normal retrieval of context fear conditioning, freezing was impaired 24 h but not 21 d later. Our findings support the idea that consolidation and reconsolidation are different memory processes and that gene rearrangement is associated specifically to the early stages of long-term information storage.
Conclusion
We postulate that DNA recombination is important for the regulation of gene expression and function only when new information is acquired and transformed into LTM. Thus, DNA recombination mechanisms could be occurring when neurons are becoming engaged in new information storage. Genomic recombination might be occurring on gene substrates associated with cell recognition or cell–cell communication, such as those encoding for cadherins (Wu and Maniatis, 2000; Hirayama et al., 2001; Yagi, 2003; Yanase et al., 2004), Ig-like cell adhesion molecules, or T-cell receptors (Syken and Shatz, 2003; Nishiyori et al., 2004), and neuropeptides (Kondo et al., 1996). Once target genes are rearranged, the novel mRNAs and proteins may be directed to any specific neuronal compartment where their function is required for memory maintenance and potentially provide for synapse specificity. Thus, somatic gene recombination may generate mRNAs and proteins that may exert neuron-wide effects if they are localized nonspecifically or mRNAs and proteins that act at specific synapses if their localization is targeted in such a manner. Thus, recombinational, transcriptional, and translational mechanisms of gene regulation in memory do not exclude one another but most likely complement each other. By restricting DNA recombination mechanisms to the initial steps of consolidation of new learning experiences, neurons may be able to generate protein diversity as a mechanism of enhancing memory storage capacity, while at the same time preventing increased mutagenic events that could occur if genomic rearrangement was also to proceed as a result of memory recall.
Footnotes
This work was supported by the National Institutes of Health (National Institute of General Medical Sciences Grant SO6GMO8102, National Institute of Neurological Disorders and Stroke Grant U54 NS39405, and National Center for Research Resources Grant 5P20 RR 15565-02 to S.P.O.). M.C.-C. and J.L. were supported by predoctoral and undergraduate research NIGMS-MBRS-RISE 5R25GM061151 fellowships, respectively. We thank Drs. Alcino J. Silva, Gregory Quirk, and Yuri Arshavsky for helpful comments and discussions regarding this manuscript.
References
- Abdel-Aziz W, Jiang HY, Hickey RJ, Malkas LH (2000). Ara-C affects formation of cancer cell DNA synthesome replication intermediates. Cancer Chemother Pharmacol 45:312–319. [DOI] [PubMed] [Google Scholar]
- Agnihotri NT, Hawkins RD, Kandel ER, Kentros C (2004). The long-term stability of new hippocampal place fields requires new protein synthesis. Proc Natl Acad Sci USA 101:3656–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini CM (2005). Mechanisms of memory stabilization: are consolidation and reconsolidation similar or distinct processes? Trends Neurosci 28:51–56. [DOI] [PubMed] [Google Scholar]
- Alberini CM, Ghirardi M, Metz R, Kandel ER (1994). C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Cell 76:1099–1114. [DOI] [PubMed] [Google Scholar]
- Anderson CN, Tolkovsky AM (1999). A role for MAPK/ERK in sympathetic neuron survival: protection against a p53-dependent, JNK-independent induction of apoptosis by cytosine arabinoside. J Neurosci 19:664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar A, Dorfman N, Dudai Y (2004). Amygdalar circuits required for either consolidation or extinction of taste aversion memory are not required for reconsolidation. Eur J Neurosci 19:1115–1118. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Kandel ER, Si K (2004). The persistence of long-term memory: a molecular approach to self-sustaining changes in learning-induced synaptic growth. Neuron 44:49–57. [DOI] [PubMed] [Google Scholar]
- Biedenkapp JC, Rudy JW (2004). Context memories and reactivation: constraints on the reconsolidation hypothesis. Behav Neurosci 118:956–964. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ (1994). Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79:59–68. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R, Abel T, Berman N, Gordon R, Lapidus K, Kandel ER (1998). Different training procedures recruit either one or two critical periods for contextual memory consolidation, each of which requires protein synthesis and PKA. Learn Mem 5:365–374. [PMC free article] [PubMed] [Google Scholar]
- Brandt MD, Jessberger S, Steiner B, Kronenberg G, Reuter K, Bick-Sander A, von der Behrens W, Kempermann G (2003). Transient calretinin expression defines early postmitotic step of neuronal differentiation in adult hippocampal neurogenesis of mice. Mol Cell Neurosci 24:603–613. [DOI] [PubMed] [Google Scholar]
- Brunzell DH, Kim JJ (2001). Fear conditioning to tone, but not to context, is attenuated by lesions of the insular cortex and posterior extension of the intralaminar complex in rats. Behav Neurosci 115:365–375. [PubMed] [Google Scholar]
- Calixto E, Thiels E, Klann E, Barrionuevo G (2003). Early maintenance of hippocampal mossy fiber–long-term potentiation depends on protein and RNA synthesis and presynaptic granule cell integrity. J Neurosci 23:4842–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarota M, Bevilaqua LR, Medina JH, Izquierdo I (2004). Retrieval does not induce reconsolidation of inhibitory avoidance memory. Learn Mem 11:572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrencik JE, Burgin AB, Pommier Y, Stewart L, Redinbo MR (2003). Structural impact of the leukemia drug 1-beta-D-arabinofuranosylcytosine (Ara-C) on the covalent human topoisomerase I-DNA complex. J Biol Chem 278:12461–12466. [DOI] [PubMed] [Google Scholar]
- Chun JM, Schatz DG, Oettinger MA, Jaenisch R, Baltimore D (1991). The recombination activating gene-1 (Rag-1) transcript is present in the murine central nervous system. Cell 64:189–200. [DOI] [PubMed] [Google Scholar]
- Cline SD, Osheroff N (1999). Cytosine arabinoside lesions are position-specific topoisomerase II poisons and stimulate DNA cleavage mediated by the human type II enzymes. J Biol Chem 274:29740–29743. [DOI] [PubMed] [Google Scholar]
- Debiec J, LeDoux JE, Nader K (2002). Cellular and systems reconsolidation in the hippocampus. Neuron 36:527–538. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM Jr (1993). Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol 123:1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessi F, Pollard H, Moreau J, Ben-Ari Y, Charriaut-Marlangue C (1995). Cytosine arabinoside induces apoptosis in cerebellar neurons in culture. J Neurochem 64:1980–1987. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Nader K (2004). Characterization of fear memory reconsolidation. J Neurosci 24:9269–9275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enokido Y, Araki T, Aizawa S, Hatanaka H (1996). p53 involves cytosine arabinoside-induced apoptosis in cultured cerebellar granule neurons. Neurosci Lett 203:1–4. [DOI] [PubMed] [Google Scholar]
- Flexner JB, Flexner LB, Stellar E (1963). Memory in mice as affected by intracerebral puromycin. Science 141:57–59. [DOI] [PubMed] [Google Scholar]
- Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho R, Alt FW (2000). DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell 5:993–1002. [DOI] [PubMed] [Google Scholar]
- Frey U, Frey S, Schollmeier F, Krug M (1996). Influence of actinomycin D, a RNA synthesis inhibitor, on long-term potentiation in rat hippocampal neurons in vivo and in vitro. J Physiol (Lond) 490:703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi V, Huang P, Chapman AJ, Chen F, Plunkett W (1997). Incorporation of fludarabine and 1-beta-D-arabinofuranosylcytosine 5′-triphosphates by DNA polymerase alpha: affinity, interaction, and consequences. Clin Cancer Res 3:1347–1355. [PubMed] [Google Scholar]
- Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, Bronson RT, Malynn BA, Bryans M, Zhu C, Chaudhuri J, Davidson L, Ferrini R, Stamato T, Orkin SH, Greenberg ME, et al (1998). A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95:891–902. [DOI] [PubMed] [Google Scholar]
- Gmeiner WH, Yu S, Pon RT, Pourquier P, Pommier Y (2003). Structural basis for topoisomerase I inhibition by nucleoside analogs. Nucleosides Nucleotides Nucleic Acids 22:653–658. [DOI] [PubMed] [Google Scholar]
- Gruest N, Richer P, Hars B (2004). Memory consolidation and reconsolidation in the rat pup require protein synthesis. J Neurosci 24:10488–10492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada A, Kawaguchi T, Nakano M (2002). Clinical pharmacokinetics of cytarabine formulations. Clin Pharmacokinet 41:705–718. [DOI] [PubMed] [Google Scholar]
- Han S, Hickey RJ, Tom TD, Wills PW, Syvaoja JE, Malkas LH (2000). Differential inhibition of the human cell DNA replication complex-associated DNA polymerases by the antimetabolite 1-beta-D-arabinofuranosylcytosine triphosphate (ara-CTP). Biochem Pharmacol 60:403–411. [DOI] [PubMed] [Google Scholar]
- Hernández PJ, Kelley AE (2004). Long-term memory for instrumental responses does not undergo protein synthesis-dependent reconsolidation upon retrieval. Learn Mem 11:748–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández PJ, Sadeghian K, Kelley AE (2002). Early consolidation of instrumental learning requires protein synthesis in the nucleus accumbens. Nat Neurosci 5:1327–1331. [DOI] [PubMed] [Google Scholar]
- Hiddemann W, Schleyer E, Unterhalt M, Zuhlsdorf M, Rolf C, Reuter C, Kewer U, Uhrmeister C, Wormann B, Buchner T (1992). Differences in the intracellular pharmacokinetics of cytosine arabinoside (AraC) between circulating leukemic blasts and normal mononuclear blood cells. Leukemia 6:1273–1280. [PubMed] [Google Scholar]
- Hirayama T, Sugino H, Yagi T (2001). Somatic mutations of synaptic cadherin (CNR family) transcripts in the nervous system. Genes Cells 6:151–164. [DOI] [PubMed] [Google Scholar]
- Igaz LM, Vianna MR, Medina JH, Izquierdo I (2002). Two time periods of hippocampal mRNA synthesis are required for memory consolidation of fear-motivated learning. J Neurosci 22:6781–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson GP, Snook MB, Wiley JS (1990). Saturation of intracellular cytosine arabinoside triphosphate accumulation in human leukemic blast cells. Leuk Res 14:475–479. [DOI] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, Peña de Ortiz S, Kogan JH, Chévere I, Masushige S, Silva AJ (2002). CREB required for the stability of new and reactivated fear memories. Nat Neurosci 5:348–355. [DOI] [PubMed] [Google Scholar]
- Kondo H, Ino M, Suzuki A, Ishizaki H, Iwami M (1996). Multiple gene copies for bombyxin, an insulin-related peptide of the silkmoth Bombyx mori: structural signs for gene rearrangement and duplication responsible for generation of multiple molecular forms of bombyxin. J Mol Biol 259:926–937. [DOI] [PubMed] [Google Scholar]
- Korzus E (2003). The relation of transcription to memory formation. Acta Biochim Pol 50:775–782. [PubMed] [Google Scholar]
- Lamballe F, Maniey D, Boscher MY, Fauchet R, le Prise PY, David JC (1988). Effects of clinical combinations of antileukemic drugs on DNA ligase from human thymocytes and normal, stimulated, or leukemic lymphocytes. Leukemia 2:363–370. [PubMed] [Google Scholar]
- Lattal KM, Abel T (2004). Behavioral impairments caused by injections of the protein synthesis inhibitor anisomycin after context retrieval reverse with time. Proc Natl Acad Sci USA 101:4667–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus HM, Herzig RH, Herzig GP, Phillips GL, Roessmann U, Fishman DJ (1981). Central nervous system toxicity of high-dose systemic cytosine arabinoside. Cancer 48:2577–2582. [DOI] [PubMed] [Google Scholar]
- LeDoux JE (2000). Emotion circuits in the brain. Annu Rev Neurosci 23:155–184. [DOI] [PubMed] [Google Scholar]
- Lee I, Kesner RP (2004). Differential contributions of dorsal hippocampal subregions to memory acquisition and retrieval in context fear-conditioning. Hippocampus 14:301–310. [DOI] [PubMed] [Google Scholar]
- Lee JL, Everitt BJ, Thomas KL (2004). Independent cellular processes for hippocampal memory consolidation and reconsolidation. Science 304:839–843. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD (2005). Epigenetic mechanisms in memory formation. Nat Rev Neurosci 6:108–118. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD (2004). Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem 279:40545–40559. [DOI] [PubMed] [Google Scholar]
- Lieber MR, Ma Y, Pannicke U, Schwarz K (2004). The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair 3:817–826. [DOI] [PubMed] [Google Scholar]
- Malkani S, Wallace KJ, Donley MP, Rosen JB (2004). An egr-1 (zif268) antisense oligodeoxynucleotide infused into the amygdala disrupts fear conditioning. Learn Mem 11:617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Fanselow MS (1995). Synaptic plasticity in the basolateral amygdala induced by hippocampal formation stimulation in vivo. J Neurosci 15:7548–7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Wallace TL, Johnson EM Jr (1990). Cytosine arabinoside kills postmitotic neurons in a fashion resembling trophic factor deprivation: evidence that a deoxycytidine-dependent process may be required for nerve growth factor signal transduction. J Neurosci 10:184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE (2000a). Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406:722–726. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE (2000b). The labile nature of consolidation theory. Nat Rev Neurosci 1:216–219. [DOI] [PubMed] [Google Scholar]
- Naghdi N, Majlessi N, Bozorgmehr T (2003). The effects of anisomycin (a protein synthesis inhibitor) on spatial learning and memory in CA1 region of rats hippocampus. Behav Brain Res 139:69–73. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER (1994). Requirement of a critical period of transcription for induction of a late phase of LTP. Science 265:1104–1107. [DOI] [PubMed] [Google Scholar]
- Nishiyori A, Hanno Y, Saito M, Yoshihara Y (2004). Aberrant transcription of unrearranged T-cell receptor beta gene in mouse brain. J Comp Neurol 469:214–226. [DOI] [PubMed] [Google Scholar]
- Ohi S (1977). Effects of actinomycin D on brain RNA synthesis and discrimination learning in the goldfish (Carassius auratus). Physiol Behav 19:261–264. [DOI] [PubMed] [Google Scholar]
- Ohno Y, Spriggs D, Matsukage A, Ohno T, Kufe D (1989). Sequence-specific inhibition of DNA strand elongation by incorporation of 9-beta-D-arabinofuranosyladenine. Cancer Res 49:2077–2081. [PubMed] [Google Scholar]
- Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM, Greene LA (1998). Multiple pathways of neuronal death induced by DNA-damaging agents, NGF deprivation, and oxidative stress. J Neurosci 18:830–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedreira ME, Dimant B, Maldonado H (1996). Inhibitors of protein and RNA synthesis block context memory and long-term habituation in the crab Chasmagnathus. Pharmacol Biochem Behav 54:611–617. [DOI] [PubMed] [Google Scholar]
- Peña de Ortiz S, Arshavsky YI (2001). DNA recombination as a possible mechanisms in declarative memory: a hypothesis. J Neurosci Res 63:72–81. [DOI] [PubMed] [Google Scholar]
- Peña de Ortiz S, Maldonado-Vlaar CS, Carrasquillo Y (2000). Hippocampal expression of the orphan nuclear receptor gene hzf-3/nurr1 during spatial discrimination learning. Neurobiol Learn Mem 74:161–171. [DOI] [PubMed] [Google Scholar]
- Peña de Ortiz S, Colon M, Carrasquillo Y, Padilla B, Arshavsky YI (2003). Experience-dependent expression of terminal deoxynucleotidyl transferase in mouse brain. NeuroReport 14:1141–1144. [DOI] [PubMed] [Google Scholar]
- Peña de Ortiz S, Colón M, Arshavsky Y (2004). Genomic theory of declarative memory. In: Dynamical genetics (Parisi V, De Fonzo V, Aluffi-Pertini F, eds) pp. 345–364. Kerala, India: Research Signpost.
- Pham K, McEwen BS, Ledoux JE, Nader K (2005). Fear learning transiently impairs hippocampal cell proliferation. Neuroscience 130:17–24. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE (1992). Differential contribution of amygdala and hippocampus to cued and context fear conditioning. Behav Neurosci 106:274–285. [DOI] [PubMed] [Google Scholar]
- Pourquier P, Takebayashi Y, Urasaki Y, Gioffre C, Kohlhagen G, Pommier Y (2000). Induction of topoisomerase I cleavage complexes by 1-beta-d-arabinofuranosylcytosine (ara-C) in vitro and in ara-C-treated cells. Proc Natl Acad Sci USA 97:1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevedo J, Vianna MR, Martins MR, Barichello T, Medina JH, Roesler R, Izquierdo I (2004). Protein synthesis, PKA, and MAP kinase are differentially involved in short- and long-term memory in rats. Behav Brain Res 154:339–343. [DOI] [PubMed] [Google Scholar]
- Ren K, Peña de Ortiz S (2002). Non-homolgous DNA end joining in the mature rat brain. J Neurochem 80:949–959. [DOI] [PubMed] [Google Scholar]
- Richmond MA, Yee BK, Pouzet B, Veenman L, Rawlins JN, Feldon J, Bannerman DM (1999). Dissociating context and space within the hippocampus: effects of complete, dorsal, and ventral excitotoxic hippocampal lesions on conditioned freezing and spatial learning. Behav Neurosci 113:1189–1203. [DOI] [PubMed] [Google Scholar]
- Santini E, Ge H, Ren K, Peña de Ortiz S, Quirk GJ (2004). Consolidation of fear extinction involves protein synthesis and c-Fos in medial prefrontal cortex. J Neurosci 24:5704–5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE (2000). Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci 20:RC96(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf MT, Woo NH, Lattal KM, Young JZ, Nguyen PV, Abel T (2002). Protein synthesis is required for the enhancement of long-term potentiation and long-term memory by spaced training. J Neurophysiol 87:2770–2777. [DOI] [PubMed] [Google Scholar]
- Schatz DG (2004). Antigen receptor genes and the evolution of a recombinase. Semin Immunol 16:245–256. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y, Gould E (2002). Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus 12:578–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulz DE, Sosnik R, Ego V, Haidarliu S, Ahissar E (2000). A neuronal analogue of state-dependent learning. Nature 403:549–553. [DOI] [PubMed] [Google Scholar]
- Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu H, Kandel ER (2003). A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in Aplysia. Cell 115:893–904. [DOI] [PubMed] [Google Scholar]
- Squire LR, Barondes SH (1970). Actinomycin-D: effects on memory at different times after training. Nature 225:649–650. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S (2004). Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci 24:4787–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syken J, Shatz CJ (2003). Expression of T cell receptor beta locus in central nervous system neurons. Proc Natl Acad Sci USA 100:13048–13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvester RK, Fisher AJ, Lobell M (1987). Cytarabine-induced cerebellar syndrome: case report and literature review. Drug Intell Clin Pharm 21:177–180. [PubMed] [Google Scholar]
- Taubenfeld SM, Milekic MH, Monti B, Alberini CM (2001). The consolidation of new but not reactivated memory requires hippocampal C/EBPβ. Nat Neurosci 4:813–818. [DOI] [PubMed] [Google Scholar]
- Tomkins CE, Edwards SN, Tolkovsky AM (1994). Apoptosis is induced in post-mitotic rat sympathetic neurons by arabinosides and topoisomerase II inhibitors in the presence of NGF. J Cell Sci 107:1499–1507. [DOI] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH (2002). Functional neurogenesis in the adult hippocampus. Nature 415:1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel H, Horoupian DS (1993). Filamentous degeneration of neurons. A possible feature of cytosine arabinoside neurotoxicity. Cancer 71:1303–1308. [DOI] [PubMed] [Google Scholar]
- von Hertzen LS, Giese KP (2005). Memory reconsolidation engages only a subset of immediate-early genes induced during consolidation. J Neurosci 25:1935–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace TL, Johnson EM Jr (1989). Cytosine arabinoside kills postmitotic neurons: evidence that deoxycytidine may have a role in neuronal survival that is independent of DNA synthesis. J Neurosci 9:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ren K, Pérez J, Flores G, Silva AJ, Peña de Ortiz S (2003). The antimetabolite ara-CTP blocks long-term memory of conditioned taste aversion. Learn Mem 10:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills PW, Hickey R, Malkas L (2000). Ara-C differentially affects multiprotein forms of human cell DNA polymerase. Cancer Chemother Pharmacol 46:193–203. [DOI] [PubMed] [Google Scholar]
- Winkelman MD, Hines JD (1983). Cerebellar degeneration caused by highdose cytosine arabinoside: a clinicopathological study. Ann Neurol 14:520–527. [DOI] [PubMed] [Google Scholar]
- Wu Q, Maniatis T (2000). Large exons encoding multiple ectodomains are a characteristic feature of protocadherin genes. Proc Natl Acad Sci USA 97:3124–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wustenberg D, Gerber B, Menzel R (1998). Short communication: long- but not medium-term retention of olfactory memories in honeybees is impaired by actinomycin D and anisomycin. Eur J Neurosci 10:2742–2745. [DOI] [PubMed] [Google Scholar]
- Yagi T (2003). Diversity of the cadherin-related neuronal receptor/protocadherin family and possible DNA rearrangement in the brain. Genes Cells 8:1–8. [DOI] [PubMed] [Google Scholar]
- Yanase H, Sugino H, Yagi T (2004). Genomic sequence and organization of the family of CNR/Pcdhalpha genes in rat. Genomics 83:717–726. [DOI] [PubMed] [Google Scholar]
- Yin H, Bardgett ME, Csernansky JG (2002). Kainic acid lesions disrupt fear-mediated memory processing. Neurobiol Learn Mem 77:389–401. [DOI] [PubMed] [Google Scholar]
- Zhang X, Kiechle FL (2004). Cytosine arabinoside decreases transcription factor-DNA binding element complex formation. Arch Pathol Lab Med 128:1364–1371. [DOI] [PubMed] [Google Scholar]
- Zittoun J, Marquet J, David JC, Maniey D, Zittoun R (1989). A study of the mechanisms of cytotoxicity of Ara-C on three human leukemic cell lines. Cancer Chemother Pharmacol 24:251–255. [DOI] [PubMed] [Google Scholar]
- Zittoun J, Marquet J, David JC (1991). Mechanism of inhibition of DNA ligase in Ara-C treated cells. Leuk Res 15:157–164. [DOI] [PubMed] [Google Scholar]