

Abstract
Various environmental and genetic factors influence the onset and progression of Alzheimer’s disease (AD). Dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis, which controls circulating levels of glucocorticoid hormones, occurs early in AD, resulting in increased cortisol levels. Disturbances of the HPA axis have been associated with memory impairments and may contribute to the cognitive decline that occurs in AD, although it is unknown whether such effects involve modulation of the amyloid β-peptide (Aβ) and tau. Using in vitro and in vivo experiments, we report that stress-level glucocorticoid administration increases Aβ formation by increasing steady-state levels of amyloid precursor protein (APP) and β-APP cleaving enzyme. Additionally, glucocorticoids augment tau accumulation, indicating that this hormone also accelerates the development of neurofibrillary tangles. These findings suggest that high levels of glucocorticoids, found in AD, are not merely a consequence of the disease process but rather play a central role in the development and progression of AD.
Keywords: corticosterone, Aβ peptide, Alzheimer’s disease, glucocorticoids, tau, stress
Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder marked by a progressive loss of memory and cognitive function. The two hallmark neuropathological features are amyloid β-peptide (Aβ) plaques and tau-laden neurofibrillary tangles. Although mutations in three different genes are known to underlie some cases of the rare, inheritable forms of the disease, the etiology of the more common sporadic cases remains unknown and likely involves complex interactions between various genetic and environmental factors, such as a stressful lifestyle or the apolipoprotein E4 allele (apoE4) (Travis, 1993). Neuroendocrine malfunctions may also be involved in the disease process, particularly because it is established that stress hormones can negatively affect neuronal survival (Stein-Behrens et al., 1994). Epidemiological evidence further supports a role for stress as a risk factor for AD because elderly individuals prone to psychological distress are more likely to develop the disorder than age-matched, nonstressed individuals (Wilson et al., 2005).
The glucocorticoid response to stressful stimuli is regulated by the hypothalamic–pituitary–adrenal (HPA) axis, which triggers the adrenal cortex to release glucocorticoids (cortisol in primates, corticosterone in mice and rats). Glucocorticoids are steroid hormones that readily cross the blood–brain barrier and bind to low-affinity glucocorticoid receptors and high-affinity mineralocorticoid receptors (Reul and de Kloet, 1985). Activity of these receptors is necessary for normal cellular metabolic activity and crucial for many CNS functions, including learning and memory (Roozendaal, 2000). There is ample evidence implicating HPA axis dysfunction in AD, reflected by markedly elevated basal levels of circulating cortisol (Davis et al., 1986; Masugi et al., 1989; Swanwick et al., 1998) and a failure to show cortisol suppression after a dexamethasone challenge (Greenwald et al., 1986; Molchan et al., 1990; Nasman et al., 1995). Of interest was the finding that, although AD patients had elevated basal cortisol levels, HPA dysfunction only seemed relevant in the early stages of the disease (Swanwick et al., 1998) because HPA dysfunction did not worsen with additional cognitive decline. In addition, more recent findings show that elevated CSF cortisol in AD patients mirrored the presence of the apoE4 allele (Peskind et al., 2001), suggesting that apoE function was influencing circulating cortisol levels. Findings from a human clinical study further suggest a detrimental role for glucocorticoids in this disorder, because AD patients treated with prednisone (a glucocorticoid used for its anti-inflammatory properties) exhibited impaired cognition compared with the placebo-treated cohort (Aisen et al., 2000). Genetic studies indicate a link between glucocorticoid function and the risk for AD, because a rare haplotype in the 5′ regulatory region of the gene encoding 11β-hydroxysteroid dehydrogenase type 1 was associated with a sixfold increased risk for sporadic AD (de Quervain et al., 2004). Furthermore, some evidence from animal studies suggests an interaction between glucocorticoids and AD pathology, including amyloid precursor protein (APP) and tau accumulation (Elliott et al., 1993; Budas et al., 1999), although the molecular mechanisms underlying these effects and the downstream consequences are unknown.
The present study sought to determine whether glucocorticoids modulate the hallmark neuropathological features of AD and, if so, the underlying mechanism. Consequently, we investigated the pathological consequences of stress-level glucocorticoid administration on Aβ formation and tau biology using both in vitro and in vivo approaches. Here we report the novel findings that levels of the β-secretase enzyme [β-APP cleaving enzyme (BACE)] and its substrate APP are selectively increased after glucocorticoid administration, resulting in increased production of Aβ. Notably, administering glucocorticoids to the triple-transgenic (3×Tg-AD) mice, which develop both Aβ and tau pathologies in an age-dependent manner (Oddo et al., 2003), exacerbated the formation of both lesions. The present findings highlight a mechanism by which stress affects AD neuropathology and suggest that stress management or pharmacological reduction of glucocorticoids warrant additional consideration in the regimen of AD therapies.
Materials and Methods
Immunoblotting.
Protein extracts were prepared from cells using M-per (Pierce, Rockford, IL) extraction buffer and Complete Mini Protease Inhibitor Tablets (Roche, Indianapolis, IN). Protein extracts were prepared from whole-brain samples by homogenizing in T-per (Pierce) extraction buffer and Complete Mini Protease Inhibitor Tablets (Roche), followed by high-speed centrifugation at 100,000 × g for 1 h. The supernatant was taken as the protein extract. Protein concentrations were determined by the Bradford method. Equal amounts of protein (20–50 μg depending on protein of interest) were separated by SDS-PAGE on a 10% Bis-Tris gel (Invitrogen, Carlsbad, CA), transferred to 0.45 μm polyvinylidene difluoride membranes, blocked for 1 h in 5% (v/v) nonfat milk in Tris-buffered saline, pH 7.5, supplemented with 0.2% Tween 20, and processed as described. Antibodies and dilutions used in this study include 6E10 (1:1000; Signet, Dedham, MA) for APP in in vivo studies, CTF20 (1:5000; Calbiochem, San Diego, CA) for C99 and C83, 22C11 (1:1000; Chemicon, Temecula, CA) and HT7 (1:3000; Innogenetics, Gent, Belgium) for APP in in vitro studies, AT8 (1:1000; Pierce), AT180 (1:1000; Pierce), anti-caspase cleaved APP (cAPP) (1:3000; Chemicon), anti-BACE (1:1000; Calbiochem), anti-glucocorticoid receptor (1:300; Affinity BioReagents, Golden, CO), and α-actin (1:10,000; Sigma, St. Louis, MO). Quantitative densitometric analyses were performed on digitized images of immunoblots with Scion Image 4.0 (Scion, Frederick, MD).
AMPA-binding protein ELISA. Aβ1–40 and Aβ1–42 were measured using a sensitive sandwich ELISA system.
Soluble and insoluble Aβ was isolated from whole-brain homogenates using T-per extraction buffer (Pierce) and 70% formic acid (FA), respectively. Soluble fractions were loaded directly onto ELISA plates, and FA fractions were diluted 1:20 in neutralization buffer (1 m Tris base, 0.5 m NaH4PO4) before loading. Secreted Aβ was measured from in vitro assays by direct addition of the cell-incubated media onto the ELISA plates. MaxiSorp immunoplates (Nunc, Rochester, NY) were coated with mAB20.1 (William Van Nostrand, Stony Bridge, NY) antibody at a concentration of 25 μg/ml in coating buffer (0.1 m NaCO3 buffer, pH 9.6) and blocked with 3% BSA. Standards of both Aβ40 and Aβ42 were made in antigen capture buffer (20 mm NaH2PO4, 2 mm EDTA, 0.4 m NaCl, 0.5 g of 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, and 1% BSA, pH 7.0), and loaded onto ELISA plates in duplicate. Samples were then loaded in duplicate and incubated overnight at 4°C. Plates were washed and then probed with either HRP-conjugated anti-Aβ35–40 (MM32-13.1.1, for Aβ1–40) or anti-Aβ35–42 (clone number MM40-21.3.4, for Aβ1–42) overnight at 4°C. 3,3′,5,5′-Tetramethylbenzidine was used as the chromogen, and the reaction was stopped by 30% O-phosphoric acid and read at 450 nm on a Molecular Dynamics (Sunnyvale, NY) plate reader. Aβ readings were then normalized to protein concentrations of the samples loaded or to the protein concentration of the cell layer that the media were incubated with in the case of the in vitro assays. This takes into account any variations of cell numbers or protein concentrations that may otherwise affect Aβ readings.
Immunostaining.
Light-level immunohistochemistry was performed using an avidin–biotin immunoperoxidase technique (ABC kit; Vector Laboratories, Burlingame, CA) and was visualized with diaminobenzidine as described previously (Oddo et al., 2003). The following antibodies were used: anti-Aβ, 6E10 (Signet), anti-tau HT7 (Innogenetics, Gent, Belgium), AT8 (Pierce), and AT180 (Innogenetics). Primary antibodies were applied at dilutions of 1:1000 for 6E10, 1:500 for AT8 and AT180, and 1:1000 for HT7. Quantification of DAB staining was performed by taking three adjacent images from each hippocampus at 20× or one image of the amygdala. These images were loaded in Scion Image 4.0 (Scion), and the DAB pixel count was measured by setting the threshold to the same value for each section. Pixel counts were averaged from the three adjacent sections for at least three animals per group, and the data were plotted.
Confocal microscopy.
Fluorescent immunolabeling followed a standard two-way technique (primary antibody followed by fluorescent secondary antibody). Free-floating sections were rinsed in TBS, pH 7.4, and then blocked (0.25% Triton X-100, 5% normal goat serum in TBS) for 1 h. Sections were incubated in primary antibody overnight (4°C), rinsed in PBS, and incubated (1 h) in either fluorescently labeled anti-rabbit or anti-mouse secondary antibodies (Alexa 488, 1:200; Invitrogen). Nuclear markers were added by incubating the slices in TOTO red (1:200 in PBS; Invitrogen) for 20 min. Antibodies were diluted as follows: HT7, 1:1000; and 6E10, 1:1000. Omission of primary antibody or use of preimmune IgG eliminated all labeling (data not shown). Confocal images were captured on a Radance 2100 (Bio-Rad, Hercules, CA) confocal system. To prevent signal bleed-through, all fluorophores were excited and scanned separately using lambda strobing.
Cell culture.
N2A cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS. Cells were passaged at 1:40 when 70% confluency had been achieved and discarded after 20 passages. For experiments, equal numbers of cells were plated down in six-well plates. Wells were treated 24 h later by removal of media and replacing with 2 ml of fresh media containing either dexamethasone (1 mm stock solution dissolved in H2O; Sigma) at a final concentration of 100 nm, 1 μm, or 10 μm, or corticosterone (1 mm stock solution dissolved in H20; Sigma) at a final concentration of 100 nm, 1 μm, or 10 μm. Control wells contained fresh media only. Media were replaced every 24 h. After the treatment period, the media were taken from the wells, and secreted Aβ measurements were taken using sandwich ELISA. Protein extracts were taken as described above.
Animal treatments.
All rodent experiments were performed in accordance with animal protocols approved by the Institutional Animal Care and Use Committee at the University of California, Irvine. The 3×Tg-AD mice have been described previously (Oddo et al., 2003). Briefly, these mice harbor a knock-in mutation of presenilin 1 (PS1M146V), the Swedish double mutation of amyloid precursor protein (APPKM670/671NL), and a frontotemporal dementia mutation in tau (tauP301L) on a 129/C57BL/6 background. Tau/PS1 mice (2×Tg-AD) harbor the same knock-in mutation of presenilin 1 (PS1M146V) and the same frontotemporal dementia mutation in tau (tauP301L) on a 129/C57BL/6 background. Tau levels are expressed to a similar level to the 3×Tg-AD mice (Oddo et al., 2003). Four-month-old male 3×Tg-AD mice were taken and given one intraperitoneal injection of either dexamethasone (dissolved in PBS at 1 mg/ml) or PBS alone daily for 7 d. Dexamethasone was administered at 1 or 5 mg/kg bodyweight or PBS vehicle. At 24 h after the final injection, the animals were killed and the brains were removed. The brains were immediately dissected in half along the coronal line; half were frozen for biochemical analysis, and the other half were fixed in 4% paraformaldehyde. At 48 h later, brains were sliced into 40 μm sections using a vibratome. Blood was also taken and stored in EDTA-coated vacu-tubes (Fisher Scientific, Pittsburgh, PA), and centrifuged at 3000 rpm on a Beckman Instruments (Fullerton, CA) bench centrifuge for 10 min. The resultant supernatant was then taken as plasma, which was then frozen at −80°C.
Measurement of corticosterone plasma levels.
A corticosterone competitive ELISA kit (Assay Systems, Ann Arbor, MI) was used to measure corticosterone levels as per the instructions of the manufacturer. Plasma samples were diluted 1:50 in the buffer provided.
Pulse chase and autoradiography.
After treatment, cells were gently washed once in PBS and placed in cysteine/methionine (Cys/Met)-free DMEM (see above, Cell culture) (Invitrogen) without serum for 1 h. The media were then removed, and cells were pulsed for 1 h in Cys/Met, serum-free DMEM containing 250 μCi/ml Tran35S Label No-Thaw Metabolic Labeling Reagent (MP Biomedicals, Irvine, CA). Cells were then washed once in PBS and placed in DMEM containing 10% FBS, and samples were chased at 0, 1, 4, and 8 h time points.
Equal amounts of protein from each sample were then immunoprecipitated overnight at 4°C with Protein-G agarose (Invitrogen) in autoclaved STEN buffer (300 mm NaCl, 100 mm Tris, 4 mm EDTA, and 2 ml of NP-40, pH 7.6) with 22C11 (1:100; Chemicon). After centrifuging the samples at 3000 × g, 4°C for 10 min, the supernatant was removed, and the pellet was washed with 0.5× STEN buffer. Washes were repeated twice using the same procedure but with SDS STEN buffer (STEN buffer containing 0.1% SDS) and 1× STEN. After the final wash, the samples were boiled for 10 min and centrifuged at 10,000 × g for 5 min. The supernatants were removed and run on a 10% Bis-Tris gel (Invitrogen).
After electrophoresis, proteins were fixed by gently agitating the gel at room temperature for 1 h in a 10% glacial acetic acid (v/v), 30% methanol (v/v) solution. The fixing solution was then discarded, and enough EN3HANCE (PerkinElmer, Groningen, The Netherlands) impregnation solution was added to the gel for it to be free floating. The entire gel container was then covered with aluminum foil and was gently agitated for 1 h at room temperature. After discarding the impregnation solution, the gel was washed with cold water for 1 h each still covered in foil to precipitate the fluorescent material. The gel was then placed on Whatman No. 3MM paper (Whatman, Maidstone, UK), covered with Saran Wrap (SC Johnson, Racine, WI), and vacuum dried at 60°C for 75 min using a Bio-Rad Gel Dryer model 583. After adequate drying, the 3MM paper containing the gel was placed in a phosphor screen (Amersham Biosciences, Piscataway, NJ) in a light-free environment for 48 h and visualized using the Storm 840 scanner (Amersham Biosciences).
Real-time PCR.
After treatment, RNA was extracted from cells using an Aurum RNA extraction kit (Bio-Rad) as per the instructions of the manufacturer. RNA (1 mg) from each sample was converted to cDNA, using the iScript cDNA synthesis kit (Bio-Rad), as per the instructions of the manufacturer. DNA (1 μg) was taken and diluted 1:100. This was mixed with 500 nm of each primer and 10 μl of SYBR Green supermix (Bio-Rad). The volume was made up to 20 μl with nuclease free water. Real-time PCR was done on a MyCycler IQ system (Bio-Rad) with the following parameters: 1× at 95.0°C for 5 min and 40× at 95.0°C for 30 s, 62.0°C for 30 s, and 72.0°C for 30 s, followed by melt–curve analysis to ensure single products.
Primers were designed as follows: actin, left, 5-ACTGTGTTGGCATAGAGGTCTTTA-3; actin, right, 5-CTAGACTTCGAGCAGGAGATGG-3; mouse APP, left 5-GGGGCCGCAAGCAGTGCAAG-3; mouse APP, right 5-CCCCACCAGACATCAGAGT-3; BACE, left, 5-TCGCTGTCTCACAGTCATCC-3; and BACE, right, 5-AACAAACGGACCTTCCACTG-3.
Results
Glucocorticoids increase Aβ formation in vitro
We first determined whether glucocorticoids adversely affect APP processing in vitro using mouse neuronal N2A cells exposed to either corticosterone or the synthetic glucocorticoid dexamethasone (100 nm to 10 μm) for 24–72 h. Both corticosterone and dexamethasone increased levels of secreted Aβ40 and the longer, more amyloidogenic Aβ42 species in a concentration- and time-dependent manner as measured by sandwich ELISA (Fig. 1a,b). Dexamethasone treatment produced significantly greater increases in Aβ levels than did corticosterone treatment (∼10-fold vs 3-fold higher after 72 h, respectively), presumably because of different receptor affinities (Reul and de Kloet, 1985). To determine whether the effect involved activation of glucocorticoid receptors and/or mineralocorticoid receptors, we used selective antagonists to identify the receptor underlying the glucocorticoid-mediated enhancement of Aβ levels. The glucocorticoid/progesterone receptor antagonist mifepristone (RU 38486; 1 μm) completely blocked the increase in Aβ levels produced by dexamethasone (Fig. 1c). In contrast, the mineralocorticoid receptor antagonist spironolactone (RU 28318; 100 nm) did not significantly affect the dexamethasone-induced increase in Aβ levels. Based on these studies, we conclude that the primary mode of action by which glucocorticoids modulate Aβ levels appears to be mediated by binding to stress-activated glucocorticoid receptors.
Figure 1.

Glucocorticoids enhance Aβ production in a time- and concentration-dependent manner. N2A cells were incubated with dexamethasone (100 nm to 10 μm; n = 3 per condition) (a) or corticosterone (100 nm to 10 μm; n = 3 per condition) (b) for 24–72 h, and Aβ levels were measured from cell media by sandwich ELISA. Aβ42 levels were expressed as a percentage of control Aβ40, shown in the thatched area. *p < 0.05, significance versus controls for either Aβ40 or Aβ42. Control Aβ40 levels average out at ∼1 pg of Aβ40 microgram of protein for a 24 h period into 1 ml of media. c, N2A cells were incubated for 72 h with or without mifepristone (Mif; 10 μm; n = 3) or spironolactone (Spiro; 100 nm; n = 3) and in the presence or absence of 1 μm dexamethasone (Dex; n = 3 per condition), and media Aβ levels were measured. *p < 0.05, significance versus controls for either Aβ40 or Aβ42. d, Treatment of N2A cells with dexamethasone (Dex; 1 μm, 48 h; n = 3) or corticosterone (Cort; 5 μm, 48 h; n = 3) or control (Con; 48 h; n = 3) increases steady-state levels of full-length APP and selectively increases in C99 as shown by Western blot. e, Quantification of d with protein levels normalized to β-actin levels as a loading control. *p < 0.05, significance versus controls. f, mRNA levels of mouse APP and BACE are increased after 72 h treatment with 10 μm dexamethasone, as measured by real-time PCR. *p < 0.05, significance versus controls. g, Pulse chase analyses of 35S-labeled APP after treatment of N2A cells with 10 μm dexamethasone for 72 h (D; n = 3) or control (C; n = 3). Cells were pulsed with 35S, after starvation, for 1 h and chased at the 0, 1, 4, and 8 h time points.
Higher APP and BACE levels underlie the glucocorticoid-mediated enhancement of Aβ
Aβ formation is mediated by the enzymatic cleavage of its precursor proteins APP and C99, as well as changes in the steady-state levels of the APP holoprotein. Because glucocorticoid exposure augmented Aβ levels, we next investigated whether APP processing was specifically affected. We found that glucocorticoid treatment increased APP steady-state levels by 50% as shown by Western blot (Fig. 1d,e). Glucocorticoid treatment also induced a ∼40% increase in the C-terminal fragment of APP, C99, which is the immediate precursor to Aβ resulting from β-secretase cleavage of APP. These findings indicate that glucocorticoids act through low-affinity glucocorticoid receptors to increase both APP and C99 levels, which may be directly responsible for the observed elevation in Aβ levels.
Although rapid glucocorticoid effects have been reported (Dallman, 2005), the glucocorticoid–glucocorticoid receptor complex typically acts as a transcription factor, making it possible that glucocorticoid treatment increased Aβ production through a transcription-mediated pathway of its precursor, APP. The APP promoter is complex and resembles a housekeeping gene, but the presence of glucocorticoid-response elements within the APP promoter sequence has been reported (Lahiri, 2004). To determine whether these elements are responsive to glucocorticoids, we used real-time PCR to monitor APP mRNA levels after dexamethasone treatment. We found that APP mRNA levels were markedly increased, indicating that the higher steady-state levels of the holoprotein likely stemmed from either enhanced transcription and/or mRNA stabilization (Fig. 1f). Pulse-chase analysis with 35S-labeled Cys/Met revealed a similar increase in APP production during the 1 h pulse but no changes in APP degradation at the 1, 4, and 8 h chase time points (Fig. 1g). Together, these results indicate that the higher APP levels observed after glucocorticoid treatment are directly attributable to increases in APP production.
To generate Aβ, APP is cleaved at the β-secretase site by the enzyme BACE, which generates two fragments: a large ectodomain, sAPPβ, and C99. Recently, Sambamurti and colleagues identified several glucocorticoid-responsive elements within the BACE promoter (Sambamuri et al., 2004) and noted that these sites occur in a region of the promoter that positively influences transcription (Ge et al., 2004). Using real-time PCR, we found that BACE transcripts were increased significantly after dexamethasone exposure (10 μm, 72 h), accounting for the selective increase in C99 (Fig. 1f). Therefore, to our knowledge, we show for the first time, using an in vitro system, that exposure to glucocorticoids increases production of both APP and BACE, which further results in higher C99 and Aβ levels.
Dexamethasone increases intraneuronal Aβ in the 3×Tg-AD mice
We next investigated whether stress levels of glucocorticoids modulate Aβ and tau pathology in vivo. To address this issue, we used the 3×Tg-AD mouse model, which develops both Aβ and tau pathologies in a progressive and region-specific manner (Oddo et al., 2003). We first determined the pathological consequences of repeated glucocorticoid exposure on both pathologies in this model. Our initial efforts focused on determining the impact of elevated glucocorticoids on the onset of pathology. Consequently, 4-month-old male 3×Tg-AD mice, which exhibit only intraneuronal Aβ in the hippocampus, cortex, and amygdala, were administered either dexamethasone (1 or 5 mg/kg) or PBS intraperitoneally daily for 7 d and killed 24 h after the last injection. This treatment regimen has been shown previously to increase glucocorticoid-response element binding activity in the rat brain (Terzic et al., 2003). The 5 mg/kg dexamethasone treatment increased levels of detergent-soluble Aβ40 and Aβ42 levels by 60% in whole-brain homogenates compared with the PBS control group (Fig. 2a). At this young age, detergent-insoluble Aβ levels were also increased by the treatment (Fig. 2b). The 1 mg/kg dexamethasone treatment also augmented detergent-soluble Aβ42 levels but had no effect on insoluble Aβ (Fig. 2a,b), demonstrating that this more physiological concentration of dexamethasone was still sufficient to show an effect.
Figure 2.
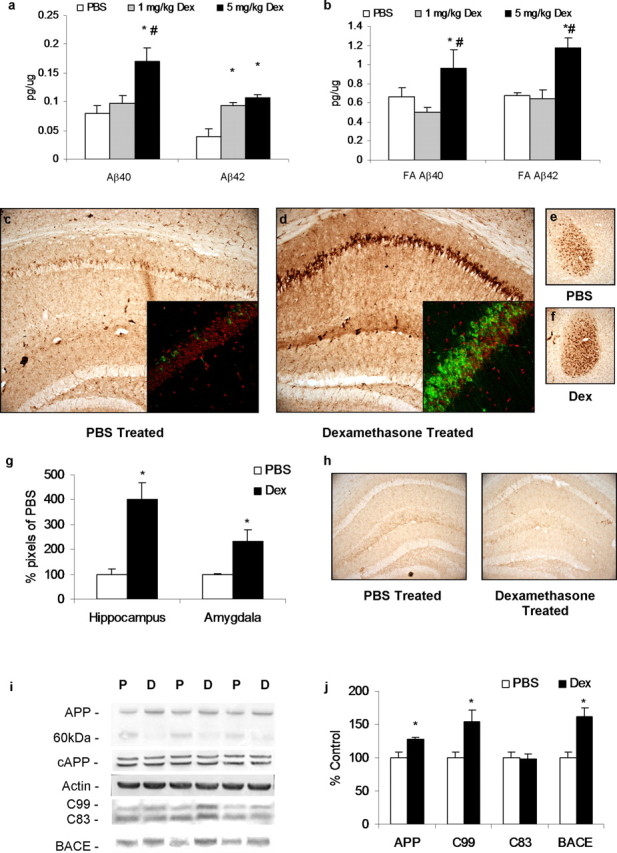
Dexamethasone treatment increases Aβ, C99, and BACE in the 3×Tg-AD mouse model. Four-month-old male 3×Tg-AD mice were treated daily for 7 d with 1 mg/kg (n = 5) or 5 mg/kg (n = 8) dexamethasone (Dex) or PBS vehicle (n = 8). a, Detergent-soluble Aβ levels were measured in whole-brain homogenates. Mice treated with 5 mg/kg dexamethasone (7 d; black bars) had significantly higher levels of Aβ40 and Aβ42 than vehicle-treated (PBS, 7 d; white bars) mice, whereas mice treated with 1 mg/kg dexamethasone (7 d; gray bars) had elevated Aβ42 compared with PBS controls. *p < 0.05, significance versus PBS controls for either Aβ40 or Aβ42; #p < 0.05, significance versus 1 mg/kg dexamethasone treatment. b, Detergent-insoluble Aβ40 and Aβ42 levels were significantly increased between 5 mg/kg dexamethasone treated and vehicle treated. c, e, DAB staining with 6E10 shows Aβ-like immunoreactivity in 40 μm sections from vehicle-treated mice. Hippocampus and amygdala regions are shown at 10× magnification. Inset demonstrates 40× confocal images with Aβ-like immunoreactivity shown in green and nuclear stain TOTO red shown in red. d, f, Same staining but in dexamethasone-treated animals. Aβ-like immunoreactivity was elevated in cell bodies of the hippocampus, as also shown by confocal imaging. g, Quantification of 6E10 DAB immunoreactivity in hippocampal and amygdala regions from PBS-treated and 5 mg/kg dexamethasone-treated groups. h, DAB staining with anti-CD45 shows that no activated microglia could be detected in the hippocampus of either PBS- or dexamethasone-treated animals. i, Western blot analyses of protein extracts from whole-brain homogenates of dexamethasone- and vehicle-treated 3×Tg-AD mice, shown as alternating lanes: P, PBS vehicle; D, 5 mg/kg dexamethasone treated. j, Quantification of protein blots from i shown normalized to β-actin levels as a loading control. Steady-state levels of APP are increased in the dexamethasone-treated group (6E10), whereas a 60 kDa band is similarly decreased, suggesting alternative processing. cAPP levels are unaltered, whereas C99 levels are increased but C83 levels remain unchanged. Furthermore, steady-state levels of BACE protein are increased concomitant to C99 levels. *p < 0.05, significance versus PBS controls.
Histological analysis of Aβ revealed that 5 mg/kg dexamethasone treatment enhanced Aβ-like immunoreactivity within cell bodies of the CA1 pyramidal neurons of the hippocampus, as well as in neurons of the cortex and amygdala (Fig. 2c–g; cortex not shown; no primary controls shown in Fig. 3f). Notably, the Aβ staining observed in these young mice was comparable with untreated 8- to 9-month-old 3×Tg-AD mice, thus demonstrating the profound consequence of glucocorticoid exposure on Aβ pathology in an in vivo setting. To determine whether the enhanced Aβ buildup was attributable to diminished clearance by microglia, because activated microglia can ingest Aβ (Webster et al., 2001), we immunostained sections with CD45, a marker of activated microglia. However, we failed to detect any differences in staining between treated and untreated mice or any activated microglia at all at this young age. Hence, it is unlikely that the increase in Aβ was attributable to a suppression of the microglial response (Fig. 2h). We reported previously that activated microglia associate with extracellular Aβ plaques in the 3×Tg-AD from ∼12 months of age (Kitazawa et al., 2005).
Figure 3.
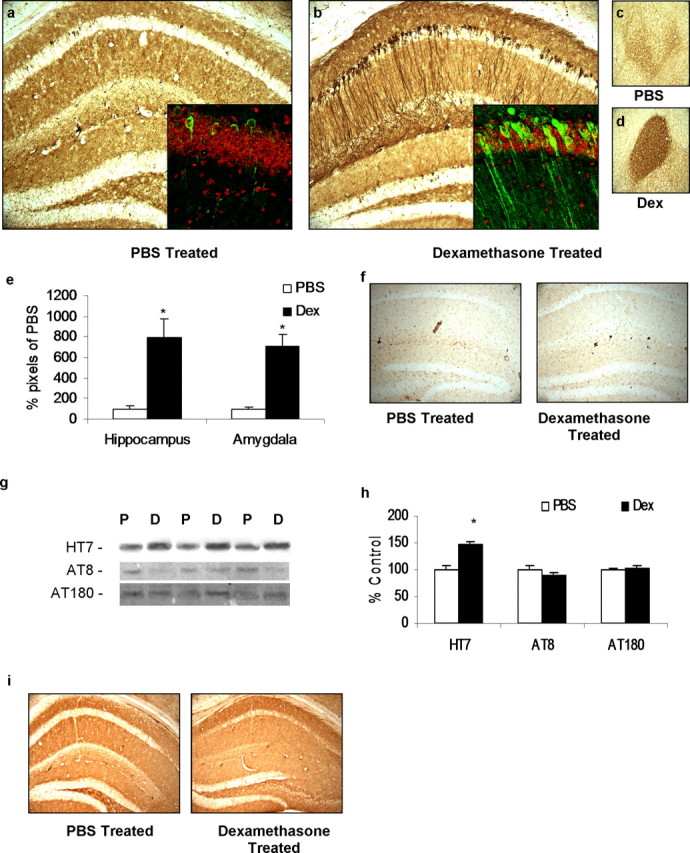
Dexamethasone (Dex) treatment in 3×Tg-AD mice increases tau accumulation. Four-month-old male 3×Tg-AD mice were treated daily for 7 d with 5 mg/kg dexamethasone (n = 8) or PBS vehicle (n = 8). a, c, Immunostaining with anti-tau antibody HT7 shows little immunoreactivity in 40 μm sections from PBS vehicle-treated mice. Hippocampus and amygdala regions are shown at 10× magnification. Inset demonstrates 40× confocal images with HT7 immunoreactivity shown in green and nuclear stain TOTO red shown in red. b, d, Same staining but in 5 mg/kg dexamethasone-treated animals. HT7 immunoreactivity was elevated in cell bodies of the hippocampus and throughout the neuronal processes, as also shown by confocal imaging. e, Quantification of HT7 DAB immunoreactivity in hippocampal and amygdala regions from PBS-treated and 5 mg/kg dexamethasone-treated groups. f, DAB staining of no primary controls shown from hippocampus of PBS-treated and 5 mg/kg dexamethasone-treated animals, 10×. g, Western blot analyses of protein extracts from whole-brain homogenates of dexamethasone- and vehicle-treated 3×Tg-AD mice, shown as alternating lanes (P, PBS vehicle; D, 5 mg/kg dexamethasone treated) showing increases in total human tau steady-state levels (HT7) but no differences in phospho-tau (AT8 and AT180). h, Quantification of protein blots from g shown normalized to β-actin levels as a loading control. i, 2×Tg mice (PS1M146VKI and tauP301L, lacking the human APP transgene) treated daily with dexamethasone (5 mg/kg, 7 d; n = 3) or vehicle (PBS, 7 d; n = 3). Staining with HT7 shows no immunoreactivity in 40 μm sections from vehicle-treated or 5 mg/kg dexamethasone-treated mice. Hippocampus region is shown at 10× magnification. DAB HT7 immunoreactivity was unchanged from vehicle.
Glucocorticoids elevate Aβ levels through increases in APP and BACE in vivo
Because glucocorticoid administration significantly increased Aβ levels in vivo, we attempted to elucidate the mechanism(s) underlying this effect. Our in vitro studies pointed to the higher Aβ levels resulting from increased production rather than diminished clearance of Aβ. Consequently, whole-brain homogenates from the 3×Tg-AD mice were analyzed by Western blot, which indicated that total APP levels were slightly increased in the 5 mg/kg dexamethasone-treated group versus the PBS-treated control mice (Fig. 2i,j). Staining with the APP antibody 6E10 (against amino acids 1–17 of Aβ) revealed a second band at ∼60 kDa, which was less abundant in the dexamethasone-treated group (corresponding to the increase in the APP holoprotein). This band was not cAPP, which has a similar weight (Gervais et al., 1999; Zhao et al., 2003), because we found no differences in cAPP steady-state levels (Fig. 2i,j). This band likely represents a breakdown product of APP, and this catabolic process may be decreased by dexamethasone treatment, resulting in an additional buildup of the holoprotein, and is not attributable to glucocorticoid-mediated transcription of the Thy1.2-driven APP transgene.
We next determined whether glucocorticoid administration altered APP processing by identifying various APP fragments by Western blot. Our results revealed a marked increase in C99 levels in the dexamethasone-treated group (Fig. 2i,j). In contrast, levels of C83, the fragment resulting from α-secretase cleavage of APP, were unaffected, suggesting a selective increase in BACE activity (Fig. 2i,j). Importantly, in the dexamethasone-treated group, steady-state BACE levels were increased to a degree that can account for the selective increase in C99 levels and subsequent Aβ production (50%) (Fig. 2i,j). Thus, these findings are consistent with our in vitro data and suggest that the BACE promoter, which has been reported to contain several glucocorticoid-response elements (Sambamurti et al., 2004), is significantly activated by glucocorticoid treatment. To our knowledge, these results provide the first in vivo demonstration that glucocorticoids potently modulate Aβ levels in a transgenic mouse model of AD, and our studies identified an upregulation of BACE and C99 as the mechanism underlying this effect.
Glucocorticoids accelerate accumulation of somatodendritic tau in the 3×Tg-AD mice
The 3×Tg-AD mice develop both amyloid plaques and neurofibrillary tangles, with early tau pathology beginning within the somatodendritic region of hippocampal neurons at ∼6 months of age, followed by conformational changes and hyperphosphorylation events at 9–12 months of age (Oddo et al., 2003). After dexamethasone treatment (5 mg/kg daily for 7 d), we assessed the consequences for tau pathology by immunostaining brain sections with antibody HT7, which recognizes all forms of human tau. We observed substantially increased HT7 staining in the somatodendritic compartment and along the axons in the hippocampus, cortex, and amygdala in the glucocorticoid-treated versus PBS-treated mice (compare with Fig. 3a–e; data for cortex not shown). To control for the possibility of dexamethasone increasing blood–brain barrier permeability to endogenous antibodies resulting in increased DAB staining, control experiments with no primary antibodies indicate that this is not the case (Fig. 3f). We also determined whether dexamethasone treatment affected the phosphorylation profile of tau in vivo. Analysis of the steady-state levels of tau phospho-epitopes failed to reveal any differences in levels of AT8- or AT180-positive tau (Fig. 3c,h), which are among the earliest phospho-tau epitopes to manifest in the 3×Tg-AD mice, although total tau levels were increased. Immunostaining with MC1, which recognizes conformationally altered tau, was similarly unaffected (data not shown). An increase in total tau, in the absence of conformational or phosphorylation alterations, suggests that dexamethasone induces increases in the steady-state levels of the protein but does not directly affect other pathways, such as those mediated by kinases and phosphatases, that are responsible for progressive tau pathology. Based on these studies, we conclude that glucocorticoid treatment causes tau mislocalization to the somatodendritic compartment, and it is plausible that longer treatments may lead to additional pathological alterations in tau, including alterations in its phosphorylation state.
Somatodendritic accumulation of tau is dependent on the presence of the APP transgene
The development of tau pathology in the 3×Tg-AD model seems to be markedly influenced by the presence of Aβ. For example, reducing Aβ levels by either immunotherapy or via γ-secretase inhibitors leads to the clearance of early pathological states of tau (Oddo et al., 2004). Likewise, genetic or pharmacological manipulations of Aβ have a direct impact on tau levels (S. Oddo and F. M. LaFerla, unpublished observations). Consequently, we sought to determine whether the observed effects on tau were a direct effect of glucocorticoids or an indirect consequence of higher Aβ levels. We treated 4-month-old male tauP301L/PS1M146V mice with dexamethasone using the same administration schedule used for the 3×Tg-AD mice. These double-transgenic mice express mutant tau to similar levels as the 3×Tg-AD mice but lack the human APP transgene and thus do not develop any Aβ pathology (Oddo et al., 2003). Importantly, in the tauP301L/PS1M146V mice, HT7 immunoreactivity was comparable in sections from the PBS- or dexamethasone-treated groups (Fig. 3i). Therefore, it appears that the accelerated tau pathology in the 3×Tg-AD mice is likely a downstream consequence of the effects of dexamethasone on Aβ. Importantly, these findings also rule out the possibility that dexamethasone nonspecifically upregulates transcription via the Thy1.2 promoter.
Glucocorticoid treatment increases the insoluble Aβ load in aged 3×Tg-AD mice
AD dementia correlates best with soluble Aβ loads (McLean et al., 1999), but the most visible lesions are the Aβ plaques and tau-laden neurofibrillary tangles. As we have shown, dexamethasone treatment in prepathological 3×Tg-AD mice increases both soluble Aβ and somatodendritic accumulation of tau but does not affect tau phosphorylation. We next determined the consequences of glucocorticoid treatment on plaque and tangle pathology in aged 3×Tg-AD mice. For this study, 13-month-old male 3×Tg-AD mice (n = 4) were injected daily with 5 mg/kg dexamethasone for 7 d and the brains were processed as before. Brains from 3×Tg-AD mice at this age harbor extracellular plaques in the cortex and immunoreactivity for specific phospho-tau epitopes. Soluble Aβ42 levels were increased in dexamethasone-treated animals compared with PBS vehicle-treated controls, although this increase did not achieve significance (Fig. 4a). Notably, insoluble Aβ40 (p < 0.05) and Aβ42 (p < 0.05) were significantly increased (Fig. 4b). Interestingly, these findings are different from our observations in prepathologic 3×Tg-AD mice, in which both soluble and insoluble Aβ levels were markedly increased. Conceivably, this could be attributable to variations in how Aβ is retained intracellularly at these different ages. In particular, the presence of increased Aβ exuded from the cells in the aged animals may explain a primary observation that insoluble Aβ plaques form with age.
Figure 4.

Dexamethasone (Dex) treatment increases insoluble Aβ in 13-month-old 3×Tg-AD mice. Thirteen-month-old male 3×Tg-AD mice were treated daily for 7 d with 5 mg/kg dexamethasone (n = 4) or PBS vehicle (n = 4). a, Detergent-soluble Aβ levels were measured in whole-brain homogenates. Mice treated with dexamethasone (5 mg/kg, 7 d; black bars) had significantly higher levels of Aβ40 and Aβ42 than vehicle treated (PBS, 7 d; white bars). b, Detergent-insoluble Aβ levels were significantly different between dexamethasone treated and vehicle treated. *p < 0.05, significance versus PBS controls for either Aβ40 or Aβ42. c, Western blot analyses of protein extracts from whole-brain homogenates of 13-month-old dexamethasone- and vehicle-treated 3×Tg-AD mice, shown as alternating lanes (P, PBS vehicle; D, 5 mg/kg dexamethasone treated) showing no differences in total or phosphorylated-tau (PHF). d, Quantification of protein blots from c shown normalized to β-actin levels as a loading control. e, Immunostaining with anti-tau antibody HT7 shows little immunoreactivity in 40 μm sections from 13-month-old PBS vehicle-treated 3×Tg-AD mice. Cortical region is shown at 10× magnification. f, Same staining but in dexamethasone-treated animals. DAB HT7 immunoreactivity was notably elevated in cell bodies and processes in the cortex, which was absent in PBS-treated vehicles.
We next investigated hyperphosphorylated tau levels in these aged, dexamethasone-treated animals. Western blot analysis revealed that there were no differences in tau hyperphosphorylation between dexamethasone- and PBS-treated mice (Fig. 4c,d), consistent with our findings in young, treated animals in which tau phosphorylation was unchanged with dexamethasone treatment. In contrast, HT7 tau immunostaining was increased in the cortex in cell bodies and axonal processes compared with the PBS vehicle controls, which had very little staining (Fig. 4e,f), whereas hippocampal staining was not further increased relative to PBS-treated mice (data not shown), presumably attributable to a “ceiling effect” because both PBS- and dexamethasone-treated hippocampi showed extensive staining. Interestingly, at this age when soluble Aβ levels are not increased by dexamethasone treatment, overall steady-state levels of tau, as measured by Western blot, are also not increased (Fig. 4c,d), strengthening the relationship between soluble Aβ and tau accumulation. These data once again demonstrate that short-term glucocorticoid treatment does not affect tau phosphorylation but does increase tau accumulation.
Endogenous plasma corticosterone levels are elevated in pathological 3×Tg-AD mice
In human subjects with sporadic AD, plasma cortisol levels are increased during both early and late stages (Umegaki et al., 2000). However, it remains unknown whether HPA axis dysfunction is an initiating factor in AD genesis or whether it arises as a result of decreased HPA axis regulation by limbic structures that are increasingly burdened with Aβ and tau pathology. To determine whether basal levels of circulating glucocorticoids are altered in a transgenic model of AD and whether Aβ and tau pathology precede or follow changes in circulating glucocorticoids, we investigated plasma corticosterone levels across a wide age range in 3×Tg-AD mice. Plasma was harvested from 2- to 18-month-old 3×Tg-AD mice, taken directly from their home cage. Until 9 months of age, basal plasma corticosterone levels were unchanged in 3×Tg-AD mice compared with age-matched nontransgenic (NonTg) mice, despite a steady accumulation of Aβ pathology over that period (Oddo et al., 2003; Billings et al., 2005). After 9 months of age, corticosterone levels were significantly elevated compared with age-matched NonTg mice (Fig. 5a), indicating that AD-like pathology in this mouse model induces dysfunction of the HPA axis. It has been shown previously that hippocampal CA3 lesions increase glucocorticoid levels, because the hippocampus plays a negative feedback role in regulating the HPA axis, which in turn causes cognitive deficits (Roozendaal et al., 2001). These higher glucocorticoid levels may then further exacerbate pathology, leading to a circle of detrimental positive feedback. We then looked at steady-state levels of glucocorticoid receptor levels in hippocampal homogenates from 4- and 15-month-old 3×Tg-AD mice and nontransgenic controls. We found no differences at either time point despite elevated corticosterone levels at 15 months (Fig. 5b,c).
Figure 5.

Plasma corticosterone levels from 3×Tg-AD mice compared with NonTg over time. a, Plasma obtained from 3×Tg-AD and NonTg mice was taken and corticosterone levels were measured at 2, 4, 6, 9, 12, 15, and 18 months of age. No significant changes are seen at 2, 4, and 6 months. At 9 months and older, 3×Tg-AD mice have significantly higher plasma corticosterone levels than NonTg mice. *p < 0.05, significance versus NonTg animals at that time point. b, Western blot analyses of protein extracts from hippocampal homogenates of 4- and 15-month-old NonTg and 3×Tg-AD mice (n = 4 per group), shown as alternating lanes (C, NonTg control; 3, 3×Tg-AD), showing no differences in glucocorticoid receptor (GR) steady-state levels. c, Quantification of protein blots from b shown normalized to β-actin levels as a loading control.
Discussion
To study the link between elevated levels of stress hormones and AD genesis, we investigated the effects of glucocorticoids on APP processing in vitro, as well as on the Aβ and tau burden in vivo. Consistent with stress being a risk factor for AD, we showed that administering glucocorticoids to young 3×Tg-AD mice increased soluble and insoluble Aβ, in which in aged mice it increased the insoluble Aβ load. In both young and aged mice, glucocorticoid administration also lead to the mislocalization of tau to the somatodendritic compartment, although phospho-tau levels were not affected. We also demonstrate that elevated glucocorticoid levels increase Aβ production by augmenting steady-state levels of APP and BACE in just 7 d. Both the APP and BACE genes contain glucocorticoid-response binding elements, making it likely that glucocorticoids directly increase transcription of the APP and BACE genes, leading to the increased Aβ production observed in vitro and in vivo. Furthermore, this effect occurs through activation of the glucocorticoid receptor, as an antagonist of this receptor type prevents glucocorticoid-mediated increases in Aβ, and the glucocorticoid receptor is widely known to mediate transcription during agonist binding, dimerization, and relocation to the nucleus (Wright et al., 1993). Increases in APP and BACE proteins lead to increased processing of APP to C99 by BACE, which is consequently cleaved by the γ-secretase to release Aβ.
We further demonstrate that glucocorticoid treatment increases tau accumulation in a manner that is dependent on the presence of the APP transgene. This finding supports our previous work showing that intraneuronal Aβ accumulation precedes the accumulation of tau (Oddo et al., 2003) and that removal of Aβ via immunotherapy also successfully clears tau provided that the tau is not yet hyperphosphorylated or the proteasome is not impaired (Oddo et al., 2004). Supporting a role for intraneuronal Aβ in AD, it has been shown that levels of soluble Aβ correlate well with cognitive decline (Lue et al., 1999; McLean et al., 1999; Naslund et al., 2000), whereas plaques correlate poorly (Braak and Braak, 1997). Furthermore, the 3×Tg-AD mouse develops intraneuronal Aβ accumulation in the cell bodies of neurons in the hippocampus before any plaque pathology develops and at an age at which both synaptic dysfunction and cognitive impairments are observed (Oddo et al., 2003; Billings et al., 2005). The sum of these findings suggests a direct relationship between intraneuronal Aβ and tau accumulation such that an increase in Aβ induces a concomitant increase in tau accumulation; likewise, a decrease in Aβ results in decreased tau accumulation. However, short-term glucocorticoid treatment does not affect the phosphorylation of tau. It is probable that the development of neurofibrillary tangles in AD results from the activation of different kinase/phosphatase pathways. Regardless, it is possible the elevated levels of tau may provide a larger pool of substrate available for hyperphosphorylation and downstream neurofibrillary tangle formation.
These findings have particular relevance to AD because it is established that early sporadic AD patients display elevated circulating cortisol (Davis et al., 1986; Masugi et al., 1989; Swanwick et al., 1998; Peskind et al., 2001). The causes underlying the higher circulating levels of cortisol are unknown and seem to be more relevant to the early stages of the disease (Swanwick et al., 1998), but clinical data suggest that a stressful lifestyle can be a risk factor for AD (Wilson et al., 2005). In addition, a strong risk factor for AD is the presence of the apoE4 allele, which has been shown to elevate CSF cortisol levels (Peskind et al., 2001) more so than the E3 or E2 allele. Taken with the present findings, these clinical data may suggest that elevated cortisol levels in early sporadic AD resulting from stress, administered exogenous glucocorticoids, or dysregulation of the HPA axis may contribute to pathology by increasing Aβ production. However it is unlikely that increased glucocorticoids lead to AD without fail, because a number of disorders result in increased cortisol, such as Cushing’s disease, yet there is no published link between these patients and AD. Therefore, it may be that increased glucocorticoids are a risk factor for AD, along with a number of other environmental and genetic risk factors or that they may exacerbate existing AD pathologies.
The underlying cause of AD is wholly unknown, although most researchers currently agree that various environmental and genetic factors are likely to act concurrently or even synergistically to trigger sporadic AD. The Aβ cascade hypothesis states that it is the buildup of Aβ that triggers AD through its downstream effects; although multiple findings support this argument, the precise mechanism(s) underlying the initial buildup of Aβ remains unknown. According to this leading hypothesis, any regimen that increases Aβ production should therefore also increase cognitive decline. Supporting this, prednisone, a glucocorticoid used to reduce inflammation, accelerated cognitive deterioration in AD patients (Aisen et al., 2000), and, although it is not yet known why this deterioration occurred, the results from the present study suggest that prednisone may have increased the pathological burden. Given our findings here, it seems plausible that increases in circulating glucocorticoids, either through pharmacological intervention or naturally through stress or AD-related increases, could lead to accelerated cognitive decline by increasing Aβ production, in line with the amyloid cascade hypothesis. However, because we were not investigating the effects of glucocorticoid-mediated increased AD pathology on cognition in the 3×Tg-AD mouse, we cannot say for certain what the cognitive outcome of this paradigm would be.
Glucocorticoid-related genetic susceptibility for AD has been identified through the 11β-hydroxysteroid dehydrogenase type 1 gene, in which a haplotype was identified with a sixfold higher risk for developing the disease (de Quervain et al., 2004). This dehydrogenase controls biologically active levels of glucocorticoids in tissues and thus links a possible increase in circulating glucocorticoids with the development of AD. Furthermore, in transgenic mouse models of AD, environmental enrichment, a paradigm that may reduce stressful housing conditions, has been shown to reduce Aβ pathology (Lazarov et al., 2005) as well as improve cognition (Jankowsky et al., 2005). It should be noted that anti-anxiety drugs are currently in clinical trials for treatment of AD, illustrating how reducing stress may be beneficial for disease prevention and treatment.
Our finding that the 3×Tg-AD mice develop elevated corticosterone levels concurrent with advancing pathology indicates that AD neuropathology could diminish negative feedback controlling glucocorticoid levels via dysregulation of the HPA axis. This increase in circulating glucocorticoids would likely increase Aβ pathology and subsequent tau accumulation, resulting in a positive feedback loop by which pathology increases circulating glucocorticoids, which further increase pathology. Because cortisol levels are elevated in early AD, the use of therapies aimed at reducing these levels or in preventing or reducing their efficacy should be investigated because they may prevent glucocorticoid-mediated increases in Aβ. The involvement of glucocorticoids in AD is universally thought to have detrimental effects on the pathology. It has been shown previously that dexamethasone treatment of microglia in culture impedes degradation of Aβ peptides (Harris-White et al., 2001), in which in vivo application resulted in more compact Aβ plaques containing more of the peptide. It has also been shown that elevated glucocorticoid levels are associated with decreased degradation of Aβ in aged macaques, through a downregulation of insulin-degrading enzyme (Kulstad et al., 2005), an Aβ-degrading enzyme. Conversely, elevated glucocorticoid levels have been shown to increase susceptibility of cholinergic neurons to Aβ42-mediated toxicity in vivo (Abraham et al., 2000) and hippocampal neuronal cultures (Goodman et al., 1996). Together with our observations, glucocorticoids mediate enhanced production of Aβ, reduce degradation, facilitate plaque formation, enhance Aβ-mediated neuronal toxicity, and increase tau accumulation. Therefore, perhaps therapies aimed at reducing glucocorticoids in the elderly and early AD patients would be beneficial, although the use of glucocorticoid-containing medications should be cautioned because they may be worsening pathology in humans. Our findings provide support for the hypothesis that elevated glucocorticoids found in AD are a consequence of the pathology and additionally, and importantly, play a significant causal role in the development of the pathology.
Footnotes
This work was supported in part by grants from Alzheimer’s Association, National Institute of Health Grant AG0212982 (F.M.L.), and National Institute of Mental Health Grant MH-12526 (J.L.M.). We thank Drs. Masashi Kitazawa, Salvatore Oddo, and Mathew Blurton-Jones for helpful discussion and Andrew Huy Tran for technical assistance.
References
- Abraham I, Harkany T, Horvath KM, Veenema AH, Penke B, Nyakas C, Luiten PG. Chronic corticosterone administration dose-dependently modulates Abeta(1–42)- and NMDA-induced neurodegeneration in rat magnocellular nucleus basalis. J Neuroendocrinol. 2000;12:486–494. doi: 10.1046/j.1365-2826.2000.00475.x. [DOI] [PubMed] [Google Scholar]
- Aisen PS, Davis KL, Berg JD, Schafer K, Campbell K, Thomas RG, Weiner MF, Farlow MR, Sano M, Grundman M, Thal LJ. A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology. 2000;54:588–593. doi: 10.1212/wnl.54.3.588. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, Laferla FM. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Diagnostic criteria for neuropathologic assessment of Alzheimer’s disease. Neurobiol Aging. 1997;18:S85–S88. doi: 10.1016/s0197-4580(97)00062-6. [DOI] [PubMed] [Google Scholar]
- Budas G, Coughlan CM, Seckl JR, Breen KC. The effect of corticosteroids on amyloid beta precursor protein/amyloid precursor-like protein expression and processing in vivo. Neurosci Lett. 1999;276:61–64. doi: 10.1016/s0304-3940(99)00790-9. [DOI] [PubMed] [Google Scholar]
- Dallman MF. Fast glucocorticoid actions on brain: back to the future. Front Neuroendocrinol. 2005;26:103–108. doi: 10.1016/j.yfrne.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Davis KL, Davis BM, Greenwald BS, Mohs RC, Mathe AA, Johns CA, Horvath TB. Cortisol and Alzheimer’s disease. I. Basal studies. Am J Psychiatry. 1986;143:300–305. doi: 10.1176/ajp.143.3.300. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Poirier R, Wollmer MA, Grimaldi LM, Tsolaki M, Streffer JR, Hock C, Nitsch RM, Mohajeri MH, Papassotiropoulos A. Glucocorticoid-related genetic susceptibility for Alzheimer’s disease. Hum Mol Genet. 2004;13:47–52. doi: 10.1093/hmg/ddg361. [DOI] [PubMed] [Google Scholar]
- Elliott EM, Mattson MP, Vanderklish P, Lynch G, Chang I, Sapolsky RM. Corticosterone exacerbates kainate-induced alterations in hippocampal tau immunoreactivity and spectrin proteolysis in vivo. J Neurochem. 1993;61:57–67. doi: 10.1111/j.1471-4159.1993.tb03537.x. [DOI] [PubMed] [Google Scholar]
- Ge YW, Maloney B, Sambamurti K, Lahiri DK. Functional characterization of the 5′ flanking region of the BACE gene: identification of a 91 bp fragment involved in basal level of BACE promoter expression. FASEB J. 2004;18:1037–1039. doi: 10.1096/fj.03-1379fje. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- Greenwald BS, Mathe AA, Mohs RC, Levy MI, Johns CA, Davis KL. Cortisol and Alzheimer’s disease. II. Dexamethasone suppression, dementia severity, and affective symptoms. Am J Psychiatry. 1986;143:442–446. doi: 10.1176/ajp.143.4.442. [DOI] [PubMed] [Google Scholar]
- Harris-White ME, Chu T, Miller SA, Simmons M, Teter B, Nash D, Cole GM, Frautschy SA. Estrogen (E2) and glucocorticoid (Gc) effects on microglia and A beta clearance in vitro and in vivo. Neurochem Int. 2001;39:435–448. doi: 10.1016/s0197-0186(01)00051-1. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Melnikova T, Fadale DJ, Xu GM, Slunt HH, Gonzales V, Younkin LH, Younkin SG, Borchelt DR, Savonenko AV. Environmental enrichment mitigates cognitive deficits in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25:5217–5224. doi: 10.1523/JNEUROSCI.5080-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulstad JJ, McMillan PJ, Leverenz JB, Cook DG, Green PS, Peskind ER, Wilkinson CW, Farris W, Mehta PD, Craft S. Effects of chronic glucocorticoid administration on insulin-degrading enzyme and amyloid-beta peptide in the aged macaque. J Neuropathol Exp Neurol. 2005;64:139–146. doi: 10.1093/jnen/64.2.139. [DOI] [PubMed] [Google Scholar]
- Lahiri DK. Functional characterization of amyloid β precursor protein regulatory elements: rationale for the identification of genetic polymorphism. Ann NY Acad Sci. 2004;1030:282–288. doi: 10.1196/annals.1329.035. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Robinson J, Tang YP, Hairston IS, Korade-Mirnics Z, Lee VM, Hersh LB, Sapolsky RM, Mirnics K, Sisodia SS. Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell. 2005;120:701–713. doi: 10.1016/j.cell.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masugi F, Ogihara T, Sakaguchi K, Otsuka A, Tsuchiya Y, Morimoto S, Kumahara Y, Saeki S, Nishide M. High plasma levels of cortisol in patients with senile dementia of the Alzheimer’s type. Methods Find Exp Clin Pharmacol. 1989;11:707–710. [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Molchan SE, Hill JL, Mellow AM, Lawlor BA, Martinez R, Sunderland T. The dexamethasone suppression test in Alzheimer’s disease and major depression: relationship to dementia severity, depression, and CSF monoamines. Int Psychogeriatr. 1990;2:99–122. doi: 10.1017/s1041610290000370. [DOI] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Nasman B, Olsson T, Viitanen M, Carlstrom K. A subtle disturbance in the feedback regulation of the hypothalamic-pituitary-adrenal axis in the early phase of Alzheimer’s disease. Psychoneuroendocrinology. 1995;20:211–220. doi: 10.1016/0306-4530(94)00054-e. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Peskind ER, Wilkinson CW, Petrie EC, Schellenberg GD, Raskind MA. Increased CSF cortisol in AD is a function of APOE genotype. Neurology. 2001;56:1094–1098. doi: 10.1212/wnl.56.8.1094. [DOI] [PubMed] [Google Scholar]
- Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- Roozendaal B. 1999 Curt P. Richter Award. Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology. 2000;25:213–238. doi: 10.1016/s0306-4530(99)00058-x. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Phillips RG, Power AE, Brooke SM, Sapolsky RM, McGaugh JL. Memory retrieval impairment induced by hippocampal CA3 lesions is blocked by adrenocortical suppression. Nat Neurosci. 2001;4:1169–1171. doi: 10.1038/nn766. [DOI] [PubMed] [Google Scholar]
- Sambamurti K, Kinsey R, Maloney B, Ge YW, Lahiri DK. Gene structure and organization of the human beta-secretase (BACE) promoter. FASEB J. 2004;18:1034–1036. doi: 10.1096/fj.03-1378fje. [DOI] [PubMed] [Google Scholar]
- Stein-Behrens B, Mattson MP, Chang I, Yeh M, Sapolsky R. Stress exacerbates neuron loss and cytoskeletal pathology in the hippocampus. J Neurosci. 1994;14:5373–5380. doi: 10.1523/JNEUROSCI.14-09-05373.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanwick GR, Kirby M, Bruce I, Buggy F, Coen RF, Coakley D, Lawlor BA. Hypothalamic-pituitary-adrenal axis dysfunction in Alzheimer’s disease: lack of association between longitudinal and cross-sectional findings. Am J Psychiatry. 1998;155:286–289. doi: 10.1176/ajp.155.2.286. [DOI] [PubMed] [Google Scholar]
- Terzic N, Vujcic M, Ristic-Fira A, Krstic-Demonacos M, Milanovic D, Kanazir DT, Ruzdijic S. Effects of age and dexamethasone treatment on glucocorticoid response element and activating protein-1 binding activity in rat brain. J Gerontol A Biol Sci Med Sci. 2003;58:297–303. doi: 10.1093/gerona/58.4.b297. [DOI] [PubMed] [Google Scholar]
- Travis J. New piece in Alzheimer’s puzzle. Science. 1993;261:828–829. doi: 10.1126/science.8346434. [DOI] [PubMed] [Google Scholar]
- Umegaki H, Ikari H, Nakahata H, Endo H, Suzuki Y, Ogawa O, Nakamura A, Yamamoto T, Iguchi A. Plasma cortisol levels in elderly female subjects with Alzheimer’s disease: a cross-sectional and longitudinal study. Brain Res. 2000;881:241–243. doi: 10.1016/s0006-8993(00)02847-x. [DOI] [PubMed] [Google Scholar]
- Webster SD, Galvan MD, Ferran E, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Antibody-mediated phagocytosis of the amyloid beta-peptide in microglia is differentially modulated by C1q. J Immunol. 2001;166:7496–7503. doi: 10.4049/jimmunol.166.12.7496. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Barnes LL, Bennett DA, Li Y, Bienias JL, Mendes de Leon CF, Evans DA. Proneness to psychological distress and risk of Alzheimer disease in a biracial community. Neurology. 2005;64:380–382. doi: 10.1212/01.WNL.0000149525.53525.E7. [DOI] [PubMed] [Google Scholar]
- Wright AP, Zilliacus J, McEwan IJ, Dahlman-Wright K, Almlof T, Carlstedt-Duke J, Gustafsson JA. Structure and function of the glucocorticoid receptor. J Steroid Biochem Mol Biol. 1993;47:11–19. doi: 10.1016/0960-0760(93)90052-x. [DOI] [PubMed] [Google Scholar]
- Zhao M, Su J, Head E, Cotman CW. Accumulation of caspase cleaved amyloid precursor protein represents an early neurodegenerative event in aging and in Alzheimer’s disease. Neurobiol Dis. 2003;14:391–403. doi: 10.1016/j.nbd.2003.07.006. [DOI] [PubMed] [Google Scholar]