

Abstract
Mitochondrial dysfunction is one of the major intracellular lesions of Alzheimer’s disease (AD). However, the causative factors involved in the mitochondrial dysfunction in human AD are not well understood. Here we report that nonglycosylated full-length and C-terminal truncated amyloid precursor protein (APP) accumulates exclusively in the protein import channels of mitochondria of human AD brains but not in age-matched controls. Furthermore, in AD brains, mitochondrially associated APP formed stable ∼480 kDa complexes with the translocase of the outer mitochondrial membrane 40 (TOM40) import channel and a super complex of ∼620 kDa with both mitochondrial TOM40 and the translocase of the inner mitochondrial membrane 23 (TIM23) import channel TIM23 in an “Nin mitochondria–Cout cytoplasm” orientation. Accumulation of APP across mitochondrial import channels, which varied with the severity of AD, inhibited the entry of nuclear-encoded cytochrome c oxidase subunits IV and Vb proteins, which was associated with decreased cytochrome c oxidase activity and increased levels of H2O2. Regional distribution of mitochondrial APP showed higher levels in AD-vulnerable brain regions, such as the frontal cortex, hippocampus, and amygdala. Mitochondrial accumulation of APP was also observed in the cholinergic, dopaminergic, GABAergic, and glutamatergic neuronal types in the category III AD brains. The levels of translocationally arrested mitochondrial APP directly correlated with mitochondrial dysfunction. Moreover, apolipoprotein genotype analysis revealed that AD subjects with the E3/E4 alleles had the highest content of mitochondrial APP. Collectively, these results suggest that abnormal accumulation of APP across mitochondrial import channels, causing mitochondrial dysfunction, is a hallmark of human AD pathology.
Keywords: mitochondria, protein import, translocational arrest, amyloid precursor protein, Alzheimer’s disease, amyloid-β
Introduction
The neuropathological hallmarks of Alzheimer’s disease (AD) include extensive neuronal loss, the presence of numerous neurofibrillary tangles (NFTs), and senile plaques in the brain, which are common to both early onset (familial) and late onset (sporadic) AD (Price and Sisodia, 1998; Selkoe, 1999). The NFTs are composed of hyperphosphorylated tau protein. The major constituent of a senile plaque is β-amyloid (Αβ), which is a 40–43 amino acid peptide produced by the action of secretory pathway-associated proteases, namely β and γ secretases, at the C terminus of a type I membrane-spanning glycoprotein termed amyloid precursor protein (APP). The γ secretase, a multimeric complex, consists of presenilin (PS), nicastrin, Aph1, and Pen2. PS is involved in the catalytic activity of the γ secretase complex (Periz and Fortini, 2004; Brunkan and Goate, 2005).
In addition to NFTs and Aβ plaques, pathobiology of AD is also associated with a number of intracellular lesions, including mitochondrial dysfunction (Reddy and Beal, 2005) and the accumulation of intracellular Aβ (Busciglio et al., 2002; Gouras et al., 2005). Decreased energy metabolism is one of the earliest detectable defects in AD. Altered mitochondrial fluidity and the activity of mitochondrial cytochrome c oxidase, which is the terminal oxidase in the respiratory chain, have been reported to be low in the AD brains (Parker, 1991; Sims, 1996; Maurer et al., 2000). Furthermore, a correlation was reported between decreased cytochrome c oxidase activity and deficits in cognitive abilities (Cada et al., 1995; Agin et al., 2001; Berndt et al., 2001). Nevertheless, the mechanisms related to the impairment of mitochondrial functions specific to AD remain unclear. Furthermore, mitochondrial dysfunction may vary with apolipoprotein E (ApoE) genotype, which is thought to be one of the major risk factors for the late onset AD (Gibson et al., 2000; Kamboh, 2004). However, it is not clear whether these factors/lesions are independent of each other or inter-related in the pathogenesis of AD.
Recently, a study from our laboratory suggested a direct relationship between mitochondrial dysfunction and mitochondrial targeting of APP (Anandatheerthavarada et al., 2003). Using a human cortical neuronal (HCN) culture system, we showed that full-length APP is targeted to both mitochondria and plasma membrane (PM) by virtue of N-terminal chimeric signals. Interestingly, mitochondrial translocation of APP was incomplete because of an internal acidic domain present between 220 and 290 amino acids. Progressive mitochondrial accumulation of incompletely translocated APP caused mitochondrial dysfunction and impaired cellular energy metabolism. In support of this study, a recent report showed the presence of full-length APP in mitochondria of PC12 cell lines stably transfected with APP cDNA (Keil et al., 2004). However, the precise role and the generality of the process of mitochondrial targeting of APP in the human brain and its role in AD pathology remain unknown. In the present study, using postmortem human brains, we investigated whether APP is accumulated in the mitochondrial compartment of AD brain and the relationship between mitochondrially accumulated APP and mitochondrial dysfunction, as well as the ApoE4 factor in the pathogenesis of AD.
Materials and Methods
Human subjects.
Individual fresh brain hemispheres of both genders from humans clinically diagnosed with dementia (n = 20) and nondementia (n = 20), obtained at autopsy, were procured from National Disease Resource Interchange (NDRI) (Philadelphia, PA). All procedures used to collect postmortem human brains from NDRI were approved by the Institutional Review Board. The macroscopic examination of hemispheres from dementia and nondementia subjects was performed according to The National Institute on Aging (NIA)–Reagan criteria (The National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease, 1997). The fresh brain hemisphere was weighed and sliced coronally to examine volumetric changes in neocortex, hippocampus, and substantia nigra to evaluate the AD-specific changes. Eleven brain regions, specifically, frontal cortex (FC), parietal cortex (PC), occipital cortex, temporal cortex, hippocampus (HP), amygdala (AG), cerebellum, brainstem, cingulum, basal ganglia, and thalamus, were separated out as described previously (Montemurro and Brunni, 1998). Small portions of each fresh brain region were fixed in 10% buffered formaldehyde and 4% buffered paraformaldehyde containing 2% glutaraldehyde to perform immunohistochemistry and immunoelectron microscopy studies, respectively. Simultaneously, another portion of each brain region was used for isolating subcellular fractions without additional delay.
Neuropathology.
The dementia cases used in the study were late onset. Neuropathological assessments of postmortem brains were performed according to the NIA–Reagan Institute recommendations (The National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease, 1997). Tissues from all 11 brain regions of dementia and nondementia cases were double stained with thioflavin S and polyclonal antibodies to Aβ (Zymed, San Francisco, CA), which detects both Aβ1–40 and Aβ1–42 species to identify amyloid plaques as described previously (Bussiere et al., 2004). NFTs were stained using polyclonal antibodies to NFTs (Serotec, Indianapolis, IN). Plaques showing both thioflavin S and Aβ staining were identified and counted from three individual serial sections under a fluorescent microscope (Olympus Optical, Thornwood, NY). The final Aβ plaque load was considered as the average number of plaques per square millimeter.
ApoE genotyping.
Apolipoprotein E genotyping was performed in the Molecular Diagnosis and Genotyping Facility at the University of Pennsylvania. Briefly, genomic DNA was isolated from brain samples using standard protocol. DNA replicates, at a concentration of 5 ng/ml, and the PCR mix were plated in a Biomek (Beckman Coulter, Fullerton, CA) multiwell plate. The plate was vortexed, centrifuged, and thermocycled (MJ Research, Watertown, MA) for 40–60 cycles, scanned on an ABI7900HT (Applied Biosystems, Foster City, CA), and analyzed using ABI Auto Call software (SDS version 2.1; Applied Biosystems). Results were confirmed by PCR-coupled restriction-length polymorphism.
Triple immunohistochemistry.
The fresh brain tissues (frontal cortex and striatum) from AD1 (category I), AD11 (category II), AD16 (category III), and nondementia control (NC17) subjects were fixed in 10% buffered Formalin. Paraffin-embedded tissues were cut into 10 μm serial sections using a microtome (Olympus Optical). Serial sections from frontal cortex [Broddman’s area 10 (BM10)] were blocked in PBS buffer (containing 0.05% Triton X-100 and 5% donkey serum) and further sequentially stained overnight at 4°C with mouse monoclonal antibodies raised to the N-terminal 18–38 amino acids of APP (APP Nt Ab) (Calbiochem, La Jolla, CA) and rabbit polyclonal anti- translocase of the outer mitochondrial membrane 40 (TOM40) (Santa Cruz Biotechnology, Santa Cruz, CA). Then sections were washed with PBS (three times for 5 min) and stained separately with either goat polyclonal anti-choline acetyltransferase (ChAT) (Chemicon, Temecula, CA) or goat polyclonal anti-glutamate decarboxylase (Santa Cruz Biotechnology) to identify cholinergic and GABAergic neurons, respectively. After the primary antibody staining, sections were washed with PBS (three times for 5 min) and stained sequentially with appropriate secondary antibodies conjugated to Alexa 488 (green), Alexa 594 (red) or Alexa 350 (blue) (Invitrogen, Carlsbad, CA) for 1 h at room temperature. Mitochondrial localization of APP in glutamatergic neurons was performed using mouse monoclonal anti-APP Nt, goat polyclonal anti-TOM40 (Santa Cruz Biotechnology) and rabbit polyclonal anti-glutamate (Chemicon). Colocalization of APP in dopaminergic neurons was investigated by staining the sections from basal ganglia (striatal area) with goat polyclonal antibodies raised to the N-terminal 44–63 amino acids of APP (Calbiochem), anti-rabbit polyclonal TOM40 (Santa Cruz Biotechnology), and mouse monoclonal-anti tyrosine hydroxylase (Sigma, St. Louis, MO) as described above. The sections were then subjected to confocal fluorescence microscopy (Meta 510; Zeiss, Oberkochen, Germany). Control staining of both AD and NC brain sections was also performed using preabsorbed APP antibodies against the antigenic peptides to examine the specificity of the APP staining.
Immunoelectron microscopy.
Triple-labeling immunoelectron microscopy was performed in the Biomedical Imaging Core Laboratory at the University of Pennsylvania as described previously (Anandatheerthavarada et al., 2003) with little modifications. Fresh tissues from the frontal cortex (BM10 area) of AD1 and NC17 were fixed in 20 mm phosphate buffer containing 4% formaldehyde and 2% glutaraldehyde immediately to avoid ice crystal formation, which will disrupt the cell morphology. Ultrathin sections were incubated with mouse monoclonal APP Nt Ab (Calbiochem) and rabbit polyclonal Aβ Ab (Zymed), as well as antibody to goat polyclonal TOM40 Ab (Santa Cruz Biotechnology) for 24 h at 4°C. The grids were rinsed with TBS with Tween 20 for 15 min and incubated with anti-goat (20 nm), anti-mouse (10 nm), and anti-rabbit (5 nm) colloidal gold-conjugated IgGs. The sections were examined and photographed in an electron microscope (JEM-1010; Jeol, Peabody, MA).
Immunoblot analysis of mitochondrial Aβ.
Mitochondrial proteins (50 μg) from the frontal cortex of AD1 and NC17 were separated on 12% tricine gels and transferred to polyvinylidene difluoride (PVDF) membrane. The blot was immunostained with rabbit polyclonal antibody to Aβ, and the bands were developed using the Pierce (Rockford, IL) Super Signal ULTRA chemiluminescent substrate kit in a Versa Doc (Bio-Rad, Hercules, CA) imaging system.
Isolation of subcellular fractions.
Approximately 1 g of brain tissue was homogenized in 4 vol of homogenization medium containing the following (in mm): 70 sucrose, 210 mannitol, 2 HEPES, and 0.1 EDTA (H medium). Homogenate was centrifuged at 2000 × g for 10 min. The pellet was resuspended in isolation buffer and processed for plasma membrane as described previously (Dasso, 1998). The resultant supernatant was centrifuged at 18,000 × g for 20 min to obtain the crude mitochondrial pellet, which was layered on 0.8 m sucrose and centrifuged at 22,000 × g for 30 min to remove myelin contamination. The pellet was further treated with digitonin, as described previously (Brown et al., 2004), to remove microsomal and synaptosomal membrane contamination. The final pellet was termed total mitochondria. Postmitochondrial supernatant was centrifuged at 100,000 × g to obtain the microsomal pellet. Subcellular fractions were resuspended in appropriate volumes of 50 mm phosphate buffer, pH 7.4, containing 10% glycerol and protease inhibitor cocktail (Roche, Indianapolis, IN) and were subjected to biochemical analysis without additional delay. Protein concentration was measured by the Lowry assay (Lowry et al., 1951). Cross-contamination of mitochondrial fractions with nonmitochondrial membranes was investigated by Western immunoblotting with antibodies to PM marker proteins Na+/K+ ATPase (Sigma), synaptosomal marker synaptophysin (Chemicon), and mitochondrial marker translocase of the inner mitochondrial membrane 44 (TIM44) (Santa Cruz Biotechnology).
Limited trypsin digestion.
One-hundred fifty micrograms of freshly isolated mitochondria was suspended in H medium and subjected to trypsin digestion (30 μg of trypsin per milligram of protein) on ice for 45 min. The reaction was terminated by adding trypsin inhibitor (300 μg/mg). The mitochondrial suspension was sedimented through 0.8 m sucrose, and the final pellet was suspended in H medium. Mitochondrial proteins were separated on a 12% tricine gel for immunoblot analysis with APP Nt and Aβ antibodies.
Glycosidase treatment.
Mitochondrial and PM proteins (150 μg) were subjected to deglycosylation in 50 μl of 100 mm Tris-HCl buffer, pH 8.6, with 2 mU of PNGase F (Roche) at 37°C for 12 h. The membranes were reisolated and suspended in 100 mm sodium citrate buffer, pH 6.0, containing 0.1% SDS, 1% NP-40, and 2 mU O-glycanase (Roche). After overnight incubation at 37°C, the reaction mixture was mixed with 0.2 vol of 5× Laemmeli’s buffer and subjected to SDS-PAGE, followed by immunoblotting using mouse monoclonal APP Nt Ab.
Blue native gel electrophoresis.
Blue native (BN)-PAGE was performed as described previously (Schagger and von Jagow, 1991) with modifications. Total mitochondria (400 μg) from the frontal cortex (BM10) of AD1, AD11, AD16, NC2, NC15, and NC17 were resuspended in extraction buffer containing 0.75 m aminocaproic acid, 50 mm BisTris, pH 7.0, and n-dodecyl-β-d-maltoside (2% w/v). The mixture was incubated for 30 min on ice. The samples were centrifuged at 72,000 × g for 30 min, and Coomassie Brilliant Blue G-250 (Serva, Garden City Park, NY) in 0.75 m aminocaproic acid was added to the supernatant to give a final concentration of 0.3% w/v. The complexes were separated on a 5–12% acrylamide gel using 50 mm BisTris as an anode buffer and 15 mm BisTris/50 mm tricine containing 0.02% Coomassie Brilliant Blue G-250 as a cathode buffer. Before blotting, the gels were soaked in Tris–glycine transfer buffer containing 0.02% SDS for 10 min, and the proteins were transferred to PVDF membranes (Bio-Rad) at 100 V for 2 h and probed with specific antibodies. The blots were developed using the Pierce Super Signal ULTRA chemiluminescent substrate kit in a Versa Doc (Bio-Rad) imaging system and quantitated using Quantity One software.
Measurement of mitochondrial cytochrome c oxidase activity and H2O2.
Cytochrome c oxidase activity in mitochondria from the frontal cortex of AD1, AD11, AD16, NC2, NC15, and NC17 was measured as described previously (Vijayasarathy et al., 1999). The rate of oxidation of ferrocytochrome c was measured by a decrease in absorbance at 550 nm. Assays were performed in 1 ml reaction mixture containing 50 mm potassium phosphate buffer, pH 7.0, 0.05% lauryl maltoside, 1 mm EDTA, and 5 μg of mitochondrial protein. The reaction was started by the addition of 80 μm ferrocytochrome c. Reduction in absorbance was monitored for 1 min. Cytochrome c oxidase activity was determined using an absorbance coefficient (reduced–oxidized) at 550 nm of 21.1 mm/cm. Values were expressed as micromoles cytochrome c oxidized/ per minute per milligram of protein.
H2O2 levels in the above mentioned AD and NC mitochondrial isolates (100 μg) were measured using the Fluoro H2O2 kit from Cell Technologies, according to protocol of the manufacturer. The final fluorescent product, resorufin, was measured at the excitation wavelength 570 nm and the emission wavelength 600 nm in a BioScan (Washington, DC) Multilabel Detection plate reader. Simultaneously, a standard curve using H2O2 was also run. The final values were expressed as micromoles of H2O2 per milligram of mitochondrial protein.
In vitro mitochondrial import of proteins.
Mitochondria from the frontal cortex (BM10) region of AD1, AD11, AD16, NC2, NC15, and NC17 were isolated essentially as described above. [35S]-labeled translation products for cytochrome c oxidase subunits IV and Vb were generated in a transcription-coupled translation system (Promega, Madison, WI) according to the protocol of the manufacturer. Import of in vitro-synthesized proteins was performed in a 200 μl reaction volume as described previously (Addya et al., 1997; Anandatheerthavarada et al., 2003). The incubation mixture was subjected to trypsin treatment (150 μg/ml reaction), and mitochondria were reisolated by layering through 0.8 m sucrose and subsequent centrifugation at 20,000 × g for 20 min. Mitochondrial proteins were separated by SDS-PAGE and subjected to fluorography to visualize the mitochondrially protected [35S]-labeled cytochrome c oxidase subunits IV and Vb.
Measurement of mitochondrial APP.
The total mitochondrial APP in various brain regions was measured by semi-quantitative Western immunoblot using mouse monoclonal APP Nt Ab and further confirmed with the quantitative Human APP Elisa kit (Biosource, Camarillo, CA) according to the protocol of the manufacturer.
MTT assay.
The conversion of MTT to formazan by mitochondria was performed as described previously (Duan et al., 1999). Mitochondria (200 μg of protein) were incubated with MTT dye (0.5 mg/ml) in a 400 μl reaction volume at 37°C for 90 min. After the incubation, mitochondria were isolated and suspended in DMSO (100%), and the absorbance of the solution was measured spectrophotometrically at 592 nm.
Statistical analysis.
Statistical analyses were performed using Origin 7.5 software (Microcal Software, Northampton, MA). The correlation between mitochondrial total APP content and MTT activity was statistically analyzed using scatter plots, which were first generated for two variables. Next, linear regression analysis was performed to measure the association between variables. Statistical analysis and the level of significance were assessed by one-way ANOVA, followed by post hoc Tukey–Kramer.
Results
Neuropathological assessment of postmortem subjects
The average postmortem intervals for dementia (supplemental Table 1, available at www.jneurosci.org as supplemental material) and nondementia (supplemental Table 2, available at www.jneurosci.org as supplemental material) subjects used in the study are 6 ± 2 and 8 ± 1 h, respectively. We performed the evaluation of amyloid plaque load and NFT in various brain regions of both dementia and nondementia subjects according to NIA–Reagan Institute neuropathological criteria (The National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease, 1997). Based on this, we divided dementia subjects into three categories (supplemental Table 1, available at www.jneurosci.org as supplemental material). Categories I, II, and III fell under moderate, moderate/high, and high probabilities of AD, respectively. Macroscopic examination showed a marked shrinkage of neocortex and hippocampus in the subjects belonging to categories II and III, but not category I, compared with nondementia subjects. However, none of the AD categoriesshowed a visible shrinkage of the substantia nigra compared with nondementia subjects, suggesting the occurrence of AD-specific abnormalities in dementia subjects. Brain regions from nondementia subjects showed no staining for Aβ-specific amyloid plaque or NFT, indicating that these subjects do not fall under AD. Dementia and nondementia subjects are referred to as AD and NC subjects, respectively, throughout the manuscript.
Purity of the isolated mitochondrial fractions
First, we investigated the cross-contamination of isolated total mitochondria from frontal cortex of AD (1 and 16), as well as age-matched NC (15 and 17) brains, with PM and synaptosomal membranes by Western immunoblot analysis using antibodies to synaptic membrane marker synaptophysin (Fig. 1A, S.P), mitochondrial marker TIM44 (Fig. 1B), and PM marker Na+/K+ ATPase (Fig. 1C). None of these marker proteins, except mitochondrial TIM44, were detected in the mitochondrial fractions isolated from both AD and NC subjects (Fig. 1B), suggesting that mitochondria isolated from AD and NC subjects were devoid of synaptosomal membrane and PM contamination.
Figure 1.
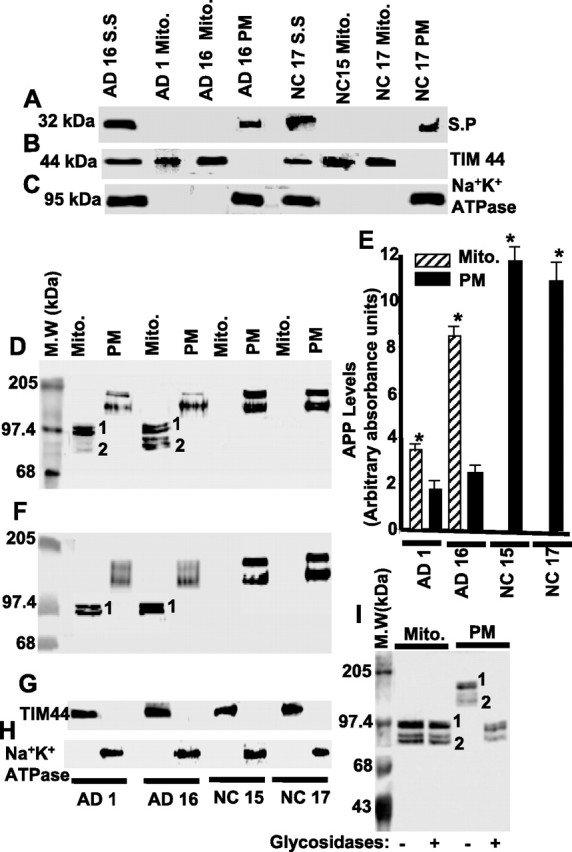
Mitochondrial targeting of APP in human frontal cortex of AD and NC brains. Immunoblot analysis of marker proteins for synaptosomal (S.S), mitochondrial (Mito.), and PM fractions from frontal cortex of AD and NC using antibodies to synaptophysin (S.P) (A), TIM44 (B), and Na+/K+ ATPase (C). Levels of APP in mitochondrial and PM fractions of AD and NC brains (frontal cortex) by immunoblot using APP Nt Ab (D) and Aβ Ab (F). E, Quantitation of immunoblot shown in D. G and H indicate the levels of TIM44 and Na+/K+ ATPase proteins in Mito and PM fractions by western immunoblotting using antibodies to TIM44 and Na+/K+ ATPase, respectively. Fifty micrograms of protein of each subcellular fraction were used in all cases. I, Immunoblot analysis of glycosidase-treated mitochondrial and PM fractions (100 μg each) from frontal cortex of AD16 brain using APP Nt Ab. M.W, Molecular weights. *p < 0.05 compared with AD1 PM fractions. Mean ± SEM calculated from three separate blots.
Accumulation of APP in mitochondria of AD brains
Next, we investigated whether APP is targeted to mitochondria in human AD and NC brains by Western immunoblot analysis using mouse monoclonal antibody specific to the APP N terminus (APP Nt Ab) that recognizes all APP isoforms. In the mitochondrial fraction from the AD16 (category III), a significant accumulation of APP forms, ranging from 80 to 97 kDa bands, migrating in two major clusters was observed (Fig. 1D). The molecular forms migrating as 90–97 kDa band formed cluster 1, whereas cluster 2 contained the 80–85 kDa forms (Fig. 1D). In contrast to AD16, the mitochondrial fraction from AD1 showed major cluster 1 and minor cluster 2 bands (Fig. 1D). Surprisingly, the immunoreactive APP forms in mitochondria of NC15 and NC17 brains were undetectable (Fig. 1D). PM fractions from AD (1 and 16) as well as NC (15 and 17) subjects showed the constitutive levels of APP forms migrating in two clusters at the range of 110–130 kDa that were recognized by APP Nt Ab (Fig. 1D). However, the level of total APP (clusters 1 and 2) in PM fractions from NC (15 and 17) subjects were approximately sixfold higher than that of AD (1 and 16) subjects (Fig. 1D,E). Interestingly, mitochondrial fractions from AD brains had significantly higher (twofold to fourfold) levels of total APP than corresponding PM fractions (Fig. 1D,E). To determine the identity of APP forms present in the two clusters of mitochondrial and PM fractions, we probed these fractions with C-terminal Aβ-specific Abs. In AD brain mitochondria, only the slower migrating bands (cluster 1) were recognized by the Aβ Ab (Fig. 1F). However, the faster migrating bands (cluster 2) did not react with Aβ Ab, indicating that they were C-terminal-truncated APP forms. On the contrary, Aβ Ab recognized both of the clusters in PM fractions from AD and NC brains (Fig. 1F). Nevertheless, the levels of mitochondrial TIM44 (Fig. 1G) and PM-associated Na+/K+ ATPase (Fig. 1H) were constant in AD and NC brains. These results suggest that the full-length and Aβ-lacking APP forms accumulate in the AD brain mitochondria. In support of this conclusion, various APP N-terminus antibodies such as goat anti-APP N-terminus 44–63, rabbit anti-APP N-terminus 99–126, and mouse anti-APP N-terminus 66–81 (Chemicon) cross-reacted with cluster 1 proteins but not with cluster 2 proteins (data not shown).
Mitochondrial accumulated APP in AD brain is nonglycosylated
We further investigated the differential mobility of AD mitochondrial and PM-associated APP forms by subjecting them to treatment with glycosidases. Western immunoblot analysis with APP Nt Ab showed that, after the treatment with glycosidases, mobility of PM-associated APP forms (clusters 1 and 2) were reduced to 90–97 kDa (Fig. 1I). In contrast, the mobility of mitochondrial APP, both full-length and C-terminal-truncated forms (clusters 1 and 2, respectively) were unaffected (Fig. 1I), suggesting that the mitochondrial-associated APP forms in AD brain are nonglycosylated, whereas those of PM are glycosylated.
Incomplete translocation of APP in AD mitochondria
We examined the orientation of mitochondrial associated APP in AD1 frontal cortex using limited trypsin digestion, followed by probing with APP Nt Ab and Aβ Ab. Results showed that only the APP Nt Ab, but not the Aβ Ab (Fig. 2B), was able to detect the mitochondrial associated ∼22 kDa APP fragment (Fig. 2A), suggesting that a part of APP (∼220 amino acid length of N terminus) is localized inside the mitochondria and the rest, which is accessible to trypsin treatment, is extra mitochondrial. After the treatment with trypsin, PM-associated APP was not recognized by either APP Nt Ab or Aβ Ab, indicating the complete accessibility of PM-associated APP forms to trypsin. To substantiate these biochemical observations, we performed triple-labeling immunoelectron microscopy of the frontal cortex (BM10; AD1) of AD brain using APP Nt Ab, Aβ Ab, and TOM40 Ab simultaneously. We then costained with colloidal gold-conjugated secondary antibodies, each specific for a given antibody. The anti-APP Nt Ab was conjugated to 10 nm gold particles (#3), the anti-Aβ Ab to 5 nm gold particles (#1), and the anti-TOM40 Ab to 20 nm gold particles (#2). The electron micrograph in Figure 2C shows that APP-specific (#1 and #3) electron-dense particles are associated with mitochondria (M) in the AD1 brain compared with that of NC17 brain, which showed only TOM40 reactivity (#2) (Fig. 2D). Interestingly, the location of N-terminal-specific (#3) and C-terminal-specific (#1) labeling suggest that the mitochondrial associated APP is organized in an “N in mitochondria–C out cytoplasm” orientation and is possibly traversing the channel-forming TOM40 protein (#2). In support of the Western blot data (Fig. 1D,F), some portions of mitochondria showed the presence of intramitochondrial APP N-terminal (#3) and TOM40 (#2) but not Aβ (#3) staining, suggesting that some APP molecules lack the Aβ-containing C terminus (Fig. 2C). In addition, we also observed the immunoreactivity for Aβ alone (#1) inside the mitochondria, which was not associated with either APP N terminus (# 3) or TOM40 (#2). As expected, endoplasmic reticulum (ER) showed only APP N-terminal (#3) and C-terminal Aβ (#1) staining but not TOM40-specific labeling in AD1 brain (Fig. 2C). These results collectively suggest that the APP protein may be incompletely translocated to mitochondria and is in direct contact with the channel-forming TOM40 protein in AD brain.
Figure 2.
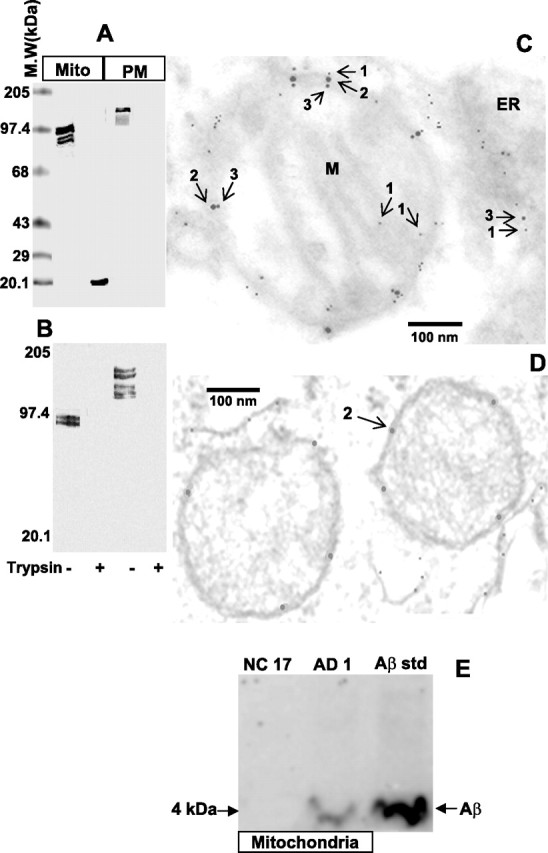
Topology of mitochondrial APP in the frontal cortex of AD brain. Limited trypsin treatment, followed by Western immunoblot analysis of mitochondrial (Mito) and PM fractions (100 μg) was performed using APP Nt Ab (A) and Aβ Ab (B) from AD16 frontal cortex. Immunoelectron microscopy of AD1 (C) and NC15 (D) frontal cortex (BM10 area) using APP Nt Ab, TOM40 Ab, and Aβ Ab. M.W, Molecular weights; M, mitochondrion; arrow 1, Aβ containing COOH terminus of APP (5 nm gold particle); arrow 2, TOM40 (20 nm gold particle); arrow 3, NH2 terminus of APP (10 nm gold particle). E, Western immunoblot analysis of Aβ in the mitochondrial fractions (50 μg) isolated from AD1 and NC17 frontal cortex. Aβstd, Synthetic Aβ peptide standard.
We further investigated the presence of Aβ in the mitochondrial fractions from AD1 and NC17 by tricine-PAGE (12%) coupled Western blot analysis. In agreement with the immunoelectron microscopy results, the mitochondrial fraction from AD1 brain, but not NC17, showed the Aβ antibody immunoreactive ∼4 kDa band, which comigrated with the synthetic Aβ peptide (Fig. 2E). Identical results were obtained when antibodies to different epitopes of Aβ, such as Aβ1–42 and Aβ1–16 (Chemicon), were used (data not shown). These results confirm the previous reports suggesting the presence of Aβ species in the mitochondria (Hirai et al., 2001; Caspersen et al., 2005; Crouch et al., 2005; Manczak et al., 2006).
APP forms complexes with mitochondrial inner and outer membrane translocases in AD brain
Previously, using a chemical cross-linking approach, we observed the interaction between mitochondrial APP and mitochondrial outer and inner membrane channel-forming TOM40 and TIM23, respectively (Anandatheerthavarada et al., 2003). Under physiological conditions, TOM40, along with other outer membrane translocases, forms an ∼380 kDa complex, whereas TIM23 interacts with TIM17 to form an ∼100 kDa inner membrane complex (Dekker et al., 1997; Moro et al., 1999). Hence, we tested whether translocationally arrested mitochondrial APP is stably associated with these membrane complexes in AD brains using BN-PAGE. Western immunoblot analysis of BN-PAGE using APP Nt Ab revealed two distinct complexes that migrated as ∼480 and ∼620 kDa in mitochondrial extracts of AD brains (Fig. 3A). The intensities of these two complexes varied among the AD brains. Highest intensity was observed in the AD subject belonging to category III, followed by categories II and I (Fig. 3B). However, APP Nt Ab failed to recognize these complexes in NC mitochondria and the microsomal fractions from both AD and NC brains (Fig. 3A), suggesting that these complexes are exclusive to AD brain mitochondria. Probing the blots with TOM40 Ab revealed the presence of outer membrane channel-forming TOM40 immunoreactivity in both of the APP-associated ∼480 and ∼620 kDa complexes (Fig. 3C). Most of the ∼380 kDa TOM40 complexes in AD mitochondria were associated with APP, because the levels of free TOM40 complex (represented as @) in AD mitochondria were less than NC mitochondria (Fig. 3C). Antibodies to TIM23 recognized an APP-associated ∼620 kDa complex and a minor free ∼100 kDa inner membrane TIM23 complex in the AD brains (Fig. 3D). However, NC mitochondria had higher amounts of the free ∼100 kDa TIM23 complex (represented as &) when compared with AD mitochondria. These results conclusively show that the accumulated mitochondrial APP forms high-molecular-weight, stable complexes with TOM40 and TIM23 protein import channels of AD brain.
Figure 3.
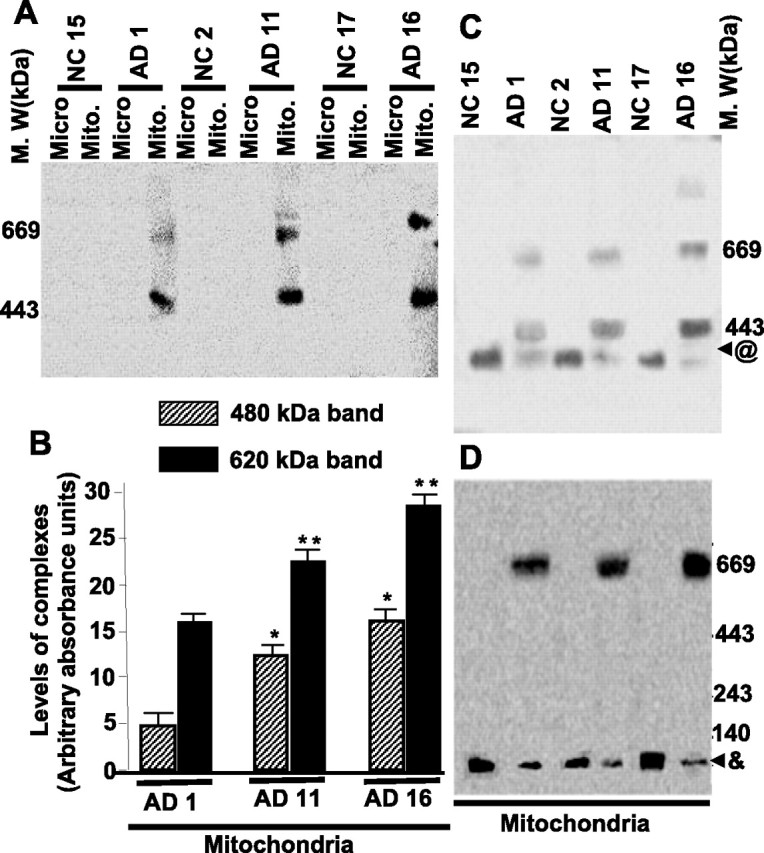
Characterization of APP-associated mitochondrial outer and inner membrane translocase complexes and the protein import efficiency of mitochondria from frontal cortex of AD brains. A, BN-PAGE-coupled Western immunoblot analysis of high-molecular-weight complexes in mitochondrial (Mito) and microsomal (Micro) fractions (200 μg) of AD and NC brains was performed using APP Nt Ab. B, Lower (480 kDa) and upper (620 kDa) complexes of AD mitochondria were quantitated by using Quantity One software from three independent experiments. M.W, Molecular weights. *p < 0.05 compared with AD1 480 kDa band; **p < 0.05 compared with AD1 620 band. BN-PAGE-coupled immunoblot analysis of AD and NC mitochondrial TOM and TIM complexes using TOM40 Ab (C) and TIM23 Ab (D), respectively. @, Free TOM40 complex; &, free TIM23 complex.
Mitochondrial accumulation of APP is associated with decreased cytochrome c oxidase activity and increased levels of H2O2 in mitochondria
Reduced mitochondrial cytochrome c oxidase activity and increased oxidative stress have been associated with the pathogenesis of AD (Parker, 1991; Maurer et al., 2000; Manczak et al., 2006). Furthermore, results from the neuronal culture system showed that incomplete mitochondrial translocation of APP was associated with decreased cytochrome c oxidase activity (Anandatheerthavarada et al., 2003). With this in mind, we determined whether the accumulation of incompletely translocated APP in the mitochondrial import channels had an adverse effect on cytochrome c oxidase activity and the generation of H2O2, which in turn produces oxy free radicals during interaction with metal ions. To address this issue, we measured the cytochrome c oxidase activity (Fig. 4A) and H2O2 levels (Fig. 4B) in the mitochondrial fractions obtained from the frontal cortex of AD1 (category I), AD11 (category II), AD16 (category III), NC2, NC15, and NC17 brains. The activity of cytochrome c oxidase was significantly decreased in AD brains belonging to all three categories (in the order of AD16 < AD11 < AD1) compared with that of NC mitochondria (Fig. 4A). In contrast, the levels of mitochondrial H2O2 increased significantly in AD brains (in the order of AD16 > AD11 > AD1) compared with that of NC brains (Fig. 4B). These results suggest that the increased mitochondrial accumulation of APP is associated with impairment of cytochrome c oxidase activity and increased H2O2 production in the AD brains.
Figure 4.
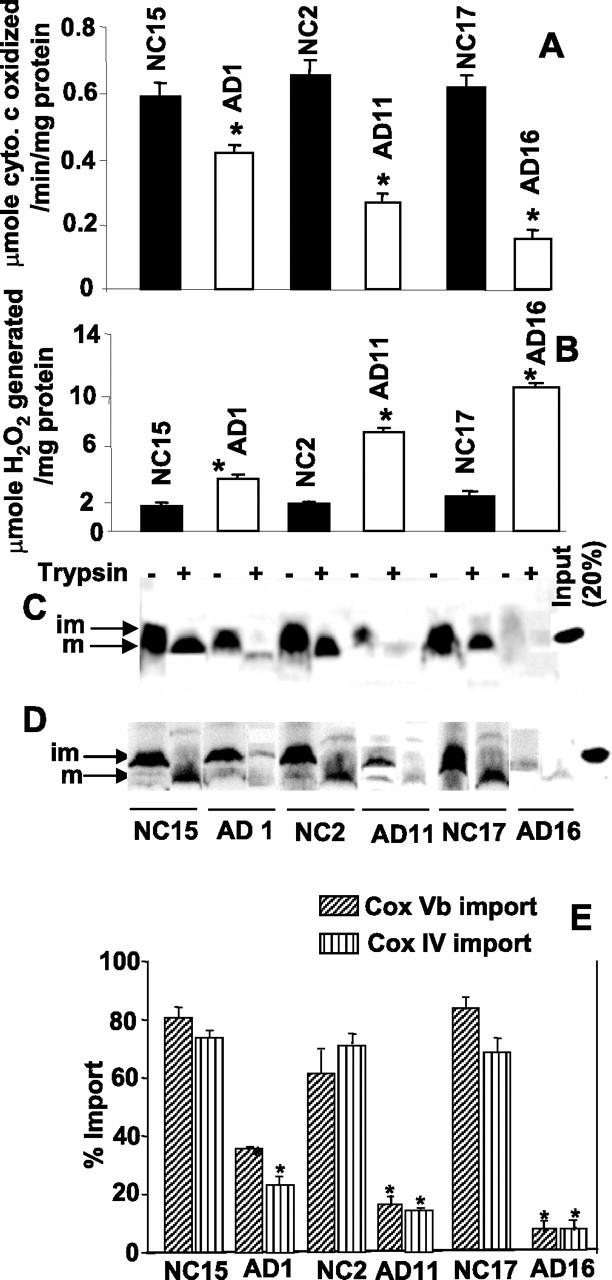
Levels of cytochrome c oxidase (A), H2O2 (B), and import of cytochrome c oxidase subunits Vb (C) and IV (D) in the isolated mitochondria from frontal cortex of AD and NC brains were measured as described in Materials and Methods using 150 μg of trypsin per milliliter of reaction (+). im, Immature; m, mature. E, Mitochondrially imported radioactive bands in C and D were quantified using a Phosphoimager. In A, B, and E (n = 3), *p < 0.05 compared with NC. Error bars indicate SEM.
Accumulation of APP in the import channels inhibits the entry of mitochondrially targeted proteins
We wondered whether accumulation of incompletely translocated APP in the mitochondrial import channels affects the mitochondrial biogenesis of cytochrome c oxidase complex (13 subunit complex) by blocking the transport of nuclear encoded proteins. These subunits are synthesized as immature precursors with N-terminal mitochondrial targeting signal sequences. After internalization into mitochondria, the signal sequences are cleaved by a matrix protease and the mature proteins are integrated into cytochrome c oxidase to form a holo complex. Therefore, we determined the AD brain mitochondrial import efficiency of some important nuclear encoded subunits (subunits IV and Vb) of cytochrome c oxidase. In vitro mitochondrial import assays using freshly isolated mitochondria from AD and NC brains were performed. This assay uses protection against limited proteolytic digestion as a criterion for the import of [35S]-labeled proteins. Results show that the levels of import of cytochrome c oxidase subunit Vb (Fig. 4C) and subunit IV (Fig. 4D) were markedly reduced in mitochondria from AD brains compared with that of NC mitochondria. Inhibition of cytochrome c oxidase subunits IV and Vb import varied among the AD brains (Fig. 4E). Lowest import was observed in the AD subject belonging to category III, which accumulated the highest amount of APP in mitochondrial import channels, followed by II and I. However, the mitochondrial import efficiency of these proteins was not significantly affected among mitochondria from age-matched NCs (Fig. 4C–E). These results suggest that accumulation of APP across the import channel blocks the entry of nuclear encoded proteins into mitochondria.
Frontal cortex, hippocampus, and amygdala of AD brains show the highest amounts of mitochondrial import channel-associated APP
We investigated the content and regional distribution of mitochondrial APP in AD subjects. The levels of APP in AD subjects selected from each category varied from region to region and subject to subject (Fig. 5A–F). Mitochondrial APP was present only in three brain regions of category I AD brain (Fig. 5A), and the highest levels were found in the frontal cortex, followed by hippocampus and amygdala (Fig. 5B). The AD subject from category II showed the accumulation of APP on mitochondria in six brain regions (Fig. 5C; Table 1), with frontal cortex, hippocampus, and amygdala showing significantly higher APP levels compared with the three other regions (Fig. 5D). Category III, which represents the most severe AD subjects, showed the highest amounts of mitochondrial APP in all 11 brain regions, including the frontal cortex, hippocampus, and amygdala, when compared with other categories (Fig. 5E,F). However, mitochondrial TIM44 protein levels were unchanged in all 11 brain regions of category III AD brain (Fig. 5G). Complete characterization of the regional distribution of mitochondrial APP for all 20 AD subjects is shown in Table 1. It should be noted that, in all 20 AD cases examined so far, the highest content of mitochondrial APP was observed in the AD-vulnerable frontal cortex, hippocampus, and amygdala. However, mitochondrial accumulation of APP in the cerebellum and brainstem regions was only seen in the category III subjects with severe AD.
Figure 5.

Levels of mitochondrial APP in various regions of AD brains carrying multiple ApoE genotypes: APP distribution in various brain regions of mitochondrial fractions (50 μg) were evaluated by immunoblot analysis using APP Nt in AD1 (A), AD11 (C), and AD16 (E) subjects. Densitometric analysis of A, C, and E are represented in B, D, and F, respectively (in each case, n = 3). *p < 0.05 compared with PC. G, Content of mitochondrial marker TIM44 in AD16 brain was analyzed by immunoblot analysis using antibodies to TIM44. H, Effect of ApoE genotyping on mitochondrial APP levels in AD brains by Western immunoblotting using APP Nt Ab. APP levels were represented as arbitrary absorbance units. I, Quantitation of the blot presented in H (n = 3). *p < 0.05 compared with E3/E3 genotype (AD1). Error bars indicate SEM. BG, Basal ganglia; CE, cerebellum; OC, occipital cortex; TC, temporal cortex; TH, thalamus.
Table 1.
Effect of ApoE genotyping on regional distribution of mitochondrial APP in AD brains
| AD subjects | Age (years) | Sex | Affected brain regions | ApoE genotype |
|---|---|---|---|---|
| Category I | ||||
| AD1 | 81 | M | FC, HP, AG | E3/E3 |
| AD2 | 74 | M | FC, HP, AG | E3/E3 |
| Category II | ||||
| AD3 | 81 | M | FC, HP, AG, BG, TH, PC | E4/E4 |
| AD4 | 76 | M | FC, HP, AG, BG, TC, CG | E4/E4 |
| AD5 | 68 | F | FC, HP, AG, BG, TH | E3/E3 |
| AD6 | 55 | F | FC, HP, TC, PC, OC | E3/E3 |
| AD7 | 80 | F | FC, HP, AG, TC, PC, OC | E3/E3 |
| AD8 | 68 | F | FC, HP, AG, TC, PC, OC | E4/E4 |
| AD9 | 79 | M | FC, HP, BG, AG, PC, OC | E3/E3 |
| AD10 | 80 | M | FC, HP, AG, PC, TC | E2/E4 |
| AD11 | 70 | M | FC, HP, AG, PC, TC, OC | E4/E4 |
| Category III | ||||
| AD12 | 80 | F | FC, HP, BG, AG, TC, PC, OC, CG, CE, BS | E3/E4 |
| AD13 | 87 | F | FC, HP, BG, AG, TC, PC, OC, CG, CE, BS | E3/E4 |
| AD14 | 80 | F | FC, HP, BG, AG, TC, PC, TH, OC, CG, CE, BS | E3/E4 |
| AD15 | 80 | M | FC, HP, AG, BG, PC, TH, CG, AG, CE, BS | E3/E4 |
| AD16 | 77 | M | FC, HP, AG, BG, TC, PC, TH, OC, CG, CE, BS | E3/E4 |
| AD17 | 85 | F | FC, HP, AG, BG, PC, TH, OC, CG, CE, BS | E3/E4 |
| AD18 | 80 | F | FC, HP, AG, BG, TC, TH, PC, OC, CE, BS | E3/E4 |
| AD19 | 75 | M | FC, HP, AG, BG, TC, OC, PC, CG, CE, | E3/E4 |
| AD20 | 80 | F | FC, HP, AG, BG, PC, TC, OC, CG, CE, BS | E3/E4 |
M, Male; F, female; FC, frontal cortex; PC, parietal cortex; OC, occipital cortex; TC, temporal cortex; HIP, hippocampus; AG, amygdala; CE, cerebellum; BS, brainstem; CG, cingulum; BG, basal ganglia; TH, thalamus.
A higher level of mitochondrial APP is associated with ApoE E3/E4 in AD
To investigate the possible relationship between mitochondrial accumulation of APP and the genetic status of ApoE, a risk factor for late onset AD (Kamboh, 2004), we performed ApoE genotyping on all AD and NC subjects. Most of the NC subjects were the bearers of the Apo E3/E3 genotype (supplemental Table 2, available at www.jneurosci.org as supplemental material). Among the AD subjects, ApoE genotype varied with mitochondrial APP levels (Table 1). Category I subjects were homozygous for the E3 allele, whereas category II, which had higher levels of mitochondrial APP when compared with category I, showed variations at this locus, displaying E4/E4, E3/E3, and E2/E4 genotypes. Interestingly, category III AD brains, which carry the E3/E4 genotype, showed the highest amount of mitochondrial APP accumulation in 11 brain regions (Table 1) when compared with categories I and II (Fig. 5H,I). These results suggest an important correlation between ApoE3/E4 heterozygosity and widespread distribution of mitochondrial APP in the AD brain.
Neuronal accumulation of mitochondrial APP
We tested whether APP is accumulated across mitochondrial membranes of cholinergic neurons, which are believed to be the most affected cell types during the pathogenesis of AD (Yan and Feng, 2004), with confocal triple-labeling immunofluorescence microscopy. Deparaffinized tissue sections from the frontal cortex (BM10) of NC17 (Fig. 6A–E) and AD16 (category III) (Fig. 5F–J) were subjected to immunostaining with APP Nt Ab (Fig. 6A,F) and TOM40 Ab (Fig. 6B,G). Sections were also stained with ChAT Abs to identify cholinergic neurons (Fig. 6C,H). In NC17 brain, a robust staining around the PM by APP Nt Ab (Fig. 6A) and TOM40-specific staining of particulate structures reminiscent of mitochondria (Fig. 6B) were observed. However, APP staining did not overlap with mitochondrial staining (Fig. 6D); notably, these cells stained positively for ChAT (Fig. 6E). In AD16 brain, the APP Nt Ab stained extranuclear granulate structures (Fig. 6F), which resembled TOM40 Ab-specific mitochondrial staining (Fig. 6G). Contrary to NC17 brain, APP staining in AD16 brain overlapped extensively with mitochondrial staining (Fig. 6I). Some of these cells were also stained with ChAT, suggesting mitochondrial accumulation of APP in cholinergic neurons (Fig. 6J). The distribution of mitochondrial APP in various AD brain regions that are rich in dopaminergic, glutamatergic, and GABAergic neurons prompted us to investigate the mitochondrial accumulation of APP in these neuronal types using the same approach as mentioned above. Triple overlaps presented in Figure 7 showed that APP staining colocalized with TOM40 mitochondrial staining in dopaminergic neurons of the basal ganglia (striatum) (Fig. 7A), glutamatergic neurons of the frontal cortex (BM10) (Fig. 7B), and GABAergic neurons of the frontal cortex (BM10) (Fig. 7C) of AD16 brain, whereas these neuronal types did not show colocalization of APP- and mitochondria-specific stains in NC17 brain (Fig. 7D–F). We further tested the mitochondrial localization of APP in the aforementioned neuronal types/brain regions in AD1 (category I) and AD11 (category II) as described above. We observed the accumulation of APP in cholinergic neuronal mitochondria of AD1, whereas the AD11 showed the accumulation of APP in the mitochondria of glutamatergic and cholinergic neurons (data not shown). However, neither AD1 nor AD11 showed accumulation of APP in dopaminergic or GABAergic neuronal mitochondria (data not shown). These results suggest that the mitochondrial accumulation of APP in cholinergic, glutamatergic, dopaminergic, and GABAergic neuronal types may be associated with the severity of AD.
Figure 6.
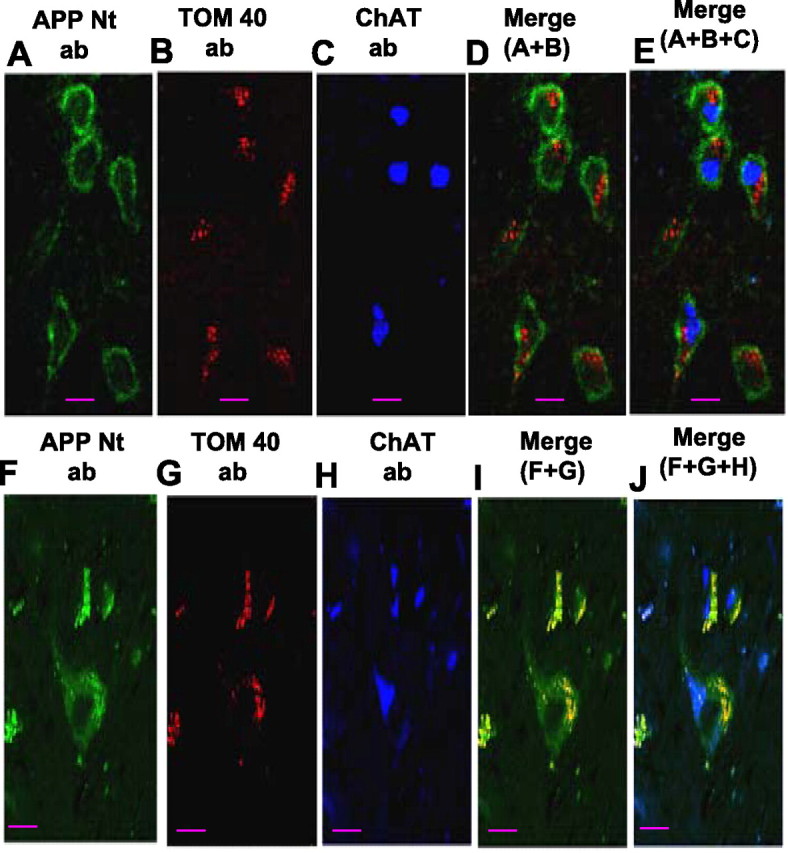
Confocal immunofluorescence microscopy analysis of mitochondrial APP in cholinergic neurons. Frontal cortex (BM10 area) tissue sections from NC17 (A–C) and AD16 (F–H) were triple immunostained with APP Nt Ab (A, F), TOM40 Ab (B, G), and ChAT Ab (C, H). Staining patterns (A–C, F–H) were developed with appropriate secondary antibodies conjugated to Alexa dyes. D and E represent overlay patterns of A + B and A + B + C, respectively, in the NC brain. In the AD brain, I and J represent overlays of F + G and F + G + H, respectively. Scale bars, 15 μm.
Figure 7.
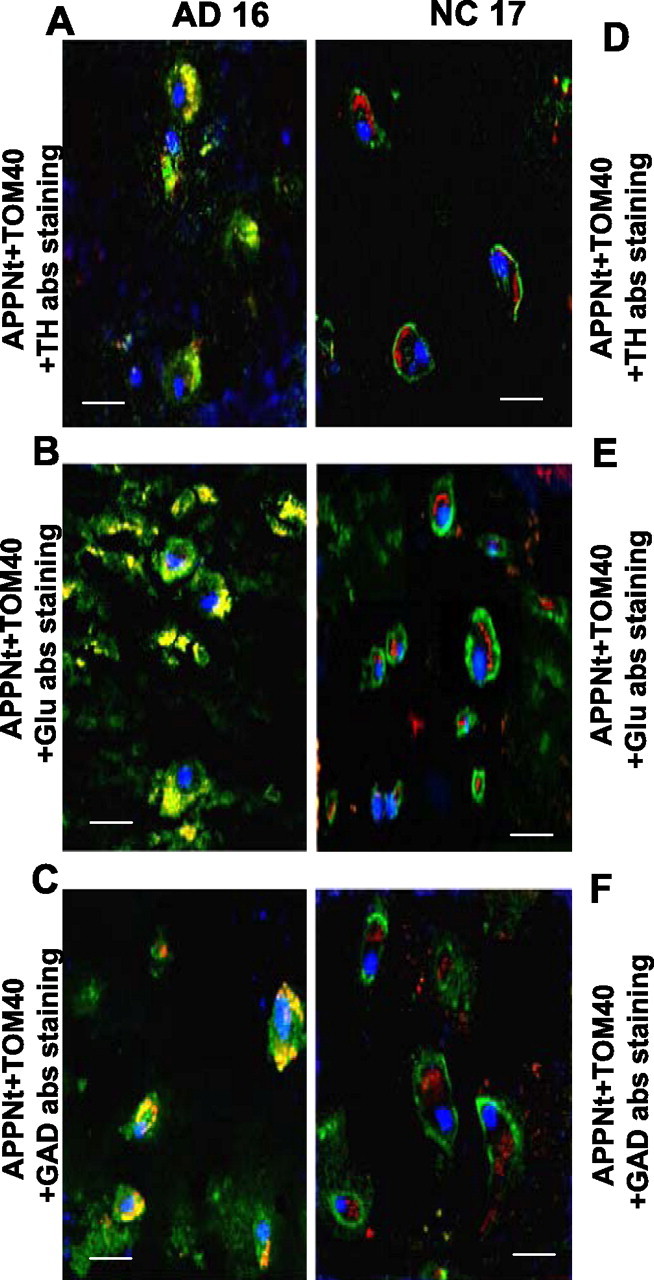
Mitochondrial accumulation of APP in noncholinergic neurons. Confocal immunofluorescence analysis of triple-stained sections from AD16 (A, C, E) and NC17 (B, D, F) brains were performed as described in Materials and Methods. A and D were from basal ganglia (striatum), and B, C, E, and F were from frontal cortex (BM10 area). Green and red staining patterns in A–F brain represent APP and mitochondrial marker TOM40, respectively. Blue staining in A,D, B,E, and C,F represent tyrosine hydroxylase (TH), glutamate (Glu), and GAD patterns, respectively. Brown–yellow staining in A–C of AD brain represent colocalization of green staining (APP) with red staining (mitochondrial TOM40). Scale bars, 15 μm.
Mitochondrial accumulation of APP directly correlates with mitochondrial dysfunction in various brain regions of AD
A progressive accumulation of APP across mitochondrial membranes has been shown to cause mitochondrial dysfunction in HCN neuronal cells (Anandatheerthavarada et al., 2003). Hence, we examined the effect of accumulation of APP in the import channels on total mitochondrial functions by performing the MTT assay (an indicator of mitochondrial redox potential) and cytochrome c oxidase as markers of mitochondrial function. These mitochondrial functions were correlated with mitochondrial total APP (as measured by quantitative ELISA) in all of the brain regions of 20 AD subjects. The linear regression plots of MTT versus mitochondrial APP in frontal cortex (Fig. 8A), hippocampus (Fig. 8B), and amygdala (Fig. 8C) yielded highly significant inverse correlations, suggesting that, in AD brains, with the accumulation of APP across mitochondrial membranes, there is a significant decrease in mitochondrial functions in various brain regions. However, MTT values in various NC brain regions were threefold higher than that of AD (data not shown). The linear regression plots of mitochondrial APP versus cytochrome c oxidase activity for FC (Fig. 8D), HP (Fig. 8E), and AG (Fig. 8F) showed a significant negative correlation with the mitochondrial APP levels.
Figure 8.

Correlation between mitochondrial APP versus MTT and cytochrome c oxidase in AD brains. Total APP levels in mitochondria (Mito) were estimated by quantitative APP ELISA in the frontal cortex, hippocampus, and amygdala of all 20 AD subjects. Scatter plots of mitochondrial APP versus mitochondrial functions measured by MTT activity were generated for FC (A), HP (B), and AG (C). Similarly, scatter plots of mitochondrial APP versus cytochrome c oxidase for FC (D), HP (E), and AG (F). Regression analysis was performed using Origin 7.5 software.
Discussion
Alzheimer’s disease is characterized by a number of chronic injuries that eventually cause progressive neurodegeneration. Mitochondrial dysfunction is one of the earliest defects that has been detected in AD patients (Valla et al., 2001; Manczak et al., 2004; Beal, 2005). In the present study, we show the accumulation of nonglycosylated APP species in mitochondrial import channels of AD brain, which is directly associated with the impairment of mitochondrial functions as judged by the decrease in cytochrome c oxidase activity (Figs. 4A, 8D,E), decreased capacity to reduce MTT dye (Fig. 8A–C), and the increased production of H2O2 (Fig. 4B). This type of translocational arrest has been observed for the first time in a disease condition in situ in the human brain.
Under normal physiological conditions, interactions between various mitochondrial translocases and translocating proteins are expected to be transient (Truscott et al., 2001). In contrast, in AD brain, interactions between APP and mitochondrial translocases appear quite stable and persistent. The stable interaction of APP with mitochondrial translocase complexes may be ascribed to the acidic domain spanning 220–290 amino acids. This domain was shown to cause translocational arrest of APP in HCN neuronal culture, thus perturbing the vital functions of mitochondria (Anandatheerthavarada et al., 2003). In addition, removal of the acidic domain caused APP to completely translocate into mitochondria and did not impair the mitochondrial functions (Anandatheerthavarada et al., 2003). In view of these observations, the present data suggest that the acidic domain-mediated mitochondrial translocational arrest of APP may contribute to mitochondrial dysfunction during the pathogenesis of AD.
Translocational arrest of APP may be influenced by many factors, such as the inability of cytoplasmic chaperones to fully unfold the polypeptide at the acidic domain during the mitochondrial import. The second possibility is that the negatively charged acidic domain fails to bind efficiently to the negatively charged TOM22 because of electrostatic repulsion. TOM22 in association with TOM40 plays an important role in the vectorial transport of proteins. The third possibility may be that the acidic domain of APP binds to many metal ions and may form a bulky structure (Hesse et al., 1994) that may cause the translocational arrest. Many studies have implicated metal ions, including zinc, copper, iron, and aluminum, in the pathogenesis of AD (McDermott et al., 1979; Lovell et al., 1998; Zatta et al., 2003). Furthermore, some studies have also shown that the cellular concentrations of these metal ions were increased in AD brain (Basun et al., 1991; Loeffler et al., 1996; Jagannatha Rao et al., 1999). Treatment with Clioquinol (a metal chelator) has been shown to reduce the accumulation of Aβ species in transgenic mouse models (Cherny et al., 2001). Using the HCN culture system, our laboratory and others have observed the association between APP-mediated mitochondrial dysfunction and the accumulation of intracellular Aβ (Busciglio et al., 2002; Anandatheerthavarada et al., 2003). However, the mechanisms involved in the association between mitochondrial dysfunction and the accumulation of intracellular Aβ is not known. Together, the above mentioned observations raise an important question whether the binding of large amounts of metal ions to the acidic domain causes increased mitochondrial translocational arrest of APP, which in turn results in the mitochondrial dysfunction and the associated Aβ accumulation during the pathogenesis of AD. Collectively, the data presented in this manuscript highlight the notion that the progressive blockade of protein import channels by APP may be one of the major causes of mitochondrial dysfunction in the human AD brain.
Well over 1500 nuclear encoded proteins are reported to be associated with mammalian mitochondria (Taylor et al., 2003; Gabaldon and Huynen, 2004). Transport of almost all of these proteins into mitochondria requires functional import channels. Accumulation of APP in such physiologically important channels may inhibit the import of proteins essential for normal mitochondrial functions. Consistent with this possibility, in the present study, we observed that the accumulation of APP in import channels is associated with the dramatic inhibition of entry of the bona fide mitochondrial cytochrome c oxidase subunits IV and Vb, which are part of the multi-subunit cytochrome c oxidase complex involved in mitochondrial respiration-coupled oxidative phosphorylation. The deficiency of these subunits causes decreased cytochrome c oxidase activity (Fig. 4A), which in turn results in mitochondrial dysfunction (Prabu et al., 2006). In support of the above, we observed widespread mitochondrial dysfunction in AD brains (Fig. 8A–C). Using the HCN culture system, we have shown previously that the accumulation of APP across mitochondrial membranes is time dependent (Anandatheerthavarada et al., 2003), which fits very well with the progressive nature of AD pathogenesis. We hypothesize that the accumulation of APP in two TOM40 antibody cross-reactive complexes in AD brain mitochondria represents two different steady-state intermediates. The ∼480 kDa complex represents translocationally arrested APP across the TOM40 complex, with an apparent size of ∼380 kDa; the ∼620 kDa complex represents APP arrested across both TOM40 and TIM23 complexes.
In support of others studies (Hirai et al., 2001; Caspersen et al., 2005; Crouch et al., 2005; Manczak et al., 2006), in the present study, we also observed Aβ species in the mitochondria of AD brain (Fig. 2). The mechanism by which mitochondrial Aβ is produced is not clear. Recently, the presence of γ secretase-like protease has been reported in mitochondria. However, the orientation of mitochondrial APP may not support the possibility of intramitochondrial generation of Aβ. It may be possible that Aβ is generated outside the mitochondria and is imported into the mitochondria. Furthermore, our finding showing the presence of C-terminal truncated (lacking Aβ) APP in the mitochondria of AD brain suggests a couple of possibilities for the genesis of extra mitochondrial Aβ. One possibility may be, with the progression of AD, there is a loss of the cytosolically exposed C terminus of mitochondrial associated APP by some unknown mechanisms, thus generating the Aβ species. Alternatively, during mitochondrial import, the C terminus of APP may be cleaved in the cytosol to generate Aβ species by an unknown mechanism, and the generated Aβ may be transported into mitochondria.
Mitochondrial dysfunction may play a role in the degeneration/cell death of neurons during the pathogenesis of AD (Sims, 1996; Zhu et al., 2004). In addition to cholinergic neurons, several other neuronal types, such as dopaminergic, GABAergic, and glutamatergic neurons, have been shown to be affected in the later stages of the pathogenesis of AD (Francis et al., 1999; Francis, 2003). As observed in the present study, mitochondrial dysfunction mediated by translocational arrest of APP in mitochondrial import channels of cholinergic, GABAergic, dopaminergic, and glutamatergic neurons in AD brain may have far reaching influence on their survival.
The cellular conditions under which the plasma-membrane-destined APP is diverted to mitochondria of AD brains, especially in sporadic AD, is not known. Results from our laboratory have demonstrated an important role for protein kinase A-mediated phosphorylation in the mitochondrial targeting of several physiologically important ER targeted proteins, such as P4502B1 and 2E1, by altering the corresponding protein levels in the ER compartment (Anandatheerthavarada et al., 1999; Robin et al., 2003; Karniely and Pines, 2005). In support of these results, our unpublished results show that mitochondrial associated APP may be a phosphoprotein. Moreover, our present results show that the mitochondrial accumulation of APP was accompanied by a corresponding reduction in plasma membrane-associated APP in the AD brain (Fig. 1D,E). Nevertheless, the control brains, which had no mitochondrial APP, showed higher levels of plasma membrane APP (Fig. 1D,E). Furthermore, studies also suggested elevated levels of protein kinases in the AD brain (Masliah et al., 1990; Lanius et al., 1997). Collectively, these observations suggest a possible role for phosphorylation of APP in the mitochondrial targeting by altering the plasma membrane-associated APP pool. Interestingly, the slow turnover of mitochondrial associated APP, as observed in the HCN neuronal culture system (Anandatheerthavarada et al., 2003), may explain the accumulation of APP in the AD brain mitochondria. With respect to the role of kinases in the mitochondrial targeting of APP, it is interesting to speculate the role of the ApoE4 allele, which is considered to be one of the risk factors in the late onset of AD, as an additional factor that may be involved in the mitochondrial targeting of APP. Recently, it has been reported that ApoE3 and ApoE4 could modulate the signal transduction pathways mediated by kinases, especially protein kinase C and protein kinase A (Cedazo-Minguez and Cowburn, 2001; Cedazo-Minguez et al., 2001; Zhu and Hui, 2003). It is therefore reasonable to hypothesize that ApoE3 and ApoE4, either individually or collectively, may influence the mitochondrial targeting of APP by modulating the intracellular levels of protein kinases C and A. Currently, experiments are underway to precisely decipher the mechanisms involved in the mitochondrial targeting and the translocational arrest of APP, which might shed light on the mechanisms underlying mitochondrial abnormalities during the pathogenesis of AD.
Footnotes
This work was supported by National Institutes of Health/National Institute on Aging Grants AG 021920 (H.K.A.) and GM49683 (N.G.A.). We thank the members of our laboratory for valuable criticisms and comments.
References
- Addya S, Anandatheerthavarada HK, Biswas G, Bhagwat SV, Mullick J, Avadhani NG. Targeting of NH2-terminal-processed microsomal protein to mitochondria: a novel pathway for the biogenesis of hepatic mitochondrial P450MT2. J Cell Biol. 1997;139:589–599. doi: 10.1083/jcb.139.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agin V, Chichery R, Chichery M. Effects of learning on cytochrome oxidase activity in cuttlefish brain. NeuroReport. 2001;12:113–116. doi: 10.1097/00001756-200101220-00030. [DOI] [PubMed] [Google Scholar]
- Anandatheerthavarada HK, Biswas G, Mullick J, Sepuri NB, Otvos L, Pain D, Avadhani NG. Dual targeting of cytochrome P4502B1 to endoplasmic reticulum and mitochondria involves novel signal activation by cyclic AMP-dependent phosphorylation at ser128. EMBO J. 1999;18:5494–5504. doi: 10.1093/emboj/18.20.5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161:41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basun H, Forssell LG, Wetterberg L, Winblad B. Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer’s disease. J Neural Transm Park Dis Dement Sect. 1991;3:231–258. [PubMed] [Google Scholar]
- Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- Berndt JD, Callaway NL, Gonzalez-Lima F. Effects of chronic sodium azide on brain and muscle cytochrome oxidase activity: a potential model to investigate environmental contributions to neurodegenerative diseases. J Toxicol Environ Health. 2001;63:67–77. doi: 10.1080/152873901750128380. [DOI] [PubMed] [Google Scholar]
- Brown MR, Sullivan PG, Dorenbos KA, Modafferi EA, Geddes JW, Steward O. Nitrogen disruption of synaptoneurosomes: an alternative method to isolate brain mitochondria. J Neurosci Methods. 2004;137:299–303. doi: 10.1016/j.jneumeth.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Brunkan AL, Goate AM. Presenilin function and gamma-secretase activity. J Neurochem. 2005;93:769–792. doi: 10.1111/j.1471-4159.2005.03099.x. [DOI] [PubMed] [Google Scholar]
- Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, Yankner BA. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron. 2002;33:677–688. doi: 10.1016/s0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- Bussiere T, Bard F, Barbour R, Grajeda H, Guido T, Khan K, Schenk D, Games D, Seubert P, Buttini M. Morphological characterization of thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Abeta immunotherapy on their clearance. Am J Pathol. 2004;165:987–995. doi: 10.1016/s0002-9440(10)63360-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cada A, Gonzalez-Lima F, Rose GM, Bennett MC. Regional brain effects of sodium azide treatment on cytochrome oxidase activity: a quantitative histochemical study. Metab Brain Dis. 1995;10:303–320. doi: 10.1007/BF02109361. [DOI] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- Cedazo-Minguez A, Cowburn RF. Apolipoprotein E: a major piece in the Alzheimer’s disease puzzle. J Cell Mol Med. 2001;5:254–266. doi: 10.1111/j.1582-4934.2001.tb00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedazo-Minguez A, Wiehager B, Winblad B, Huttinger M, Cowburn RF. Effects of apolipoprotein E (apoE) isoforms, beta-amyloid (Abeta) and apoE/Abeta complexes on protein kinase C-alpha (PKC-alpha) translocation and amyloid precursor protein (APP) processing in human SH-SY5Y neuroblastoma cells and fibroblasts. Neurochem Int. 2001;38:615–625. doi: 10.1016/s0197-0186(00)00128-5. [DOI] [PubMed] [Google Scholar]
- Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-β1–42. J Neurosci. 2005;25:672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasso M. In vitro reconstitution. In: Bonifacino JS, Dasso M, Harford JB, Lippincot-Schawartz J, Yamada KM, editors. Current protocols in cell biology. New York: Wiley; 1998. pp. 11.0.1–11.0.2. [Google Scholar]
- Dekker PJ, Martin F, Maarse AC, Bomer U, Muller H, Guiard B, Meijer M, Rassow J, Pfanner N. The Tim core complex defines the number of mitochondrial translocation contact sites and can hold arrested preproteins in the absence of matrix Hsp70-Tim44. EMBO J. 1997;16:5408–5419. doi: 10.1093/emboj/16.17.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Rangnekar VM, Mattson MP. Prostate apoptosis response-4 production in synaptic compartments following apoptotic and excitotoxic insults: evidence for a pivotal role in mitochondrial dysfunction and neuronal degeneration. J Neurochem. 1999;72:2312–2322. doi: 10.1046/j.1471-4159.1999.0722312.x. [DOI] [PubMed] [Google Scholar]
- Francis PT. Glutamatergic systems in Alzheimer’s disease. Int J Geriatr Psychiatry. 2003;18:S15–S21. doi: 10.1002/gps.934. [DOI] [PubMed] [Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabaldon T, Huynen MA. Shaping the mitochondrial proteome. Biochem Biophys Acta. 2004;1659:212–220. doi: 10.1016/j.bbabio.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Haroutunian V, Zhang H, Park LC, Shi Q, Lesser M, Mohs RC, Sheu RK, Blass JP. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann Neurol. 2000;48:297–303. [PubMed] [Google Scholar]
- Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Hesse L, Beher D, Masters CL, Multhaup G. The beta A4 amyloid precursor protein binding to copper. FEBS Lett. 1994;349:109–116. doi: 10.1016/0014-5793(94)00658-x. [DOI] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannatha Rao KS, Shanmugavelu P, Shankar SK, Rukmini Devi RP, Rao RV, Sanjay P. Trace elements in the cerebrospinal fluid of Alzheimer’s disease. Alzheimers Rep. 1999;2:333–338. [Google Scholar]
- Kamboh MI. Molecular genetics of late-onset Alzheimer’s disease. Ann Hum Genet. 2004;68:381–404. doi: 10.1046/j.1529-8817.2004.00110.x. [DOI] [PubMed] [Google Scholar]
- Karniely S, Pines O. Single translation–dual destination: mechanisms of dual protein targeting in eukaryotes. EMBO Rep. 2005;6:420–425. doi: 10.1038/sj.embor.7400394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil U, Bonert A, Marques CA, Scherping I, Weyermann J, Strosznajder JB, Muller-Spahn F, Haass C, Czech C, Pradier L, Muller WE, Eckert A. Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J Biol Chem. 2004;279:50310–50320. doi: 10.1074/jbc.M405600200. [DOI] [PubMed] [Google Scholar]
- Lanius RA, Wagey R, Sahl B, Beattie BL, Feldman H, Pelech SL, Krieger C. Protein kinase C activity and protein levels in Alzheimer’s disease. Brain Res. 1997;764:75–80. doi: 10.1016/s0006-8993(97)00422-8. [DOI] [PubMed] [Google Scholar]
- Loeffler DA, LeWitt PA, Juneau PL, Sima AA, Nguyen HU, DeMaggio AJ, Brickman CM, Brewer GJ, Dick RD, Troyer MD, Kanaley L. Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders. Brain Res. 1996;738:265–274. doi: 10.1016/s0006-8993(96)00782-2. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004;5:147–162. doi: 10.1385/NMM:5:2:147. [DOI] [PubMed] [Google Scholar]
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of Abeta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- Masliah E, Cole G, Shimohama S, Hansen L, DeTeresa R, Terry RD, Saitoh T. Differential involvement of protein kinase C isozymes in Alzheimer’s disease. J Neurosci. 1990;10:2113–2124. doi: 10.1523/JNEUROSCI.10-07-02113.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer IS, Zierz HJ, Moller A selective defect of cytochrome c oxidase is present in brain of Alzheimer’s disease patients. Neurobiol Aging. 2000;21:455–462. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- McDermott JR, Smith AI, Iqbal K, Wisniewski HM. Brain aluminum in aging and Alzheimer disease. Neurology. 1979;29:809–814. doi: 10.1212/wnl.29.6.809. [DOI] [PubMed] [Google Scholar]
- Montemurro DG, Brunni JE. The human brain in dissection. Ed 2. Philadelphia: Saunders; 1998. [Google Scholar]
- Moro F, Sirrenberg C, Schneider HC, Neupert W, Brunner M. The TIM17.23 preprotein translocase of mitochondria: composition and function in protein transport into the matrix. EMBO J. 1999;18:3667–3675. doi: 10.1093/emboj/18.13.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker WD., Jr Cytochrome oxidase deficiency in Alzheimer’s disease. Ann NY Acad Sci. 1991;640:59–64. doi: 10.1111/j.1749-6632.1991.tb00191.x. [DOI] [PubMed] [Google Scholar]
- Periz G, Fortini ME. Functional reconstitution of gamma-secretase through coordinated expression of presenilin, nicastrin, Aph-1, and Pen-2. J Neurosci Res. 2004;77:309–322. doi: 10.1002/jnr.20203. [DOI] [PubMed] [Google Scholar]
- Prabu SK, Anandatheerthavarada HK, Raza H, Srinivasan S, Spear JF, Avadhani NG. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J Biol Chem. 2006;281:2061–2070. doi: 10.1074/jbc.M507741200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DL, Sisodia SS. Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Beal MF. Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res Brain Res Rev. 2005;49:618–632. doi: 10.1016/j.brainresrev.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Robin MA, Prabu SK, Raza H, Anandatheerthavarada HK, Avadhani NG. Phosphorylation enhances mitochondrial targeting of GSTA4–4 through increased affinity for binding to cytoplasmic Hsp70. J Biol Chem. 2003;278:18960–18970. doi: 10.1074/jbc.M301807200. [DOI] [PubMed] [Google Scholar]
- Schagger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature. 1999;399:A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- Sims NR. Energy metabolism, oxidative stress and neuronal degeneration in Alzheimer’s disease. Neurodegeneration. 1996;5:435–440. doi: 10.1006/neur.1996.0059. [DOI] [PubMed] [Google Scholar]
- Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, Murphy AN, Gaucher SP, Capaldi RA, Gibson BW, Ghosh SS. Characterization of the human heart mitochondrial proteome. Nat Biotechnol. 2003;21:281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- The National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- Truscott KN, Pfanner N, Voos W. Transport of proteins into mitochondria. Rev Physiol Biochem Pharmacol. 2001;143:81–136. doi: 10.1007/BFb0115593. [DOI] [PubMed] [Google Scholar]
- Valla J, Berndt JD, Gonzalez-Lima F. Energy hypometabolism in posterior cingulate cortex of Alzheimer’s patients: superficial laminar cytochrome oxidase associated with disease duration. J Neurosci. 2001;21:4923–4930. doi: 10.1523/JNEUROSCI.21-13-04923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayasarathy C, Damle S, Lenka N, Avadhani NG. Tissue variant effects of heme inhibitors on the mouse cytochrome c oxidase gene expression and catalytic activity of the enzyme complex. Eur J Biochem. 1999;266:191–200. doi: 10.1046/j.1432-1327.1999.00843.x. [DOI] [PubMed] [Google Scholar]
- Yan Z, Feng J. Alzheimer’s disease: interactions between cholinergic functions and beta-amyloid. Curr Alzheimer Res. 2004;1:241–248. doi: 10.2174/1567205043331992. [DOI] [PubMed] [Google Scholar]
- Zatta P, Lucchini R, van Rensburg SJ, Taylor A. The role of metals in neurodegenerative processes: aluminum, manganese, and zinc. Brain Res Bull. 2003;62:15–28. doi: 10.1016/s0361-9230(03)00182-5. [DOI] [PubMed] [Google Scholar]
- Zhu X, Smith MA, Perry G, Aliev G. Mitochondrial failures in Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2004;19:345–352. doi: 10.1177/153331750401900611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Hui DY. Apolipoprotein E binding to low density lipoprotein receptor-related protein-1 inhibits cell migration via activation of cAMP-dependent protein kinase A. J Biol Chem. 2003;278:36257–36263. doi: 10.1074/jbc.M303171200. [DOI] [PubMed] [Google Scholar]