

Abstract
Much evidence indicates that extinction training does not erase memory traces but instead forms inhibitory learning that prevents the expression of original memory. Fear conditioning induces long-term potentiation and drives synaptic insertion of AMPA receptors into the amygdala. Here we show that extinction training applied 1 h after training reversed the conditioning-induced increase in surface glutamate receptor subunit 1 (GluR1) in parallel with the inhibition of startle potentiation. However, if applied 24 h after training, extinction training reduced startle potentiation without influencing the GluR1 increase. We infused d-cycloserine (DCS), a partial agonist of the glycine site on the NMDA receptor, bilaterally into the amygdala 30 min before extinction training. This augmented the extinction training-elicited reduction in startle and reversed the conditioning-induced increase in GluR1. Delivery of five sets of tetanic stimulation (TS) to the external capsule produced a robust enhancement of synaptic responses in the lateral amygdala neurons that persisted for >2 h. Low-frequency stimulation applied 1 h after TS had no long-lasting effect on synaptic responses. The same treatments, however, induced depotentiation in the presence of DCS and reversed TS-induced increase in surface GluR1. Together, this study has two important findings: (1) whether a memory trace remains intact or is erased depends on the interval between conditioning and extinction training and (2) DCS facilitates the reversal of memory trace. DCS-induced augmentation of extinction and reversal of GluR1 surface expression are likely mediated by DCS-facilitated endocytosis of AMPA receptors.
Keywords: amygdala, extinction, fear, glutamate receptor, depotentiation, learning and memory
Introduction
The treatment of choice for a number of anxiety disorders is exposure-based psychotherapy (Yehuda, 2002). Homologously, studies of experimental extinction show that if animals are exposed to the conditioned stimulus (CS), without pairing with the unconditioned stimulus (US), previously acquired responses will gradually decline (Pavlov, 1927; Falls et al., 1992; Rescorla, 2001). Unfortunately, successful reduction in fear through exposure therapy is often followed by a return of fear that is attributable to the persistence of original memory (Quirk et al., 2000; Herry and Garcia, 2002; Myers and Davis, 2002; Maren and Quirk, 2004). As a result, testing animals in a context different from that used for extinction would lead to the renewal of learned responses (Bouton and King, 1983). Re-exposing animals to the US before the test would restore its representation and reinstate the learned responses (Rescorla and Heth, 1975). Furthermore, responses to the CS spontaneously recover after some time after extinction training (Bouton and Peck, 1989). On the other hand, the theory of memory storage that relies on bidirectional synaptic modification suggests that changes in synaptic weights must be reversible (Heynen et al., 2000; Rogan et al., 2005). If the mechanisms underlying long-term potentiation (LTP) and depotentiation were distinct and irreversible, eventually saturation would occur because synapses were subjected to repeated stimulation. Thus, theoretically, there may be a way to erase memory, because the brain needs a mechanism to prevent itself from being saturated with useless information.
d-Cycloserine (DCS), a partial agonist at the glycine-binding site of the NMDA receptor, augments the extinction of conditioned fear when administered immediately before or after extinction training (Walker et al., 2002; Ledgerwood et al., 2003). These results suggest that DCS might act at either the acquisition or the consolidation, or both, of extinction. If DCS merely augmented extinction, then re-exposure of DCS-treated animals to the US should reinstate fear to an extinguished CS. In fact, this was not the case (Ledgerwood et al., 2004), thus suggesting that extinction after DCS treatment is more robust and less susceptible to subsequent US reinstatement. Alternatively, the effect of DCS could be accounted for by the reversal of synaptic change that underlies fear conditioning. Neuronal changes mediating the association between the CS and the US occur in the lateral amygdala (LA) and basolateral amygdala (BLA) (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997). We (Yeh et al., 2006) reported that fear conditioning results in an increase in surface expression of glutamate receptor subunit 1 (GluR1) of AMPA receptors in the amygdala, a result we arrived at by labeling surface receptors with biotin or using membrane fractionation. The purpose of this study was to determine whether a conditioning-induced increase in GluR1 was altered after the extinction of fear responses.
Materials and Methods
Surgery
Male Sprague Dawley rats weighing between 175 and 225 g were anesthetized using sodium pentobarbital (50 mg/kg, i.p.) and were subsequently mounted on a stereotaxic apparatus. Two cannulas made of 22 gauge stainless steel tubing were implanted bilaterally into the LA or BLA (anteroposterior, −2.8 mm; mediolateral, ±4.5 mm; dorsoventral, −7.0 mm) (Paxinos and Watson, 1986). A 28 gauge dummy cannula was inserted into each cannula to prevent clogging. Three jewelry screws were inserted into the top of the skull to act as anchors for the entire assembly, which was affixed to the skull using dental cement. The rats were monitored, handled daily, and given 7 d to recover. (±)-3-Amino-1-hydroxy-2-pyrrolidone [(±)-HA-966] (6 mg/kg; Sigma, St. Louis, MO) was dissolved in saline and injected intraperitoneally 30 min before DCS or extinction training. DCS (10 μg/side, dissolved in saline), d-2-amino-5-phosphonovalerate (d-APV; 12.5 nmol/side, dissolved in saline), and wortmannin (2.5 μg/side, dissolved in 20% DMSO) (Sigma) were administered bilaterally into the amygdala at a volume of 1 μl and a rate of 0.1 μl/min. The infusion cannulas were left in place for 2 min before being withdrawn.
Fear conditioning
The rats were trained and tested in a stabilimeter device. The acoustic startle stimulus was a 50 ms, 95 dB white noise. The visual CS was a 3.7 s light produced by an 8 W fluorescent bulb attached to the back of the stabilimeter. The US was a 0.6 mA, 0.5 s footshock.
Acclimation.
On 3 consecutive days, rats were placed in the startle test boxes for 10 min and returned to their home cages.
Matching.
On 2 consecutive days, the rats were placed in the startle box and 3 min later presented with 10 startle stimuli at 2 min intertrial intervals (ITIs). Based on their mean startle amplitudes in the second of these two sessions, the rats were sorted into groups with similar response levels.
Training.
The rats were placed in the startle boxes and received 10 light–footshock pairings at an ITI of 2 min. Unpaired controls received the same number of light and footshock presentations, but in a pseudorandom manner in which the US could occur at anytime except 3.2 s after the CS.
Test.
Twenty-four hours after training, the rats were tested for fear-potentiated startle. This involved 10 startle-eliciting noise bursts presented alone (noise-alone trial) and 10 noise bursts presented 3.2 s after the onset of the 3.7 s light (light–noise trials). The two trial types were presented in a balanced mixed order (ITI, 30 s). The percentage of fear-potentiated startle was computed as follows: [(startle amplitude on CS-noise − noise-alone trials)/(noise-alone trials)] × 100.
Extinction training.
One hour or 24 h after the training, the rats were returned to the stabilimeter and given three sessions of 10 presentations of the 3.7-s light with neither a shock nor a startle-elicited noise burst (light-alone trials). Each session was separated by 10 min with an ITI of 1 min.
DCS experiments.
On day 1, rats were trained with 10 pairings of light and footshocks and randomly assigned to one of six experimental groups: no extinction, extinction, DCS before extinction, DCS after extinction, vehicle before extinction, and DCS no-extinction controls. On day 2, rats from the extinction group received three sessions of 10 presentations of light-alone trials, whereas no-extinction rats were exposed to the conditioning chamber for an equivalent amount of time without receiving light-alone trials. Rats from the DCS-before-extinction and vehicle-before-extinction groups were infused with either DCS or vehicle bilaterally into the amygdala. The infusion rate was 0.1 μl/min. After the infusion, a delay of 20 min was interposed before extinction training. DCS-after-extinction rats received extinction training first, followed 30 min later by DCS infusion. Rats in the DCS-no-extinction group were given DCS infusion without extinction training. On day 3, all groups were tested for fear-potentiated startle.
Slice preparation and electrophysiological recordings
Male Sprague Dawley rats (175–200 g) were decapitated, and their brains were rapidly removed and placed in cold, oxygenated artificial CSF (ACSF) solution. Subsequently, each brain was hemisected and cut transversely posterior to the first branch and anterior to the last branch of the superior cerebral vein. The resulting section was glued to the chuck of a Vibroslice (Campden Instruments, Sileby, UK) tissue slicer. Transverse slices of 450 μm thickness were cut, and the appropriate slices were placed in a beaker of oxygenated ACSF at room temperature for at least 1 h before recording. The ACSF solution had the following composition (in mm): 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, and 11 glucose. The ACSF was bubbled continuously with 95%O2/5%CO2 and had a pH of 7.4.
A single slice was transferred to the recording chamber where it was held submerged between two nylon nets and maintained at 32 ± 1°C. The chamber consisted of a circular well of low volume (1–2 ml) and was perfused constantly at a rate of 2–3 ml/min. Extracellular field potentials were made by electrical stimulation of the external capsule (EC), which contained fibers from the auditory cortex to the LA, with a concentric bipolar stimulating electrode. Electrical stimuli (150 μs in duration) were delivered at a frequency of 0.05 Hz. Baseline field potentials were adjusted to ∼30–40% of the maximal responses. LTP was elicited by five trains of tetanus (100 Hz, 1 s at 1 min intervals) at the same stimulation intensity used for baseline. Depotentiation was elicited by a 5 Hz stimulation for 3 min. Bicuculline (2 μm) was present in the perfusion solution.
Surface biotinylation and Western blot analysis of surface GluR1
Brain slices containing only LA and BLA were placed on ice and washed twice with ice-cold ACSF. The slices were then incubated with ACSF containing 0.5 mg/ml sulfo-NHS-LC-biotin (Pierce, Rockford, IL) for 1 h on ice. Next, the slices were rinsed in TBS buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl) to quench the biotin reaction and sonicated briefly in homogenizing buffer (1% Triton X-100, 0.1% SDS, 50 mm Tris-HCl, pH 7.5, 0.3 m sucrose, 5 mm EDTA, 2 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride, 20 μg/ml leupeptin, and 4 μg/ml aprotinin). After sonication, the samples were centrifuged at 14,000 rpm for 30 min at 4°C and the supernatant was obtained. Protein concentration in the soluble fraction was then measured using a Bradford assay, with bovine serum albumin as the standard. Biotinylated protein (400 μg) from the supernatant was precipitated with 50 μl of 50% Neutravidin agarose (Pierce) for 16 h at 4°C and washed four times with homogenizing buffer. Bound protein was resuspended in 4 μl of SDS sample buffer and boiled. Biotinylated protein was resolved in 8.5% SDS-polyacrylamide gels, blotted electrophoretically to polyvinylidene difluoride membrane, and blocked overnight in TBS buffer containing 5% nonfat milk. Surface GluR1 receptors and pan-cadherin (surface protein control) were labeled with a biotinylation assay, followed by Western blot analysis that used a GluR1 antibody (1:4000; Santa Cruz Biotechnology, Santa Cruz, CA), pan-cadherin antibody (1:2500; Sigma), or actin (1:4000; Santa Cruz Biotechnology), and the addition of HRP-conjugated secondary antibodies for 1 h. An enhanced chemiluminescence kit was used for detection of the labeled proteins. Western blots were developed in the linear range used for densitometry. GluR1 levels in paired or unpaired animals were expressed as a percentage of those in naive controls.
Synaptoneurosome preparation
Brain slices containing only LA and BLA were homogenized in 70 μl of ice-cold lysis buffer in an Eppendorf tube. The buffer consisted of the following (in mm): 118.5 NaC1, 4.7 KC1, 1.18 MgSO4, 2.5 CaCl2, 1.l8 KH2PO4, 24.9 NaHCO3, 10 dextrose, and 10 μg/ml adenosine deaminase, and its pH was adjusted to 7.4 by bubbling with 95% O2/5% CO2. Proteinase inhibitors (0.0l mg/ml leupeptin, 0.005 mg/ml pepstatin A, 0.l mg/ml aprotinin, and 5 mm Benzamide) were included in the buffer to minimize proteolysis. The homogenate was diluted with 350 μl of additional ice-cold lysis buffer. This mixture was loaded into a l ml tuberculin syringe attached to a 13-mm-diameter syringe filter holder (Millipore, Bedford, MA). The diluted filtrate was forced over three layers of nylon (100 μm pores; Nytex: Tetko, Elmsford, NY) prewetted with 150 μl of lysis buffer and collected in a 1.5 ml Eppendorf tube. The nylon-prefiltered mixture was loaded into another 1 ml tuberculin syringe and forced through a prewetted 5 μm nitrocellulose filter (Millipore). The homogenate was kept ice cold at all times to minimize proteolysis. The filtered particulate was then spun at l000 × g for l5 min at 4°C. The supernatant was removed, and the pellet (synaptoneurosome) was resuspended in 80 μl of lysis buffer for Western blot analysis.
Data analysis
The percentage of fear-potentiated startle was used as a measure of conditioned fear. ANOVA followed by Bonferroni tests were performed to analyze the degree of significance among multigroup experiments. A paired t test was used to analyze differences in field EPSPs (fEPSPs) before and after tetanic stimulation (TS) or low-frequency stimulation (LFS). All values are mean ± SEM.
Results
Light-alone trials conducted 1 h, but not 24 h, after training reverse conditioning-induced increase in surface GluR1
Rats were conditioned with 10 light–shock pairings, and memory was assessed 24 h later. Trained rats learned to associate the conditioning light with shock. This was manifested with an increase in acoustic startle (175.8 ± 11.2%; n = 7) relative to naive (22.9 ± 1.5%; n = 6) and unpaired (33.1 ± 6.1%; n = 6) controls (F(2,16) = 119.7; p < 0.0001). Thirty minutes after training, the rats were infused with NMDA receptor antagonist d-APV (12.5 nmol/side), phosphatidylinositol 3-kinase (PI-3 kinase) inhibitor wortmannin (2.5 μg/side), or vehicle bilaterally into the amygdala, followed 30 min later by three sessions of light-alone trials. Extinction training significantly reduced fear-potentiated startle to 67.3 ± 6.6% (n = 7; p < 0.001) (Fig. 1A). Treatment with d-APV (176.1 ± 6.8%; n = 6) or wortmannin (177.3 ± 10.4%; n = 6) blocked extinction training-induced reduction in fear-potentiated startle. ANOVA indicated a main effect of group (F(2,16) = 119.7; p < 0.0001), with vehicle rats showing significantly less startle potentiation than d-APV and wortmannin rats (p < 0.001) and equivalent startle potentiation to extinction controls (p > 0.1). The photomicrograph illustrating the placement of the infusion needle tip into the LA is shown in supplemental Figure 1 (available at www.jneurosci.org as supplemental material), and the diagram of the infusion cannula tip locations is shown in Figure 1B. Only rats with cannula tips at or within the boundaries of LA and BLA were included in the data analysis.
Figure 1.

Light-alone trials 1 h after training reverse conditioning-induced increase in the surface expression of GluR1. Rats were conditioned and randomly assigned to the extinction, d-APV, wortmannin, and vehicle groups. One hour later, they were given three sessions of 10 presentations of light without footshock. d-APV (12.5 nmol per side) or wortmannin (2.5 μg per side) were given 30 min before light-alone trials. A, Plot of percentage of startle potentiation in unpaired control, conditioned, wortmannin-treated, and d-APV-treated animals. ***p < 0.001 versus the wortmannin and d-APV groups. B, Cannula tip placements from paired rats infused with d-APV (●), wortmannin (▵), or vehicle (★, 1 μl of 50% DMSO) in experiment A. C, Representative blots and mean ± SEM of GluR1 immunoreactivity from rats decapitated 24 h after extinction training. LA and BLA tissues were dissected out, and surface GluR1 levels were determined by biotin labeling. Biotinylated surface proteins were precipitated using immobilized avidin and analyzed using Western blot analysis with anti-GluR1 or anti-pan-cadherin (surface protein control) antibodies. ***p < 0.001 versus the d-APV group. D, Light-alone trials reversed the conditioning-induced increase in GluR1 in synaptoneurosomes; both wortmannin treatment and d-APV treatment inhibited this effect. **p < 0.01 versus the d-APV and wortmannin groups. Wort, Wortmannin; Unp, unpaired; Ext, extinction; Veh, vehicle.
In parallel with behavioral performance, LA and BLA tissues were dissected out, and surface receptors were labeled with biotin 24 h after training. Biotinylated receptors were precipitated, and surface GluR1 was determined by immunoblotting. As shown in Figure 1C, fear conditioning significantly increased surface expression of GluR1 compared with naive and unpaired controls (F(2,15) = 21.3; p < 0.0001). Conditioning-induced increase in GluR1 was inhibited in the extinction group, and such effect was blocked by d-APV. ANOVA indicated a main effect of group (F(3,20) = 29.6; p < 0.0001), with d-APV rats showing significantly higher GluR1 levels than vehicle and extinction rats (p < 0.001) as well as equivalent GluR1 levels to paired controls (p > 0.1). Similar experimental procedures were performed to determine GluR1 levels in the synaptic sites using synaptoneurosomes that resemble isolated glutamatergic synapses located on the dendritic spines with resealed presynaptic and postsynaptic compartments (Hollingsworth et al., 1985; Hampson et al., 1992; Heynen et al., 2000). The GluR1 level was significantly increased after conditioning (F(2,15) = 19.6; p < 0.0001), and the increase was reversed by extinction training. The reduction in GluR1 increase could be blocked by wortmannin or d-APV (F(3,20) = 19.6; p < 0.01). Wortmannin and d-APV rats exhibited significantly higher GluR1 levels than vehicle and extinction rats (p < 0.01) and equivalent GluR1 levels to paired controls (p > 0.5) (Fig. 1D).
Light-alone trials applied 1 h after fear training may not induce fear extinction, but it may instead block consolidation of memory. Therefore, a separate set of experiments was performed in which light-alone trials were applied 24 h after training. Behavioral assessment revealed that light-alone trials caused a significant reduction in the fear-potentiated startle (F(2,19) = 33.9; p < 0.0001). Context control rats were returned to the startle box at the equivalent time without receiving light-alone trials. The startle potentiations in paired, extinction, and context control groups were 175.8 ± 11.2% (n = 7), 82.0 ± 7.1% (n = 9), and 165.6 ± 10.1% (n = 6), respectively. Treatment with d-APV (12.5 nmol/side; n = 6) or wortmannin (2.5 μg/side, n = 6) 30 min before light-alone trials blocked extinction training-induced reduction in startle. ANOVA indicated a main effect of group (F(3,20) = 31.1; p < 0.0001), with d-APV and wortmannin rats showing significantly higher startle potentiation than vehicle rats (p < 0.001) and equivalent startle potentiation to context-alone controls (p > 0.5).
In contrast to training applied after 1 h, light-alone trials 24 h after training did not significantly influence the conditioning-induced increases in surface expression of GluR1 (Fig. 2A). In addition, wortmannin and d-APV rats exhibited comparable GluR1 levels to the extinction group (F(4,37) = 0.71; p = 0.59). A lack of the effect of extinction training applied 24 h after training on conditioning-induced increase in GluR1 was also observed in synaptoneurosomes (p = 0.12) (Fig. 2B). In summary, light-alone trials applied 1 h after training attenuated the increase in surface GluR1 in parallel with the inhibition of startle potentiation. Light-alone trials applied 24 h after training also attenuated startle potentiation but did not influence the GluR1 increase.
Figure 2.
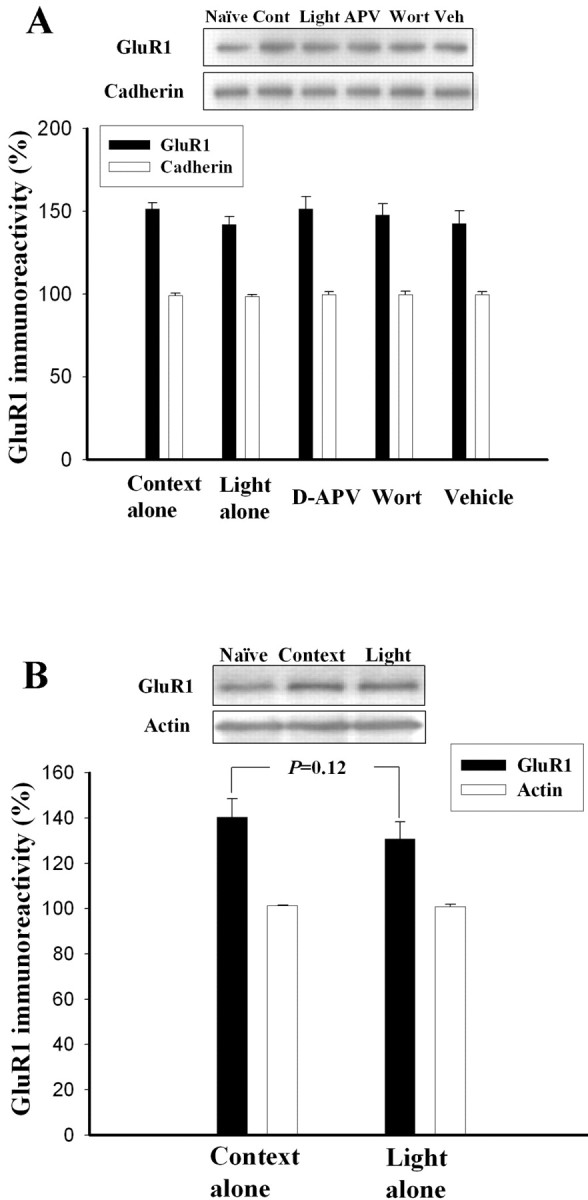
Light-alone trials 24 h after training extinguish fear memory but do not reverse the conditioning-induced increase in the surface expression of GluR1. Rats were conditioned with 10 light–shock pairings; they were given light-alone trials 24 h after training. Context control rats were returned to the startle box at the equivalent time without light-alone trials. Memory retention was assessed 24 h after light-alone trials. A, Representative blots and mean ± SEM of GluR1 immunoreactivities from rats decapitated 24 h after extinction training. LA and BLA tissues were dissected out, and the surface GluR1 levels were determined by biotin labeling. B, GluR1 levels in the context control and extinction groups were determined in synaptoneurosomes. Cont, Context; Wort, wortmannin; Veh, vehicle.
Coapplication of light-alone trials with DCS reverses conditioning-induced increase in surface expression of GluR1
DCS facilitates extinction training-induced reduction in fear response (Walker et al., 2002; Ledgerwood et al., 2003) and enhances treatment responses (Ledgerwood et al., 2004). Interestingly, extinction training combined with DCS treatment does not show reinstatement by unpaired US presentation in rats. We therefore investigated the mechanism behind the facilitation of extinction of DCS. On day 1, rats were conditioned. On day 2, they were infused with DCS (10 μg per side) bilaterally into the amygdala either 30 min before or 30 min after light-alone trials. Vehicle-control rats were given saline 30 min before extinction training. The rats were tested for fear-potentiated startle 24 h after extinction training. Fear-potentiated startle was significantly lower in both DCS-treated rats than in vehicle-treated and extinction controls (F(3,23) = 4.45; p < 0.05) (Fig. 3A). The effect of DCS might reflect reduction in fear response or a disruption of fear-potentiated startle independent of fear reduction. To distinguish these possibilities, rats were treated with DCS without extinction training and tested for fear memory 24 h later. The startle reflex in these rats (DCS without extinction) was comparable with those without extinction training (paired without extinction) (p = 0.52), suggesting that DCS augmented extinction training-induced reduction in startle potentiation and did not exert a general effect on startle reflex.
Figure 3.

DCS augments extinction training-induced reduction in startle reflex. A, Rats were conditioned and 24 h later were given light-alone trials. The percentage of fear-potentiated startle (mean ± SEM) was measured 24 h after light-alone trials. DCS (10 μg per side) was infused bilaterally into the amygdala 30 min before or after light-alone trials. B, Cannula tip placements in rats infused with DCS before extinction (●), DCS after extinction (▴), vehicle before extinction (○), or DCS without extinction (▵) in experiment A. C, HA-966 (6 mg/kg, i.p.) was given 30 min before DCS; 30 min later, light-alone trials were given. HA-966 blocked the effect of DCS, but HA-966 alone did not affect extinction training. D, Cannula tip placements in rats infused with DCS plus HA-966 (●) or DCS plus vehicle (○) in experiment C. Ext, Extinction; Veh, vehicle.
We tested whether HA-966, an antagonist for the glycine-binding site of NMDA receptors, blocked the effect of DCS. Twenty-four hours after conditioning, the rats were given intraperitoneal injections of saline or HA-966 (6 mg/kg); 30–60 min later, they were treated with DCS. After 30 min, the rats were given extinction training. Twenty-four hours later, they were tested for fear-potentiated startle. HA-966-treated rats given extinction training, but no DCS treatment, were the controls. HA-966 blocked the effect of DCS. Startle potentiation in rats treated with HA-966 plus DCS was significantly higher than in those treated with saline plus DCS or DCS alone (F(2,14) = 5.25; p < 0.05) and was comparable to those that received extinction training only (p = 0.96) (Fig. 3C). Furthermore, HA-966-treated rats given extinction training, but no DCS treatment, had startle potentiation equivalent to extinction rats (p > 0.5), suggesting that HA-966 by itself did not influence extinction.
We then examined whether DCS affected GluR1 expression after extinction training. On day 1, rats were conditioned; 24 h later, they were treated with DCS (10 μg per side) either before or after extinction training. Twenty-four hours after extinction training, LA and BLA tissues were dissected out, and the surface receptors were labeled with biotin. Extinction training did not significantly influence the conditioning-induced increases in GluR1 (p = 0.10), but DCS treatment either 30 min before or after light-alone trials reversed them (F(2,15) = 6.8; p < 0.01) (Fig. 4A). The reversal of GluR1 increase after extinction training plus DCS could be blocked by HA-966. HA-966 plus DCS rats exhibited significantly higher GluR1 levels than DCS plus vehicle, DCS-before-extinction, and DCS-after-extinction rats (F(3,20) = 5.1; p < 0.01) and equivalent GluR1 levels to extinction controls (p = 0.77).
Figure 4.
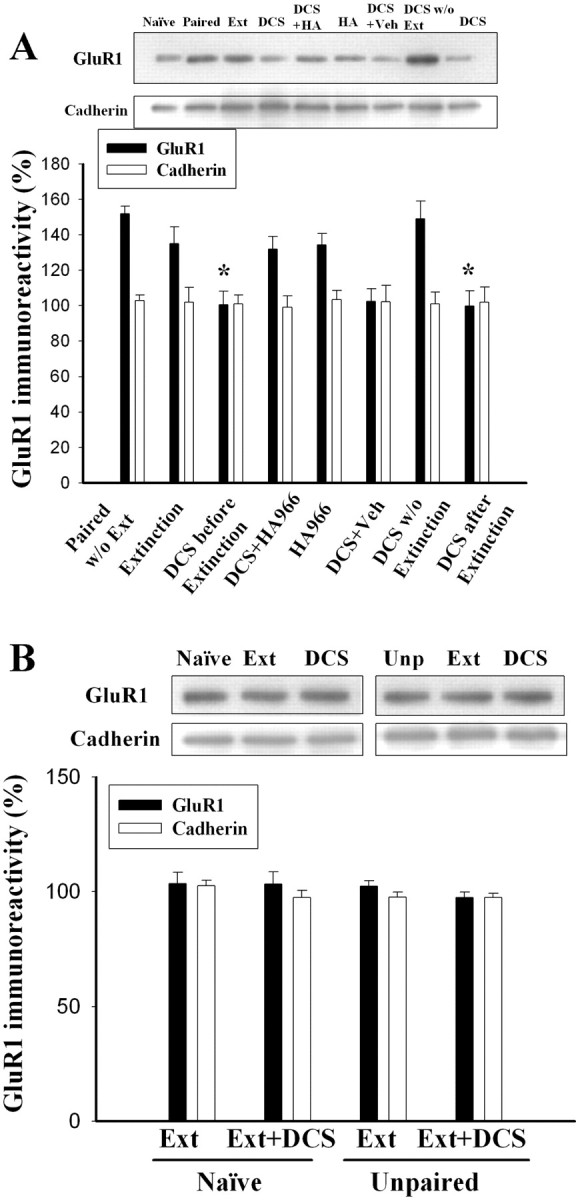
Extinction training plus DCS reverses the conditioning-induced increase in the surface expression of GluR1. A, Representative blots and mean ± SEM of GluR1 immunoreactivity. Rats were conditioned, and 24 h later they were infused with DCS (10 μg per side) bilaterally into the amygdala 30 min before or after extinction training. Twenty-four hours after extinction training, LA and BLA tissues were dissected out and the surface GluR1 levels were determined using biotin labeling. Cotreatment of DCS with light-alone trials blocked the conditioning-induced increases in surface expression of GluR1. Intraperitoneal HA-966 (6 mg/kg) 30 min before DCS blocked the effect of DCS. *p < 0.05 versus extinction. B, Extinction training plus DCS treatment did not influence surface GluR1 levels in control rats that were given light and footshock in an unpaired manner (unpaired) or received neither light nor footshock (naive). Ext, Extinction; Veh, vehicle; Unp, unpaired; HA, HA-966.
The effects of extinction training and treatment of DCS on the unconditioned rats were examined. As shown in Figure 4B, extinction training and DCS treatment did not affect surface GluR1 levels in both naive (F(2,9) = 0.30; p > 0.5) and unpaired rats (F(2,9) = 1.65; p > 0.1).
The decrease in surface GluR1 could represent internalization or catalysis of GluR1. Therefore, we performed Western blot analysis of GluR1 in tissue homogenates from LA and BLA. The result revealed that the total levels of GluR1 were not different among the rats in naive, fear-conditioned, and after-extinction training plus DCS treatment (F(2,9) = 1.65; p = 0.25) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
DCS facilitates depotentiation and reduction in surface levels of GluR1
Previous studies indicate that LTP of synapses from the auditory thalamus and cortex to the LA underlies the encoding of fear memory (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997). LTP of synaptic responses in the amygdala could be reversed by LFS of afferent fibers (Aroniadou-Anderjaska et al., 2001), a phenomenon termed depotentiation (Barrionuevo et al., 1980; Staubli and Chun, 1996). Reversal of LTP is time dependent; LFS erases LTP when delivered within 20 min after the induction of LTP, but not at the time when LTP is consolidated (Staubli and Chun, 1996). We examined whether DCS facilitated LFS-induced depotentiation in the amygdala slices. fEPSPs in the LA were elicited by stimulation of the EC. Delivery of five sets of TS (100 Hz for 1 s) at an interstimulus interval of 1 min to the EC produced a robust enhancement of synaptic responses in the LA neurons that persisted for >2 h. The slopes of fEPSP were 212.0 ± 7.6% and 195.9 ± 7.7% (n = 8) of the pretetanus level at 1 and 2 h after the stimulation (Fig. 5A). We first tested whether LFS (5 Hz for 3 min) applied at 10 min after TS (LFS 10 min) was able to elicit depotentiation. In six experiments, the slopes of fEPSP declined to 117.8 ± 9.4% and 103.0 ± 10.2% (n = 6) of the pretetanus level at 1 and 2 h after TS. In the next experiments, LFS was applied 60 min after TS (LFS 60 min). As shown in Figure 5A, LFS induced an initial decline in fEPSP that partially recovered to 166.1 ± 10.2% (n = 7) of baseline at 2 h after TS. In contrast, when DCS was applied 5 min before and during the LFS (DCS plus LFS 60 min), fEPSP declined to 100.2 ± 1.7% (n = 7) of baseline at 2 h after TS. ANOVA indicated a main effect of group (F(2,19) = 14.21; p < 0.001), with DCS plus LFS 60 min slices showing significantly less fEPSP slopes than LFS 60 min (p < 0.01) and no-LFS (p < 0.001) slices as well as an equivalent fEPSP slope to LFS 10 min slices (p > 0.05).
Figure 5.
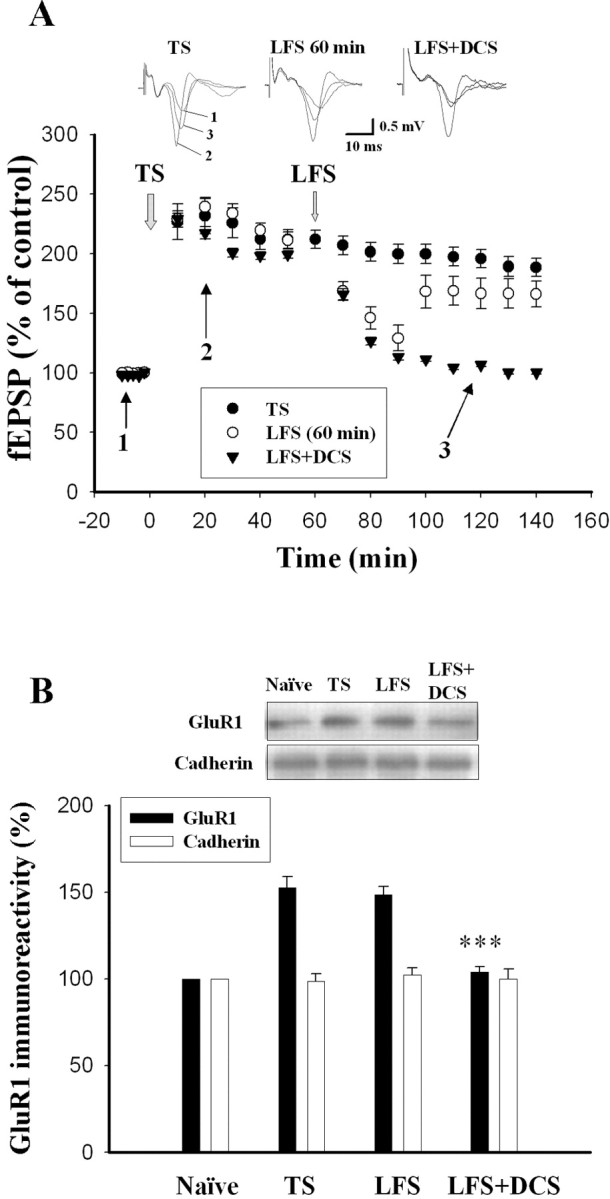
DCS facilitates LFS-induced depotentiation and reduction in surface levels of GluR1. A, The graph represents the mean ± SEM slope of fEPSPs plotted against time. LFS applied 60 min after the TS had no lasting effect on the LTP. However, in the presence of DCS (10 μm), LFS induced depotentiation. B, Amygdala slices were randomly assigned to the naive, TS, LFS 60 min, and LFS 60 min plus DCS groups. Slices were given five sets of TS at an interstimulus interval of 1 min to the EC in the TS group. In the LFS 60 min group, LFS was delivered at 60 Hz after the onset of TS. DCS was applied 5 min before and during the LFS in the DCS plus LFS 60 min group. Two hours after the onset of TS, a relatively small portion of the LA was dissected from each slice. Four to six slices from each animal were pooled together for biochemical analysis. Naive control slices were incubated in ACSF for 2 h. ***p < 0.001 versus TS.
Using a biotinylation assay, we tested whether DCS facilitated LFS-induced reduction in surface GluR1. Rats were killed, and amygdala slices were prepared and assigned randomly to the naive, TS, LFS 60 min, and LFS 60 min plus DCS groups with the same protocols as the electrophysiological experiments. Thus, amygdala slices were given five sets of TS at an interstimulus interval of 1 min to the EC in the TS group. In the LFS 60 min group, LFS was delivered at 60 Hz after the onset of TS. DCS was applied 5 min before and during the LFS in the LFS 60 min plus DCS group. Two hours after the onset of TS, a relatively small portion of the LA was dissected from each slice. Four to six slices from each animal were pooled together for biochemical analysis. Biotinylated receptors were precipitated, and surface GluR1 was determined by quantitative immunoblotting. Figure 5B shows that the GluR1 level was significantly elevated in the LA (152.5 ± 6.8%; n = 6 rats) when compared to naive slices without receiving TS. LFS applied 60 min after TS did not significantly influence the TS-induced increases in GluR1 (148.6 ± 4.8%; n = 6 rats), but DCS treatment 5 min before LFS reversed them (F(2,15) = 32.73; p < 0.001) (Fig. 5B).
Discussion
Understanding how learned fear might be diminished or abolished has important implications for the treatment of anxiety disorders. Psychological data and animal studies indicate memory extinction as an active learning process that inhibits expression of the original association rather than erasing it. We tested this hypothesis by labeling surface receptors with biotin or using membrane fractionation. There are two important findings. First, the interval between conditioning and extinction training determines whether a memory trace remains intact or is erased. Extinction training applied 1 h after training reversed the conditioning-induced increase in surface GluR1 in parallel with the inhibition of startle potentiation. However, if applied 24 h after training, extinction training reduced startle potentiation without influencing GluR1 increase. Second, DCS facilitates the reversal of memory trace. Neither extinction training applied 24 h after conditioning nor DCS alone altered surface expression of GluR1. However, coadministration of DCS with extinction training facilitated extinction and reversed the increase in GluR1. Similarly, in the in vitro amygdala slices, application of LFS 1 h after the TS had no long-lasting effect on synaptic responses. The same treatment, however, induced depotentiation in the presence of DCS and reversed TS-induced increase in surface GluR1. Recent studies have shown that encoding of fear memory in the amygdala is associated with AMPA receptor trafficking (Rumpel et al., 2005; Yeh et al., 2006). More importantly, blocking synaptic GluR1 receptor incorporation inhibited amygdala LTP as well as fear conditioning (Rumpel et al., 2005), suggesting an essential contribution of this molecular process to memory formation. Thus, our findings here raise the possibility of erasing memory by application of extinction training shortly after conditioning or by combining extinction training with DCS infusion of the amygdala.
We have shown previously that fear conditioning induced Akt (a serine/threonine kinase) phosphorylation in the amygdala. However, when light-alone trials were subsequently applied within 1 h after training, phosphorylated Akt was reduced and accompanied by an increase in enzymatic activity and protein level of calcineurin (Lin et al., 2003). Therefore, adding light-alone trials 1 h after training increased phosphatase activity that counteracted consolidation of memory. Complementary to this result, Myers et al. (2006) recently demonstrated that rats receiving extinction training within 1 h after conditioning exhibited little or no reinstatement, renewal, and spontaneous recovery. On the other hand, rats that were killed 24–72 h after acquisition exhibited reinstatement, renewal, and spontaneous recovery. The differential effects with varying intervals between acquisition and extinction training were also reflected on the sensitivity of extinction training to the l-type calcium channel blocker nifedipine. For example, extinction was blocked by nifedipine when extinction training was given 1 h or longer but not immediately after conditioning (Cain et al., 2005).
Extinction 24 h after acquisition is also sensitive to NMDA receptor and PI-3 kinase inhibitors (Falls et al., 1992; Lee and Kim, 1998; Lin et al., 2003; Chen et al., 2005). Converging evidence indicates that the medial prefrontal cortex (mPFC) and the intercalated cells within the amygdala play important roles in the extinction of fear memory (Milad and Quirk, 2002; Myers and Davis, 2002; Royer and Paré, 2002; Maren and Quirk, 2004; Sotres-Bayon, 2004). Intercalated GABAergic neurons receive input from the BLA and exhibit NMDA-dependent LTP (Royer and Pare, 2002). During extinction training, mPFC neurons activate and cause sufficient depolarization of intercalated neurons to allow their expression of NMDA-dependent LTP from BLA inputs. Subsequently, in response to CS-alone trials, BLA neurons would produce relatively more inhibition on the central nucleus of the amygdala leading to the suppression of conditioned fear (Likhtik et al., 2005). PI-3 kinase signaling is required for LTP in several areas of the brain, including hippocampal CA1 (Sanna et al., 2002; Opazo et al., 2003), dentate gyrus (Kelly and Lynch, 2000), and amygdala (Lin et al., 2001). It is possible that intercalated GABAergic neurons also exhibit PI-3 kinase-dependent LTP from the BLA because extinction 24 h after acquisition is blocked by PI-3 kinase inhibitor (Chen et al., 2005).
AMPA receptors can be removed from synapses by briefly treating them with glutamate receptor agonists (Shi et al., 1999; Ehlers, 2000; Lin et al., 2000). Protein kinase A (PKA) is targeted to the synapse by an interaction with the A kinase-anchoring protein AKAP79/150 (Colledge et al., 2000). Disruption of PKA–AKAP interaction is sufficient to induce reduction in synaptic AMPA receptors. DCS facilitates activation of NMDA receptors that induces Ca2+ influx resulting in the activation of calcineurin. Calcineurin activation leading to local reorganization of F-actin, which causes displacement of PKA from the synapse that may initiate endocytosis of receptor and trigger a loss of GluR1 from the surface of neurons (Gomez et al., 2002; Snyder et al., 2005). Assuming that the DCS mechanisms in vivo and in vitro are similar, it suggests that DCS augments depotentiation of synaptic responses and extinction of fear memory via facilitation of NMDA-mediated disruption of PKA–AKAP interaction, resulting in receptor endocytosis.
Extinction training usually consisted of CS-alone trials that induced memory retrieval and reconsolidation (Nader et al., 2000; Duvarci and Nader, 2004). Activation of PKA in the amygdala enhanced reconsolidation, whereas inhibition of PKA impaired reconsolidation (Tronson et al., 2006). Thus, facilitation of NMDA-induced activation of calcineurin and inhibition of PKA by DCS could lead to a block on reconsolidation. This result could provide a mechanistic explanation for the retardation of reinstatement by the treatment of extinction training plus DCS (Ledgerwood et al., 2004). In the present study, the complete reversal of GluR1 by extinction training plus DCS suggests that DCS may transform the effect of light-alone trials from inhibitory learning (extinction) to erasure (blockage of reconsolidation). In contrast, in behavioral tests, HA-966 blocked only the additional reduction caused by DCS treatment, rather than returning fear potentiation levels to those of fear-conditioned animals without extinction, implicating that inhibitory learning is intact and DCS only adds a bit of erasure. It is possible that DCS does change the nature of the extinction learning, but the HA-966 experiment cannot reveal it, because it blocks rather than reverses the effect of DCS.
In summary, encoding fear memory is thought to require long-lasting change in the strength of synaptic transmission, likely via the expression of surface AMPA receptors in the amygdala (Rumpel et al., 2005; Yeh et al., 2006). Our data provide the first evidence that extinction training plus DCS reverses conditioning-induced increases in the synaptic expression of GluR1. DCS facilitates LFS-induced depotentiation and reduction in surface expression of GluR1. If DCS mechanisms in vivo and in vitro are similar, these results suggest that facilitation of NMDA-mediated endocytosis of AMPA receptors might be the underlying mechanism. A schematic diagram illustrating fear conditioning, extinction, and the effect of DCS is shown in supplemental Figure 3 (available at www.jneurosci.org as supplemental material). The present study demonstrates that DCS plus extinction training might erase memory trace, and it explains the clinical benefit of using DCS with extinction-based psychotherapy to treat post-traumatic stress disorder.
Footnotes
This work was supported by Grant NSC94-2752-B-006-001-PAE from the National Science Council, Taiwan. We thank Drs. Min-Der Lai and A-Min Huang for critical comments on this manuscript.
References
- Aroniadou-Anderjaska V, Post RM, Rogawski MA, Li H. Input-specific LTP and depotentiation in the basolateral amygdala. NeuroReport. 2001;12:635–640. doi: 10.1097/00001756-200103050-00041. [DOI] [PubMed] [Google Scholar]
- Barrionuevo G, Schottler F, Lynch G. The effect of repetitive low frequency stimulation on control and “potentiated” synaptic responses in the hippocampus. Life Sci. 1980;27:2385–2391. doi: 10.1016/0024-3205(80)90509-3. [DOI] [PubMed] [Google Scholar]
- Bouton ME, King DA. Contextual control of the extinction of conditioned fear: tests for the associative value of the context. J Exp Psychol Anim Behav Process. 1983;9:248–265. [PubMed] [Google Scholar]
- Bouton ME, Peck CA. Spontaneous recovery in cross-motivational transfer (counterconditioning) Anim Learn Behav. 1989;20:313–321. [Google Scholar]
- Cain CK, Godsil BP, Jami S, Barad M. L-type calcium channel blocker nifedipine impairs extinction, but not reduced contingency effects, in mice. Learn Mem. 2005;12:277–284. doi: 10.1101/lm.88805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Li V, Athos J, Storm D. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8:925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Nader K. Characterization of fear memory reconsolidation. J Neurosci. 2004;24:9269–9275. doi: 10.1523/JNEUROSCI.2971-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Falls WA, Miserendino JD, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci. 2002;22:7027–7044. doi: 10.1523/JNEUROSCI.22-16-07027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson DR, Huang XP, Oberdorfer MD, Goh JW, Auyeung A, Wenthold RJ. Localization of AMPA receptors in the hippocampus and cerebellum of the rat using an anti-receptor monoclonal antibody. Neuroscience. 1992;50:11–22. doi: 10.1016/0306-4522(92)90378-f. [DOI] [PubMed] [Google Scholar]
- Herry C, Garcia R. Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J Neurosci. 2002;22:577–583. doi: 10.1523/JNEUROSCI.22-02-00577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heynen AJ, Quinlan EM, Bae DC, Bear MF. Bidirectional, activity-dependent regulation of glutamate receptors in the adult hippocampus in vivo. Neuron. 2000;28:527–536. doi: 10.1016/s0896-6273(00)00130-6. [DOI] [PubMed] [Google Scholar]
- Hollingsworth EB, McNeal ET, Burton JL, Williams RJ, Daly JW, Creveling CR. Biochemical characterization of a filtered synaptoneurosome preparation from guinea pig cerebral cortex: cyclic adenosine 3′,5′-monophosphate-generating systems, receptors, and enzymes. J Neurosci. 1985;5:2240–2253. doi: 10.1523/JNEUROSCI.05-08-02240.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, Lynch MA. Long-term potentiation in dentate gyrus of the rat is inhibited by the phosphoinositide 3-kinase inhibitor, wortmannin. Neuropharmacology. 2000;39:643–651. doi: 10.1016/s0028-3908(99)00169-0. [DOI] [PubMed] [Google Scholar]
- Ledgerwood L, Richardson R, Cranney J. Effects of d-cycleserine on the extinction of conditioned freezing. Behav Neurosci. 2003;117:341–349. doi: 10.1037/0735-7044.117.2.341. [DOI] [PubMed] [Google Scholar]
- Ledgerwood L, Richardson R, Cranney J. D-cycleserine and the facilitation of extinction of conditioned fear: consequences for reinstatement. Behav Neurosci. 2004;118:505–513. doi: 10.1037/0735-7044.118.3.505. [DOI] [PubMed] [Google Scholar]
- Lee H, Kim JJ. Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J Neurosci. 1998;18:8444–8454. doi: 10.1523/JNEUROSCI.18-20-08444.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, Paz R, Paré D. Prefrontal control of the amygdala. J Neurosci. 2005;25:7429–7437. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Yeh HW, Lin CH, Lu KT, Leu TH, Chang WC, Gean PW. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–851. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Gean PW. The similarities and diversities of signal pathways leading to consolidation and extinction of fear memory. J Neurosci. 2003;23:8310–8317. doi: 10.1523/JNEUROSCI.23-23-08310.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee HS, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–610. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- Myers KM, Ressler KJ, Davis M. Different mechanisms of fear extinction dependent on length of time since fear acquisition. Learn Mem. 2006;13:216–223. doi: 10.1101/lm.119806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SGN, O'Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23:3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov IP. London: Oxford UP; 1927. Conditioned reflex: an investigation of the physiological activity of the cerebral cortex. [Google Scholar]
- Paxinos G, Watson C. New York: Academic; 1986. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla RA. Experimental extinction. In: Mowrer RR, Klein S, editors. Handbook of contemporary learning theories. Mahwah, NJ: Erlbaum; 2001. pp. 119–154. [Google Scholar]
- Rescorla RA, Heth CD. Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behav Process. 1975;1:88–96. [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Rogan MT, Leon KS, Perez DL, Kandel ER. Distinct neural signatures for safety and danger in the amygdala and striatum of the mouse. Neuron. 2005;46:309–329. doi: 10.1016/j.neuron.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Royer S, Paré D. Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience. 2002;115:455–462. doi: 10.1016/s0306-4522(02)00455-4. [DOI] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci. 2002;22:3359–3365. doi: 10.1523/JNEUROSCI.22-09-03359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Colledge M, Crozier RA, Chen WS, Scott JD, Bear MF. Role for A kinase-anchoring proteins (AKAPS) in glutamate receptor trafficking and long-term synaptic depression. J Biol Chem. 2005;280:16962–16968. doi: 10.1074/jbc.M409693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Bush DEA, LeDoux JE. Emotional perseveration: an update on prefrontal-amygdala interactions in fear extinction. Learn Mem. 2004;11:525–535. doi: 10.1101/lm.79504. [DOI] [PubMed] [Google Scholar]
- Staubli U, Chun D. Factors regulating the reversibility of long-term potentiation. J Neurosci. 1996;16:853–860. doi: 10.1523/JNEUROSCI.16-02-00853.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Wiseman SL, Olausson P, Taylor JR. Bidirectional behavioral plasticity of memory reconsolidation depends on amygdalar protein kinase A. Nat Neurosci. 2006;9:167–169. doi: 10.1038/nn1628. [DOI] [PubMed] [Google Scholar]
- Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of d-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh SH, Mao SC, Lin HC, Gean PW. Synaptic expression of glutamate receptor after encoding of fear memory in the rat amygdala. Mol Pharmacol. 2006;69:299–308. doi: 10.1124/mol.105.017194. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Current concepts: post-traumatic stress disorder. N Engl J Med. 2002;346:108–114. doi: 10.1056/NEJMra012941. [DOI] [PubMed] [Google Scholar]