

Abstract
Lithium is used as treatment for bipolar disorder with particular efficacy in the treatment of mania. Lithium inhibits glycogen synthase kinase 3β (GSK-3β) directly or indirectly via stimulation of the kinase Akt-1. We therefore investigated the possibility that transgenic mice overexpressing GSK-3β could be of relevance to model bipolar disorder. Transgenic mice showed hypophagia, an increased general locomotor activity, and decreased habituation as assessed in an open field, an increased acoustic startle response, and again decreased habituation. The forced swim test revealed a reduced immobility in transgenic mice, but this is probably related to the hyperactivity of the animals. There were no differences in baseline and stress-induced increases of plasma adrenocorticotrophic hormone and corticosterone levels. Molecular analysis suggests compensatory mechanisms in the striatum of these transgenic mice for the overload of active GSK-3β by dimming the endogenous GSK-3β signaling pathway via upregulation of Akt-1 expression. Brain-derived neurotrophic factor protein levels were increased in the hippocampus of the transgenic mice. This suggests some kind of compensatory mechanism to the observed reduction in brain weight, which has been related previously to a reduced size of the somatodendritic compartment. Together, in mice overexpressing GSK-3β, specific intracellular signaling pathways are affected, which is accompanied by altered plasticity processes and increased activity and reactivity, whereas habituation processes seem to be decreased. The behavioral observations led to the suggestion that the model at hand recapitulates hyperactivity as observed in the manic phase of bipolar disorder.
Keywords: Akt-1, mania, lithium, bipolar disorder, schizophrenia, dopamine
Introduction
Glycogen synthase kinase 3 (GSK-3) is a serine/threonine kinase that was first isolated and purified as an enzyme capable of phosphorylating and inactivating the enzyme glycogen synthase. In vertebrates, GSK-3 is found as two isoforms: GSK-3α and GSK-3β. Beyond its role in glycogen metabolism, GSK-3 acts as a downstream regulatory switch that determines the output of numerous signaling pathways coupled to receptors, e.g., growth factors, neurotransmitters, and neurotrophins (Frame and Cohen, 2001; Grimes and Jope, 2001; Doble and Woodgett, 2003). Signaling pathways act by inhibiting GSK-3 activity by phosphorylation of serine at positions 21 and 9 for GSK-3α and GSK-3β, respectively. Examples of such kinases are Akt-1 and protein kinases C and A (Cross et al., 1995; Doble and Woodgett, 2003; Gould et al., 2004b).
The pathways in which GSK-3 acts as a key regulator have been implicated in the development of a wide variety of psychiatric and nonpsychiatric human diseases, such as diabetes, cancer, inflammation, Alzheimer’s disease, schizophrenia, attention deficit hyperactivity disorder (ADHD), and bipolar disorder (Manji et al., 2003; Van Wauwe and Haefner, 2003; Beaulieu et al., 2004; Emamian et al., 2004; Gould et al., 2004b). The role of GSK-3-mediated signaling in the pathophysiology of bipolar disorder received special attention over recent years, in particular because of the effects of lithium on GSK-3 activity (Klein and Melton, 1996; Beaulieu et al., 2004; Gould et al., 2004b). Bipolar disorder is characterized by both manic and depressed states, and lithium is in particular a potent therapeutic drug for the acute treatment of mania. Although the mechanism of its therapeutic effect remains unknown, lithium was found to be a selective inhibitor of a limited number of enzymes, including GSK-3, as well as four related phosphomonoesterases (Berridge et al., 1989; Klein and Melton, 1996; Gould et al., 2004a). Along these lines, it has been demonstrated recently that pharmacologic inhibition of GSK-3 activity produces antidepressant-like effects in rodents (Redrobe and Bourin, 1999; Gould et al., 2004c; Kaidanovich-Beilin et al., 2004).
In light of the emerging role of GSK-3 in the pathophysiology of psychiatric disorders such as bipolar disorder, we investigated the possibility that transgenic mice overexpressing GSK-3β could be of relevance to model in particular depressive or manic states of bipolar disorder. These transgenic mice express a constitutively active mutated form of GSK-3β, i.e., GSK-3β[S9A], under the transcriptional regulation of a Thy-1 promotor (Spittaels et al., 2000, 2002). Postnatal expression of GSK-3β[S9A] was shown to interfere with neuronal development, resulting in a volume loss in the brain that has been related to a reduced size in the somatodendritic compartment (Spittaels et al., 2002). At the behavioral level, only subtle changes in psychomotor function have been reported in these mice thus far (Spittaels et al., 2002). In the present study, we assessed the striatal Akt-1/GSK-3β pathway in heterozygous GSK-3β[S9A]-overexpressing (GSK) mice. Animals were tested on activity (open field), reactivity (startle response), and affect (forced swimming). Hypothalamus–pituitary–adrenal axis activation was measured [adrenocorticotrophic hormone (ACTH) and corticosterone (CORT)], as well as plasticity (brain-derived neurotrophic factor protein levels and cell proliferation) in the neurogenic hippocampus and subventricular zone (SVZ).
Materials and Methods
Animals and experiments
Heterozygous GSK mice on FVB/NTac background and FVB/NTac littermate wild-type (WT) mice were bred and reared in-house. The generation of these transgenic mice has been described in detail previously (Spittaels et al., 2000, 2002). The animals were housed individually in ventilated cages on sawdust bedding in an air-conditioned room (∼24°C). The animals were kept under a normal 12 h light/dark cycle (lights on from 6:00 A.M. to 6:00 P.M.) and had access to food and water ad libitum. Experimental animals were 4–5 months old. All tests were performed during the light phase. All experiments were performed in accordance with the European Communities Council Directives (86/609/EEC) and were approved by the local ethical committee.
Molecular protocols
Quantitative reverse transcription (RT)-PCR analysis was used to determine expression levels of human and murine GSK-3β, murine GSK-3α, Akt-1, protein phosphatase 2, regulatory subunit β″, alpha (PPP2R3A), phosphoinositide-dependent kinase 1 (PDK1), and dopamine and cAMP-regulated phosphoprotein-32 (DARPP-32). Total RNA was isolated from striatum and cortex of six WT and six GSK male littermates using RNeasy (Qiagen, Hilden, Germany), and first-strand cDNA synthesis was performed on 0.5 μg of total RNA using random hexamer primers and SuperscriptII RT (Invitrogen, Carlsbad, CA). Quantitative PCR was performed on an ABI Prism 7700 cycler (Applied Biosystems, Foster City, CA) using a Taqman PCR kit. Serial dilutions of cDNA were used to generate standard curves of threshold cycles versus the logarithms of concentration for β-actin and the respective genes of interest. A linear regression line calculated from the standard curves allowed the determination of transcript levels in RNA samples from mice. The primer–probe pair of the respective genes of interest (see below) relative to actin was used to asses expression levels. For human and murine GSK-3β, the respective “assay on demand” primer–probe sets (human GSK-3β, Hs00275656_m1; murine GSK-3β, Mm00444911_m1) were obtained from Applied Biosystems. The following sequences were used: actin, 5′ CATCTTGGCCTCACTGTCCAC3′, 5′ GGGCCGGACTCATCGTACT3′, and 5′ TGCTTGCTGATCCACATCTGCTGGA3′ ([5′] 6-carboxyfluorescein (FAM), [3′] 6-carboxytetramethylrhodamine (TAMRA)); GSK-3α, 5′CAAGTTCCCCCAGATCAAAGC3′, 5′GGCTAGAGCAGAGTGCAATGG3′, and 5′CCCTTGGACAAAGGTGTTCAAATCTTCAAAG3′ ([5′]FAM, [3′]TAMRA); Akt-1, 5′GAACGACGTAGCCATTGTGAAG3′, 5′TGGCCGCCAGGTTTTAATAT3′, and 5′CCTCGTTTGTGCAGCCAGCCCT3′ ([5′]FAM, [3′]TAMRA); PPP2R3A, 5′GATGAACCTGTATCTCCCTCTGAAC3′, 5′TGAAGCTTTCCACATTTCTCTGAA3′, and 5′GGCTTGAAGTTACTATTTTATTGCCCTTGTTTCCA3′ ([5′]FAM, [3′]TAMRA); PDK1, 5′TGCTATGGCAACTACGACAATCTC3′, 5′TGGGAAGAGGAGGAGGATGA3′, and 5′CCTGCATGAAGCCAAACTGGCTCA3′ ([5′]FAM, [3′]TAMRA); and DARPP-32, 5′ACGCCCCCATCACTGAAA3′, 5′GGCCTGGTTCTCACTCAAGTTG3′, and 5′TGTGCAGCACCTGCAGACCATT3′ ([5′]FAM, [3′]TAMRA).
Biochemical protocols
GSK-3β protein.
ELISAs were used to determine the levels of total GSK-3β protein (catalog #KHO0451; Biosource, Camarillo, CA) and phosphorylated GSK-3β Ser9 protein (catalog #KHO0461; Biosource) in the striatum of 10 WT and 10 GSK male littermates. Samples were prepared and measured as described by the manufacturer.
DNA synthetic rate.
First, to reduce tracer injection stress, animals were habituated to the injection by a daily saline injection (0.25 ml, i.p.) in the week preceding the experiment. Eight WT and seven GSK male littermates were used to assess the DNA synthetic rate and were injected intraperitoneally with 8 μCi/g [3H]thymidine (20 Ci/mmol, 1 mCi/ml; PerkinElmer, Zaventem, Belgium), returned to their home cage, and rapidly decapitated after 1 h. After decapitation, the entire hippocampal formation and the SVZ (contained within the rostral forebrain) were dissected using the method of Wagner et al. (1999). After meninges and surface blood were removed, the dissected regions were weighed, quickly snap frozen in liquid nitrogen, and stored at −70°C.
Brain regions were homogenized in 350 μl (hippocampus) or 1 ml (SVZ) of lysis buffer [137 mm NaCl, 20 mm Tris-HCl, pH 8.0, 1% NP-40, 10% glycerol, and a complete EDTA-free protease inhibitor tablet (Roche, Vilvoorde, Belgium)] using a Mini-Bead beater (BioSpec, Bartlesville, OK). Samples were homogenized at least three times for 30 s each, with cooling of the samples on ice between runs. Aliquots of the homogenates were stored at −70°C.
The assay used to measure DNA synthetic rate in specific brain regions by means of [3H]thymidine incorporation into DNA as an index of mitotic activity has been previously described (Tao et al., 1997; Wagner et al., 1999; Scheepens et al., 2003). Briefly, one aliquot of the homogenate was precipitated using a standard trichloroacetic precipitation protocol to measure the incorporation of the amount of [3H]thymidine into newly synthesized DNA during the 1 h exposure. Essentially, this measure represents the product of the number of S-phase cells within the sample times the DNA synthetic rate of these S-phase cells. However, incorporation of the labeled precursor into DNA depends on the amount of precursor taken up by the tissue. As a consequence, differences in brain penetration of the precursor or differences in blood flow between experimental groups might result in experimental effects on incorporation that do not reflect differences in DNA synthetic rate. Therefore, the incorporation was calculated as the fraction of the total radioactivity of the tissue sample, which was measured in one aliquot of the same size. The DNA and tissue samples were solubilized in 1 ml of Soluene-350 (Packard, Groningen, The Netherlands) at 50°C for 2–24 h or until the samples were completely dissolved. A 5 ml volume of Hionic-Flour scintillation cocktail (Packard) was then added, and the samples were read for 15 min in a liquid scintillation counter. Radioactivity measures (in disintegrations per minute) were corrected for the wet weight of tissue (in milligrams), and the percentage of [3H]thymidine incorporated into DNA relative to the complete radioactivity counted in the appropriate tissue of each brain region was calculated.
BDNF protein.
BDNF was determined in hippocampal samples of animals, which had been used 3 d earlier in the forced swim test (see below, Behavioral protocols). Dissections, homogenizing, and storage was the same as for the DNA synthetic rate measurements. BDNF protein levels were measured using the Emax ImmunoAssay system from Promega (Madison, WI) according to the protocol of the manufacturer. Samples were acid treated because this reliably increased the detectable BDNF in a dilution-dependent way. Corning (Corning, NY) high-affinity enzyme immunoassay/radioimmunoassay (RIA) 96-well plates (catalog #9018) were used. Samples were diluted 10 times, and the resulting absorbency was read in duplicate using a Bio-Rad (Hercules, CA) Benchmark microplate reader at 450 nm. BDNF protein levels were expressed in nanograms, corrected for the tissue wet weight (in grams).
ACTH and CORT.
Three weeks after the open-field study (see below, Behavioral protocols), eight WT and six GSK male mice were placed in a glass cylinder (diameter of 10 cm) filled with 10 cm of water (25°C) for a period of 6 min. Ten minutes after this forced swimming, animals were decapitated and trunk blood was collected in EDTA-coated blood collection tubes (Microtainer EDTA; Becton Dickinson, Basal, Switzerland). Basal plasma ACTH and CORT levels were also measured in seven unstressed WT and six unstressed GSK mice. All tubes were kept on ice and then centrifuged at 3200 rpm for 5 min at 4°C. Next, the plasma was frozen down to −70°C for subsequent ACTH or CORT determination (both in duplicate) using the RSL 125I ACTH RIA kit and ImmuChem double antibody CORT 125I kit from MP Biomedicals (Morgan Irvine, CA) following the protocol of the manufacturer. ACTH and CORT levels were expressed in picograms per milliliter and nanograms per milliliter plasma, respectively.
Behavioral protocols
Food and water intake.
Body weight and food and water intake (absolute and relative to body weight) were determined twice a day for 5 consecutive days in 15 WT and 16 GSK female littermate mice. To not disturb the animals during the active dark phase, measurements were performed at ∼1 h after lights turned on and before lights turned off. Water bottles and the amount of food on the cage were weighed, and data were subtracted per phase (light or dark). Furthermore, the body weight value at the end of the phase was used to calculate the relative intake over that particular phase.
Open-field test.
Locomotor activity was measured using 17 WT and 15 GSK male littermate mice. Each mouse was subjected to a 60 min testing session in an automated open-field system (Tru-scan system; Coulbourn Instruments, Allentown, PA). The open field consisted of a transparent Perspex cage (width × depth × height, 260 × 260 × 400 mm) equipped with two photobeam sensor rings to register horizontal and vertical activity. The illumination level was 55–85 lux at the floor of the open field. An animal was placed in the center of the field, and the total distance traveled (in centimeters), time spent moving (in seconds), velocity (in centimeters per second), and number of moves were recorded in 5 min time bins with a sample rate of two samples per second. Per bin habituation of the distance in percentage was calculated as 100 − [(distance in bin/mean distance in bin 1) × 100].
Acoustic startle response.
Behavioral reactivity (startle responsivity) was measured in 4 male WT, 13 female WT, 10 male GSK, and 11 female GSK littermates. The startle response was measured in a noise burst intensity function test, which was done in a fully automated setup (Med Associates, Georgia, VT). To acclimatize, the mice were placed into the test tubes and into the chamber for 10 min on 3 consecutive days. On the day of testing (day 4), mice were placed into the test tubes and chambers, and 30 startle stimuli were presented (10 of each intensity of 100, 105, and 110 dB, 20 ms duration per stimulus, at 1 min intertrial intervals). The startle response was defined as the largest peak amplitude during the first 100 ms from the onset of the startle stimulus. The percentage of habituation of the startle response was calculated as 100 − [(mean startle stimuli 27–30/mean startle stimuli 1–4) × 100].
Forced swim test.
Three weeks after the startle response study, all mice of both sexes were used for a forced swim test (FST). Mice were placed in a glass cylinder (diameter of 10 cm) filled with 10 cm of water (25°C). Over a period of 6 min, all movements were registered with a video tracking system (Viewpoint, Champagne au Mont d’Or, France). The total immobility time (in seconds), i.e., the time that no movements of the animal were registered, was calculated from these data.
Statistics
Overall, data were expressed as mean ± SEM and were analyzed parametrically. However, hippocampal BDNF protein levels were not normally distributed as tested with a one-sample Kolmogorov–Smirnov test, and, therefore, rank scores were used for parametric analyses. The open-field test data were analyzed using an ANOVA (genotype × time) with repeated measures (time). Furthermore, a repeated-measures design was used to analyze parts of the startle data (genotype × trial), as well as the food and water intake (genotype × phase). For the remaining analyses, one-way ANOVAs or Student’s t tests were used. When appropriate, post hoc comparisons were made using least significant difference post hoc tests. For all analyses, significance was accepted at p < 0.05.
Results
GSK-3 signaling
We analyzed the Akt-1/GSK-3 signaling pathway at a molecular level. As established by quantitative RT-PCR, expression of murine GSK-3β was upregulated in the cortex of GSK mice (t(9) = 3.32; p < 0.01) compared with WT mice, although it was not changed in striatum. Murine GSK-3α was downregulated in the striatum of GSK mice compared with WT (t(8) = 3.54; p < 0.01), although it was not changed in cortex (Table 1). The constitutively active human GSK-3β was expressed specifically in the brain of the GSK transgenic mice. Expression of Akt-1 was upregulated in striatum of GSK mice (t(9) = 6.66; p < 0.001). Akt-1 is mostly regulated via the phosphoinositol-dependent phosphorylation of both its Thr308 [by PDK1 (Alessi et al., 1996)] and Ser473 [by the elusive PDK2 (Doble and Woodgett, 2003)], resulting in its activation. The heterotrimeric serine/threonine phosphatase protein phosphatase 2A (PP2A) mediates this dephosphorylation and, as such, inactivation of Akt-1 (Andjelkovic et al., 1996; Beaulieu et al., 2005). Expression of PPP2R3A, a subunit of PP2A, was downregulated in striatum of the GSK mice (t(9) = 3.73; p < 0.01). In the cortex of GSK mice, the same trend for an upregulation of Akt-1 expression and a downregulation of PPP2R3A expression was present, but this was not significant. Expression of PDK1 and DARPP-32, a cAMP/protein kinase A pathway-dependent regulatory subunit of PP1 (Greengard et al., 1999), was not different in GSK versus WT striatum and cortex (Table 1).
Table 1.
Expression analysis in WT (n = 6) and GSK (n = 6) male mouse brain
| WT, cortex | GSK, cortex | WT, striatum | GSK, striatum | |
|---|---|---|---|---|
| Human GSK-3β | 0.00 ± 0.00 | 3.01 ± 0.21 ** | 0.00 ± 0.00 | 1.39 ± 0.17** |
| Murine GSK-3β | 1.17 ± 0.10 | 1.68 ± 0.11* | 1.60 ± 0.24 | 1.61 ± 0.70 |
| Murine GSK-3α | 2.21 ± 0.15 | 1.90 ± 0.11 | 1.73 ± 0.07 | 1.23 ± 0.14* |
| Murine Akt-1 | 3.35 ± 0.70 | 5.37 ± 1.47 | 2.70 ± 0.14 | 5.61 ± 0.46** |
| Murine PPP2R3A | 1.51 ± 0.16 | 1.42 ± 0.11 | 2.00 ± 0.17 | 1.26 ± 0.08* |
| Murine PDK1 | 1.56 ± 0.04 | 1.43 ± 0.13 | 2.08 ± 0.20 | 1.96 ± 0.28 |
| Murine DARP32 | 2.56 ± 0.35 | 2.30 ± 0.41 | 25.26 ± 3.44 | 27.96 ± 1.48 |
Values are shown as means ± SEM.
*p < 0.01 and
**p < 0.001 (t test; different between WT and GSK mice for the same region).
As established by ELISA, protein levels of total GSK-3β (murine and human) were significantly higher in the striatum of GSK mice compared with WT (t(18) = 15.53; p < 0.001) (Table 2). Protein levels of GSK-3β phosphorylated at the Ser9 position were significantly higher in the striatum of GSK mice compared with WT (t(18) = 5.00; p < 0.001) (Table 2). Because the human-derived GSK-3β is mutated at position Ser9, the levels of phosphorylated GSK-3β are mouse derived.
Table 2.
Protein analysis in WT (n = 10) and GSK (n = 10) male mouse striatum
| WT, brain | GSK, brain | |
|---|---|---|
| GSK-3β (human and murine) | 54.46 ± 1.75 | 89.91 ± 1.27 * |
| P-GSK-3β (Ser9) (murine) | 24.10 ± 0.94 | 30.29 ± 0.55 * |
Values are shown as means in picograms per milliliter ± SEM.
*p < 0.001 (t test; different between WT and GSK mice).
Food and water intake
Food and water intake was measured over 5 consecutive days. All mice showed increased food intake in the dark phase compared with the light phase (phase effect, F(1,29) = 285.9; p < 0.001) (Fig. 1A). Compared with WT mice, food intake in the GSK mice was significantly lower in both the dark and light phases (genotype effect, F(1,29) = 8.92; p < 0.01). After adjustment for the body weight, which was not different between the GSK and WT mice (data not shown, but this applied to all of the experiments done), the observed differences in food intake were even more pronounced (genotype effect, F(1,29) = 347.71; p < 0.001) (Fig. 1B), with a differentially affected food intake in the two phases between the two genotypes (genotype × phase effect, F(1,29) = 8.92; p < 0.01). Thus, the reduction in relative food intake in GSK mice was stronger in the dark compared with the light phase. All mice consumed more water in the dark phase compared with the light phase (phase effect, F(1,29) = 224.2; p < 0.001) (Fig. 1C). After correction for body weight, a significant interaction between phase and genotype was found (F(1,29) = 4.47; p < 0.05) (Fig. 1D). This shows that GSK mice also exhibit reduced water intake per gram body weight during the dark phase but not during the light phase in which water intake is low.
Figure 1.

Averaged food intake (means ± SEM) (A), food intake relative to body weight (B), water intake (C), and water intake relative to body weight (D) over 5 consecutive days in female WT mice (n = 15) and female GSK mice (n = 16) during the light and dark phase. #p < 0.05 (genotype × phase effect); *p < 0.01 and **p < 0.001 (overall genotype effect).
DNA synthetic rate
On average, all brain region wet weights were reduced by 15% in GSK mice, although the olfactory bulb and SVZ weight did not reach significance (Fig. 2A). This weight loss is in accordance with the previous reports that the transgenic mice expressing a constitutively active mutant GSK-3β display a relative loss in the brain volume (Spittaels et al., 2002).
Figure 2.

Brain region wet weights (means ± SEM; **p < 0.01, t tests) (A) and DNA synthetic rate as an index of proliferation rate expressed as a percentage [3H]thymidine incorporation in the hippocampus and SVZ (no differences between groups) (B) of male WT mice (n = 8) and male GSK mice (n = 7). OB, Olfactory bulb.
Investigation of [3H]thymidine incorporation revealed an average of 15,908 ± 249 dpm/mg hippocampal wet weight in WT and 15,972 ± 668 dpm/mg hippocampal wet weight in GSK mice. In the SVZ tissue fraction, radioactivity was 14,425 ± 711 and 15,090 ± 849 dpm/mg wet weight in WT and GSK mice, respectively. These radioactivity measures of the tissue fractions of the hippocampus and SVZ were not different between the two experimental groups (t(13) = 0.10 and t(12) = 0.60, respectively; NS). This indicates that there were no differences in the amount of labeled precursor tissue uptake between WT and GSK mice.
The percentages of [3H]thymidine incorporation reflecting DNA synthetic rate in the hippocampus and SVZ were also not different between WT and GSK mice (t(13) = 0.74 and t(12) = 0.50, respectively; NS) (Fig. 2B). This indicates that the cell proliferation rate was not affected in GSK mice.
Locomotor activity
Locomotor activity in response to a novel environment was measured in the open field. Overall locomotor activity was higher in the GSK group than in the WT group, as evident from the total distance traveled (17,582 ± 1207 cm in GSK and 12,096 ± 318 cm in WT; genotype effect, F(1,30) = 21.55; p < 0.01), total time traveled (2179 ± 63 s in GSK and 1865 ± 26 s in WT; genotype effect, F(1,30) = 23.05; p < 0.01), and the velocity (7.95 ± 0.37 cm/s in GSK and 6.47 ± 0.11 cm/s in WT; genotype effect, F(1,30) = 16.39; p < 0.01). Compared with their WT littermates, the GSK mice were observed not only to travel longer and a larger distance but to do so using slightly less moves (2153 ± 44 in GSK and 2280 ± 19 in WT mice; genotype effect F(1,30) = 7.69; p < 0.01).
As exemplified by the distance traveled, both genotypes showed reduced locomotor activity over a period of 1 h (time effect, F(11,330) = 37.32; p < 0.01) (Fig. 3A), suggesting habituation. The slope of the curves did not differ between genotypes, i.e., both groups decreased activity in parallel (genotype × time, F(11,330) = 1.12; NS). However, habituation expressed as percentage relative to the initial activity during the first bin was different for GSK versus WT littermates, i.e., GSK mice showed less habituation in time than the WT mice did (genotype × time effect, F(11,330) = 2.02; p < 0.05) (Fig. 3B).
Figure 3.
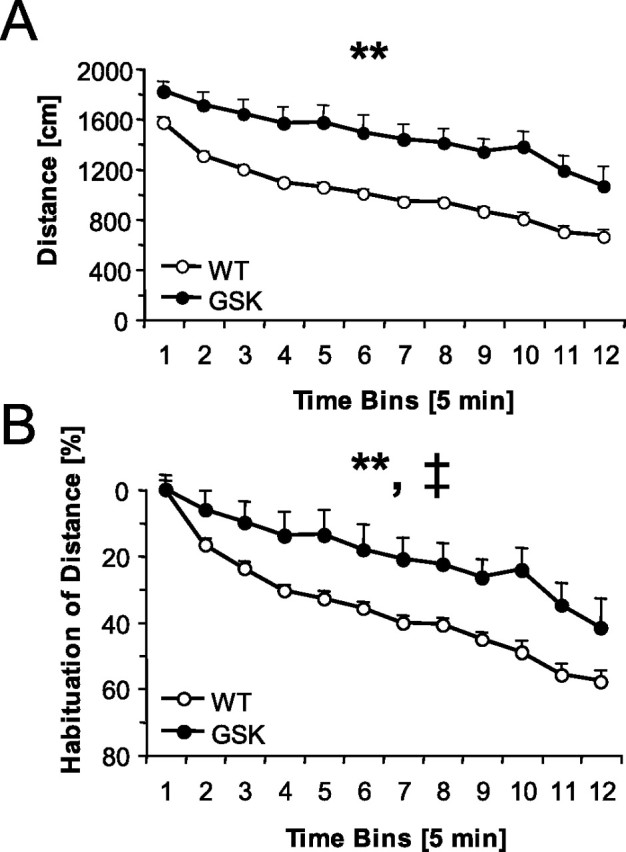
Locomotor activity (means ± SEM) in an open field during 1 h (in 5 min bins). A, Male GSK mice (n = 15) traveled a longer distance than male WT mice (n = 17). B, Percentage habituation of the distance traveled. **p < 0.01 (overall genotype effect); ‡p < 0.05 (genotype × time bin effect).
Plasma ACTH and corticosterone
Six minutes of forced swimming was used to assess the stress hormone response in the animals, which had been used previously for the assessment of locomotor activity. ACTH and CORT plasma concentrations were increased in both genotypes 10 min after forced swimming (stress effect, F(1,21) = 16.57 and F(1,23) = 14.33, respectively; both p < 0.01) (Table 3). There was no genotype effect on either baseline or post-stress ACTH and CORT levels (all F values <0.58; NS).
Table 3.
Plasma ACTH and CORT concentrations 10 min after 6 min of forced swimming
| WT, no stress | WT, swim stress | GSK, no stress | GSK, swim stress | |
|---|---|---|---|---|
| ACTH (pg/ml) | 313.5 ± 39.2 | 489.9 ± 32.8* | 264.5 ± 48.5 | 468.1 ± 70.8* |
| CORT (ng/ml) | 133.0 ± 44.0 | 246.8 ± 10.0* | 143.7 ± 46.5 | 276.1 ± 18.4* |
Values are shown as means ± SEM. There were no differences between male WT mice (no stress, n = 7; swim stress, n = 6) and male GSK mice (no stress, n = 8; swim stress, n = 6) in the ACTH and CORT response after forced swim stress.
*p < 0.05 (least significant difference post hoc test; different from the no-stress group of the same genotype).
Behavioral reactivity
Potential differences in sensorimotor responsivity were evaluated by measuring the acoustic startle response (ASR). GSK-3β transgenic mice were hyper-reactive compared with their WT littermates, as evident from the increased startle response at each stimulus intensity tested (genotype effects, all F values >25.77; p values <0.01) (Fig. 4A). In contrast to the previous experiments, both male and female mice were tested, although there was no gender difference (all F values <1.20; NS).
Figure 4.

Startle responses (means ± SEM) in the noise burst intensity function test with 20 ms startle stimuli of 100, 105, and 110 dB (A), immobility time in the FST for 6 min (B), and BDNF protein concentrations in homogenates of the hippocampus (C) of male WT mice (n = 4), female WT mice (n = 13), male GSK mice (n = 10), and female GSK mice (n = 11). †p < 0.05 and ††p < 0.01 (overall sex effect); **p < 0.01 (overall genotype effect).
To gain insight in potential differences in habituation of the startle response, pooled responses of both genders per experimental group measured after the first four and the last four stimuli administered were compared (the first four stimuli consisted of two stimuli with a intensity of 100 dB and two stimuli with an intensity of 105 dB, the last four stimuli consisted of two stimuli with a intensity of 100 dB and one of 105 and 110 dB). Again, GSK mice showed overall increased startle responses compared with their WT littermates (genotype effect, F(1,300) = 105.59; p < 0.001). Both the GSK mice and their WT littermates demonstrated overall habituation (period effect, F(1,300) = 11.57; p < 0.001), but no significant genotype by period effect was observed (F(1,300) = 0.33; NS). However, when the percentage habituation relative to the first four stimuli was calculated, GSK mice showed significantly less habituation than did the WT littermates (t(36) = 2.33; p < 0.01) (Table 4).
Table 4.
Habituation of acoustic startle responses in male and female WT mice (n = 17) and male and female GSK mice (n = 21)
| WT, first four stimuli | WT, last four stimuli | GSK, first four stimuli | GSK, last four stimuli | |
|---|---|---|---|---|
| Startle response | 8.5 ± 0.7 | 6.2 ± 0.4* | 14.1 ± 0.5 | 12.4 ± 0.6* |
| Percent habituation | 26.6 ± 5.1 | 12.0 ± 4.2** |
Values are shown as means ± SEM.
*p < 0.05 (t test; different from first stimuli of the same genotype);
**p < 0.01 (t test; different between GSK and WT mice).
Forced swim test
When both genders were tested in the FST, GSK mice had lower immobility than WT mice (genotype effect, F(1,34) = 9.48; p < 0.01) (Fig. 4B). In addition, immobility was lower in females than in males (gender effect, F(1,34) = 5.58; p < 0.05) (Fig. 4B), whereas the genotype effect was not differently affected by gender (genotype × gender effect, F(1,34) = 1.24; NS).
BDNF protein
Three days after the FST, mice were decapitated and fresh tissue was collected. There was an effect of genotype on hippocampal wet weights (F(1,34) = 50.01; p < 0.01; WT male, 35.0 ± 0.7 mg; WT female, 33.8 ± 0.7 mg; GSK male, 27.6 ± 0.8 mg; GSK female, 28.3 ± 0.8 mg). There was no gender effect (F(1,34) = 0.06; NS).
Hippocampal BDNF protein levels were higher in female than in male mice (gender effect, F(1,34) = 14.60; p < 0.01) (Fig. 4C). Furthermore, hippocampal BDNF protein levels were increased in GSK mice when compared with WT mice (genotype effect, F(1,34) = 17.36; p < 0.01) (Fig. 4C). Hippocampal BDNF levels of the sexes were not differently affected by genotype (genotype × gender effect, F(1,34) = 0.56; NS).
Discussion
We investigated the possibility that transgenic mice overexpressing GSK-3β could be of relevance to model-specific aspects of bipolar disorder. Our molecular analysis of the Akt-mediated signaling pathway suggests that the GSK overexpression leads to compensation in the striatum and to a lesser extend in the cortex, as indicated by an upregulation of the expression of Akt-1 and a downregulation of the expression of PPP2R3A, a regulatory subunit of the phosphatase PP2A that inactivates Akt-1. This combination should result in an increased level of phosphorylated, and thus active, Akt-1. In turn, this should lead to increased levels of phosphorylated, and thus inactive, murine GSK-3α and GSK-3β, which for the latter was confirmed by ELISA on striatum.
The expression of constitutively active GSK-3β in the brain triggered locomotor hyperactivity, which is associated with many animal models of mania (Lyon, 1999; Machado-Vieira et al., 2004). Among the neurotransmitter systems that might play a role in the pathophysiology of mania, changes in dopaminergic neurotransmission have received much attention recently (Machado-Vieira et al., 2004), and locomotor hyperactivity has been associated with the activation of the mesolimbic dopaminergic system (D’Aquila et al., 2000). Mouse mutants lacking a functional dopamine transporter [DAT knock-out (KO) or DAT knockdown] are hyperactive (Giros et al., 1996; Ralph et al., 2001; Zhuang et al., 2001; Ralph-Williams et al., 2003). In addition, both of these DAT mouse mutants displayed impaired response habituation in novel environments (Gainetdinov et al., 1999; Zhuang et al., 2001; Ralph-Williams et al., 2003), just as the GSK mice. Interestingly, recent evidence from DAT KO mice suggests that GSK-3 partly mediates locomotor hyperactivity via the striatal Akt-1/GSK-3 signaling pathway, i.e., both Akt-1 and GSK-3β were less phosphorylated than in WT mice (Beaulieu et al., 2004, 2005). The Akt-1/GSK-3β pathway is similarly affected by treatment with amphetamine (Beaulieu et al., 2004, 2005). Lithium as well as specific GSK-3 inhibitors were reported to attenuate the locomotor hyperactivity in mice with increased dopamine neurotransmission as a result of absence of the DAT or administration of amphetamine. Likewise, administration of the selective GSK-3 inhibitor AR-A014418 [N-(4-methoxybenzyl)-N′-(5-nitro-1,3-thiazol-2-yl)urea] in rats was found to decrease amphetamine-induced hyperactivity (Gould et al., 2004c). Furthermore, heterozygote GSK-3β knock-out mice showed attenuated hyperlocomotion after amphetamine administration (Beaulieu et al., 2004). Thus, our findings indicate that GSK-3β overexpression in the brain of the transgenic mice results in increased locomotor activity, which shows similarity with dopamine-induced, Akt-1/GSK-3β-mediated hyperactivity.
Recently, it has been observed that dopamine overactivity as seen in DAT KO mice resulted in locomotor hyperactivity that became evident after more than 5 min (Pogorelov et al., 2005). This is comparable with our data with the GSK-3 mice. The same DAT KO mice displayed anxiety-like behavior only during the first 5 min in a zero maze, i.e., the relative time spent in the less safe open arms was decreased. After 5 min, these mice became stereotypically activated, making the behavior inflexible. The relative time spent in the center of the open field during the first 5 min was decreased in our GSK mice (21 ± 1% in GSK and 28 ± 2% in WT; t(30) = 3.50; p < 0.01). This might be indicative of altered anxiety-related behavior, although additional studies are required to further evaluate this type of behavior.
In addition to increased activity in an open-field arena, GSK-3β-overexpressing mice also showed an increased ASR and a decreased habituation of the ASR. The amplitude of the ASR is under control of various modulatory neural circuits that mediate the enhancement of the ASR by fear or anxiety and the inhibition of the ASR by prepulse (Koch, 1999). A consequence of the latter may be an impaired habituation of the ASR because deficits in habituation of ASR have been reported in manic patients with psychotic symptoms (Perry et al., 2001) and schizophrenics (Braff et al., 1978; Perry et al., 2001). An increased ASR has been observed in the DAT knockdown mouse (Ralph-Williams et al., 2003). Likewise, activation of the dopaminergic system by amphetamine or cocaine is known to increase the ASR in man and rodents (Swerdlow et al., 1990; Zhang et al., 2000; Lewis and Gould, 2003). In addition, habituation of the ASR in rodents is disrupted by amphetamine (Klamer et al., 2004). Thus, our ASR data are also in agreement with dopamine-induced changes in sensory information processing.
GSK-3 has a role in the regulation of energy metabolism (Gould and Manji, 2005). In particular, peripherally, GSK-3 is mediating the insulin/Akt pathway, i.e., GSK-3 inhibits glycogen synthesis. In the hypothalamus, insulin has food intake-lowering effects via Akt signaling (Niswender et al., 2003). However, the role of the two isoforms GSK-3α and GSK-3β in food, but also water, intake is to our knowledge not known. Expression of constitutively active GSK-3β in the brain of our GSK mice resulted in a decreased intake of food and water. Changes in eating patterns, both hyperphagia and hypophagia, have been described in a wide variety of psychiatric illnesses, including bipolar disease and schizophrenia (Foulon, 2003; Kishi and Elmquist, 2005; Krishnan, 2005). The dopamine system also plays a key role in the regulation of food and water intake (Bray, 2000), and it is suggested that, next to hypothalamic catecholamines, regulated striatal dopamine signaling is required for adequate feeding (Cannon et al., 2004). Acute systemic administration of drugs that prolong normal dopamine signaling, including amphetamine, methamphetamine, cocaine, DAT blockers, monoamine oxidase inhibitors, and dopamine receptor agonists, can produce hypophagia (Cannon et al., 2004). Consequently, our finding of hypophagia in GSK mice suggests that striatal dopamine regulation of food and water intake involves GSK-3β regulation.
Lithium has modest clinical antidepressant effects, but it can potentiate the action of antidepressants (De Montigny et al., 1981; Gould and Manji, 2005). It has been speculated that some mood stabilizers and antidepressants might share common mechanisms influencing GSK-3 activity (Li et al., 2002). The cAMP regulatory element-binding protein transcription factor (CREB), which has been shown to be activated by antidepressant drug activity, is inhibited by GSK-3 (Salas et al., 2003). CREB activation results in elevated levels of BDNF protein, and the latter is assumed to produce at least some of the antidepressant drug activity in animal models (Nibuya et al., 1996). Lithium administration has been reported to increase BDNF content in the frontal cortex and hippocampus of rats (Jacobsen and Mork, 2004). Conversely, BDNF inhibits GSK-3 in vitro (Mai et al., 2002), probably through Akt activation (Gould and Manji, 2005). Pharmacologic inhibition of GSK-3 activity was shown to produce antidepressant-like effects in rodents (Gould et al., 2004c; Kaidanovich-Beilin et al., 2004). We therefore investigated whether overexpression of the constitutively active GSK-3β resulted in the induction of depression-like behavior in the FST. However, an expected increase in immobility was not observed, although the behavioral readout is probably compromised by the locomotor hyperactivity. Alternatively, this finding could imply that GSK-3β overexpression is more related to a state of mania than a state of depression.
ACTH and CORT levels, which have been reported to be altered in psychiatric disorders such as depression and bipolar disorder (Steckler et al., 1999), were not altered in the GSK-3β-overexpressing mice. The observed volume loss in different brain regions including the hippocampus has been reported previously, whereas the total number of neurons in GSK-3β-overexpressing mice remained unaltered and no necrosis and apoptosis were observed (Spittaels et al., 2002). This is corroborated by our finding that the cell proliferation rate was not affected in the neurogenic hippocampus. Of note, inhibition of GSK-3 with lithium increases hippocampal cell proliferation in mice (Chen et al., 2000). It might be speculated that GSK-3β overexpression in the mice was not high enough to influence cell proliferation. However, upregulated hippocampal BDNF levels were observed in GSK-3β-overexpressing mice. This increase in BDNF protein levels is possibly a compensatory response to the reduction of the dendritic compartment and reduced size of neuronal cell bodies that is responsible for the reduction in brain volume in the GSK-3β-overexpressing mice (Spittaels et al., 2002). The mechanism by which GSK-3β causes this reduction is unknown. Circumstantial evidence suggested that the Akt-1/GSK-3 pathway is involved (Beck et al., 1995; Cheng et al., 2000; Spittaels et al., 2002). Recently, it has been shown that Akt-1 and GSK-3β indeed have multiple roles in axonal and dendritic development and establishment and maintenance of neuronal polarity (Jiang et al., 2005).
Based on the current data, we suggest that GSK-3β-overexpressing mice can be used as a model for studying hyperactivity, hyperreactivity, and a disturbed eating pattern as seen in the manic phase of bipolar disorder, but also as in dopamine-related disorders such as ADHD and schizophrenia because of the overlap with dopamine-induced behavioral changes as observed previously in mice (Manji et al., 2003; Beaulieu et al., 2004, 2005; Emamian et al., 2004; Gould et al., 2004b). Thus, overexpression of the constitutively active GSK-3β in the transgenic mouse might reflect a state of constitutive activation of the dopaminergic system in the brain. An important future challenge is to determine the signaling pathway(s) downstream of activated GSK-3β that is (are) responsible for the induction of the hyper(re)activity in this mouse model. Delineation of this pathway could well deliver potential novel drug targets for the treatment of psychiatric disorders involving deregulated Akt-1/GSK-3β signaling, such as bipolar disorder, ADHD, or schizophrenia.
Footnotes
GSK-3β-overexpressing mice were originally generated by Prof. Fred Van Leuven (Belgium). J.P. is supported by European Union Framework 6 Integrated Project NEWMOOD Grant LSHM-CT-2004-503474.
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Andjelkovic M, Jakubowicz T, Cron P, Ming XF, Han JW, Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc Natl Acad Sci USA. 1996;93:5699–5704. doi: 10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Beck KD, Powell-Braxton L, Widmer HR, Valverde J, Hefti F. Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron. 1995;14:717–730. doi: 10.1016/0896-6273(95)90216-3. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Downes CP, Hanley MR. Neural and developmental actions of lithium: a unifying hypothesis. Cell. 1989;59:411–419. doi: 10.1016/0092-8674(89)90026-3. [DOI] [PubMed] [Google Scholar]
- Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L. Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology. 1978;15:339–343. doi: 10.1111/j.1469-8986.1978.tb01390.x. [DOI] [PubMed] [Google Scholar]
- Bray GA. A concise review on the therapeutics of obesity. Nutrition. 2000;16:953–960. doi: 10.1016/s0899-9007(00)00424-x. [DOI] [PubMed] [Google Scholar]
- Cannon CM, Abdallah L, Tecott LH, During MJ, Palmiter RD. Dysregulation of striatal dopamine signaling by amphetamine inhibits feeding by hungry mice. Neuron. 2004;44:509–520. doi: 10.1016/j.neuron.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK. Enhancement of hippocampal neurogenesis by lithium. J Neurochem. 2000;75:1729–1734. doi: 10.1046/j.1471-4159.2000.0751729.x. [DOI] [PubMed] [Google Scholar]
- Cheng CM, Reinhardt RR, Lee WH, Joncas G, Patel SC, Bondy CA. Insulin-like growth factor 1 regulates developing brain glucose metabolism. Proc Natl Acad Sci USA. 2000;97:10236–10241. doi: 10.1073/pnas.170008497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- D’Aquila PS, Collu M, Gessa GL, Serra G. The role of dopamine in the mechanism of action of antidepressant drugs. Eur J Pharmacol. 2000;405:365–373. doi: 10.1016/s0014-2999(00)00566-5. [DOI] [PubMed] [Google Scholar]
- De Montigny C, Grunberg F, Mayer A, Deschenes JP. Lithium induces rapid relief of depression in tricyclic antidepressant drug non-responders. Br J Psychiatry. 1981;138:252–256. doi: 10.1192/bjp.138.3.252. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- Foulon C. Schizophrenia and eating disorders. Encephale. 2003;29:463–466. [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG. Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science. 1999;283:397–401. doi: 10.1126/science.283.5400.397. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Gould TD, Manji HK. Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology. 2005;30:1223–1237. doi: 10.1038/sj.npp.1300731. [DOI] [PubMed] [Google Scholar]
- Gould TD, Chen G, Manji HK. In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology. 2004a;29:32–38. doi: 10.1038/sj.npp.1300283. [DOI] [PubMed] [Google Scholar]
- Gould TD, Zarate CA, Manji HK. Glycogen synthase kinase-3: a target for novel bipolar disorder treatments. J Clin Psychiatry. 2004b;65:10–21. [PubMed] [Google Scholar]
- Gould TD, Einat H, Bhat R, Manji HK. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int J Neuropsychopharmacol. 2004c;7:387–390. doi: 10.1017/S1461145704004535. [DOI] [PubMed] [Google Scholar]
- Greengard P, Allen PB, Nairn AC. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–447. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Jacobsen JP, Mork A. The effect of escitalopram, desipramine, electroconvulsive seizures and lithium on brain-derived neurotrophic factor mRNA and protein expression in the rat brain and the correlation to 5-HT and 5-HIAA levels. Brain Res. 2004;1024:183–192. doi: 10.1016/j.brainres.2004.07.065. [DOI] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol Psychiatry. 2004;55:781–784. doi: 10.1016/j.biopsych.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Kishi T, Elmquist JK. Body weight is regulated by the brain: a link between feeding and emotion. Mol Psychiatry. 2005;10:132–146. doi: 10.1038/sj.mp.4001638. [DOI] [PubMed] [Google Scholar]
- Klamer D, Palsson E, Revesz A, Engel JA, Svensson L. Habituation of acoustic startle is disrupted by psychotomimetic drugs: differential dependence on dopaminergic and nitric oxide modulatory mechanisms. Psychopharmacology (Berl) 2004;176:440–450. doi: 10.1007/s00213-004-1901-z. [DOI] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M. The neurobiology of startle. Prog Neurobiol. 1999;59:107–128. doi: 10.1016/s0301-0082(98)00098-7. [DOI] [PubMed] [Google Scholar]
- Krishnan KR. Psychiatric and medical comorbidities of bipolar disorder. Psychosom Med. 2005;67:1–8. doi: 10.1097/01.psy.0000151489.36347.18. [DOI] [PubMed] [Google Scholar]
- Lewis MC, Gould TJ. Nicotine and ethanol enhancements of acoustic startle reflex are mediated in part by dopamine in C57BL/6J mice. Pharmacol Biochem Behav. 2003;76:179–186. doi: 10.1016/s0091-3057(03)00213-2. [DOI] [PubMed] [Google Scholar]
- Li X, Bijur GN, Jope RS. Glycogen synthase kinase-3beta, mood stabilizers, and neuroprotection. Bipolar Disord. 2002;4:137–144. doi: 10.1034/j.1399-5618.2002.40201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon M. Animal models for the symptoms of mania. In: Boulton AA, editor. Animal models in psychiatry. Clifton, NJ: Humana; 1999. pp. 197–244. [Google Scholar]
- Machado-Vieira R, Kapczinski F, Soares JC. Perspectives for the development of animal models of bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:209–224. doi: 10.1016/j.pnpbp.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3beta and mood stabilizing agents. J Neurochem. 2002;82:75–83. doi: 10.1046/j.1471-4159.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- Manji HK, Gottesman II, Gould TD. Signal transduction and genes-to-behaviors pathways in psychiatric diseases. Sci STKE 2003. 2003:pe49. doi: 10.1126/stke.2003.207.pe49. [DOI] [PubMed] [Google Scholar]
- Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16:2365–2372. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG, Jr, Seeley RJ, Schwartz MW. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes. 2003;52:227–231. doi: 10.2337/diabetes.52.2.227. [DOI] [PubMed] [Google Scholar]
- Perry W, Minassian A, Feifel D, Braff DL. Sensorimotor gating deficits in bipolar disorder patients with acute psychotic mania. Biol Psychiatry. 2001;50:418–424. doi: 10.1016/s0006-3223(01)01184-2. [DOI] [PubMed] [Google Scholar]
- Pogorelov VM, Rodriguiz RM, Insco ML, Caron MG, Wetsel WC. Novelty seeking and stereotypic activation of behavior in mice with disruption of the Dat1 gene. Neuropsychopharmacology. 2005;30:1818–1831. doi: 10.1038/sj.npp.1300724. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, Paulus MP, Fumagalli F, Caron MG, Geyer MA. Prepulse inhibition deficits and perseverative motor patterns in dopamine transporter knock-out mice: differential effects of D1 and D2 receptor antagonists. J Neurosci. 2001;21:305–313. doi: 10.1523/JNEUROSCI.21-01-00305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph-Williams RJ, Paulus MP, Zhuang X, Hen R, Geyer MA. Valproate attenuates hyperactive and perseverative behaviors in mutant mice with a dysregulated dopamine system. Biol Psychiatry. 2003;53:352–359. doi: 10.1016/s0006-3223(02)01489-0. [DOI] [PubMed] [Google Scholar]
- Redrobe JP, Bourin M. The effect of lithium administration in animal models of depression: a short review. Fundam Clin Pharmacol. 1999;13:293–299. doi: 10.1111/j.1472-8206.1999.tb00348.x. [DOI] [PubMed] [Google Scholar]
- Salas TR, Reddy SA, Clifford JL, Davis RJ, Kikuchi A, Lippman SM, Menter DG. Alleviating the suppression of glycogen synthase kinase-3beta by Akt leads to the phosphorylation of cAMP-response element-binding protein and its transactivation in intact cell nuclei. J Biol Chem. 2003;278:41338–41346. doi: 10.1074/jbc.M302972200. [DOI] [PubMed] [Google Scholar]
- Scheepens A, Van De Waarenburg M, Van Den Hove HD, Blanco CE. A single course of prenatal betamethasone in the rat alters postnatal brain cell proliferation but not apoptosis. J Physiol (Lond) 2003;552:163–175. doi: 10.1113/jphysiol.2003.043414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spittaels K, Van den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K, Lasrado R, Vandezande K, Laenen I, Boon T, Van Lint J, Vandenheede J, Moechars D, Loos R, Van Leuven F. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. 2000;275:41340–41349. doi: 10.1074/jbc.M006219200. [DOI] [PubMed] [Google Scholar]
- Spittaels K, Van den Haute C, Van Dorpe J, Terwel D, Vandezande K, Lasrado R, Bruynseels K, Irizarry M, Verhoye M, Van Lint J, Vandenheede JR, Ashton D, Mercken M, Loos R, Hyman B, Van der Linden A, Geerts H, Van Leuven F. Neonatal neuronal overexpression of glycogen synthase kinase-3 beta reduces brain size in transgenic mice. Neuroscience. 2002;113:797–808. doi: 10.1016/s0306-4522(02)00236-1. [DOI] [PubMed] [Google Scholar]
- Steckler T, Holsboer F, Reul JM. Glucocorticoids and depression. Baillieres Best Pract Res Clin Endocrinol Metab. 1999;13:597–614. doi: 10.1053/beem.1999.0046. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Mansbach RS, Geyer MA, Pulvirenti L, Koob GF, Braff DL. Amphetamine disruption of prepulse inhibition of acoustic startle is reversed by depletion of mesolimbic dopamine. Psychopharmacology (Berl) 1990;100:413–416. doi: 10.1007/BF02244616. [DOI] [PubMed] [Google Scholar]
- Tao Y, Black IB, DiCicco-Bloom E. In vivo neurogenesis is inhibited by neutralizing antibodies to basic fibroblast growth factor. J Neurobiol. 1997;33:289–296. [PubMed] [Google Scholar]
- Van Wauwe J, Haefner B. Glycogen synthase kinase-3 as drug target: from wallflower to center of attention. Drug News Perspect. 2003;16:557–565. doi: 10.1358/dnp.2003.16.9.829337. [DOI] [PubMed] [Google Scholar]
- Wagner JP, Black IB, DiCicco-Bloom E. Stimulation of neonatal and adult brain neurogenesis by subcutaneous injection of basic fibroblast growth factor. J Neurosci. 1999;19:6006–6016. doi: 10.1523/JNEUROSCI.19-14-06006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Forkstam C, Engel JA, Svensson L. Role of dopamine in prepulse inhibition of acoustic startle. Psychopharmacology (Berl) 2000;149:181–188. doi: 10.1007/s002130000369. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Oosting RS, Jones SR, Gainetdinov RR, Miller GW, Caron MG, Hen R. Hyperactivity and impaired response habituation in hyperdopaminergic mice. Proc Natl Acad Sci USA. 2001;98:1982–1987. doi: 10.1073/pnas.98.4.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]