

Abstract
Arrestins constitute a family of small cytoplasmic proteins that mediate deactivation of G-protein-coupled receptors (GPCRs) and are known to be essential for cascade inactivation and receptor desensitization. Alternative splicing produces an array of arrestin gene products that have widely different specificities for their cognate receptors in vitro, but the differential functions of these splice variants in vivo are essentially unknown. Bovine rod photoreceptors express two splice variants of visual arrestin (p44 and p48) that display different affinities for the GPCR rhodopsin. To determine the functions of these splice variants in intact cells, we expressed a transgene encoding either a truncated form of murine arrestin (mArr1–369, or m44) or the long (p48) isoform in mouse rods lacking endogenous arrestin (Arr–/–). Morphological analysis showed that expression of either variant attenuated the light-induced degeneration that is thought to result from excessive cascade activity in Arr–/–rods. Suction electrode recordings from individual rods indicated that the expression of either m44 or p48 splice variants could restore normal kinetics to Arr–/– dim flash responses, indicating that both isoforms can bind to and quench phosphorylated rhodopsin rapidly. To our surprise, only the full-length variant was able to alter the kinetics of responses in rods lacking both arrestin and rhodopsin kinase, indicating that p48 can also quench the activity of nonphosphorylated rhodopsin.
Keywords: phototransduction, photoreceptor, retina, GPCR, G-protein cascade, retinal degeneration
Introduction
In photoreceptor cells, light activation of rhodopsin drives the phototransduction cascade that initiates vision. A single photoexcited rhodopsin (R*) activates many copies of the G-protein transducin, providing one of the primary mechanisms of cascade amplification that allows rods to signal the absorption of single photons. Amplification persists as long as R* remains active; defects in rhodopsin deactivation cause photoresponses to be greatly prolonged (Nakatani and Yau, 1988; Chen et al., 1995; Xu et al., 1997; C. K. Chen et al., 1999) and produce a form of stationary night blindness called Oguchi disease in humans (Fuchs et al., 1995; Yamamoto et al., 1997).
Rhodopsin deactivation is initiated when serine and threonine residues at the COOH terminus of rhodopsin are phosphorylated by rhodopsin kinase (Maeda et al., 2003). Multiple phosphorylation of R* at its COOH terminus (R*-P) is required for the high-affinity binding of visual arrestin (Wilden, 1995; Brannock et al., 1999; Gibson et al., 2000; Vishnivetskiy et al., 2000; Kennedy et al., 2001) that completes the quench of the activity of rhodopsin and permits full recovery of the light response (Xu et al., 1997).
Several splice variants of the arrestin gene family have been described previously (Parruti et al., 1993; Sterne-Marr et al., 1993), including variants of rod arrestin (Palczewski, 1994; Smith et al., 1994). All of these variants are produced by alternative mRNA splicing at the 3′ end, producing truncated isoforms that have different biochemical characteristics than their full-length counterparts (Gurevich and Benovic, 1993; Gurevich et al., 1993). The C-terminal truncation of all of these variants is thought to destabilize the basal conformation, reducing selectivity for the light-activated, phosphorylated receptor R*-P (Gurevich and Gurevich, 2004).
Splice variants of visual arrestins were first described and characterized in bovine rod outer segments (ROS). In this system, alternative splicing results in a truncated form of arrestin in which the C-terminal 35 residues are replaced by a single alanine (Smith et al., 1994). This truncated isoform has a faster on-rate than full-length (p48) visual arrestin for binding R* and R*-P in vitro (Palczewski et al., 1994; Pulvermuller et al., 1997) and is more efficient than p48 at turning off R* in some in vitro assays (Palczewski et al., 1994; Langlois et al., 1996). Although p48 is ∼10 times more abundant than p44 in retinal homogenates (Palczewski et al., 1994), p48 is primarily excluded from darkadapted rod outer segments (Palczewski et al., 1994; Mendez et al., 2003; Elias et al., 2004; Nair et al., 2004). These observations have lead to the hypothesis that p44 is more important than p48 in deactivating rhodopsin under dark-adapted conditions. To test the ability of each arrestin splice variant to quench lightactivated rhodopsin activity in intact rods, we used transgenic techniques in mice to selectively express p44 and p48 in the absence of endogenous arrestin.
Materials and Methods
Generation of transgenic mice. The use of mice in these experiments was in accordance with the guidelines established by the National Institutes of Health and the animal care and use guidelines of our respective universities. DNA constructs for the specific expression of arrestin and its truncated form in photoreceptor cells were obtained by fusing the murine arrestin cDNAs to the mouse opsin promoter and the mouse protamine 1 (MP1) polyadenylation signal, as detailed below. To generate the p48 construct, murine arrestin cDNA (nucleotides 11–1482; GenBank accession number M24086) was amplified by PCR with the following primers: mArr1(+) XhoI, 5′ggtatctcgagggattgcaccaggtc3′; and mArr1483(–) XbaI, 5′cctctagatctgggaggaatgctcatgc3′. The 1.5 kb DNA fragment encompassing the whole coding sequence of murine arrestin was cloned into pBl-II SK (Stratagene, La Jolla, CA), followed by a XbaI-ClaI 0.6 kb DNA fragment containing the MP1 polyadenylation signal. The 4.4 kb KpnI-XhoI mouse opsin promoter region was subsequently inserted to yield the 9.4 kb expression vector.
The mouse p44 (m44) expression vector encodes a form of murine arrestin in which the last 35 amino acids of arrestin were substituted by a single Ala. The DNA sequence encoding the m44 transgene was obtained by two step PCR amplication of murine arrestin cDNA using two pairs of primers: 5′cacaggatccctctcgtgacttc3′ (forward) and 5′ccttgtcggtgttgttggtcacg3′ (reverse); and 5′gacaagcccctgaacctctcag3′ (forward) and 5′gatcagatcttcaggcactttcttttgctggatcc3′ (reverse). The primers converted the KpnI site at position 66 into a BamHI site and introduced a BglII site that converted Val369 into Ala and introduced a stop codon immediately after. This 1.2 kb fragment was inserted into a preassembled pBl-II SK-based vector containing the 4.4 kb mouse opsin promoter and MP1 polyadenylation signal separated by a BamHI site.
The coding regions of p48 and m44 arrestins were verified by sequencing. The resulting fusion genes, p48 (6.4 kb) and m44 (6.2 kb), were excised from the cloning vectors, purified, and microinjected into the pronuclei of C57BL6/J × DBA/2J F1 hybrid mouse embryos. Mice were screened for the presence of the transgene by PCR with the following primers: Rh1.1, 5′gtgcctggagttgcgctgtggg3′ (forward); and M44, 5′gctcagggtccaccagcacaacac3′ (reverse). Mice expressing the transgene (founders) were bred to C57BL6/J and to arrestin knock-out mice to establish transgenic lines that express the heterologous arrestins in the presence or absence of the endogenous form.
Characterization of transgene expression by Western blot. For the Western blot analysis in Figure 1 B, the indicated mice (m44Arr+/+ or a littermate negative control) were killed after 3 h of light adaptation. Retinas were dissected and homogenized in 100 μl of homogenization buffer (80 mm Tris-HCl, pH 8, 10 mm EDTA, 4 mm MgCl2 containing leupeptin, pepstatin, and aprotinin to 40 μg/ml, and PMSF to 0.5 mm final concentration). Retinal homogenates were centrifuged at 13,000 × g for 15 min at 4°C, and equivalent amounts of the pellet and supernatant fractions were mixed with Laemmli buffer, boiled, and resolved in a 10% Trisglycine SDS-PAGE. Proteins were transferred to nitrocellulose membranes (Protran; Schleicher and Schuell BioScience, Keene, NH), and arrestin was immunodetected with C10C10 monoclonal antibody, which recognizes residues 290–297 of arrestin (Knospe et al., 1988). For Figure 1C, mice were processed under room light, and retinas were homogenized in 80 μl of homogenization buffer [80 mm Tris-HCl, pH 8, and 4 mm MgCl2 containing 10 μl of a freshly prepared 2 mg/ml solution of calpain II inhibitor and one-eighth of a tablet of Complete Mini Protease Inhibitor Cocktail (Roche Applied Science, Basel, Switzerland)]. Protein concentration in retinal homogenates was determined by Bradford's assay (Bio-Rad protein assay; Bio-Rad, Hercules, CA), and equal protein amounts were resolved by SDS-PAGE. Blotting conditions and antibodies used for arrestin detection were as described above.
Figure 1.

Expression of the p48 or p44 forms of arrestin in transgenic mice. A, Expression vectors used for the generation of transgenic mice. The cDNAs encoding the p48 or m44 (murine Arr1–369,V369A,Q370ter) forms of arrestin were flanked by the murine rhodopsin promoter (MOP) and the mouse protamine 1 (MP1) polyadenylation signal. Gray boxes indicate 3′ end untranslated regions.B, m44 transgene expression in wild-type mice. Pellet (P) and supernatant (S) fractions of total retinal homogenates from light-adapted m44-positive and control mice were analyzed by Western blot using C10C10 antibody (specific for residues 290–297 of arrestin). The m44 protein in transgenic mice comigrated with p44 in bovine rod outer segment preparations (bROS), between two bands of ∼46 and 42 kDa (arrowheads), which are likely proteolyzed forms of arrestin. C, Expression of p48 and m44 transgenes in the Arr–/– background. Equivalent fractions of total retinal homogenates from the indicated mice were compared by Western blot. D, The relative abundance of arrestin isoforms in ROS from dark-adapted p48Arr–/–, m44Arr–/–, and wild-type mice determined by Western blot using C10C10 antibody. Numbers above each lane indicate total protein loaded in nanograms. Note that the range of dilutions loaded in the m44Arr–/– sample differed from those of the wild type and p48Arr–/–. E, Extent of p48 proteolysis was independent of lighting conditions. Retinal homogenates from dark- or light-adapted p48Arr–/– mice were subjected to serial dilutions and loaded in the same gel. Sample loaded (in micrograms of total protein) is indicated above each lane. The ratio of p48 to its proteolyzed forms is not affected by light conditions. F, Comparison of pellet and supernatant fractions of total retinal homogenates from dark- or light-adapted p48Arr–/– mice. The proteolyzed forms are enriched in the supernatant fraction of dark-adapted mice and the pellet fraction of light-exposed animals.
Preparation of dark-adapted ROS and quantification of transgenic protein expression. For Figure 1 D, rod outer segment preparations were obtained from dark-adapted wild-type, p48Arr–/–, and m44Arr–/– mice. Briefly, retinas were dissected in darkness in Ringer's solution (in mm: 130 NaCl, 3.6 KCl, 1.2 MgCl2, 10 HEPES, and 0.02 EDTA). Six retinas were pooled per genotype in 120 μl of 8% iodixanol in Ringer's solution (prepared from a 60% w/v Optiprep solution; Nycomed Pharma, Roskilde, Denmark). Samples were vortexed at maximum speed for 2 min and centrifuged at 1000 rpm for 1 min at 4°C, and the upper 100 μl of the supernatant was transferred to a new tube kept in ice. A total of 100 μlof 8% iodixanol solution was added to the original sample, and the process was repeated four times, for collection of 500 μl of supernatant. Collected supernatant was loaded on top of a discontinuous Optiprep gradient in Ringer's solution (1.5 ml, 18% to 1.4 ml, 10%) and centrifuged at 70,000 rpm for 1 h at 4°C. The band corresponding to ROS was collected and diluted four times in Ringer's solution. ROS were pelleted by high-speed centrifugation (50,000 rpm, 20 min, 4°C), washed, and resuspended in Ringer's solution. Protein concentration was determined by Bradford's assay, and 0.24 mg/ml stocks were prepared in homogenization buffer/Laemmli buffer (1:1) and boiled. Twofold serial dilutions of samples were resolved by 10% Tris-glycine SDS-PAGE. Arrestin was immunodetected by Western blot with C10C10 antibody, and the blot was analyzed by densitometry using the Quantity One software (Bio-Rad).
Comparison of the extent of p48 proteolysis in dark- or light-adapted mice. p48Arr–/– mice were either dark-adapted overnight or exposed to 2000 lux constant white light for 30 min with their pupils dilated with 0.5% tropicamide and 2.5% phenylephrine hydrochloride. Retinas were homogenized, and protein concentration was determined as for Figure 1C. Serial fourfold dilutions were loaded from whole retinal homogenates (Fig. 1 E) or samples were centrifuged at 13,000 rpm for 15 min, 4°C, and equal fractions were loaded from the pellet or supernatant (Fig. 1 F).
Histological analysis. For analysis of retinal morphology by light microscopy, eyecups were dissected, fixed, embedded in epoxy resin, and sectioned as described previously (Mendez et al., 2000). For Figure 4 B, dark-reared mice from the indicated genotype were exposed to diffuse white light (2000 lux at cage level) for 72 h before processing. Measurements of the outer nuclear layer thickness were taken at four distinct regions of each retinal section: A, B, C, and D, as shown in Figure 4 B schematically, using the SlideBook 3.0 software (Intelligent Imaging Innovations, Denver, CO).
Figure 4.
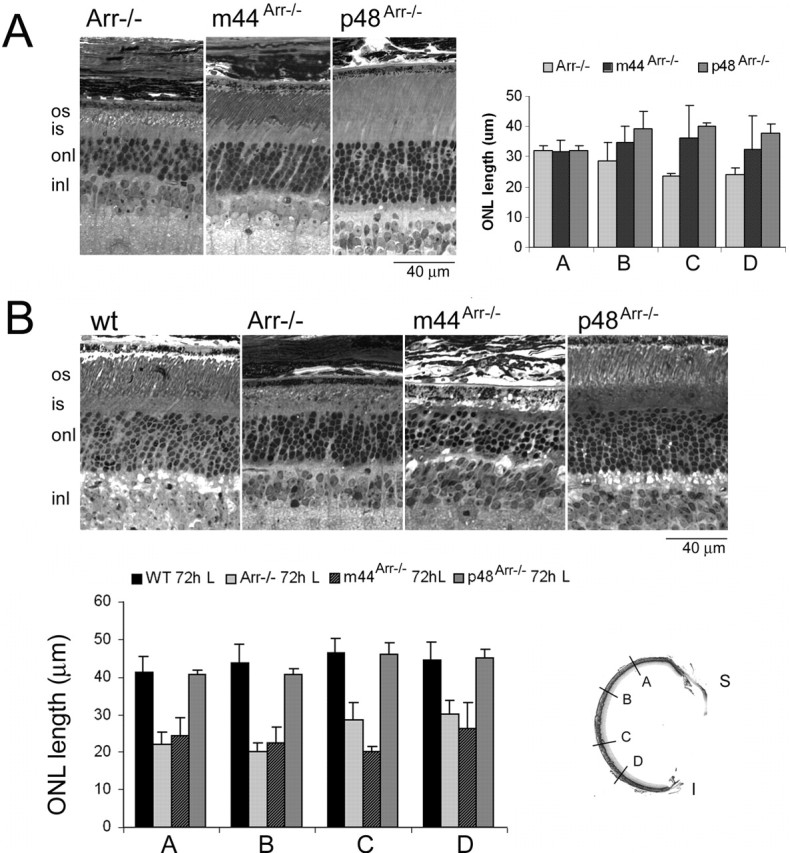
Effect of m44 or p48 transgene expression on the light-dependent degeneration of Arr–/– retinas. A, Transgene expression attenuated (m44) or prevented (p48) the photoreceptor cell death observed in Arr–/– mice under standard cyclic light conditions. Left, Light micrographs of retinal sections from 1-year-old Arr–/–, m44Arr–/–, and p48Arr–/– mice reared under standard 12 h dark/light cycles. Right, Outer nuclear layer thickness at four different regions of the retina (see A, B, C, D regions in retinal section inset) was determined in 1-year-old Arr–/–, m44Arr–/–, and p48Arr–/– mice. Two mice were analyzed per phenotype, and error bars represent the range of measurements. B, Protective effect of p48, but not m44, to damage induced by a bright light regimen. Dark-reared mice from the indicated genotypes were exposed to constant 2000 lux white fluorescent light for 72 h. The eyes were then processed immediately for morphological analysis. Top, Representative light micrographs. Bottom, Measurements of the outer nuclear layer thickness taken at four different regions (A, B, C, D, inset) of central retinal sections. Error bars indicate SD for wild type (n = 4), Arr–/– (n = 4), and m44Arr–/– (n = 4) and the range of measurements for p48Arr–/– (n = 2). os, Outer segment; is, inner segment; onl, outer nuclear layer; inl, inner nuclear layer.
Electrophysiology. Mice of the RK–/– or Arr–/– backgrounds were reared in continuous darkness to prevent light-dependent retinal degeneration (Xu et al., 1997; J. Chen et al., 1999). On the day of the recordings, an adult mouse that had been dark adapted for a minimum of 12 h was anesthetized by CO2 narcosis and killed in darkness. Under infrared light, the eyes were dissected and retinas were removed. Retinas were kept on ice in L-15 solution (Invitrogen, Carlsbad, CA) containing 10 mm glucose and 0.1 mg/ml bovine serum albumin. Small pieces of retina were finely chopped using a razor blade and placed in the recording chamber. The chamber was perfused with a bicarbonate-buffered Locke's solution, pH 7.4, which was oxygenated and maintained at physiological temperature (34–37°C). Infrared light (>700 nm wavelength) was used to visualize retinal pieces using an infrared-sensitive CCD camera (Stanford Photonics, Palo Alto, CA). Suction electrodes were made from borosilicate glass pulled (Brown and Flaming) and polished to a final diameter of ∼1.5 μm. The electrode was filled with a HEPES-buffered Ringer's solution at pH 7.4 (36°C). Single intact rod outer segments were sucked into the electrode, and the membrane current was amplified (Axopatch 1B; Molecular Devices, Union City, CA), filtered via an eight-pole Bessel filter (cutoff frequency of 20 or 30 Hz), and recorded continuously with IGOR NiDAQ acquisition software (WaveMetrics, Oswego, OR). Flashes (500 nm; 10 ms) were delivered by a stepper-motordriven shutter, controlled by the acquisition computer. Lamp power was calibrated daily using a silicon photodiode (United Detector Technology, Baltimore, MD).
The form of the single-photon response of each rod was estimated by squaring the mean response to at least 25 dim flashes of fixed strength and comparing the squared mean with the time-dependent ensemble variance. The time-dependent ensemble variance was also set to zero over the 1 s time interval preceding the flash. The scaling factor that brings the rising phase of the mean squared response into alignment with the rising phase of the timedependent variance provides an estimate of 1/n, where n is the mean number of photoisomerizations per flash. The flash strength was divided by n to calculate the effective collecting area (Ac) for our recordings, which on average was 0.36 ± 0.04 μm2 (n = 14). For brighter flash responses (see Figs. 3, 6), this mean Ac was used to estimate the mean number of R* per flash (n), as given in Results. The observed mean response was divided by n to estimate the mean amplitude and form of the single-photon response. The time integral of the singlephoton response was divided by the peak amplitude to obtain the integration time (Baylor and Hodgkin, 1973).
Figure 3.

Bright flash responses of m44 rods recover abnormally slowly. A, Family of bright flash responses of a p48 (blue) and m44Arr–/– (red) rod. Flash strengths varied from 8600 to 114,00 photons/μm2 by factors of ∼2. B, The time that a bright flash response from a m44Arr–/– rod remained in saturation (Tsat) increased dramatically at a flash strength equivalent to ln i ∼9.5, or 13,360 photons/μm2.
Figure 6.
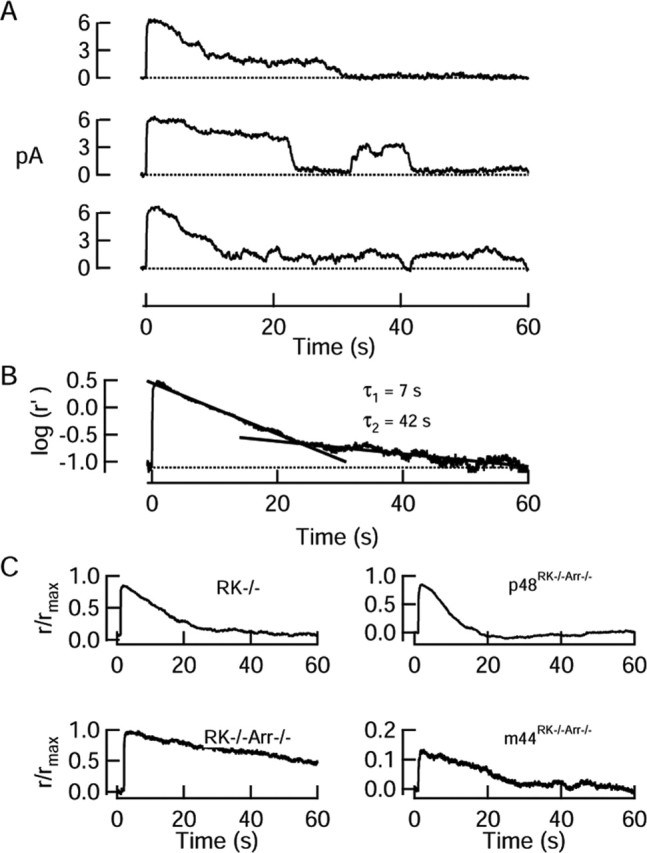
Responses to moderate flash strengths in rods lacking rhodopsin phosphorylation. A, Representative recordings from a RK–/– rod to three flashes that each activated ∼15 R* on average. Flashes were delivered at t = 0. Responses to individual flashes recovered in a series of steps; these step-like events frequently reactivated, typically for several tens of seconds after the flash. B, Mean response to 17 flashes (same rod as in A), plotted on a semilog scale. Response has been decompressed to allow for exponential saturation of the current (see Materials and Methods). Dark current was 8.2 pA. C, Mean responses to flashes that activated ∼15 R* on average from representative rods (RK–/– from A, B; RK–/–Arr–/–; p48RK–/–Arr–/–) and mean response of a m44RK–/–Arr–/– rod to a flash that activated 0.92 R* per flash. Dark currents were (in pA) 8.2 (RK–/–), 15.2 (RK–/–Arr–/–),13.4 (p48RK–/–Arr–/–), and 15.4 (m44RK–/–Arr–/–).
The time constants of response recovery were obtained by fitting the final falling phases of the dim flash responses by single- and doubleexponential functions. If the two time constants obtained from a double-exponential fit were <30% different from each other, the time constant of the single-exponential function was recorded.
Results
Expression of m44 or p48 transgenes in the arrestin knock-out background
We engineered mice to express transgenes encoding p48 or the murine equivalent of bovine p44 (mArr1-369 or m44) in rod photoreceptor cells of transgenic mice using the murine rod opsin promoter (Lem et al., 1991) (Fig. 1A). The m44 cDNA was generated by PCR-mediated mutagenesis of the mouse p48 cDNA and encodes a truncated protein that lacks the 35 C-terminal amino acids of arrestin (mArr1–369,V369A,Q370ter).
To compare the expression of endogenous splice variants and transgenic isoforms, we performed subcellular fractionation of homogenates of wild-type retinas and retinas expressing each of the transgene products in the presence of endogenous arrestin. In homogenates of light-adapted retinas from wild-type mice, the endogenous p48 protein was predominantly localized to the membrane fraction (Fig. 1B, P). In homogenates of m44 transgenic retinas, the m44 isoform was also predominant in the membrane fraction. The m44 transgene product migrated at the predicted molecular weight of 44 kDa and could be distinguished from endogenous p48, as well as two other less abundant endogenous forms of arrestin that were observed at ∼46 and 42 kDa (short arrows; see below).
Mice expressing the p48 or m44 transgenes were bred with arrestin knock-out mice (Xu et al., 1997) to express each arrestin variant separately. The levels of arrestin in equal fractions of total retinal homogenates from wild-type, Arr–/–, m44Arr–/–, and p48Arr–/– mice were compared by Western blot (Fig. 1C). Serial dilutions of wild-type and m44Arr–/– retinal homogenates showed that the expression level of m44 in the transgenic retinas was ∼6% that of p48 in wild-type retinas (Fig. 1C, and quantitative Western blot not shown) and similar to the level of endogenous truncated forms observed in wild-type retinas. Quantitative Western blotting also showed that the expression of the p48 transgene in the p48Arr–/– line was ∼140% that of the endogenous levels of p48 in wild-type mice (Fig. 1C, and quantitative Western blot not shown). Bands corresponding to 46 and 42 kDa were also present in retinas expressing only the p48 cDNA transgene (Fig. 1C, short arrows), indicating that the lower molecular weight species observed in wild-type homogenates originated not from alternative splicing but probably from proteolysis of fulllength arrestin. Although murine-truncated splice variants of arrestin have been described (Palczewski and Smith, 1996) and were readily detectable by Northern blot or reverse transcription-PCR amplification (data not shown), their expression levels in wild-type retinas were below the detection limit of our Western blot analysis (Fig. 1C).
Arrestin is not homogeneously distributed throughout the photoreceptor cytoplasm. In the dark, arrestin resides primarily in the inner segment and translocates to the outer segment during steady, bright illumination. Thus, to compare the functional roles of the arrestin isoforms, it was important to estimate the relative abundance of arrestin isoforms in purified, dark-adapted ROS from wild-type, p48Arr–/–, and m44Arr–/– mice (Fig. 1D). The ratio of p48 in wild-type ROS to m44 in m44Arr–/– ROS was ∼8:1 (∼12%), which is similar to the reported ratio of truncated and short splice variants found in bovine ROS (Palczewski et al., 1994). Under conditions of dark adaptation, ROS of transgenic p48Arr–/– mice contained approximately twice the amount of p48 present in ROS of wild-type mice.
Proteolysis of p48 has been reported previously in purified bovine ROS, particularly when p48 binds to R*-P (Azarian et al., 1995). To determine whether the presence of truncated forms of p48 observed in our studies was dependent on the binding of p48 to R*-P, we compared the relative abundance of the truncated forms in retinal homogenates from dark-adapted and lightadapted p48Arr–/– mice. The ratio of the total truncated forms to p48, ∼1:16, did not increase during illumination (Fig. 1E). After illumination, arrestin shifted from the soluble to the membrane fraction, presumably as a consequence of arrestin binding to R*-P (Fig. 1F). However, the extent of proteolysis was not increased in the light condition. Thus, the binding of p48 to R*-P did not affect the levels of the truncated forms of arrestin.
Deactivation of phosphorylated rhodopsin
To test the ability of each arrestin variant to deactivate R* in intact cells, we used suction electrodes to measure the flash responses of the m44Arr–/– and p48Arr–/– rods, as well as wild-type and Arr–/– rods. Consistent with previous findings (Xu et al., 1997), the dim flash responses of Arr–/– rods showed two components of recovery: one that occurred on a timescale of hundreds of milliseconds, and one that occurred on a timescale of tens of seconds (Xu et al., 1997) (Fig. 2). The initial recovery phase in the Arr–/– responses results from the partial loss of catalytic activity, which results from R* phosphorylation alone, whereas the final recovery phase likely reflects the thermal decay of R* to free opsin (Xu et al., 1997). In our experiments, the final recovery phase of Arr–/– responses was well fitted by a singleexponential function, with a time constant (τrec) of 26.0 ± 9.1 s (mean ± SEM; n = 6) (Table 1). Expression of either the m44 or the p48 transgene in the Arr–/– background restored normal kinetics to dim flash responses (Fig. 2B). The final time constants of recovery of dim flash responses of the transgenic rods (τrec of ∼250 ms) (Table 1) was not significantly different from that of wild-type responses (two-tailed t test, p = 0.33 for p48 and p = 0.58 for m44). Expression of the transgenes produced no detectable changes in any aspects of the light responses; rods of m44Arr–/– and p48Arr–/– mice had normal mean dark currents and sensitivities (Table 1).
Figure 2.

Expression of p48 or m44 restores normal recovery to flash responses. A, Families of responses to increasing flash strengths from representative wild-type, knock-out, and transgene-positive rods. Responses have been normalized by the maximal response amplitudes, which were (in pA) 11.4 (WT), 10.8 (Arr–/–), 17.0 (m44Arr–/–), and 14.6 (p48Arr–/–). Flash strengths ranged from (in photons/μm2) 20.4 to 2612 by factors of 4 (WT); 6.2 to 2621 by factors of 4 (m44Arr–/–); 20.5 to 2621 by factors of 4 (p48Arr–/–) and for Arr–/– were 8.9, 17.6, 24, 227, and 1154. B, Population mean single-photon responses from WT (n = 21), m44Arr–/– (n = 13), and p48Arr–/– (n = 11) rods. A representative scaled dim flash response from an Arr–/– rod is shown for comparison. Right panel is same traces as on the left, shown on an expanded timescale. Error bars represent SEM. In all panels, flashes (10 ms, 500 nm) were delivered at t = 0.
Table 1.
Characteristics of flash responses of rods with and without arrestin splice variants
|
|
Dark current (pA) |
Time-to-peak (ms) |
Elementary amplitude (pA) |
τrec (s) |
Integration time (ms) |
Ioa (photons/μm2) |
|---|---|---|---|---|---|---|
| WT | 12.2 ± 0.6 (31) | 133 ± 8 (23) | 0.43 ± 0.05 (20) | 0.23 ± 0.02 (23) | 317 ± 28 (23) | 77.9 ± 5.9 (21) |
| m44Arr—/— | 11.3 ± 0.9 (22) | 144 ± 15 (20) | 0.48 ± 0.09 (16) | 0.24 ± 0.02 (20) | 315 ± 39 (20) | 69.2 ± 6.3 (19) |
| p48Arr—/— | 11.4 ± 0.7 (23) | 130 ± 12 (20) | 0.44 ± 0.05 (18) | 0.30 ± 0.09 (19) | 407 ± 56 (20) | 77.8 ± 5.4 (21) |
| Arr—/— |
9.6 ± 1.1 (9) |
220 ± 56 (6) |
ND |
26.0 ± 9.1 (6) |
ND |
ND |
ND, Not determined.
Flash strength that elicited a half-maximal response.
The ability of the p48 transgene to restore normal recovery kinetics persisted across all flash strengths examined (5–100,000 photons/μm2), indicating that the level of expression of p48 and/or its ability to bind to R*-P was sufficient to rapidly deactivate thousands of simultaneously activated rhodopsin molecules. However, responses of m44Arr–/– rods slowed as the flash strength increased (Fig. 3). For bright flashes (∼13,000 photons/μm2, corresponding to ∼5000 R*/flash), the responses of the m44Arr–/– rods returned to baseline over many tens of seconds, much like the responses of Arr–/– rods.
Rescue of light-dependent degeneration in m44Arr–/– and p48Arr–/– retinas
Mice lacking arrestin show a progressive loss of photoreceptors when they are reared in standard cyclic light conditions. This degeneration has been attributed to excessive signaling of the phototransduction cascade. It can be dramatically accelerated by exposing mice to constant light or prevented by raising the mice in constant darkness (Fain et al., 1989; Xu et al., 1997; J. Chen et al., 1999). To assess the ability of either p48 or m44 to prevent light damage in Arr–/– retinas, we compared the retinal morphology of mice reared under normal 12 h dark/light cycles. Figure 4A (left) shows retinal sections from Arr–/–, m44Arr–/–, and p48Arr–/– mice at 1 year of age. Arr–/– retinas showed disrupted outer segment structures and a 50% reduction of photoreceptor cell nuclei in the outer nuclear layer. In comparison, both p48Arr–/– and m44Arr–/– retinas had longer and more orderly rod outer segment structure and more remaining photoreceptor cell nuclei, as measured by outer nuclear layer thickness (Fig. 4A, right). Similar results were observed in four independent groups of mice ranging from 8 to 12 months of age. Therefore, both m44 and p48 were able to attenuate photoreceptor degeneration under cyclic light-rearing conditions.
Exposure of Arr–/– mice to constant illumination causes accelerated retinal degeneration (J. Chen et al., 1999). To test the ability of the transgenes to prevent retinal damage caused by constant light, dark-reared pigmented wild-type, Arr–/–, m44Arr–/–, and p48Arr–/– mice were exposed to 2000 lux white fluorescent light for 72 h. A comparison of the thickness of the outer nuclear layer at this time point revealed that Arr–/– and m44Arr–/– retinas had ∼50% fewer photoreceptor cell nuclei and extensively disrupted outer segments (Fig. 4B). In contrast, wild-type and p48Arr–/– retinas were not affected by the constant light exposure (Fig. 4B). The retinal thickness and ordered outer segment structures were similar to those of wild-type mice that were housed in the 12 h cyclic light. These results show that p48 can prevent light-induced retinal degeneration. The inability of m44 to prevent retinal light damage is consistent with the lower expression levels of m44 observed in the transgenic rods (Fig. 1) and the slowed recovery of m44 rods to bright flashes (Fig. 3).
Deactivation of unphosphorylated rhodopsin by arrestin
Unlike the responses of wild-type and Arr–/– rods, singlephoton responses from rods lacking rhodopsin phosphorylation typically maintain a steady amplitude for several seconds before turning off abruptly, producing step-like responses (Chen et al., 1995; C. K. Chen et al., 1999; Mendez et al., 2000). The durations of these responses are distributed exponentially, suggesting that a slow, first-order process deactivates rhodopsin in the absence of phosphorylation. This deactivation appears to be reversible: for tens of seconds after a bright flash, there are frequent spontaneous events resembling single-photon responses. Because p44 can bind unphosphorylated R* in vitro (Palczewski et al., 1994; Pulvermuller et al., 1997; Schröder et al., 2002), this reversible deactivation of the single-photon responses in RK–/– rods has been proposed to reflect the binding and unbinding of p44 to unphosphorylated R* (C. K. Chen et al., 1999).
To test this idea directly, we recorded spontaneous steplike events in darkness and compared the durations of the events of RK–/– rods with those of the RK–/–Arr–/–, m44RK–/–Arr–/–, and p48RK–/–Arr–/– transgenic rods. To our surprise, the RK–/–Arr–/– double-knock-out rods, like the RK–/– rods, displayed step-like responses that turned off abruptly. However, these responses were frequently of longer duration than those of RK–/– rods (Fig. 5). A histogram of the durations of RK–/–Arr–/– responses could not be fitted by a single-exponential function but rather by a double exponential (A1 e–t /τ 1 + A2 e–t /τ 2 ; Α1 = 0.16, τ1 = 4.1 s, Α2 = 1.0, and τ2 = 24.3 s, with 1328 step-like responses from 37 rods). Thus, endogenous arrestin promotes the deactivation of nonphosphorylated rhodopsin, although its binding per se is not the sole mechanism capable of quenching nonphosphorylated R* activity.
Figure 5.

Elementary, step-like responses in rods lacking rhodopsin phosphorylation. A, Records from knock-out and transgenic rods showing the variable nature of the elementary responses recorded spontaneously in darkness. Dark currents were (in pA) 10.5 (RK–/–), 14.4 (RK–/–Arr–/–), 17.0 (p48RK–/–Arr–/–), and 14.5 (m44RK–/–Arr–/–). B, Frequency distributions of elementary response durations. Distribution of responses from RK–/– and p48 rods were very similar, whereas that of the m44Arr–/– responses resembled that of RK–/–Arr–/– responses. Solid curves are the best fits of scaled exponential functions (e–t/τ or A1e–t/τ1 + A2e–/τ2) with the following parameters: RK–/–, τ = 3.2 s; p48RK–/–Arr–/–, τ = 5.4 s; RK–/–Arr–/–, A1 = 0.16, A2 = 1.0, τ1 = 4.1 s, τ2 = 24.3 s; m44RK–/–Arr–/–, A1 = 0.47, A2 = 1.0, τ1 = 3.7 s, τ2 = 24.2 s.
To determine whether the facilitated quench of R* activity in RK–/– rods was mediated by p48 and/or m44, we performed similar analysis on the elementary responses of m44RK–/–Arr–/– and p48RK–/–Arr–/– rods. Single-photon responses in both of these transgenic lines were also step-like in form (Fig. 5A). On average, expression of the p48 transgene resulted in responses that were more similar in duration to the RK–/–, with a mean duration of 5.4 s (Fig. 5B) (1082 step-like responses from 34 cells). In contrast, responses of the m44RK–/–Arr–/– rods were not markedly different from those of RK–/–Arr–/– rods (Α1 = 0.47, τ1 = 3.7 s, Α2 = 1.0, and τ2 = 24.2 s, with 287 step-like responses from 26 rods). This suggests that p48, but not m44, can deactivate unphosphorylated R* in RK–/– rods.
Role for p48 in preventing reactivation of unphosphorylated rhodopsin
Measuring the mean duration of elementary events in RK–/– rods provides a measure of the catalytic lifetime of an unphosphorylated R* molecule. However, it does not reflect in any way the occasional reactivation of rhodopsin that is often observed in RK–/– rods after brighter flashes. Because it is impossible to distinguish reactivation of a previously photoexcited rhodopsin from the spontaneous activation of a new rhodopsin, we analyzed the responses to moderately bright flashes, which then represent the summed activity of multiple rhodopsin molecules. When RK–/– rods were presented with moderately bright flashes (∼15 R*/flash) they generated responses that were also long lasting and turned off in a series of stochastic, step-like transitions that occasionally reactivated (Fig. 6A). These step-like events are likely to originate from transitions in individual rhodopsin molecules, because their form resembles single-photon responses from these rods (C. K. Chen et al., 1999). In all RK–/– rods, the average response to moderately bright flashes recovered initially, with a time constant of a few seconds. However, in 11 of 28 cells, the response recovery showed an additional slower component of recovery (Fig. 6B). On average, the parameters of these two kinetically distinct recovery modes of recovery were as follows: Α1 = 0.96 ± 0.06, τ1 = 6.0 ± 1.0 s; A2 = 0.57 ± 0.18, τ2 = 21.4 ± 2.8 s. This first time constant (τ1) is similar to the mean time required for the initial quench of unphosphorylated single R* molecules (Fig. 5B) (Chen et al., 1995; C. K. Chen et al., 1999). The second (τ2) is similar to the final recovery time constant of Arr–/– rods (see above), which has been attributed to the thermal decay of metarhodopsin II to free opsin.
To further test the hypothesis that arrestin participates in quenching unphosphorylated R*, we examined moderately bright flash responses from mouse rods that expressed neither RK nor arrestin (RK–/–Arr–/–). Unlike all RK–/– rods examined, none of the RK–/–Arr–/– rods showed a rapid component of recovery, nor could their responses be well fitted by a double-exponential function (Fig. 6C). In 7 of 15 rods, the recovery could not be approximated by an exponential but rather recovered incompletely or proceeded along a nearly linear time course (Fig. 6C). However, in all rods that did display responses with full, exponential recovery (n = 8), the average responses were fitted by a single exponential, with a time constant (τ = 37.7 ± 10.2 s) that was similar to that of the recovery of Arr–/– responses (τ = 26.0 ± 9.1 s).
To determine whether p48 and m44 are equally capable of preventing reactivation of R*, we recorded the responses to moderately bright flashes in RK–/–Arr–/– rods expressing either the m44 or the p48 transgenes (Fig. 6 C). In most p48RK–/–Arr–/– rods (21 of 23 cells), the average response to a moderate flash recovered like those of RK–/– rods, along a single-exponential time course with a time constant of 5.1 ± 2.5 s. In the remaining two cells, the recovery was fitted by an exponential function with two time constants of 2.3 and 4.4 s (τ1) and 21 and 44 s (τ2) of approximately equal amplitude. Thus, p48 quenched unphosphorylated R* in a manner similar to that of endogenous arrestin.
Responses of m44RK–/–Arr–/– rods to moderately bright flashes were much more variable but in general resembled those of the RK–/–Arr–/– rods. In four cells, the recovery was fitted by a singleexponential function with a time constant of 26.7 ± 4.8 s. However, in one cell, the average response recovery was fitted by a single exponential with a time constant of 6.9 s. In two other cells, the recovery time constant was fitted by a double exponential of approximately equal amplitudes, having time constants of 1.3 and 2.8 s (τ1) and 27.1 and 9.8 s (τ2), respectively. Because m44 was expressed at levels comparable with that of the endogenous truncated form in wild-type rods, the inability of m44 to influence the deactivation of unphosphorylated R* supports the conclusion (Fig. 5) that the truncated splice variant of arrestin plays little role in normally quenching the rare “rogue” rhodopsins that escape phosphorylation by rhodopsin kinase (Baylor et al., 1984).
Discussion
It has been more than a decade since different splice variants of the arrestin genes have been described, yet until now there has been no functional test of the ability of the splice variants to deactivate rhodopsin in intact cells. In this study, we have investigated which splice form of arrestin is involved in rhodopsin deactivation in dark-adapted rods under conditions of both dim and bright light stimulation. The levels of p48 and m44 in the transgenic rods closely resembled those of the endogenous splice variants reported in bovine (Palczewski et al., 1994) and human (Smith, 1996) retina. Therefore, the ability of p48 and m44 to quench R* and R*-P in the transgenic rods likely reflects the relative functional contributions of p48 and p44 under darkadapted in vivo conditions.
Ability of p48 and p44 arrestin isoforms to quench R*-P
In recent years, numerous crystallographic and mutagenesis studies have provided a mechanistic model for the structural rearrangements of arrestin after its encounter with rhodopsin (Gurevich and Gurevich, 2004). In its basal state, rod arrestin has an “activation-recognition” domain and a “phosphorylationrecognition” domain that exhibit low-affinity binding to R* and R-P, respectively (Gurevich and Gurevich, 2004). The shift to the high-affinity state is termed “arrestin activation” and is thought to occur when an encounter with R*-P disrupts interactions that stabilize the basal state of arrestin, causing a global conformational change (Hirsch et al., 1999). This conformational change exposes a secondary binding site that confers high-affinity interaction with R*-P (Vishnivetskiy et al., 2000).
Alternative splicing of the arrestin gene is thought to produce a COOH-terminal truncated protein with a destabilized basal conformation and reduced selectivity (Palczewski et al., 1994; Pulvermuller et al., 1997). In our experiments, expression of either the p48 or m44 transgene in the arrestin knock-out genetic background was able to restore normal kinetics to flash responses across a wide range of light intensities, and there were no differences in the overall response time courses mediated by the two arrestin isoforms, except in response to very bright flashes. The similarity of p48Arr–/– and m44Arr–/– single-photon responses indicates that either form of arrestin can mediate timely rhodopsin deactivation in dark-adapted rods. These results are consistent with previous in vitro studies demonstrating that both short and long splice variants bind avidly to R*-P (Palczewski et al., 1994; Pulvermuller et al., 1997; Schröder et al., 2002). In our single-cell recordings, small differences in the rate at which m44 and p48 mediate R*-P deactivation may be masked by negative feedback mechanisms (Burns et al., 2002) and deactivation of slower downstream cascade reactions (Sagoo and Lagnado, 1997; Krispel et al., 2003). Nevertheless, the similarity of p48Arr–/– and m44Arr–/– responses indicates a functional redundancy of arrestin isoforms in dark-adapted rods.
In response to bright flashes, however, only p48 supported normal recovery. The brightest brief (10 ms) flash that we were able to elicit from our optical bench (∼30,000 R*/flash) yielded responses with normal recovery kinetics. Assuming a 1:1 binding stoichiometry, this indicates that there were at least 30,000 instantaneously available p48 molecules present in the outer segment at the time of the flash. In an attempt to cumulatively deplete the outer segment of available p48 molecules, we delivered bright flashes repeatedly to p48Arr–/– rods and monitored the time course of response recovery. We were unable to produce responses that resembled those of the Arr–/– rods (data not shown). This suggests that the dissociation and reuse of a given p48 molecule is very rapid, or that translocation of p48 into the outer segment can occur continuously to replenish the available arrestin pool. In contrast, single bright flashes readily depleted available m44 molecules from m44 transgenic rods. Responses of m44Arr–/– rods to flashes exciting ∼5000 R*/flash (∼13,000 photons/μm2; see Materials and Methods) resembled those of Arr–/– rods. From this result, we infer that there were ∼5000 available molecules of m44 per dark-adapted outer segment, which is lower than the levels of m44 estimated by Western blot analysis of purified ROS. The higher levels detected by Western blot may include a pool of m44 that is unavailable for binding R*-P, such as a pool that is aggregated (Pulvermuller et al., 1997) or bound to microtubules (Nair et al., 2004, 2005).
The differences in the ability of m44 and p48 to mediate recovery from bright flash responses are consistent with the observed differential ability of m44 and p48 to prevent lightinduced photoreceptor cell death (Fig. 4). Expression of either m44 or p48 in the Arr–/– rods prevented the dim, cyclic-lightinduced degeneration (Fig. 4) characteristic of Arr–/– retinas (J. Chen et al., 1999). However, only the expression of p48 was able to prevent photoreceptor cell death caused by exposure to 72 h of constant bright light (2000 lux). These results are consistent with the physiological results and the differences in expression level that we observed in the two transgenic lines.
In Drosophila photoreceptors, a low-abundance visual arrestin similar to p44 (Arr1) mediates light-induced rhodopsin endocytosis and cell survival (Satoh and Ready, 2005). The absence of Arr1 causes retinal degeneration as a result of defects in rhodopsin endocytosis when Arr2, the abundant visual arrestin responsible for deactivating rhodopsin, is expressed. In our experiments, however, we did not observe retinal degeneration in p48Arr–/– mice, suggesting that, unlike in Drosophila, expression of a truncated arrestin is not essential for photoreceptor viability.
Arrestin deactivates unphosphorylated rhodopsin (R*)
In the absence of rhodopsin phosphorylation and endogenous arrestin (RK–/–Arr–/–), expression of m44 was unable to quench unphosphorylated R*. This result may seem surprising because p44 can quench R* activity in phosphodiesterase assays (Palczewski et al., 1994) and because p44 enhances the formation of unphosphorylated metarhodopsin II in light-scattering studies (Pulvermuller et al., 1997; Schröder et al., 2002). Because p44 also shows prominent membrane association (Palczewski et al., 1994; Schröder et al., 2002) and self-aggregation, particularly at higher concentrations (Pulvermuller et al., 1997), we cannot rule out that the m44 in our studies may have been simply unavailable for quenching R*. However, it is important to note that the relative expression level of m44 in the m44Arr–/– rods was comparable with that of truncated arrestins generated by alternative splicing (Palczewski and Smith, 1996; Smith, 1996) or proteolysis (Fig. 1), supporting the idea that m44 plays little functional role in quenching unphosphorylated R* that may occur in normal rods.
Indeed, although R* is normally rapidly phosphorylated, steplike responses that resemble the single-photon responses of RK–/– and RK–/– Arr–/– rods are also occasionally observed in wild-type mouse rods (in this study, 26 step-like responses were observed in 17 wild-type rods to flashes that cumulatively activated 25,840 rhodopsins). Healthy primate rods also display similar infrequent events, which have been proposed to result from a small fraction of rhodopsin molecules (∼1 in 1000) that are incapable of being phosphorylated (Baylor et al., 1984). These long-lasting events may dominate the responses to extensive bleaches, producing one component of human dark adaptation (Baylor et al., 1984; Lamb and Pugh, 2004). The ability of p48 to promote the deactivation of unphosphorylated R* in our experiments suggests that p48 may be functionally important for decreasing the time required for dark adaptation.
The ability of p48 to quench the activity unphosphorylated R*, albeit slowly (after an average time of ∼5 s) (Fig. 5), is consistent with the fact that p48 binds poorly to unphosphorylated R* in vitro (for review, see Gurevich and Gurevich, 2004). Unlike p44, p48 does not enhance the formation of metarhodospin II in light-scattering studies (Pulvermuller et al., 1997; Schröder et al., 2002), which perhaps might arise if p48 binds equally well to metarhodopsin I and metarhodopsin II (thereby not stabilizing one over the other), or if p48 binds to a post-metarhodopsin II decay product (Sommer et al., 2005). This latter possibility is supported indirectly by the fact that elementary responses of rods that lacked both rhodopsin phosphorylation and arrestin (RK–/–Arr–/– rods) also showed step-like elementary responses, but their durations were, on average, considerably longer than those of RK–/– rods. Thus, the abrupt transitions that turn off these steps cannot arise exclusively from the binding and unbinding of arrestin but may include transitions to postmetarhodospin II decay products (metarhodopsin III) with much lower catalytic activity. The ability of p48 to decrease the overall duration of the steps suggests that such a decay product is stabilized by arrestin binding.
In summary (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), we found that both truncated and full-length splice variants of visual arrestin can rapidly quench phosphorylated rhodopsin (R*-P) in dark-adapted rod outer segments. In a normal rod, phosphorylation of R* by RK partially reduces the catalytic activity of R* (Xu et al., 1997; C. K. Chen et al., 1999), with complete deactivation requiring the subsequent binding of either splice variant of arrestin. This deactivation of R* occurs within ≤200 ms, the time constant of recovery of wild-type photoresponses. In the absence of phosphorylation, a backup mechanism exists to abruptly deactivate R* on a timescale of a few seconds, a process that is reversible and is facilitated by p48, but not p44, arrestin. Finally, in the absence of arrestin, gradual deactivation of R* and R*-P proceeds by thermal decay to free opsin over many tens of seconds.
Footnotes
This work was supported by the National Eye Institute of the National Institutes of Health in the form of Vision Core Grants EY12576 (to University of California, Davis) and EY03040 (to Doheny Eye Institute) and Grants EY14047 (M.E.B.),EY12155(J.C.),and EY05750(D.A.B.).We thank Claudia Krispel and Will Rubin for helpful comments on this manuscript and technical assistance in its preparation.
Correspondence should be addressed to either of the following: Marie E. Burns, Center for Neuroscience and Department of Psychiatry and Behavioral Sciences, University of California, Davis, 1544 Newton Court, Davis, CA 95616, E-mail: meburns@ucdavis.edu; or Jeannie Chen, Departments of Ophthalmology and Cell and Neurobiology, Keck School of Medicine, University of Southern California, 1501 San Pablo Street, Los Angeles, CA 90089-2821, E-mail: jeannie@usc.edu.
DOI:10.1523/JNEUROSCI.3301-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/261036-09$15.00/0
References
- Azarian SM, King AJ, Hallett MA, Williams DS (1995) Selective proteolysis of arrestin by calpain. Molecular characteristics and its effect on rhodopsin dephosphorylation. J Biol Chem 270: 24375–24384. [DOI] [PubMed] [Google Scholar]
- Baylor DA, Hodgkin AL (1973) Detection and resolution of visual stimuli by turtle photoreceptors. J Physiol (Lond) 234: 163–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor DA, Nunn BJ, Schnapf JL (1984) The photocurrent, noise and spectral sensitivity of rods of the monkey Macaca fascicularis J Physiol (Lond) 357: 575–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannock MT, Weng K, Robinson PR (1999) Rhodopsin's carboxylterminal threonines are required for wild-type arrestin-mediated quench of transducin activation in vitro. Biochemistry 38: 3770–3777. [DOI] [PubMed] [Google Scholar]
- Burns ME, Mendez A, Chen J, Baylor DA (2002) Dynamics of cyclic GMP synthesis in retinal rods. Neuron 36: 81–91. [DOI] [PubMed] [Google Scholar]
- Chen CK, Burns ME, Spencer M, Niemi GA, Chen J, Hurley JB, Baylor DA, Simon MI (1999) Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci USA 96: 3718–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Makino CL, Peachey NS, Baylor DA, Simon MI (1995) Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science 267: 374–377. [DOI] [PubMed] [Google Scholar]
- Chen J, Simon MI, Matthes MT, Yasumura D, LaVail MM (1999) Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness). Invest Ophthalmol Vis Sci 40: 2978–2982. [PubMed] [Google Scholar]
- Elias RV, Sezate SS, Cao W, McGinnis JF (2004) Temporal kinetics of the light/dark translocation and compartmentation of arrestin and alphatransducin in mouse photoreceptor cells. Mol Vis 10: 672–681. [PubMed] [Google Scholar]
- Fain GL, Lamb TD, Matthews HR, Murphy RL (1989) Cytoplasmic calcium as the messenger for light adaptation in salamander rods. J Physiol (Lond) 416: 215–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S, Nakazawa M, Maw M, Tamai M, Oguchi Y, Gal A (1995) A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet 10: 360–362. [DOI] [PubMed] [Google Scholar]
- Gibson SK, Parkes JH, Liebman PA (2000) Phosphorylation modulates the affinity of light-activated rhodopsin for G protein and arrestin. Biochemistry 39: 5738–5749. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL (1993) Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward lightactivated phosphorylated rhodopsin. J Biol Chem 268: 11628–11638. [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV (2004) The molecular acrobatics of arrestin activation. Trends Pharmacol Sci 25: 105–111. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Richardson RM, Kim CM, Hosey MM, Benovic JL (1993) Binding of wild type and chimeric arrestins to the m2 muscarinic cholinergic receptor. J Biol Chem 268: 16879–16882. [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV, Sigler PB (1999) The 2.8 A crystal structure of visual arrestin: a model for arrestin's regulation. Cell 97: 257–269. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Lee KA, Niemi GA, Craven KB, Garwin GG, Saari JC, Hurley JB (2001) Multiple phosphorylation of rhodopsin and the in vivo chemistry underlying rod photoreceptor dark adaptation. Neuron 31: 87–101. [DOI] [PubMed] [Google Scholar]
- Knospe V, Donoso LA, Banga JP, Yue S, Kasp E, Gregerson DS (1988) Epitope mapping of bovine retinal S-antigen with monclonal antibodies. Curr Eye Res 7: 1137–1147. [DOI] [PubMed] [Google Scholar]
- Krispel CM, Chen CK, Simon MI, Burns ME (2003) Novel form of adaptation in mouse retinal rods speeds recovery of phototransduction. J Gen Physiol 122: 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb TD, Pugh Jr EN (2004) Dark adaptation and the retinoid cycle of vision. Prog Retin Eye Res 23: 307–380. [DOI] [PubMed] [Google Scholar]
- Langlois G, Chen CK, Palczewski K, Hurley JB, Vuong TM (1996) Responses of the phototransduction cascade to dim light. Proc Natl Acad Sci USA 93: 4677–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lem J, Applebury ML, Falk JD, Flannery JG, Simon MI (1991) Tissuespecific and developmental regulation of rod opsin chimeric genes in transgenic mice. Neuron 6: 201–210. [DOI] [PubMed] [Google Scholar]
- Maeda T, Imanishi Y, Palczewski K (2003) Rhodopsin phosphorylation: 30 years later. Prog Retin Eye Res 22: 417–434. [DOI] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, Baylor DA, Chen J (2000) Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron 28: 153–164. [DOI] [PubMed] [Google Scholar]
- Mendez A, Lem J, Simon M, Chen J (2003) Light-dependent translocation of arrestin in the absence of rhodopsin phosphorylation and transducin signaling. J Neurosci 23: 3124–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Kennedy MJ, Hurley JB, Gurevich VV, Slepak VZ (2004) Direct binding of visual arrestin to microtubules determines the differential subcellular localization of its splice variants in rod photoreceptors. J Biol Chem 279: 41240–41248. [DOI] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ (2005) Light-dependent redistribution of arrestin in vertebrate rods is an energyindependent process governed by protein-protein interactions. Neuron 46: 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani K, Yau KW (1988) Guanosine 3′,5′-cyclic monophosphateactivated conductance studied in a truncated rod outer segment of the toad. J Physiol (Lond) 395: 731–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K (1994) Structure and functions of arrestins. Protein Sci 3: 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Smith WC (1996) Splice variants of arrestins. Exp Eye Res 63: 599–602. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Buczylko J, Ohguro H, Annan RS, Carr SA, Crabb JW, Kaplan MW, Johnson RS, Walsh KA (1994) Characterization of a truncated form of arrestin isolated from bovine rod outer segments. Protein Sci 3: 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parruti G, Ambrosini G, Sallese M, De Blasi A (1993) Molecular cloning, functional expression and mRNA analysis of human beta-adrenergic receptor kinase 2. Biochem Biophys Res Commun 190: 475–481. [DOI] [PubMed] [Google Scholar]
- Pulvermuller A, Maretzki D, Rudnicka-Nawrot M, Smith WC, Palczewski K, Hofmann KP (1997) Functional differences in the interaction of arrestin and its splice variant, p44, with rhodopsin. Biochemistry 36: 9253–9260. [DOI] [PubMed] [Google Scholar]
- Sagoo MS, Lagnado L (1997) G-protein deactivation is rate-limiting for shut-off of the phototransduction cascade. Nature 389: 392–395. [DOI] [PubMed] [Google Scholar]
- Satoh AK, Ready DF (2005) Arrestin1 mediates light-dependent rhodopsin endocytosis and cell survival. Curr Biol 15: 1722–1733. [DOI] [PubMed] [Google Scholar]
- Schröder K, Pulvermuller A, Hofmann KP (2002) Arrestin and its splice variant Arr1–370A (p44). Mechanism and biological role of their interaction with rhodopsin. J Biol Chem 277: 43987–43996. [DOI] [PubMed] [Google Scholar]
- Smith WC (1996) A splice variant of arrestin from human retina. Exp Eye Res 62: 585–592. [DOI] [PubMed] [Google Scholar]
- Smith WC, Milam AH, Dugger D, Arendt A, Hargrave PA, Palczewski K (1994) A splice variant of arrestin. Molecular cloning and localization in bovine retina. J Biol Chem 269: 15407–15410. [PubMed] [Google Scholar]
- Sommer ME, Smith WC, Farrens DL (2005) Dynamics of arrestinrhodopsin interactions: arrestin and retinal release are directly linked events. J Biol Chem 280: 6861–6871. [DOI] [PubMed] [Google Scholar]
- Sterne-Marr R, Gurevich VV, Goldsmith P, Bodine RC, Sanders C, Donoso LA, Benovic JL (1993) Polypeptide variants of beta-arrestin and arrestin3. J Biol Chem 268: 15640–15648. [PubMed] [Google Scholar]
- Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez MG, Gurevich VV (2000) An additional phosphate-binding element in arrestin molecule. Implications for the mechanism of arrestin activation. J Biol Chem 275: 41049–41057. [DOI] [PubMed] [Google Scholar]
- Wilden U (1995) Duration and amplitude of the light-induced cGMP hydrolysis in vertebrate photoreceptors are regulated by multiple phosphorylation of rhodopsin and by arrestin binding. Biochemistry 34: 1446–1454. [DOI] [PubMed] [Google Scholar]
- Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J (1997) Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature 389: 505–509. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Sippel KC, Berson EL, Dryja TP (1997) Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet 15: 175–178. [DOI] [PubMed] [Google Scholar]