

Abstract
Myelin-associated inhibitors (MAIs) signal through a tripartate receptor complex on neurons to limit axon regeneration in the CNS. Inhibitory influences ultimately converge on the cytoskeleton to mediate growth cone collapse and neurite outgrowth inhibition. Rho GTPase and its downstream effector Rho kinase are key signaling intermediates in response to MAIs; however, the links between Rho and the actin cytoskeleton have not been fully defined. We found that Nogo-66, a potent inhibitory fragment of Nogo-A, signals through LIM (LIM is an acronym of the three gene products Lin-11, Isl-1, and Mec-3) kinase and Slingshot (SSH) phosphatase to regulate the phosphorylation profile of the actin depolymerization factor cofilin. Blockade of LIMK1 activation and subsequent cofilin phosphorylation circumvents myelin-dependent inhibition in chick dorsal root ganglion neurons, suggesting that phosphorylation and inactivation of cofilin is critical for neuronal inhibitory responses. Subsequent activation of SSH1 phosphatase mediates cofilin dephosphorylation and reactivation. Overexpression of SSH1 does not mimic the neurite outgrowth inhibitory effects of myelin, suggesting an alternative role in MAI inhibition. We speculate that SSH-mediated persistent cofilin activation may be responsible for maintaining an inhibited neuronal phenotype in response to myelin inhibitors.
Keywords: Nogo, Lim kinase, Slingshot phosphatase, CNS regeneration, myelin inhibition, rho kinase
Introduction
After injury in the adult CNS, myelin-associated inhibitors (MAIs) such as myelin-associated glycoprotein (MAG), oligodendrocyte-myelin glycoprotein (OMgp), and Nogo signal to injured nerve fibers to block spontaneous axon regeneration (He and Koprivica, 2004). Binding of MAIs to a tripartate receptor complex (Fournier et al., 2001; Wang et al., 2002; Wong et al., 2002; Mi et al., 2004; Park et al., 2005; Shao et al., 2005) on neuronal growth cones stimulates intracellular signaling pathways (He and Koprivica, 2004) that converge to rearrange the actin cytoskeleton causing growth cone collapse and outgrowth inhibition. A key regulatory molecule in this pathway is Rho GT-Pase, which is activated in response to MAIs (Dergham et al., 2002; Niederost et al., 2002; Fournier et al., 2003), and is known to play a role in mediating actin cytoskeletal rearrangements (Hall, 1998). Beyond Rho, the molecular links between receptor engagement and the actin cytoskeleton have not been fully defined.
Actin dynamics are orchestrated by a network of actin regulatory proteins, which affect actin polymerization, depolymerization, stability, severing, and filament capping (Pollard and Borisy, 2003). Cofilin is a major regulatory protein of the actin depolymerization factor/cofilin family that regulates actin dynamics by increasing the off-rate of actin monomers from the pointed actin filament end and by severing actin filaments (Bamburg and Wiggan, 2002). Cofilin is a good candidate molecule for mediating MAI-dependent responses, because it has been implicated previously in basal neurite outgrowth (Kuhn et al., 2000; Meberg and Bamburg, 2000) and in modifying growth cone dynamics in response to repulsive guidance cues (Aizawa et al., 2001). We hypothesized that MAIs may regulate actin dynamics by signaling to cofilin via Rho GTPases.
Cofilin activity is regulated by phosphorylation at serine 3. LIM (LIM is an acronym of the three gene products Lin-11, Isl-1, and Mec-3) (Arber et al., 1998) and testicular (TES) (Toshima et al., 2001) kinases [LIM kinase (LIMK) and testicular kinase (TESK)] phosphorylate and inactivate cofilin, whereas Slingshot (SSH) (Niwa et al., 2002) and Chronophin (Gohla et al., 2005; Wiggan et al., 2005) phosphatases dephosphorylate and reactivate cofilin. Intriguingly, LIMK itself is positively regulated via phosphorylation by Rho kinase (ROCK) (Edwards et al., 1999; Ohashi et al., 2000), a Rho effector that is activated in response to stimulation with MAIs (Dergham et al., 2002; Niederost et al., 2002; Fournier et al., 2003). Members of the p21-activated kinase (PAK) protein family, downstream effectors of Rac GTPase, can also regulate phosphorylation of LIMK and SSH. Phosphorylation of SSH1 can reduce its activity toward downstream substrates by enhancing its association with 14-3-3 adaptor proteins (Nagata-Ohashi et al., 2004).
To study the impact of MAIs on cofilin phosphorylation, we examined cellular responses to Nogo-66, a potent inhibitory fragment of Nogo-A (GrandPre et al., 2000). We found that cofilin phosphorylation is regulated in neurons treated with Nogo-66 via sequential activation of LIMK1 and SSH1. Treatment of dorsal root ganglion (DRG) neurons with dominant-negative LIMK1 (DN-LIMK1) protects neurons from myelin-dependent outgrowth inhibition, suggesting that phosphorylation and inactivation of cofilin is necessary for inhibitory responses. Catalytically inactive SSH1 blocks Nogo-dependent cofilin dephosphorylation and reactivation. Together, these findings suggest that LIMK1-mediated phosphorylation of cofilin is a critical step in MAI-dependent neuronal inhibition. We speculate that SSH1-mediated dephosphorylation of cofilin may play a role in the maintenance of an inhibitory neuronal phenotype.
Materials and Methods
Plasmids and antibodies. Human wild-type (WT) SSH1 (WTSSH1) was amplified by PCR using fully sequenced full-length human cDNAs from the mammalian gene collection (Open Biosystems, Huntsville, AL) as template and ligated into pcDNA3.1V5His (Invitrogen, Burlingame, CA). Phosphatase inactive SSH1 [SSH1(CS)] was generated by site-directed mutagenesis using the ExSite PCR-based site-directed mutagenesis kit (Stratagene, La Jolla, CA). Cysteine 393 of SSH1 was mutated to serine generating a catalytically inactive mutant as described previously (Niwa et al., 2002) using human SSH1 pcDNA3.1V5His as template. Dominant-negative LIMK1 (DN-LIMK1) was amplified from pSinRep5DN-LIMK1 (Aizawa et al., 2001) (generously provided by Dr. Hiroyuki Aizawa, Johns Hopkins University, Baltimore, MD) and ligated into pcDNA3.1V5His. DN-LIMK1 is a mutant in which Thr-508 is substituted with Valine, thereby preventing LIMK1 phosphorylation and activation in response to upstream regulators such as Rho kinase and p21-activated kinase. To construct herpes simplex virus HSVWTSSH1, HSVSSH1(CS), and HSVDN-LIMK1, the corresponding cDNA with the V5 and His tags was amplified by PCR and ligated into pHSVPrPUC.
All antibodies and probes are from commercial sources: antiphospho-LIMK and anti-phospho-cofilin (Cell Signaling Technology, Pickering, Ontario), anti-cofilin (Cytoskeleton, Denver, CO), anti-V5 (Sigma-Aldrich, St. Louis, MO), anti-actin (MP Biomedicals, Aurora, OH), anti-BIII tubulin (Covance, Berkeley, CA), and rhodamine-phalloidin (Invitrogen, Eugene, OR).
Preparation of myelin and recombinant proteins. To purify alkaline-phosphatase (AP)-conjugated Nogo-66 (AP-Nogo-66) or AP, conditioned medium was collected from human embryonic kidney 293A (HEK293A) cells stably transfected with pcDNA3.1AP-Nogo-66-His (generously provided by Dr. Stephen Strittmatter, Yale University, New Haven, CT) or HEK293T cells transiently transfected with pcDNA-AP-His. Secreted protein was purified by Ni2+ affinity chromatography (Nakamura et al., 1998). Glutathione S-transferase (GST)-Nogo-66 (GST-Nogo-66) and GST-Reticulon1–66 was expressed in Escherichia coli and purified on glutathione-Sepharose (Amersham Biosciences, Piscataway, NJ) as described previously (GrandPre et al., 2000). For all cellular treatments, AP-Nogo-66-His (8 nm) or AP (8 nm) were preaggregated with 100 ng/ml anti-human AP (Niederost et al., 2002). GST-Nogo-66 was added at a final concentration of 50 nm. Myelin extracts were prepared from the bovine brain as described previously (Igarashi et al., 1993). After clarification of the myelin extract by centrifugation at 400,000 × g, the detergent was removed by dialyzing with PBS. For growth cone collapse assays, myelin was briefly sonicated before application.
Preparation of recombinant viral preparations. Recombinant HSV plasmids were transfected into 2-2 cells (3 × 105) with lipofectamine and superinfected with 5dl1.2 HSV helper virus after 1 d of incubation. The recombinant virus was amplified through three passages and stored at –80°. Helper virus stocks and 2-2 cells were generously provided by Dr. Rachel Neve (Harvard Medical School, Cambridge, MA).
Primary cell culture, growth cone collapse, and neurite outgrowth assays. To prepare postnatal day 8 (P8) to P10 rat cerebellar neurons, cerebella were dissected, dissociated with trypsin and mechanical trituration, and cultured for 18 h on poly-l-lysine-coated dishes in Sato media (DMEM with 0.1 μg/ml l-thyroxine, 0.08 μg/ml tri-iodothyronine, and N2 supplement) before ligand treatment. Rat DRGs were dissected at P4 and plated as dissociated cells after dissociation by sequential treatment with collagenase (1 μg/ml) and trypsin/EDTA (0.25%) followed by mechanical trituration. DRGs were grown in DRG outgrowth media (F-12, 10% fetal bovine serum, penicillin/streptomycin, 1% glutamine, 50 ng/ml nerve growth factor).
For growth cone collapse experiments, embryonic day 13 (E13) chick DRG explants were cultured in a minimal amount of DRG outgrowth media in four-well chamber slides sequentially coated with poly-l-lysine and laminin until they adhered to the substrate. Once explants adhered, additional medium supplemented with recombinant HSV preparations was added. Twenty-four hours later, infected explants were treated for 60 min with myelin and fixed in 4% paraformaldehyde (PFA), 0.1 m phosphate buffer, and 20% sucrose. Explants were double stained with rhodamine-phalloidin, and anti-V5 antibody was detected with an FITC-conjugated secondary antibody. Control HSV-infected explants were stained with rhodamine phalloidin and anti-β-III tubulin. Growth cone collapse was assayed by established criteria (Luo et al., 1993).
For neurite outgrowth assays, Nogo or myelin was dried on poly-l-lysine-coated wells of 96-well plates. Substrates were washed and coated with 10 μg/ml laminin for 1 h. Dissociated E13 chick DRG neurons were grown in the presence of virus for 18 h and double stained with anti-βIII tubulin and anti-V5 antibody. Neurite outgrowth lengths per cell were assessed using Image J software as described previously (http://rsb.info.nih.gov/ij/) (Fournier et al., 2003).
Assessment of cofilin phosphorylation. PC12 cells were differentiated in RPMI/1% BSA/50 ng/ml NGF for 24 h before treatment with recombinant proteins. Cerebellar neurons were grown for 18 h before treatment. Cell lysates were analyzed by 15% SDS-PAGE and immunoblotting with anti-cofilin, anti-phospho-cofilin, anti-actin, or anti-V5 antibodies. Phospho-cofilin levels were analyzed by densitometry and were normalized to total cofilin levels for each treatment.
For immunohistochemical analysis, cerebellar neurons were plated on eight-well chamber slides for 18 h before treatment. Cells were fixed in 4% PFA/0.1 m PO4 buffer/20% sucrose for 30 min and stained with anti-cofilin or anti-phospho-cofilin antibodies. Fluorescence intensity was analyzed using Image J image analysis program by outlining the entire cell body and neurite and calculating average fluorescence intensity per neuron. Independent analysis was performed separately outlining cell bodies versus neurites. False color images were generating by creating a stack of images in ImageJ and false coloring the images together using the fire look-up table. Brightness and contrast of the images were adjusted in the stack to ensure that each image was adjusted equally. The brightness and contrast were adjusted to highlight intensity differences along the length of the neurite.
LIMK1 and SSH1 phosphorylation studies. To analyze SSH1-actin association, PC12 cells were transfected with pcDNA3.1SSH1V5His for 48 h using lipofectamine 2000 (Invitrogen, Burlingame, CA). For the final 24 h of transfection, PC12 cells were differentiated in RPMI/1% BSA/50 ng/ml NGF. Transfected cells were treated with AP-Nogo-66-His, washed, and lysed in radioimmunoprecipitation assay buffer (20 mm HEPES, 150 mm NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% Triton X-100) with a protease inhibitor cocktail (Roche, Mannheim, Germany) and phosphatase inhibitors. Cell lysates were precleared for 30 min with protein A/G-agarose (Santa Cruz Biotechnology, Santa Cruz, CA), and V5-tagged SSH1 was immunoprecipitated with V5-agarose (Sigma-Aldrich). Immune complexes were analyzed by SDS-PAGE and immunoblotting with anti-V5 or anti-actin antibodies. LIMK phosphorylation was assessed in nontransfected PC12 cells with an anti-phospho-LIMK antibody that recognizes the phosphorylated forms of LIMK1 and LIMK2.
Reverse transcription-PCR. For reverse transcription (RT)-PCR experiments, total RNA from PC12 cells or from total cerebellum, cultured cerebellar neurons, total brain, or DRGs from P8 rat pups was prepared using an RNeasy kit (Qiagen, Mississauga, Ontario, Canada). For NgR, p75NTR, and LINGO analyses, the RNA was treated with DNaseI (Amersham Biosciences, Baie d'Urfe, Quebec) for 1 h at 30°C (50 mm Tris acetate, pH 8.4, 75 mm potassium acetate, 8 mm magnesium acetate), phenol/chloroform was extracted, and ethanol was precipitated. cDNA was prepared with a Thermoscript Reverse Transcriptase kit (Invitrogen, Burlington, Ontario). Primers for PCR detection were designed to span an intron boundary with the exception of LINGO-1. SSH1F 5′atc ggg atg gtc ctt cga ct, SSH1R 5′gtc cat ctg gcc caa gat gag; LIMK1F 5′tgg agt cag agg gtc agc tt, LIMK1R 5′acc acc acg ttc ctg ttc tc; ChronophinF 5′gct acg acg agc att ttt cc, ChronophinR 5′gtc ctg agg ctg tct cca ct; TESK1F 5′agg tgt gtt tca cc gaga caa, TESK1R 5′ cga tga gct cac aga gga ca; NgRF 5′atg aag agg gcg tcc tcc, NgRR 5′gca ttt gag tgc agc cac; p75F 5′cga caa cct cat ccc tgt ct, p75R 5′ tcg ctg tgg agt ttt tct cc; and Lingo F 5′atc tcc cac tgg ccc tact tt Lingo R 5′ ttg cca gag aca ttg agc ac.
Results
Nogo regulates cofilin phosphorylation
MAIs increase Rho activation in responsive neurons; however, the intracellular substrates transducing Rho signals to the actin cytoskeleton have not been fully defined. To examine whether Nogo regulates the activity of cofilin, an important regulator of actin dynamics, we assessed the phosphorylation profile of cofilin in response to stimulation with Nogo-66, an inhibitory fragment of Nogo-A (Grand-Pre et al., 2000). Differentiated PC12 cells were treated with AP or AP-Nogo-66 for 30 s to 60 min, a time course over which Nogo-66 or myelin stimulate significant growth cone collapse in primary neurons. PC12 cells express NgR, p75NTR, and LINGO1 (Fig. S1, available at www.jneurosci.org as supplemental material) and activate Rho in response to Nogo stimulation (Fournier et al., 2003), supporting their suitability as a model system for MAI signaling. AP-Nogo-66 stimulation leads to a modest but significant increase in cofilin phosphorylation 30 s after AP-Nogo-66 treatment followed by a reduction in cofilin phosphorylation to ∼40% of control levels by 30 min (Fig. 1A,C). The specificity of the Nogo-66 effect is supported by the similar effect of bacterially purified GST-Nogo-66 on the cofilin phosphorylation profile and the failure of the corresponding region of reticulon-1 (GST-RTN1–66) to modify cofilin phosphorylation (GrandPre et al., 2000) (Fig. 1A,C). The efficacy of the phosphocofilin antibody was confirmed by verifying that treatment of cerebellar neurons with calyculin, a serine-threonine phosphatase inhibitor, increases phospho-cofilin levels (Fig. 1B,D).
Figure 1.

Nogo-66 regulates cofilin phosphorylation. A, Total cofilin and phospho-cofilin levels in PC12 cells treated with AP-Nogo-66, GST-Nogo-66, AP, or GST-RTN1–66. B, Total cofilin and phospho-cofilin levels in P8 rat cerebellar neurons treated with AP-Nogo-66, AP, calyculin (Cal), or myelin. C, D, Quantification of cofilin phosphorylation. Bands were analyzed by densitometry and normalized to total cofilin levels for each treatment. Values are expressed as a percentage of control (0 min) ± SEM. Determinations are from a minimum of three experiments, each performed in duplicate. *p < 0.01 compared with controls by Student's t test. Error bars represent SEM.
To examine the regulation of cofilin phosphorylation in primary neurons, cerebellar neurons were treated with AP-Nogo-66. In cerebellar neurons, cofilin phosphorylation is unaffected 30 s after Nogo treatment; however, it is dephosphorylated commencing by 30 min (Fig. 1B,D). Cerebellar neurons were also stimulated with myelin membranes, which contain all three MAIs to assess the effect on cofilin phosphorylation. Results, to date, demonstrate that multiple MAIs, including FcMAG, Nogo-66, or OMgp, stimulate common intracellular signaling pathways. Myelin also stimulates cofilin dephosphorylation in dissociated cerebellar neurons (Fig. 1B). Together, these results demonstrate that MAIs affect cofilin phosphorylation in PC12 cells and in primary neurons.
To assess the subcellular regulation of cofilin phosphorylation, dissociated cerebellar neurons were stained with antiphospho-cofilin antibody (Fig. 2). The suitability of the total cofilin and phospho-cofilin antibodies for immunofluorescence analysis was determined by Western blotting (Fig. 2C). The cofilin antibody recognizes a single band at 18 kDa in cerebellar lysates. The phospho-cofilin antibody recognizes a major band at 18 kDa, which is regulated by treatment with Calyculin. Immunofluorescence analysis indicates that cofilin phosphorylation is regulated in both the cell body and in the neurite/growth cone compartment of dissociated cerebellar neurons in response to bath application of AP-Nogo-66 (Fig. 2A). Analyses of phosphocofilin fluorescence intensity over the surface of dissociated neurons revealed a 60% decrease in cofilin phosphorylation after 60 min of AP-Nogo-66 treatment (Fig. 2D), which could be attributed to changes in the cell body and neurite compartments (Fig. 2E); however, concentrations of phospho-cofilin immunofluorescence could be seen decorating the tips of cerebellar processes 30 s after Nogo stimulation but were mostly absent by 60 min (Fig. 2B). Total cofilin levels were not affected by AP-Nogo-66 treatment (Fig. 2A,D).
Figure 2.

Nogo induces cofilin dephosphorylation in cell bodies and neurites. A, Total cofilin and phospho-cofilin levels in P8 rat cerebellar neurons treated with AP-Nogo-66 or AP. Scale bar, 200 μm. B, Higher magnification view of cerebellar neurons treated with AP-Nogo-66 for 0, 0.5, or 60 min. Each image is accompanied by a false color image [colder colors (blue) representing low intensity to warmer colors (red) representing high intensity] to demonstrate concentrations of cofilin phosphorylation at neurite tips only 0.5 min after AP-Nogo-66 treatment. Green arrowheads signify neurite tips. C, Western blot of nonstimulated (–) or calyculin-stimulated (+) cerebellar lysates probed with anti-cofilin (Cof) or anti-phospho-cofilin antibody. D, E, Quantification of total cofilin or phospho-cofilin fluorescence intensity over cell bodies and neurites together (D) or independently (E) using ImageJ analysis software. Fluorescence intensity was normalized to control levels (0 min) for each experiment. Determinations are from three experiments with a minimum of 50 neurons per data point. *p < 0.01 compared with controls by Student's t test. Error bars represent SEM.
Slingshot and LIMK are expressed in neurons
When considering kinases and phosphatases that could be responsible for Nogo-dependent changes in cofilin phosphorylation, we focused on LIM kinase and SSH phosphatase, because both proteins have been previously implicated in regulating growth cone motility (Endo et al., 2003). We performed RT-PCR to evaluate the expression of these regulators in cell types that have been commonly used to study the inhibitory effects of MAIs. Expression was analyzed in PC12 cells and in the brain, cerebellar neurons, and dorsal root ganglion neurons from a P8 rat, a developmental stage when neurons from cerebella and dorsal root ganglia are sensitive to Nogo stimulation. We found that SSH1 and LIMK1 are present in these tissues (Fig. S1, available at www.jneurosci.org as supplemental material).
Effect of LIMK1 and SSH1 on growth cone collapse and neurite outgrowth
To study the functional significance of LIMK1 and SSH1 activity in neurons treated with MAIs, we generated recombinant HSV preparations of DN-LIMK1, SSH1(CS), and WTSSH1 with the goal of examining their effects on myelin-dependent growth cone collapse and neurite outgrowth inhibition from DRG neurons. Robust viral infection in cell bodies and neurites was validated by analyzing expression of V5-tagged proteins in DRG explants, axons, and growth cones for each virus (Fig. 3A,B).
Figure 3.

DN-LIMK1 blocks myelin-dependent growth cone collapse. A, Detection of recombinant proteins in a low-magnification view of DRG explants by immunofluorescence in HSV-, V5DN-LIMK1-, V5WTSSH1-, or V5SSH1(CS)-infected E13 chick DRG explants. Scale bar, 100 μm. B, Detection of recombinant protein in a high-magnification view of axons and growth cones from a V5DN-LIMK1-infected DRG explant. Scale bar, 40 μm. C, Growth cone collapse in nonstimulated (control) or AP-Nogo-66- or myelin-stimulated DRG explants infected with HSV, V5DN-LIMK1, V5WTSSH1, or V5SSH1(CS). Arrows indicate spread growth cones. Arrowheads indicate collapsed growth cones. Scalebar, 40 μm. D, Quantification of growth cone collapse from DRG explants infected with HSV, V5DN-LIMK1, V5WTSSH1, or V5SSH1(CS). Determinations are mean ± SEM from four experiments, each performed in duplicate. *p < 0.01 compared with HSV infection by Student's t test. E, F, Quantification of growth cone collapse from noninfected DRG explants (Control) or explants infected with HSV or V5DN-LIMK1 and treated with AP or AP-Nogo-66 (E) or myelin (F). Determinations are mean ± SEM from at least three experiments, each performed in duplicate. *p < 0.05 compared with no treatment by Student's t test. Error bars represent SEM.
Infection with SSH1(CS) dramatically limits neurite outgrowth from E13 chick DRG explants (data not shown). To circumvent the inhibitory outgrowth effects of SSH1(CS), DRG explants were infected 12 h after plating to allow DRG neurites to extend before infection. Using this experimental paradigm, SSH1(CS) causes robust growth cone collapse compared with control explants infected with HSVGFP or HSV control virus (Fig. 3C,D). This varies from the well spread growth cone phenotype reported in E7 chick DRG neurons infected with SSH1(CS) (Endo et al., 2003), suggesting that the neuronal response to SSH1(CS) may be age dependent. Infection with WT-SSH1 also enhances growth cone collapse (Fig. 3C), indicating that regulated SSH levels are critical for the elaboration of E13 DRG growth cones.
Infection of E13 DRG explants with DN-LIMK1 has no effect on growth cone collapse (Fig. 3C,D); therefore, we examined their sensitivity to AP-Nogo-66-dependent growth cone collapse. DRG neurons infected with DN-LIMK1 are significantly less sensitive to the growth cone collapse effect of AP-Nogo-66 (Fig. 3C,E). Additionally, explants infected with DN-LIMK1 are significantly less sensitive to myelin-dependent growth cone collapse (Fig. 3C,F), indicating that DN-LIMK1 attenuates the inhibitory responses of multiple myelin-associated inhibitors.
To examine the effects of LIMK1 and SSH1 on myelin-dependent inhibition of neurite outgrowth, dissociated E13 DRG neurons were infected with HSVDN-LIMK1, HSVSSH1(CS), or HSVWTSSH1 at the time of seeding on control or myelin substrates (Fig. 4). By analyzing infection with HSVGFP viral preparations, we estimate that recombinant proteins are detectable ∼6 h after plating (data not shown). Consistent with the growth cone collapse assay, infection of dissociated DRG neurons with DN-LIMK1 significantly attenuates myelin-dependent outgrowth inhibition (Fig. 4A,B). There is no significant difference in neurite outgrowth on myelin substrates for neurons infected with DN-LIMK1. Overexpression or disruption of SSH activity differentially affects basal neurite outgrowth by promoting (WT-SSH1) or inhibiting [SSH1(CS)] DRG neurite outgrowth, respectively. On the myelin substrate, neurite extension from WT-SSH1-infected DRG neurons is improved compared with HSV-infected neurons; however, when considering the effect of WT-SSH-1 on basal outgrowth, it appears that WT-SSH-1-infected neurons remain sensitive to myelin inhibition.
Figure 4.

DN-LIMK1 circumvents myelin-dependent neurite outgrowth inhibition. A, Neurite outgrowth from E13 chick dissociated DRG neurons infected with HSV, V5DN-LIMK1, V5SSH1(CS), or V5WTSSH1 on laminin (control) or myelin substrates. Scale bar, 100 μm. B, Quantification of neurite outgrowth/cell for each experiment. Determinations are from a minimum of three experiments. *p < 0.01 by Student's t test compared with 0 μg of treatment for each virus. Error bars represent SEM.
Blockade of LIMK1 and SSH1 prevents Nogo-dependent changes in cofilin phosphorylation
To determine whether LIMK1 activation is responsible for the early Nogo-dependent increase in cofilin phosphorylation, PC12 cells transfected with DN-LIMK1 were studied. Introduction of DN-LIMK1 blocks increased cofilin phosphorylation 30 s after Nogo treatment (Fig. 5A,C), indicating that LIMK activity is necessary for the early cofilin response. Additionally, introduction of SSH1(CS) into PC12 cells inhibits cofilin dephosphorylation 60 min after Nogo treatment (Fig. 5A,C). AP treatment has no effect on cofilin phosphorylation in PC12 cells transfected with DN-LIMK1 or SSH1(CS) (data not shown). Together, this is consistent with a model whereby LIMK1 and SSH1 are sequentially activated in response to Nogo resulting in a transient increase and subsequent decrease in cofilin phosphorylation.
Figure 5.
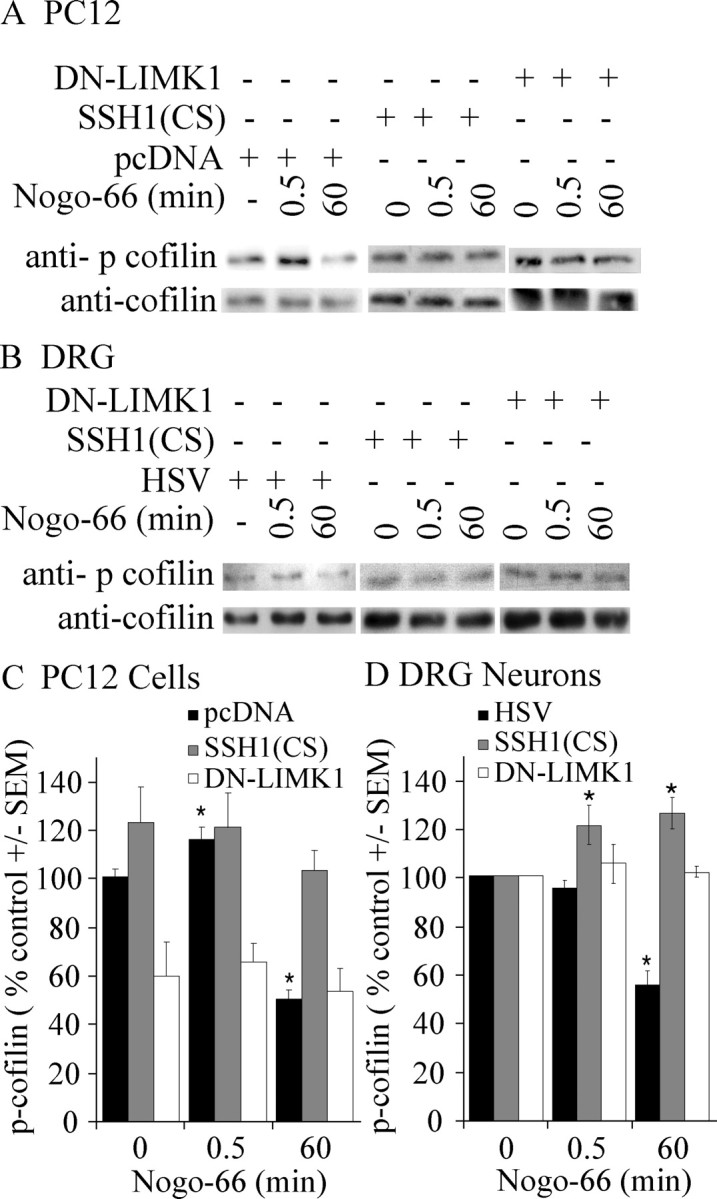
DN-LIMK1 and SSH1(CS) attenuate Nogo-dependent changes in cofilin phosphorylation. A, B, Phospho-cofilin levels in AP-Nogo-66-stimulated PC12 cells transfected with pcDNA, V5DN-LIMK1, or SSH1 (CS) (A) or dissociated P4 rat DRG neurons infected with HSV, HSVV5SSH1(CS), or HSVV5DN-LIMK1 (B). C, D, Quantification of phospho-cofilin levels in AP-Nogo-66 stimulated PC12 cells or DRG neurons. For DRG neurons, values were normalized to 0 min treatment for each virus. Determinations are from a minimum of three experiments. *p < 0.01 by Student's t test compared with 0 μg of treatment. Error bars represent SEM.
To determine whether this mechanism is conserved in primary neurons, Nogo responses were examined in dissociated rat DRGs infected with HSV, DN-LIMK1, or SSH1(CS) (Fig. 5B,D). In DRGs, early changes in cofilin phosphorylation are only detected in the presence of SSH1(CS), and subsequent cofilin dephosphorylation is blocked by this dominant-negative virus. In both PC12 cells and DRG neurons, cofilin dephosphorylation 60 min after Nogo treatment is also attenuated by introducing DN-LIMK1 raising the possibility that SSH activation is dependent on LIMK.
Nogo-66 sequentially activates LIMK1 and SSH1
The ability of DN-LIMK1 to block myelin-dependent inhibition suggests that LIMK1 may be directly activated by MAI stimulation. To examine the activity of LIMK1, we studied its phosphorylation state in response to Nogo-66 (Fig. 6). Analysis was performed in PC12 cells with an anti-phospho-LIMK antibody, which recognizes both LIMK1 and LIMK2. LIMK phosphorylation enhances its activity toward downstream substrates, including cofilin; therefore, we hypothesized that MAIs may positively regulate LIMK phosphorylation. AP-Nogo-66 rapidly and transiently enhances LIMK phosphorylation (Fig. 6A,C). LIMK is phosphorylated 30 s and 10 min after AP-Nogo-66 stimulation and returns to basal levels by 60 min.
Figure 6.

SSH1 and LIMK are regulated by Nogo-66 in a ROCK-dependent manner. A, Phospho-LIMK levels in PC12 cells stimulated with Nogo-66. B, V5-SSH and actin content in immune complexes isolated with V5 agarose after Nogo-66 stimulation of PC12 cells. C, Quantification of phospho-LIMK levels in response to AP-Nogo-66 stimulation. Values were normalized to total actin in the lysates. D, Quantification of actin content in V5-SSH1 immune complexes in response to AP-Nogo-66. Actin levels were normalized to the V5SSH1 content for each immunoprecipitate. E, Phospho-cofilin levels in PC12 cells treated with AP-Nogo-66. For A, B, and E, dependence on ROCK was assessed with a 30 min pretreatment of the cells with 10 μm Y-27632. F, Quantification of phospho-cofilin levels in Y-27632-treated PC12 cells and in PC12 cells treated with AP-Nogo-66 after a 30 min pretreatment with 10 μm Y-27632. IP, Immunoprecipitation. Error bars represent SEM.
We were also interested in examining the regulation of SSH1 in response to Nogo stimulation. V5-SSH1 was immunoprecipitated from transfected PC12 cells treated with AP-Nogo-66, and immune complexes were analyzed for actin content (Fig. 6B,D). Actin weakly associates with V5-SSH1 in basal conditions and is recruited to V5-SSH1 by 10 min after AP-Nogo-66 stimulation (Fig. 6B,D). Increased SSH1-actin association is consistent with the idea that Nogo activates SSH, based on a previous report demonstrating that actin association enhances SSH1 phosphatase activity by ∼10-fold (Nagata-Ohashi et al., 2004). The time course of this enhanced association is consistent with the time course of Nogo-dependent cofilin dephosphorylation.
Nogo signaling to LIMK1, SSH1, and cofilin is ROCK dependent
We also studied the contribution of ROCK to Nogo-dependent changes in LIMK1, SSH1, and cofilin activity (Fig. 6). LIMK1 is a known substrate for ROCK, and experiments demonstrating that the ROCK inhibitor Y-27632 attenuates myelin-dependent inhibition suggest that ROCK is activated by MAIs. PC12 cells were pretreated for 30 min with Y-27632 before examining the effects of Nogo-66 stimulation. Both LIMK1 phosphorylation (Fig. 6A,C) and enhanced SSH1-actin association (Fig. 6B,D) are blocked by Y-27632 pretreatment, indicating that activation of both molecules is downstream of ROCK.
Pretreatment with Y-27632 also reduces basal cofilin phosphorylation in PC12 cells (Fig. 6D,F), likely resulting from inhibition of LIMK. In response to Nogo-66, the Nogo-dependent increase in cofilin phosphorylation is blocked 30 s after stimulation, as would be expected by blockade of LIMK1. Intriguingly, the late Nogo-dependent decrease in cofilin phosphorylation is not only blocked but converted to a robust increase in the presence of Y-27632 (Fig. 6D,F). This late increase in cofilin phosphorylation could be attributable to complete Y-27632-mediated blockade of SSH1 activity combined with persistent low-level LIMK activity or activity of other cofilin-directed kinases such as TESK. Consistent with the idea that other cofilin-directed kinases and phosphatases may contribute to these effects, we found that TESK1 and chronophin are expressed in tissues with myelin-responsive neurons (Fig. S1, available at www.jneurosci.org as supplemental material).
Discussion
Together, these data suggest a model whereby Nogo-66 stimulation sequentially stimulates LIMK1 and SSH1 activity in a ROCK-dependent manner. Activation of LIMK1 and SSH1 mediate predicted changes in cofilin phosphorylation in PC12 cells. Activation of LIMK1 is necessary for myelin-dependent inhibition as introduction of DN-LIMK circumvents myelin-dependent inhibition in DRG neurons. Intriguingly, SSH1 activation, an activity that is generally associated with increases in neurite outgrowth, is enhanced subsequent to LIMK activation, suggesting that the role of SSH1 is modulated by the intracellular signaling context. We speculate that SSH1-mediated cofilin dephosphorylation may mediate maintenance of an inhibited neuronal phenotype.
LIMK1 is a key regulator of neuronal inhibition
LIMK1 activation has been implicated previously in growth cone collapse in response to the repulsive guidance cue Sema3A. Aizawa et al. (2001) demonstrated that DN-LIMK1 can protect DRG neurons from Sema3A-dependent growth cone collapse. The idea that LIMK1 may serve as a convergent mediator of inhibitory signals from multiple repulsive guidance cues and growth inhibitory proteins is attractive, because LIMK1 is a substrate for both Rho kinase and p21-activated kinase (Edwards et al., 1999; Ohashi et al., 2000). In the case of Nogo stimulation, we demonstrate that LIMK1 phosphorylation is dependent on ROCK, a molecule that has been clearly implicated in MAI signaling both in vitro and in vivo (Dergham et al., 2002; Niederost et al., 2002; Fournier et al., 2003). In contrast, Sema3A signaling is attenuated by introduction of dominant-negative Rac, suggesting a PAK-mediated event (Jin and Strittmatter, 1997). A previous study demonstrated that E6 chick motor neurons infected with constitutively active Rac1, which stimulates the PAK-LIMK signaling axis, are less sensitive to myelin-dependent growth cone collapse and neurite outgrowth inhibition (Kuhn et al., 1999). One possible explanation for this apparent discrepancy is that activation of additional Rac effectors mediates this protective effect. Additional differences in the developmental stage and neuronal cell type used in the two studies are likely confounding factors. For example cAMP levels are downregulated between E6 and E13, and this is known to impact neuronal responses to multiple extracellular cues (Song et al., 1998; Cai et al., 2001; Qiu et al., 2002).
Notably, the time course of LIMK phosphorylation correlates with relatively modest increases in cofilin phosphorylation. Although an obvious possibility is that local changes in cofilin phosphorylation are difficult to detect in biochemical assays, it is also possible that coincident phosphatase activity results in a modest net change in cofilin phosphorylation. However, if this is the case, it is surprising that Nogo stimulation of PC12 cells infected with DN-LIMK does not stimulate cofilin dephosphorylation at early time points. Another possibility is that LIMK1 acts on additional downstream substrates to mediate its effects. This idea is supported by the observation that diminished growth cone motility in neurons overexpressing LIMK1 is not completely rescued by coexpression of a phospho-mutant cofilin (S3A) or of SSH1 (Endo et al., 2003).
SSH1 activity is regulated by Nogo
The increased association between SSH1 and actin, together with the ability of SSH1(CS) to block Nogo-dependent cofilin dephosphorylation, strongly supports a model in which SSH1 activity is enhanced in response to Nogo-66. It is reasonable to speculate that Nogo may promote SSH1 dephosphorylation, consequently enhancing its translocation from 14-3-3 sequestering proteins to actin. To date, SSH1 has been shown to associate with 14-3-3β, γ, and ζ (Nagata-Ohashi et al., 2004; Soosairajah et al., 2005), and this association is enhanced by SSH1 phosphorylation at Ser-937 and Ser-978. We were unable to detect reproducible changes in V5-SSH1 phosphorylation with an anti-phospho-serine antibody (data not shown); however, this question should ultimately be reexamined with a specific phospho-SSH1 antibody. PAK4, an effector of Rac GTPase, is the only known SSH1-directed kinase. Intriguingly, Rac activity is reportedly decreased in cerebellar granule cells treated with MAIs (Niederost et al., 2002), raising the possibility that down-regulation of the Rac pathway could reduce PAK4-mediated phosphorylation of SSH. The predicted outcome would be reduced SSH affinity for 14-3-3 adapter proteins and an increased concentration of free SSH available for actin binding. A similar mechanism has been demonstrated to mediate responses to neuregulin-1β in the MCF-7 breast carcinoma cell line (Nagata-Ohashi et al., 2004). However, it is equally plausible that SSH1 recruitment to actin is regulated by an unknown mechanism that is independent of SSH1 phosphorylation.
In addition to the known phosphatase activity of SSH1 on cofilin, LIMK1 has also been reported to be an SSH1 substrate (Soosairajah et al., 2005). The time course of SSH1-actin association and of LIMK1 dephosphorylation is consistent with a model whereby strongly activated SSH1 may dephosphorylate and inactivate LIMK1 by 60 min after Nogo treatment.
How does regulation of cofilin mediate myelin-dependent inhibition?
How disruption of cofilin phosphorylation mediates neurite outgrowth inhibition remains an open question. It seems likely that disrupted cofilin phosphorylation–dephosphorylation cycling ultimately disrupts outgrowth. This is supported by a report that phosphorylation of cofilin by LIMK1 is necessary but not sufficient for Sem3A-dependent growth cone collapse (Aizawa et al., 2001). The authors suggest that other candidate proteins such as myosin or a cofilin phosphatase may be critical for Sema3A-dependent growth cone collapse. Cofilin cycling may be required for properly regulated actin polymerization to mediate neurite outgrowth. Activation of cofilin by phosphatases is required to create new filamentous F-actin ends and to generate actin monomers, whereas the phosphorylated form of cofilin could stimulate actin turnover by promoting the release of free actin and cofilin from actin-cofilin complexes.
If cofilin cycling is critical, it is particularly interesting that SSH1(CS) inhibits neurite outgrowth, whereas DNLIMK does not have a parallel effect. One might expect that both interventions would block outgrowth. We are interested in the possibility that other kinases, including TES kinases (Toshima et al., 2001) may be capable of regulating cofilin phosphorylation in neurons. On a similar note, the function of chronophin phosphatase (Gohla et al., 2005; Wiggan et al., 2005) in neurons is unknown. Intriguingly, both of these transcripts are present in DRGs and cerebellum (Fig. S1, available at www.jneurosci.org as supplemental material).
A model of Nogo-dependent regulation of Cofilin
We propose a model whereby Nogo-dependent activation of ROCK stimulates sequential activation of LIMK1 and SSH1 (Fig. S2, available at www.jneurosci.org as supplemental material). Initial activation of LIMK1 is rapid, occurring by 30 s after Nogo stimulation, and is mediated by ROCK-dependent phosphorylation. Phosphorylation and activation of LIMK1 mediates enhanced cofilin phosphorylation, consequently inactivating the molecule. This early phase is critical to myelin-dependent inhibition. Subsequent to LIMK activation, SSH1 activity is enhanced ∼10 min after Nogo stimulation. The detailed mechanism of SSH1 activation remains to be determined; however, translocation to the actin cytoskeleton may be involved. Activated SSH1 also targets cofilin leading to its dephosphorylation and reactivation. In control neurons, WT-SSH1 enhances neurite outgrowth by severing actin and creating new filament ends and by generating new actin monomers from the pointed end of the actin filament. In control conditions, neurite extension is then favored through enhanced actin elongation through actin-profilin complexes. We speculate that in the context of Nogo signaling, early activation of LIMK may inhibit the activity of actin polymerization factors such as profilin, perhaps by favoring the formation of actin-thymosin complexes. Subsequent reactivation of the actin depolymerization and severing activity of cofilin without coincident enhancement of actin polymerization strategies may mediate a persistent inhibited neuronal phenotype.
Footnotes
This study was supported by a grant from the Canadian Institutes of Health Research. We thank Drs. Phil Barker, Wayne Sossin, and Tim Kennedy for comments and suggestions on this manuscript, Dr. Rachel Neve for providing herpes simplex virus reagents, and Dr. Hiroyuki Aizawa for providing LIM kinase reagents.
Correspondence should be addressed to Alyson Fournier, Montreal Neurological Institute, BT-109, 3801 Rue University, Montreal, Quebec, Canada H3A 2B4. E-mail: alyson.fournier@mcgill.ca.
DOI:10.1523/JNEUROSCI.2806-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/261006-10$15.00/0
S.H.-K.H. and G.B.F. contributed equally to this work.
References
- Aizawa H, Wakatsuki S, Ishii A, Moriyama K, Sasaki Y, Ohashi K, Sekine-Aizawa Y, Sehara-Fujisawa A, Mizuno K, Goshima Y, Yahara I (2001) Phosphorylation of cofilin by LIM-kinase is necessary for semaphorin 3A-induced growth cone collapse. Nat Neurosci 4: 367–373. [DOI] [PubMed] [Google Scholar]
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P (1998) Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393: 805–809. [DOI] [PubMed] [Google Scholar]
- Bamburg JR, Wiggan OP (2002) ADF/cofilin and actin dynamics in disease. Trends Cell Biol 12: 598–605. [DOI] [PubMed] [Google Scholar]
- Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT (2001) Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci 21: 4731–4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L (2002) Rho signaling pathway targeted to promote spinal cord repair. J Neurosci 22: 6570–6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN (1999) Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol 1: 253–259. [DOI] [PubMed] [Google Scholar]
- Endo M, Ohashi K, Sasaki Y, Goshima Y, Niwa R, Uemura T, Mizuno K (2003) Control of growth cone motility and morphology by LIM kinase and Slingshot via phosphorylation and dephosphorylation of cofilin. J Neurosci 23: 2527–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, GrandPre T, Strittmatter SM (2001) Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 409: 341–346. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Takizawa BT, Strittmatter SM (2003) Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci 23: 1416–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohla A, Birkenfeld J, Bokoch GM (2005) Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat Cell Biol 7: 21–29. [DOI] [PubMed] [Google Scholar]
- GrandPre T, Nakamura F, Vartanian T, Strittmatter SM (2000) Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403: 439–444. [DOI] [PubMed] [Google Scholar]
- Hall A (1998) Rho GTPases and the actin cytoskeleton. Science 279: 509–514. [DOI] [PubMed] [Google Scholar]
- He Z, Koprivica V (2004) The Nogo signaling pathway for regeneration block. Annu Rev Neurosci 27: 341–368. [DOI] [PubMed] [Google Scholar]
- Igarashi M, Strittmatter SM, Vartanian T, Fishman MC (1993) Mediation by G proteins of signals that cause collapse of growth cones. Science 259: 77–79. [DOI] [PubMed] [Google Scholar]
- Jin Z, Strittmatter SM (1997) Rac1 mediates collapsin-1-induced growth cone collapse. J Neurosci 17: 6256–6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn TB, Brown MD, Wilcox CL, Raper JA, Bamburg JR (1999) Myelin and collapsin-1 induce motor neuron growth cone collapse through different pathways: inhibition of collapse by opposing mutants of rac1. J Neurosci 19: 1965–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn TB, Meberg PJ, Brown MD, Bernstein BW, Minamide LS, Jensen JR, Okada K, Soda EA, Bamburg JR (2000) Regulating actin dynamics in neuronal growth cones by ADF/cofilin and rho family GTPases. J Neurobiol 44: 126–144. [PubMed] [Google Scholar]
- Luo Y, Raible D, Raper JA (1993) Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell 75: 217–227. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Bamburg JR (2000) Increase in neurite outgrowth mediated by overexpression of actin depolymerizing factor. J Neurosci 20: 2459–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, Crowell T, Cate RL, McCoy JM, Pepinsky RB (2004) LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci 7: 221–228. [DOI] [PubMed] [Google Scholar]
- Nagata-Ohashi K, Ohta Y, Goto K, Chiba S, Mori R, Nishita M, Ohashi K, Kousaka K, Iwamatsu A, Niwa R, Uemura T, Mizuno K (2004) A pathway of neuregulin-induced activation of cofilin-phosphatase Slingshot and cofilin in lamellipodia. J Cell Biol 165: 465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura F, Tanaka M, Takahashi T, Kalb RG, Strittmatter SM (1998) Neuropilin-1 extracellular domains mediate semaphorin D/III-induced growth cone collapse. Neuron 21: 1093–1100. [DOI] [PubMed] [Google Scholar]
- Niederost B, Oertle T, Fritsche J, McKinney RA, Bandtlow CE (2002) Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. J Neurosci 22: 10368–10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T (2002) Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108: 233–246. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K (2000) Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem 275: 3577–3582. [DOI] [PubMed] [Google Scholar]
- Park JB, Yiu G, Kaneko S, Wang J, Chang J, He Z (2005) A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron 45: 345–351. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112: 453–465. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT (2002) Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34: 895–903. [DOI] [PubMed] [Google Scholar]
- Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, Thill G, Levesque M, Sah D, McCoy JM, Murray B, Jung V, Pepinsky RB, Mi S (2005) TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron 45: 353–359. [DOI] [PubMed] [Google Scholar]
- Song H, Ming G, He Z, Lehmann M, McKerracher L, Tessier-Lavigne M, Poo M (1998) Conversion of neuronal growth cone responses from repulsion to attraction by cyclic nucleotides. Science 281: 1515–1518. [DOI] [PubMed] [Google Scholar]
- Soosairajah J, Maiti S, Wiggan O, Sarmiere P, Moussi N, Sarcevic B, Sampath R, Bamburg JR, Bernard O (2005) Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J 24: 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima J, Toshima JY, Amano T, Yang N, Narumiya S, Mizuno K (2001) Cofilin phosphorylation by protein kinase testicular protein kinase 1 and its role in integrin-mediated actin reorganization and focal adhesion formation. Mol Biol Cell 12: 1131–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z (2002) P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420: 74–78. [DOI] [PubMed] [Google Scholar]
- Wiggan O, Bernstein BW, Bamburg JR (2005) A phosphatase for cofilin to be HAD. Nat Cell Biol 7: 8–9. [DOI] [PubMed] [Google Scholar]
- Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM (2002) A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci 5: 1302–1308. [DOI] [PubMed] [Google Scholar]