

Abstract
Nerve cells that re-enter a cell cycle will die rather than divide, a fact that likely underlies the neurodegeneration in Alzheimer's disease (AD). Several mouse models of familial AD have been created, and although many display amyloid plaques in their brains, none captures the extensive pattern of nerve cell death found in the human disease. Using both immunocytochemistry and fluorescent in situ hybridization, we show that neurons in three different mouse models reproduce the ectopic cell cycling found in human AD. The temporal and spatial appearance of the cell cycle events in the mouse closely mimics the human disease progression. The cell-cycle events are evident 6 months before the first amyloid deposits and significantly precede the appearance of the first CD45+ microglia. These data suggest that the ectopic initiation of cell-cycle processes in neurons is an early sign of neuronal distress in both human and mouse AD. The close phenotypic correspondence indicates a previously unsuspected level of fidelity of the mouse model to the human disease. Finally, the relative timing suggests that neither the activated microglia nor the amyloid plaques themselves are necessary to initiate the pathogenic events in AD.
Keywords: PCNA, cyclin A, FISH, neuronal death, APP, β-amyloid
Introduction
Alzheimer's disease (AD) is a late-onset human neurodegenerative disorder marked by a progressive dementia and a spectrum of behavioral abnormalities. Neuropathological investigation of the AD brain reveals plaques of β-amyloid peptide and tangles of hyperphosphorylated tau protein. Neuronal density (cell bodies and synapses) is reduced in the frontal, entorhinal, and hippocampal cortices. In addition, certain subcortical populations of neurons undergo substantial amounts of neurodegeneration (Lyness et al., 2003). These include the locus ceruleus (Zweig et al., 1988; Zarow et al., 2003), the dorsal raphe (Zweig et al., 1988; Chen et al., 2000), and the basal nucleus of Meynart (Whitehouse et al., 1982).
Several transgenic mouse models have been generated that recreate the genetic changes found in familial AD (Lamb, 1995; Hsiao et al., 1996; Sturchler-Pierrat et al., 1997; Holcomb et al., 1998; Oddo et al., 2003). Transgenic mice expressing mutant human amyloid precursor protein (APP) genes exhibit an age-related development of diffuse and neuritic plaques, with plaque burdens often approaching those found in advanced cases of AD. This has proven to be a valuable resource in the exploration and design of disease therapies (Schenk, 2002). In addition, the AD mice show microglial activation, astrocytosis, and changes in neuronal cytoskeletal proteins including tau (Games et al., 1995; Hsiao et al., 1996; Stalder et al., 1999). Many of these model organisms have also been shown to have significant memory deficits (Hsiao et al., 1996; Holcomb et al., 1998; Oddo et al., 2003). Despite these parallels to the human disease state, however, none of the mouse models has yet been shown to develop the typical neurofibrillary tangles or to suffer any significant loss of neuronal cell bodies (for review, see Hock and Lamb, 2001). The reason for the discrepancy between the human and mouse neurodegenerative phenotype is unclear. It is conceivable that the models are inaccurate, yet the genetic mimicry is excellent.
Recently, we and others have shown a close association between the neuronal cell death in AD and cellular processes that normally only occur during a mitotic cell cycle. Cell-cycle-related proteins are expressed in neurons that are “at risk” in AD but not in age-matched controls or in regions of the AD brain itself where degeneration is not prevalent. This ectopic re-expression of cell-cycle markers is functional as shown by fluorescent in situ hybridization (FISH) (Yang et al., 2001). For a more complete discussion, see the studies by Arendt (2000), Liu et al. (2001), and Herrup and Arendt (2002).
Because an aberrant neuronal cell cycle is closely related to the neuronal degeneration in human brain, we wanted to explore whether this phenotype was preserved in the neurons of the AD mouse model. We report here that cell-cycle processes have begun in three different models. These cell-cycle alterations are found before β-amyloid (Aβ) plaques in a pattern that recapitulates the selective neuronal vulnerability observed in AD. Thus, the fidelity of the mouse models of AD, although not perfect, may be better than had been appreciated previously.
Materials and Methods
Transgenic mouse models of AD. Four different transgenic mouse models of AD were used in the current studies. In each line, the APP-induced amyloid plaques develop at different ages. Detailed information about the mouse strains are summarized in Table 1. Animals (R1.40, Tg2576, and Tg2576/PSEN1) were genotyped by PCR and housed at Case Western Reserve University Medical School Animal Resource Center, a facility full accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All procedures for animals were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University. Paraffin-embedded, 10 μm sections of the APP23 mouse brain were a generous gift from Dr. M. Staufenbiel (Novartis Institute for Biomedical Research, Basel, Switzerland).
Table 1.
Transgenic mouse lines used in this study
|
Transgenic lines |
Strain |
Approach |
Mutation |
Promoter |
Aβ deposits (age of onset) |
Neuronal cell loss |
NFT |
|---|---|---|---|---|---|---|---|
| R1.40-YAC | C57BL/6 | Genomic | Swedish | Human APP | 14 months | N.R. | A-T 8(+) |
| Tg2576 | C56BL/6-SJL | cDNA (695) | Swedish | Hamster PrP | 9-12 months | N.D. | A-T8(+) |
| Tg2576/PSEN1 | C56BL/6-SJL/2C57BL/6 | cDNA (695) | Swedish/human PS1 | Hamster PrP | 9-12 months | N.R. | A-T8(+) |
| TgAPP23 |
C56BL/6-DBA/2, C57BL/6 |
cDNA (751) |
Swedish |
Murine Thy-1 |
6 months |
25% in CA1 at 14 months |
A-T8(+) |
NFT, Neurofibrillary tangle; N.R., not reported; N.D., not detectable; PrP, prion protein; PS1, presenilin 1.
Histology. Animals were deeply anesthetized with Avertin (0.02 cc/mg body weight); they were perfused transcardially with PBS, followed by 4% paraformaldehyde in 0.1 m sodium phosphate buffer (PB). The brain was dissected, immediately removed from the cranium, and transferred to fresh 4% paraformaldehyde at 4°C overnight. The brains were then cryoprotected by sinking in 30% sucrose in 0.125 m PB at 4°C overnight. After bisecting along the midline, the brains were embedded in OCT compound. Then cryostat sections were cut and allowed to air dry on SuperPlus glass slides overnight.
Single and double immunocytochemistry. The proliferating cell nuclear antigen (PCNA) mouse monoclonal antibody recognizing the PCNA p36 protein (Dako, High Wycombe, UK) was diluted 1:250 in 10% goat serum/PBS blocking buffer before use. The rabbit polyclonal cyclin A antibody (ab 7956; working dilution, 1:200; Abcam, Cambridge, UK) was raised against the C-terminal domain of cyclin A2. The mouse monoclonal NeuN antibody (dilution, 1:500; Chemicon, Temecula, CA) was used as a neuronal marker. The rat monoclonal CD45 antibody (catalog #MCA1388; working dilution, 1:500; Serotec, Raleigh, NC) was used as a microglial maker.
To perform double fluorescence immunocytochemistry, sections were first rinsed in PBS, followed by pretreatment in citrate acid buffer (0.1 m citrate acid and 0.1 m sodium citrate, pH 7.4) for 6–8 min at 95°C. After the slides had cooled in buffer for 30 min at room temperature, they were rinsed in PBS. Sections were incubated for 1 h at room temperature in 10% goat serum in PBS to block nonspecific binding. All primary antibodies were diluted in PBS containing 0.4% Triton X-100 and 10% goat serum and were applied to sections and incubated overnight at 4°C. After rinsing in PBS, they were incubated for 2 h with a secondary antibody, which was conjugated with various fluorescent Alexa dyes (dilution, 1:500; Molecular Probes, Eugene, OR). The sections were then rinsed in PBS and reincubated in 10% goat serum blocking solution for 1 h, followed by the addition of the second primary antibody (raised in a different species from the first primary antibody) for a second overnight incubation at 4°C. Sections were then rinsed in PBS, and the second secondary antibody, conjugated with a different fluorescent dye, was applied to the sections for 2 h at room temperature. After rinsing, all sections were mounted in PBS/glycerol under a glass coverslip.
All paraffin-embedded APP23 mouse sections were deparaffinized in xylene and rehydrated through graded ethanols to water. For staining using HRP-conjugated secondary antibodies, the sections were pretreated in 0.3% hydrogen peroxide in methanol for 30 min to remove endogenous peroxidase activity, rinsed in Tris-buffered saline (TBS), and treated with 0.1 m citrate buffer in a microwave at sufficient power to keep the solution at 95–100°C for 10 min. Sections were cooled in the same buffer at room temperature for 30 min and rinsed in TBS. Slides were incubated in 10% goat serum in PBS blocking solution for 1 h at room temperature, after which a primary antibody was applied to the sections that were then incubated at 4°C overnight. The sections were washed three times in TBS before applying the secondary antibody (Vector Laboratories, Burlingame, CA), which was diluted in blocking solution at 1:300. The secondary antibody was applied for 1 h at room temperature. Afterward, sections were rinsed three times in TBS. Rinsed sections were then incubated in Vectastain ABC Elite reagent for 1 h and developed using diaminobenzidine (DAB), according to the protocol of the manufacturer. The sections were counterstained with hematoxylin, and after dehydration, all sections were mounted in Permount under a glass coverslip. Control sections were subjected to the identical staining procedure, except for the omission of the primary antibody.
FISH. Three mouse-specific DNA probes were generated from bacterial artificial chromosomes (BACs), which carried individual specific mouse genomic DNA sequences. One of the DNA probes (480C6) was made from the region that encodes the APP located on mouse chromosome 16. The other two probes were generated from overlapping BACs (170L21 and 566) containing the structural gene for aldolase C on mouse chromosome 11. Each of the three probes covers 100–300 kb of unique genomic sequence. They were labeled by standard nick translation protocols using digoxygenin-labeled dUTP. After labeling, probes were concentrated with mouse Cot-1 DNA (Invitrogen, San Diego, CA) to block hybridization to repetitive sequences. Before hybridization, all sections were rinsed in PBS and pretreated with 30% pretreatment powder (Oncor, Gaithersburg, MD) for 15 min at 45°C, followed by treatment with protease (0.25 mg/ml; Oncor) for 25 min at 45°C. After rinsing in 2× SSC [for details, see Sambrook et al. (1989)], the slides were dehydrated through graded alcohols and allowed to air dry. The labeled probe was applied to the individual sections, which were then covered with a glass coverslip and sealed with rubber cement. To denature DNA, slides were heated at 90–92°C on a heated block for 12 min and hybridized with probe overnight at 37°C. After rinsing in 50% formamide and 2×SSC at 37°C for 15 min, the slides were transferred to 0.1× SSC buffer for 30 min at 37°C and rinsed in PN buffer (0.5 m PB with 0.5% NP-40) at room temperature. To block nonspecific antibody binding, 10% goat serum in PBS was applied to the sections, followed by incubation in mouse anti-digoxygenin primary antibody (dilution, 1:200; Boehringer Mannheim, Indianapolis, IN) for 30 min at 37°C. After rinsing, a secondary anti-mouse antibody conjugated with Alexa 488 (dilution, 1:250) was applied to the sections for 30 min at 37°C. Slides were rinsed three times in PN buffer, and the sections were counterstained with either 4–6-diamidino-2-phenylindole (DAPI) or propidium iodide and covered with a coverslip. The number of spots of hybridization in each nucleus was determined at 1000× under fluorescent illumination. Images were captured on a Leitz (Wetzlar, Germany) research microscope equipped with a digital camera (Prog C14). Neurons were counterstained and identified on the basis of the size of their nucleus and their position either in the front cortex or hippocampus.
Results
Cell-cycle events in R1.40 animals
We chose as our mouse model a line carrying the entire human βAPP gene on a yeast artificial chromosome. The transgene was engineered to mimic the double mutation known as the Swedish mutant (K670M/N671L), and the line of mice carrying the construct is known as R1.40. As reported previously, this line develops classic Aβ plaques in cortical regions beginning at 13 postnatal months. By 22 months of age, there are substantial numbers of 6E10-immunoreactive deposits in both the cortex and hippocampus. The density of these plaques is higher in the cortex than in the hippocampus, and antibody staining for the presence of hyperphosphorylated tau epitopes (AT8) is found around the plaques (Kulnane and Lamb, 2001).
R1.40 brain tissue was examined for the presence of cell-cycle proteins by immunocytochemistry. Figure 1 shows images from a 22-month-old R1.40 homozygous transgenic mouse. Marked amyloid plaque pathology is found in both the hippocampus and cortex by this age (data not shown). Figure 1, A–F, shows a typical pattern of immunostaining of the PCNA in the frontal cortex (Fig. 1A–C) and hippocampus (Fig. 1D–F) of transgenic R1.40 animals. As indicated by the arrows, many neuronal nuclei stained positive for the presence of this DNA polymerase/replication fork subunit. Age-matched nontransgenic mice from the same genetic background (C57BL/6) had no or only an occasional PCNA-positive neuron in these regions, as illustrated here in the frontal cortex (Fig. 1G–I). A similar pattern of cell-cycle protein staining was found with the S-phase cyclin, cyclin A. Cyclin A is a regulatory subunit of Cdk2 and is normally elevated only during the S phase in dividing cells. We observed substantial numbers of cyclin A-positive cells in the R1.40 transgenic neocortex (Fig. 1J–L) and hippocampus (Fig. 1M–O) but not in nontransgenic, age-matched controls (Fig. 1P–R). Figure 1, J–L, taken at a higher magnification, clearly illustrates the neuronal morphology of the “cycling” cells. Cells located in cortical layers II/III and V were most frequently stained for cell-cycle proteins. In contrast to the images of PCNA-positive neurons in Figure 1, A–F, however, cyclin A protein was found localized to the neuronal cytoplasm of the cortex or hippocampus of transgenic R1.40 animals.
Figure 1.

Appearance of cell-cycle proteins in the neurons of 22-month-old transgenic R1.40 mice. A–F, PCNA (green) was re-expressed in the nuclei of cortical (A–C) and hippocampal (D–F) neurons. G–I, No PCNA expression was found in the age-matched nontransgenic control cortex (G–I) or hippocampus (data not shown). J–O, Expression of cyclin A was also detected in the cytoplasm of cortical (J–L) and hippocampal CA1 pyramidal (M–O) neurons of the R1.40 mouse. P–R, There was no cyclin A found in comparable neuronal populations in the controls. Nuclei were counterstained with DAPI (blue; B, E, H, K, N, Q). The arrows indicate examples of cell-cycle-positive cells. Scale bar, 10 μm.
To insure that the presence of both PCNA and cyclin A proteins was indicative of neurons undergoing a true cell cycle, we used FISH on our material. Probing with a digoxygenin-labeled 300 kb BAC carrying unique sequences from mouse chromosome 11, we observed the situation illustrated in Figure 2. Many cells in the region where PCNA staining was found were labeled with more than two hybridization signals in their nuclei. This was true of neurons in both the neocortex (Fig. 2A,C) and hippocampus (Fig. 2B,D). No such cells were found in the aged-matched nontransgenic controls (Fig. 2E,F), and no hyperploid neuronal cells were found in the cerebellum (data not shown). Two different autosomal probes were used to achieve these images, one on chromosome 11 and a second on chromosome 16. The results from both probes were identical: substantial evidence of hyperploidy in cells of the R1.40 transgenic brain with a few examples of aneuploid neurons in comparable regions of nontransgenic, age-matched controls. Fields such as these are representative of the situation found in our material. Because of the relatively large areas that we routinely scan, we rely on a standard fluorescent microscope rather than a confocal microscope. The photographs of hyperploid cells taken on such a microscope will often give the impression that the spots of hybridization are actually coming from two overlapping nuclei rather than one. Part of this impression derives from the fact that the nuclei of the aneuploid cells are often misshapen. This is common experience in the field of cancer biology, in which the pathologist will use the deviation from spherical of the nuclear shape of a cell to obtain an estimate of the degree of aneuploidy in a tumor (Boone et al., 1992). For each field illustrated, however, the observer was careful to focus through the entire depth of the section (at 1000× using an oil-immersion objective) to ensure that there was only one nucleus. Furthermore, reanalysis of the same sections on a confocal microscope resulted in the same conclusions. These images regularly reveal three (Fig. 3A) and four (Fig. 3B) hybridization spots in transgenic but not control animals.
Figure 2.
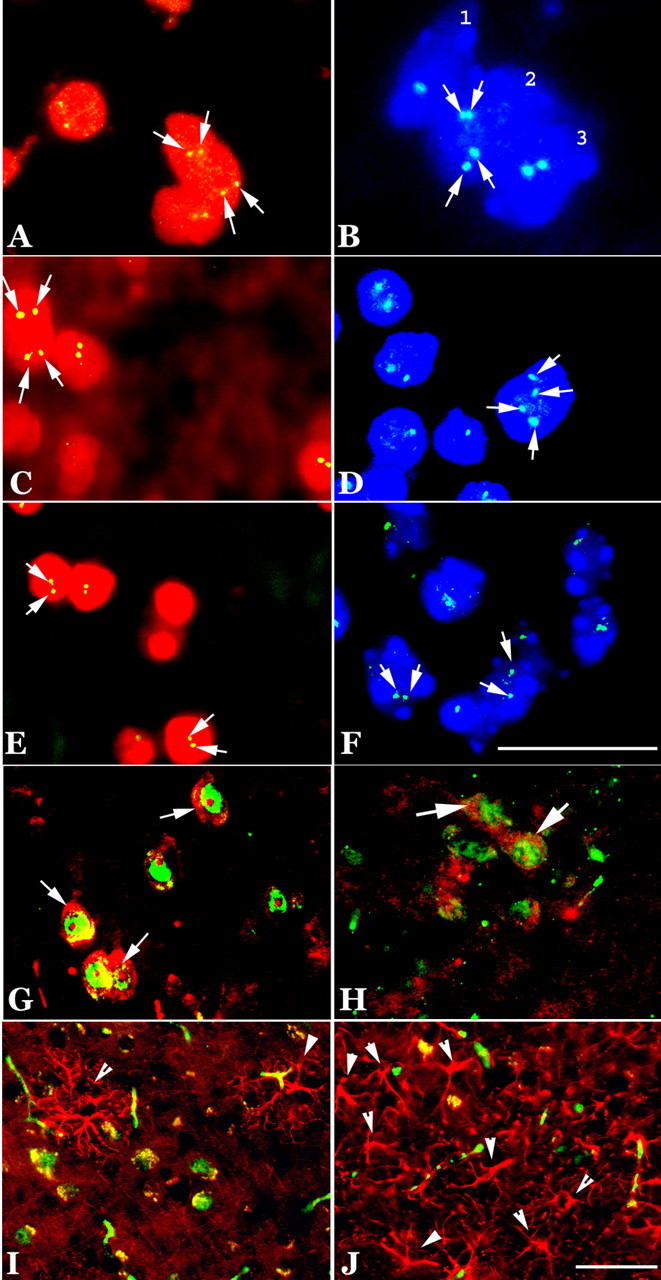
DNA replication in neurons of transgenic R1.40 mice. A, FISH with unique genomic sequences reveals four bright hybridization signals for mouse chromosome 11 (170L21) in the cortex. The arrows indicate the hybridization signals (green); nuclei are counterstained with propidium iodide (red). B, Hippocampal neurons show a similar pattern. The cell labeled 2 is shown with arrows to indicate the hybridization signals (green). In this image, the nuclei have been counterstained with DAPI (blue). C, D, A second BAC probe for sequences on mouse chromosome 16 (4806C) shows four hybridized signals (green) merged with propidium iodide (red) counter stain in the neuronal nuclei of the cortex (C; arrows) and hippocampus (D) of R1.40 mice. E, F, DAPI counterstain. Only two hybridized signals were found in the nuclei of age-matched nontransgenic controls (arrows). G, H, Double immunostaining showed coexpression of cyclin A (red) with a neuronal marker, NeuN (green), in both the frontal cortex (G) and hippocampal pyramidal neurons (H). GFAP (red) as a marker for astrocytes shows that astrocyte activation (arrowheads) occurred in the neuropil surrounding the cell-cycle-positive neuronal cells (PCNAs; green) of R1.40 mice in both the cortex (I) and hippocampus (J). Only scattered GFAP-positive cells were also cell cycle positive (data not shown). Scale bar, 10 μm.
Figure 3.

Confocal images of increased ploidy in some of the neurons of the adult R1.40 transgenic mouse brain. These images were taken with a Zeiss (Thornwood, NY) 510 confocal microscope of neurons in the frontal cortex of a 22-month-old R1.40 mouse. The cell nucleus is revealed with a propidium iodide (red) counterstain. A, B, The hyperploid DNA content of both neurons is illustrated by the three (A, arrows) and four (B, arrows) spots of hybridization of a BAC genomic probe complementary to sequences on mouse chromosome 16. Note that despite the elongated shape of the nucleus shown in A, the three hybridization signals are clearly contained within a single nuclear profile.
Thus, by both immunocytochemistry and FISH, many neurons in the 22-month R1.40 transgenic mouse brain show evidence of having re-entered a cell cycle. We wanted to determine whether the evidence indicated an active process of cell division or instead a situation more similar to the human AD brain in which the neurons enter the cycle only to stall at a point either near the end of S phase or during G2. Bromodeoxyuridine (BrdU) was injected into the transgenic mice 2 h before they were killed. If cycling were on-going, the modified DNA precursor should be taken up into the dividing cells and incorporated into their DNA. Immunocytochemical labeling of the CNS should then reveal the location of the actively dividing cells. Despite repeated attempts, and examination of thousands of neurons in dozens of sections, no BrdU-labeled neuronal nuclei were ever observed. The only exceptions were cells in the known neurogenic regions lining the lateral ventricle and in the inferior blade of the hippocampal dentate gyrus (data not shown).
The size, cytological features, and position of the cycle-positive cells strongly implicates their identity as neuronal. We used a panel of antibodies to validate this impression. As a neuronal marker, we used the nuclear antigen NeuN. GFAP was used as an astrocyte marker. The non-neuronal (GFAP-positive) cells in the transgenic brain were more often reactive than in controls (Fig. 2I,J). This is in keeping with the previously described reactive gliosis and inflammatory processes at work in these animals (Games et al., 1995; Rossner et al., 2001; Hartlage-Rubsamen et al., 2003). Nonetheless, most of the cyclin A-positive nuclei were double labeled with NeuN antibody, indicating their identity as neurons (Fig. 2G,H).
Other mouse models of AD
To validate our findings in the R1.40 mouse, we chose to examine additional transgenic mouse models of AD. The Tg2576 mouse is a commonly used model in which a cDNA for human APP695, containing the Swedish mutation (K670M/N671L) is driven by the hamster prion promoter. The animals develop an early behavioral phenotype, and plaques appear by 9 postnatal months. Examination of the immunostained cortex and hippocampus of 12-month-old Tg2576 mice revealed a substantial number of PCNA- or cyclin A-positive neurons in both the cortex (Fig. 4A) and hippocampus (Fig. 4B). Neurons from control mice were negative for cell-cycle proteins in these same regions (Fig. 4C,D). We were also able to obtain sections from APP23 mice. These animals carry a cDNA for human APP751 driven by the murine Thy-1 promoter (Bornemann and Staufenbiel, 2000). They normally develop plaques at 6 months of age and have ∼25% neuronal cell loss in the CA1 region of the hippocampus by 14 months of age. When we examined these animals for evidence of cell-cycle-related proteins, we found PCNA and cyclin A staining in both the cortex (Fig. 4E) and hippocampus (Fig. 4F) by 1 year of age. Indeed, consistent with the more virulent nature of the pathology in these animals, the density of immunopositive cells in APP23 was greater than in any of the other models we examined. Wild-type littermates had no such staining (Fig. 4G,H). We note that Gartner et al. (2003) have previously examined the APP23 for cell-cycle protein immunoreactivity, finding no difference in staining between transgenic and age-matched controls at 21.5 months of age. Our findings differ from theirs primarily in that we are unable to find evidence for cell-cycle protein expression in 22-month-old C57BL/6 nontransgenic controls. The direct overlap between this study and ours is restricted to the expression of cyclin A, and we would suggest that any one of several points could explain the discrepancies in the two studies. The previous study used floating 30 μm sections as opposed to 10 μm paraffin sections in the current work. Our staining regimen used an antigen-retrieval protocol, whereas the previous work did not. Finally, Gartner et al. (2003) used a polyclonal cyclin A antibody (Santa Cruz Biotechnology, Santa Cruz, CA) that would recognize both cyclin A1 and A2. The antibody used to obtain the results in Figure 4 was a monoclonal antibody raised against a cyclin A2-specific peptide. A high background of cyclin A1 might have obscured an increase in the mitotic cyclin A2 protein. Because the previous study provided no evidence for the presence or absence of DNA replication, we propose that in the aggregate, our data provide strong evidence that three of the current mouse models of AD each develop cell-cycle-positive neurons in the cortex and hippocampus.
Figure 4.

CCEs are observed in multiple AD transgenic mouse models. A, B, Cyclin A (red, arrows) is expressed in the cytoplasm of frontal cortical (A) and hippocampal (B) neurons of the Tg2576 AD transgenic mouse. The neuronal antigen NeuN (green) was used as a marker for neurons. C, D, No expression of PCNA (green) is found in the either cortical or hippocampal neurons of littermate animals from the same genetic background. The red fluorescence is a propidium iodide counterstain. Neuronal nuclei in the APP23 mouse, another AD transgenic mouse model, also exhibited CCEs. E–H, Immunostaining revealed PCNA-positive neurons (arrows) in the frontal cortex (E) and hippocampus (F) in APP23 mice but not in the nontransgenic controls (G, H). Scale bars, 10 μm.
Regional variation
The plaque pathology of the R1.40 mouse does not develop uniformly throughout the CNS. Rather, the neocortex is heavily involved with the hippocampus, and the olfactory bulb is also affected. Similarly, the presence of cell-cycle-positive neurons also varied substantially from region to region of the transgenic nervous system. As in AD, the R1.40 transgenic mouse cerebellar cortex is spared the effects of the disease. No cell evidence of neuronal cell cycling was found in Purkinje, granule, basket, or stellate cells (Fig. 5O). Nearly one-third of the neurons of the deep nuclei, however, were brightly positive for both PCNA and cyclin A (data not shown). The neocortex was the site with the highest density of “dividing” neurons (Fig. 1A–C,J–L) although even here there was consistent variation among the regions of the neocortex, with frontal regions showing the highest density of cycle-positive cells, followed by occipital. Parietal regions had the lowest density of neurons.
Figure 5.
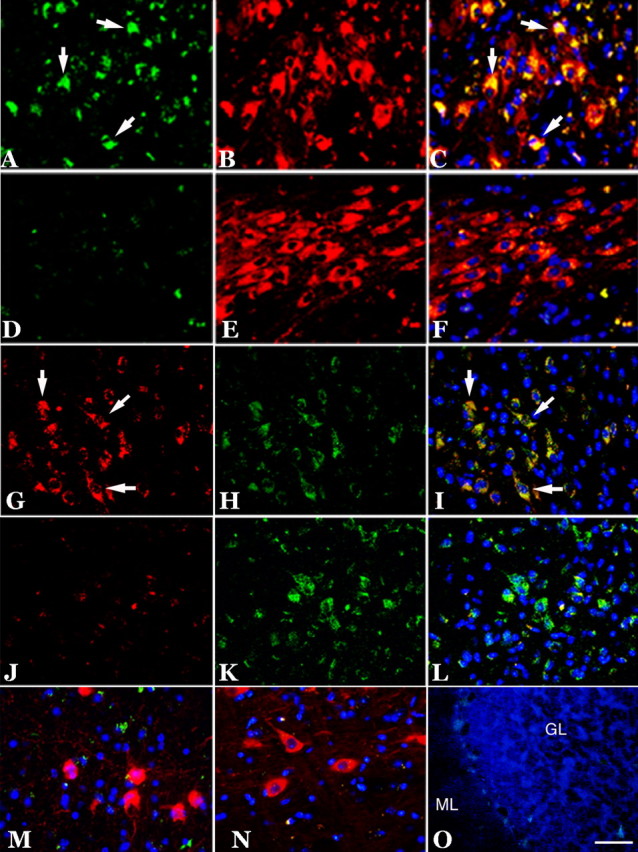
Expression of cell-cycle proteins in subcortical structures in 22-month-old R1.40 mice. A–F, Cyclin A (green) was expressed in TH-positive (red) neurons of the locus ceruleus (A–C, arrows), whereas an aged-matched wild-type control showed only nonspecific cyclin A immunoreactivity (D–F). G–L, Cyclin A (red) was also expressed in the tryptophan hydroxylase-positive (green) neurons of the dorsal raphe (G–I, arrows), whereas an aged-matched wild-type control showed only background cyclin A immunoreactivity (J–L). M–O, No cell-cycle expression was observed in the ChAT-positive (red) neurons of the basal nucleus (M), the TH-positive (red) neurons of the substantia nigra (N), or in the neurons of the cerebellar cortex (O). Cell nuclei counterstained with DAPI (blue) are shown throughout. Scale bars, 10 μm. GL, Granule cell layer; ML, molecular layer.
Subcortical structures were also examined carefully. The thalamus and hypothalamus were negative, as were most dorsal midbrain structures. We paid special attention to the cholinergic neurons in the basal forebrain as well as the neurons of the dorsal raphe and the locus ceruleus. The two brainstem nuclei were strongly immunoreactive for cell-cycle protein (Fig. 5). Tyrosine hydroxylase (TH) immunoreactivity was used to identify the neurons of the locus ceruleus. Double immunostaining clearly shows the presence of cyclin A protein (Fig. 5A) in these noradrenergic neurons (Fig. 5B) in R1.40 animals but not in nontransgenic mice (Fig. 5D–F). The characteristic shape and position of the raphe nuclei were used as the identifiers for the serotonergic neurons of the dorsal raphe. A significant number of neurons in these regions were positive for the cell-cycle marker cyclin A (Fig. 5G) and tryptophan hydroxylase (Fig. 5H), which is a marker of serotonergic neurons. Cell-cycle markers were absent from nontransgenic controls (Fig. 5J–L). No evidence of neuronal cell cycling was found among the TH-positive neurons of the substantia nigra (Fig. 5N) in any of the 22-month-old transgenic and wild-type animals (data not shown) we examined. This finding was especially significant because in the sagittal sections we examined, cells of the locus ceruleus (cell cycle positive) and substantia nigra (cell cycle negative) were often encountered in the same section. The basal nucleus of Meynart is the source of the forebrain cholinergic projection fibers to the hippocampus. The majority of these neurons are lost during the course of AD, and previous work from our laboratory has shown evidence for both cell-cycle protein reexpression and DNA replication in the human neurons (Yang et al., 2001). Curiously, none of the animals that we examined had any instances of cell-cycle proteins re-expressed in the ChAT-positive neurons of the basal forebrain (Fig. 5M).
Neuronal cell cycles initiate before plaques
One value of a mouse model of a human disease is the opportunity it offers to perform precise longitudinal studies of genetically identical animals. Previous work has shown that the R1.40 mice develop their first dense plaque deposits at 13 months of age. We wanted to determine the relationship between the timing of the appearance of these deposits with the initiation of the ectopic cell-cycle events (CCEs). To accomplish this, we killed animals at various ages (3, 6, and 10 months) and examined their CNS for signs of cell-cycle processes. As expected from previous studies, no evidence of either Aβ deposits or neurofibrillary tangles could be found in these younger brains (data not shown). In contrast, when 10-month-old R1.40 transgenic animals were examined for the presence of cell-cycle proteins, we found numerous examples of cycling neurons in both the neocortex (Fig. 6I) and hippocampus (Fig. 6K). At 3 months of age, we found no examples of CCEs in any of the neurons of the R1.40 mouse brain (Fig. 6A,C, 7A,B). At 6 months, however, we were reliably able to detect cell-cycle proteins in both the cortex and hippocampus (Fig. 6E,G). No evidence of cell-cycle proteins (Fig. 6B,D,F,H,J,L) or DNA replication (Fig. 7C,F,I) were observed in the non-transgenic controls.
Figure 6.

Expression of cell-cycle proteins at various ages of R1.40 mice. A–D, At 3 months of age, no expression of cyclin A (data not shown) or PCNA (green) was found in any region of the R1.40 brain (A, frontal cortex; C, hippocampus) or in nontransgenic controls (B, frontal cortex; D, hippocampus). E–L, At 6 and 10 months of age, PCNA (green) appeared in the cortical (E, I; arrows) and hippocampal (G, K; arrows) neurons of R1.40 mice, but no immunoreactivity was found in controls (F, H, J, L). Nuclei were counterstained with propidium iodide (red; A–D, F–L) or DAPI (blue; E). Scale bar, 10 μm.
Figure 7.

DNA replication in neurons at various ages in the R1.40 mouse model. A–C, At 3 months of age, only two hybridized signals (arrows) were found in the nuclei of cortical and hippocampal neurons of both R1.40 (A, B) and control mice (C). D, E, G, H, However, there were four bright hybridization signals (arrows) for mouse chromosome 11 (170L21) or 16 (480C6) at 6 months of age (D, cortex; E, hippocampus) and 10 months of age (G, cortex; H, hippocampus). F, I, Nontransgenic controls exhibited only two hybridization signals. Scale bar, 10 μm.
In addition to this immunocytochemical evidence, the same relationship between cell-cycle proteins and DNA replication was found at this age. FISH with probes for loci on mouse chromosome 11 and chromosome 16 applied to adjacent sections demonstrated significant numbers of cells in the 6-month-old (Fig. 7D, E) and 10-month-old (Fig. 7G,H) R1.40 animals with three or four “spots” of hybridization in their nucleus. Thus, the neurons in the R1.40 transgenic mouse initiate a cell cycle before the appearance of the Aβ deposits form anywhere in the brain.
Reactive microglia do not associate with locations of neuronal cell cycles
To examine the relationship between sites of the neuroinflammatory response and neuronal cell-cycle involvement, R1.40 animals were examined for activated microglia. We examined adjacent brain sections for cell-cycle markers and reactive microglial cells, using CD45 immunostaining as an indicator of the latter. In adult R1.40 transgenic mice, cyclin A- and PCNA-immunopositive cells were found at relatively high densities in the frontal cortex, with the greatest concentration located in cortical layers II and III. In addition to these Alzheimer's-like pathological features, when we examined 22-month-old R1.40 animals, CD45-immunoreactive microglia were observed throughout (Fig. 8A,B). However, this coincidence of microglial markers with the CCEs did not extend to other regions. The most extreme example of this was the brainstem nuclei in which, as shown in Figure 5, CCEs are easily detected in both the dorsal raphe and locus ceruleus. Examination of adjacent sections, stained for CD45 antigen, however, revealed no reactive microglial cells in either of these regions. This is illustrated by the absence of DAB reaction product in the R1.40 locus ceruleus shown in Figure 8C. In addition to this spatial dissociation between the CCEs and the microglial cell response, there was also a significant temporal dissociation. The first CD45+ cells are observed at close to 1 year of age in the R1.40 cortex. Yet, as illustrated in Figures 6 and 7, the CCEs are apparent 6 months earlier. Thus, in both space and time, the inflammatory response in the R1.40 mouse model does not appear to predict or even coincide with the occurrence of the cell-cycle markers.
Figure 8.
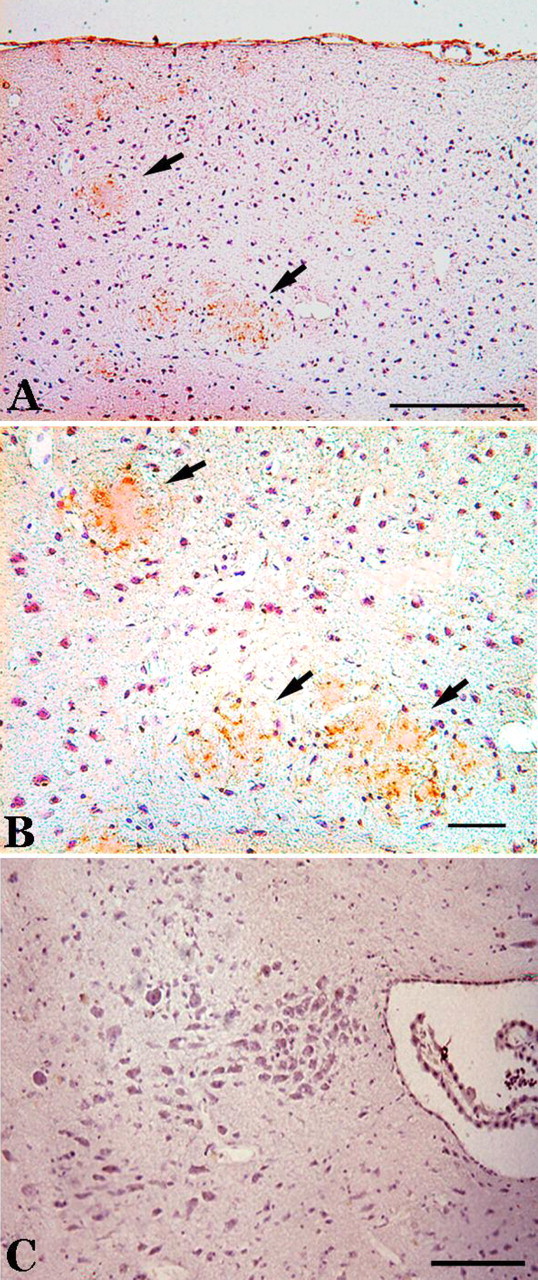
An inflammatory response is observed in the cortex of 22-month-old R1.40 mice. Aβ plaques (arrows) are surrounded by activated microglial cells. A, An antibody for CD45 reveals the presence of activated microglia in the frontal cortex. B, A higher-magnification image shows microglia with robust processes. C, The locus ceruleus remains devoid of CD45-positive microglia at this age. Scale bars: A, 100 μm; B, C, 10 μm.
Discussion
The results presented here document the initiation of a cell-cycle process in the neurons of several mouse models of familial AD. By both the expression of cell-cycle proteins and the documentation of DNA replication, we have shown that these normally postmitotic cells attempt a process that strongly resembles a mitotic cell division. There are a growing number of conditions in the mouse in which neuronal cell death is associated with re-entrance into a lethal cell cycle. These include the retinoblastoma-deficient mouse (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992), cerebellar target-related cell death (Herrup and Busser, 1995), oncogene expression in maturing neurons (Feddersen et al., 1992), oxidative stress as suggested by the harlequin mouse (Klein et al., 2002), as well as models of stroke (Katchanov et al., 2001) and the superoxide dismutase-1 mouse model of amyotrophic lateral sclerosis (Ranganathan et al., 2001; Ranganathan and Bowser, 2003). The weight of this in vivo evidence, combined with elegant studies in cell culture (Farinelli and Greene, 1996; Giovanni et al., 1999; Greene et al., 2004) strongly implicates the initiation of cell cycling as a causative factor in the death of these neurons. A recent addition to this correlation between neuronal CCEs and cell death is the demonstration by our laboratory that in the mouse model of ataxia-telangiectasia (atm–/–), Purkinje cells and striatal neurons also initiate a cell-cycle-like process (Yang and Herrup, 2005). The current report illustrates that CCEs are correlated with neuronal death in several different mouse models of human AD just as in the human condition itself. But viewed in conjunction with the previous work, it is clear that the CCEs should not be interpreted as unique to Alzheimer's disease or any other disease or disease model.
Our findings also emphasize the unexpected conclusion that although the CCEs may be a necessary first step (as suggested by the cell-culture findings cited above), they are not an immediate cause of neuronal cell death, at least in mouse models. In the R1.40 mice, DNA replication leading to “hyperploid” cells is apparent as early as 6 months, yet cell death is not observed at any age. This means the initiation of the cell cycle, including DNA replication, can be followed by many months during which the aneuploid neurons persist in what we can assume is a near functional state [only mild behavioral abnormalities are reported (Hock and Lamb, 2001), and these can be resolved rapidly (Dodart et al., 2002)]. This is consistent with the calculations of Busser et al. (1998) and Yang et al. (2001) in human Alzheimer's disease brains. Using different techniques, both studies reached nearly identical conclusions: the death of the neurons in the Alzheimer's brain occurs months after the CCEs. There would seem to be two basic interpretations of these observations. The first is that the CCEs themselves are necessary but not sufficient to induce the death of adult CNS neurons. Perhaps, just as in cancer, a “second hit” is needed (Zhu et al., 2004). One such triggering event could be hypoxia–ischemia, as two recent studies implicate CCEs in the neuronal death after hypoxia–ischemia in rodents (Kuan et al., 2004; Wen et al., 2004). In addition, recent studies in transgenic mice producing wild-type human tau but no endogenous mouse tau have documented the formation of neurofibrillary tangles and neuronal cell death associated with altered CCEs (Andorfer et al., 2005). The suggestion is that different tau isoforms or tau expression patterns could be another potential triggering event. A final intriguing possibility is that, for unknown reasons, the CCEs we observe are actually protective responses of a neuron under stress and that much as in certain developmental situations (Claycomb et al., 2004) the neuron benefits from having multiple copies of each allele. The end result of a neuronal cell cycle is the death of the cells, but the long times required between initiation of the cycle and death suggests that in vivo there are other factors that must be involved in the death process.
Putting aside the consequences of the CCEs, it is noteworthy that in a wide variety of different neurodegenerative conditions, these events are reliable and early markers of neuronal distress. Viewed through this lens, the R1.40 mouse model of AD becomes a more accurate model of the human disease than might have been expected. All extant mouse models have been viewed as compromised because they lack the severe behavioral and neurological phenotypes of the human disease. Furthermore, although the mimicry of the amyloid plaque pathology is excellent, it has been somewhat disappointing that neurofibrillary tangles and the neuronal cell death, both central to the human phenotype, are found only occasionally in the numerous mouse models of AD. If we take the cell-cycle abnormalities as surrogates for the early stages of cell death, however, the work reported here suggests that the R1.40 mouse is actually a significant and relatively accurate disease model. As has already been reported, the evolution of the plaque pathology, early tau phosphorylation events and inflammatory markers, are both spatially and temporally correct in their expression (Kulnane and Lamb, 2001). What is remarkable about the CCEs is that they are similarly correct in their anatomical appearance. The markers appear most strongly in the hippocampal formation (including the entorhinal cortex) and in frontal cortical regions. Furthermore, just as in the human disease, the adrenergic neurons of the locus ceruleus and the serotonergic neurons of the dorsal raphe are also affected by the abnormalities in cell-cycle regulation. Equally important, the neurons of the substantia nigra show no evidence of CCEs. These neurons are spared in Alzheimer's disease but represent a focal point of the neurodegeneration in Parkinson's disease, and their normalcy in the R1.40 animals provides yet another indication of the anatomical specificity of the disease model.
Standing in contrast to these human/mouse analogies are the cholinergic neurons of the basal forebrain. In humans, the basal nucleus of Meynert suffers substantial cell losses by the end stage of the disease (Whitehouse et al., 1982), and signs of distress are present in early stages (Chu et al., 2001; Mufson et al., 2002; Mesulam et al., 2004) including the presence of cell-cycle markers (Yang et al., 2003). In the mouse, however, we find no evidence of CCEs in the ChAT-positive neurons of R1.40 transgenic mice at any age. One might want to dismiss the discordance as an example of the difficulties of modeling a complex human disorder in a mouse. A mouse, even with a perfect genetic reproduction of a human disease is, after all, still only a model. But the discordance might also suggest a somewhat different interpretation of the human situation. The basal forebrain neurons have a chronic trophic dependency on the hippocampal pyramidal neurons, and it is possible that their dysfunction and death in Alzheimer's disease represents not a primary disease event, but rather a secondary consequence of the hippocampal damage. The mouse hippocampus, spared the atrophy that is found in the human, can continue to support a full complement of basal forebrain cholinergic neurons. Because the above-cited human basal nucleus studies document biochemical changes in the neurons rather than cell death, this may yet prove to be the situation in early Alzheimer's disease as well.
A final consideration raised by our findings is the perspective it offers on the amyloid hypothesis of AD. Summarized broadly, the hypothesis is that abnormal processing of the APP protein leads to the accumulation of the highly insoluble Aβ peptide. The Aβ peptides form aggregates, in either micromolecular or macromolecular size, leading to the death of neurons and thus the clinical symptoms of Alzheimer's disease. The findings of the current study both strengthen and weaken this linear view of the dementia. The finding that the CCEs precede Aβ plaque deposition by 6 months means that the plaques per se cannot be the immediate cause of the neuronal death. Small, micromolecular aggregates are still a plausible factor, however, and our findings are consistent with a growing consensus that these may be the truly toxic agents. Our findings strengthen this hypothesis in a different way as well. The strong anatomical reproduction of the sites of cell death in the human represented by the location of the CCEs in the mouse means that the overexpression of a single human disease gene has closely reproduced a complex anatomical pattern of neuronal distress. This can hardly be a random finding and deepens the suggestion that imperfections in APP processing (though not necessarily Aβ) can produce the neurological problems of AD.
Footnotes
This work was supported by grants from the National Institute on Aging and the National Institute of Neurological Disorders and Stroke (AG08012 and NS20591 to K.H., AG023012 to B.T.L.), the Alzheimer's Association (K.H. and B.T.L.), and The Fidelity Foundation (B.T.L.) and by a gift from the Blanchette Hooker Rockefeller Fund. We thank Dr. Matthias Staufenbiel (Novartis Institute for Biomedical Research, Basel, Switzerland) for providing the paraffin sections of the APP23 transgenic mice.
Correspondence should be addressed to Dr. Yan Yang, Alzheimer Research Laboratory, Case Western Reserve University School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106. E-mail: yxy33@case.edu.
DOI:10.1523/JNEUROSCI.3707-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260775-10$15.00/0
Y.Y. and N.H.V. contributed equally to this work.
References
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P (2005) Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci 25: 5446–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T (2000) Alzheimer's disease as a loss of differentiation control in a subset of neurons that retain immature features in the adult brain. Neurobiol Aging 21: 783–796. [DOI] [PubMed] [Google Scholar]
- Boone CW, Kelloff GJ, Steele VE (1992) Natural history of intraepithelial neoplasia in humans with implications for cancer chemoprevention strategy. Cancer Res 52: 1651–1659. [PubMed] [Google Scholar]
- Bornemann KD, Staufenbiel M (2000) Transgenic mouse models of Alzheimer's disease. Ann NY Acad Sci 908: 260–266. [DOI] [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K (1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci 18: 2801–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Eastwood SL, Hope T, McDonald B, Francis PT, Esiri MM (2000) Immunocytochemical study of the dorsal and median raphe nuclei in patients with Alzheimer's disease prospectively assessed for behavioural changes. Neuropathol Appl Neurobiol 26: 347–355. [DOI] [PubMed] [Google Scholar]
- Chu Y, Cochran EJ, Bennett DA, Mufson EJ, Kordower JH (2001) Down-regulation of trkA mRNA within nucleus basalis neurons in individuals with mild cognitive impairment and Alzheimer's disease. J Comp Neurol 437: 296–307. [DOI] [PubMed] [Google Scholar]
- Clarke A, Maandag E, van Roon M, van der Lugt N, van der Valk M, Hooper M, Berns A, te Riele H (1992) Requirement for a functional Rb-1 gene in murine development. Nature 359: 328–330. [DOI] [PubMed] [Google Scholar]
- Claycomb JM, Benasutti M, Bosco G, Fenger DD, Orr-Weaver TL (2004) Gene amplification as a developmental strategy: isolation of two developmental amplicons in Drosophila Dev Cell 6: 145–155. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM (2002) Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci 5: 452–457. [DOI] [PubMed] [Google Scholar]
- Farinelli S, Greene L (1996) Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J Neurosci 16: 1150–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT (1992) Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron 9: 955–966. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, Mcconlogue L, et al. (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373: 523–527. [DOI] [PubMed] [Google Scholar]
- Gartner U, Bruckner MK, Krug S, Schmetsdorf S, Stanfenbiel M, Arendt T (2003) Amyloid deposition in APP23 mice is associated with the expression of cyclins in astrocytes but not in neurons. Acta Neuropathol (Berl) 106: 535–544. [DOI] [PubMed] [Google Scholar]
- Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS (1999) Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. J Biol Chem 274: 19011–19016. [DOI] [PubMed] [Google Scholar]
- Greene LA, Biswas SC, Liu DX (2004) Cell cycle molecules and vertebrate neuron death: E2F at the hub. Cell Death Differ 11: 49–60. [DOI] [PubMed] [Google Scholar]
- Hartlage-Rubsamen M, Zeitschel U, Apelt J, Gartner U, Franke H, Stahl T, Gunther A, Schliebs R, Penkowa M, Bigl V, Rossner S (2003) Astrocytic expression of the Alzheimer's disease beta-secretase (BACE1) is stimulus-dependent. Glia 41: 169–179. [DOI] [PubMed] [Google Scholar]
- Herrup K, Arendt T (2002) Re-expression of cell cycle proteins induces neuronal cell death during Alzheimer's disease. J Alzheimers Dis 4: 243–247. [DOI] [PubMed] [Google Scholar]
- Herrup K, Busser JC (1995) The induction of multiple cell cycle events precedes target-related neuronal death. Development 121: 2385–2395. [DOI] [PubMed] [Google Scholar]
- Hock Jr BJ, Lamb BT (2001) Transgenic mouse models of Alzheimer's disease. Trends Genet 17: S7–12. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: 97–100. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99–102. [DOI] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt E, Bronson R, Goodell M, Weinberg R (1992) Effects of an Rb mutation in the mouse. Nature 359: 295–300. [DOI] [PubMed] [Google Scholar]
- Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R, Bruck W, Hortnagl H, Dirnagl U, Bhide PG, Endres M (2001) Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci 21: 5045–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL (2002) The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 419: 367–374. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Schloemer AJ, Lu A, Burns KA, Weng WL, Williams MT, Strauss KI, Vorhees CV, Flavell RA, Davis RJ, Sharp FR, Rakic P (2004) Hypoxia–ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci 24: 10763–10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulnane LS, Lamb BT (2001) Neuropathological characterization of mutant amyloid precursor protein yeast artificial chromosome transgenic mice. Neurobiol Dis 8: 982–992. [DOI] [PubMed] [Google Scholar]
- Lamb BT (1995) Making models for Alzheimer's disease. Nat Genet 9: 4–6. [DOI] [PubMed] [Google Scholar]
- Lee EY-HP, Chang C-Y, Hu N, Wang Y-CJ, Lai C-C, Herrup K, Lee W-H, Bradley A (1992) Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359: 288–294. [DOI] [PubMed] [Google Scholar]
- Liu T, Zwingman T, Li R, Pan T, Wong BS, Petersen RB, Gambetti P, Herrup K, Sy MS (2001) Differential expression of cellular prion protein in mouse brain as detected with multiple anti-PrP monoclonal antibodies. Brain Res 896: 118–129. [DOI] [PubMed] [Google Scholar]
- Lyness SA, Zarow C, Chui HC (2003) Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta-analysis. Neurobiol Aging 24: 1–23. [DOI] [PubMed] [Google Scholar]
- Mesulam M, Shaw P, Mash D, Weintraub S (2004) Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol 55: 815–828. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Ma SY, Dills J, Cochran EJ, Leurgans S, Wuu J, Bennett DA, Jaffar S, Gilmor ML, Levey AI, Kordower JH (2002) Loss of basal forebrain P75(NTR) immunoreactivity in subjects with mild cognitive impairment and Alzheimer's disease. J Comp Neurol 443: 136–153. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39: 409–421. [DOI] [PubMed] [Google Scholar]
- Ranganathan S, Bowser R (2003) Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am J Pathol 162: 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan S, Scudiere S, Bowser R (2001) Hyperphosphorylation of the retinoblastoma gene product and altered subcellular distribution of E2F-1 during Alzheimer's disease and amyotrophic lateral sclerosis. J Alzheimers Dis 3: 377–385. [DOI] [PubMed] [Google Scholar]
- Rossner S, Apelt J, Schliebs R, Perez-Polo JR, Bigl V (2001) Neuronal and glial beta-secretase (BACE) protein expression in transgenic Tg2576 mice with amyloid plaque pathology. J Neurosci Res 64: 437–446. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning, a laboratory manual, p B13. Cold Spring Harbor, NY: Cold Spring Harbor.
- Schenk D (2002) Amyloid-beta immunotherapy for Alzheimer's disease: the end of the beginning. Nat Rev Neurosci 3: 824–828. [DOI] [PubMed] [Google Scholar]
- Stalder MPA, Probst A, Sommer B, Staufenbiel M, Jucker M (1999) Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol 154: 1673–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94: 13287–13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen TC, Rogido M, Genetta T, Sola A (2004) Permanent focal cerebral ischemia activates erythropoietin receptor in the neonatal rat brain. Neurosci Lett 355: 165–168. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR (1982) Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science 215: 1237–1239. [DOI] [PubMed] [Google Scholar]
- Yang Y, Herrup K (2005) Loss of neuronal cell cycle control in ataxia-telangiectasia: a unified disease mechanism. J Neurosci 25: 2522–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K (2001) DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci 21: 2661–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mufson EJ, Herrup K (2003) Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci 23: 2557–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA, Chui HC (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 60: 337–341. [DOI] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Perry G, Smith MA (2004) Alzheimer's disease: the two-hit hypothesis. Lancet Neurol 3: 219–226. [DOI] [PubMed] [Google Scholar]
- Zweig RM, Ross CA, Hedreen JC, Steele C, Cardillo JE, Whitehouse PJ, Folstein MF, Price DL (1988) The neuropathology of aminergic nuclei in Alzheimer's disease. Ann Neurol 24: 233–242. [DOI] [PubMed] [Google Scholar]