

Abstract
T-cell recruitment into the brain is critical in inflammatory and autoimmune diseases of the CNS. We use intracerebral antigen microinjection and tetramer technology to track antigen-specific CD8+ T-cells in the CNS and to clarify the contribution of antigen deposition or traumatic injury to the accumulation of T-cells in the brain. We demonstrate that, after intracerebral microinjection of ovalbumin, ovalbumin-specific CD8+ T-cells expand systemically and then migrate into the brain where they complete additional proliferation cycles. T-cells in the brain are activated and respond to in vitro secondary antigen challenge. CD8+ T-cells accumulate and persist in sites of antigen in the brain without replenishment from the periphery. Persistent survival of CD8+ T-cells at sites of cognate antigen is significantly reduced by blocking CD154 molecules. A small traumatic injury itself does not lead to recruitment of CD8+ T-cells into the brain but attracts activated antigen-specific CD8+ T-cells from cognate antigen injection sites. This process is presumably antigen independent and cannot be inhibited by blocking CD154 molecules. These data show that activated antigen-specific CD8+ T-cells accumulate in the CNS at both cognate antigen-containing and traumatic injury sites after intracerebral antigen delivery. The accumulation of activated antigen-specific T-cells at traumatic injury sites, in addition to antigen-containing areas, could amplify local inflammatory processes in the CNS. Combination therapies in neuroinflammatory diseases to block both of these processes should be considered.
Keywords: antigen specificity, CD8+ T-cell homing, traumatic brain injury, neuroimmunology, ovalbumin antigen, intracerebral injection
Introduction
The CNS is considered an immune-privileged site because of the blood–brain barrier, the lack of conventional lymphatic drainage, and low levels of major histocompatibility complex (MHC) expression (Vass et al., 1986; Fontana et al., 1987; Cserr et al., 1992); however, both CD4+ and CD8+ cells can migrate into the CNS (Hickey, 1991; Williams and Hickey, 1995; Brabb et al., 2000; Qing et al., 2000) and cause inflammation and neurodegeneration (Xiao and Link, 1999; Ransohoff et al., 2003). The severity of inflammatory damage in the CNS is dependent on the density and localization of infiltrated T-cells (McGavern et al., 2002; McGavern and Truong, 2004). The regulation of these processes is not yet fully understood.
It is well accepted that infections can induce homing of T-cells into the CNS. In models of myelin-induced CNS autoimmune disease, however, CD8+ and CD4+ T-cells can infiltrate the CNS even in the absence of an active infection. CD8+ T-cells distribute diffusely through the brain parenchyma, whereas CD4+ T-cells localize to perivascular inflammatory cuffs (Gay et al., 1997; Babbe et al., 2000). Although mechanisms that regulate this distribution pattern are not clear, it has been proposed that proinflammatory cytokines, chemokines, and costimulatory molecules that are upregulated in traumatic injuries play a role in these processes (Arand et al., 2001; Lenzlinger et al., 2001; Morganti-Kossmann et al., 2001, 2002; Swartz et al., 2001; Hensler et al., 2002; Ray et al., 2002; Fee et al., 2003, 2004; Long et al., 2003). Recently, it was also suggested that in the absence of infections, antigen expression and antigen-presenting cells could also direct the preferential localization of antigen-specific T-cells to nonlymphoid tissue (Ingulli et al., 2002; Reinhardt et al., 2003). Understanding the effects of antigen expression and traumatic injury-related focal inflammation on the localization of CD8+ T-cells in the brain could help us to define the mechanism of localized lesion formation in CNS inflammatory diseases.
We demonstrated that, after intracerebral ovalbumin (IC-OVA) microinjection, antigen drainage and transport by dendritic cells to the cervical lymph nodes are crucial in directing antigen-specific accumulation of CD8+ T-cells in the brain (Ling et al., 2003; Karman et al., 2004a). Here we established an experimental system to address the contribution of antigen localization and traumatic injury to the preferential distribution of antigen-specific CD8+ T-cells in the CNS. We focused on CD8+ T-cells, because they often undergo much more vigorous clonal expansion than CD4+ T-cells in the CNS during autoimmune diseases including multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (Booss et al., 1983; Woodroofe et al., 1986; Hayashi et al., 1988; Gay et al., 1997; Babbe et al., 2000; Brabb et al., 2000; Sun et al., 2001). We demonstrated that antigen-specific CD8+ T-cells were recruited to antigen-containing sites as well as to sites of trauma in the brain. The mechanisms of CD8+ T-cell accumulation at these two different sites are regulated differentially. The process of antigen-specific T-cell accumulation at traumatic injury sites can contribute to the pathogenesis of T-cell-mediated inflammatory diseases in the CNS.
Materials and Methods
Experimental animals. C57BL/6 female mice, 5–7 weeks old, were purchased from The Jackson Laboratory (Bar Harbor, ME). OT-I mice transgenic for a T-cell receptor recognizing OVA257–264 (SIINFEKL) peptide bound to H2-Kb (OT-I) on the C57BL/6 background (Kelly et al., 1993; Hogquist et al., 1994) were provided by Dr. Kris Hogquist (University of Minnesota, Minneapolis, MN) and used at 6–10 weeks of age. All mice were housed in a pathogen-free facility at the University of Wisconsin, Medical School Animal Care Unit. All experiments involving mice were performed in accordance with published National Institutes of Health guidelines and approved by the Committee on Animal Care, University of Wisconsin–Madison.
Tetramer, antibodies, and protein antigen reagents. MHC I-Kb/SIINFEKL tetramer complexes conjugated with streptavidin–allophycocyanin (APC) (SIINFEKL-APC) were provided by the MHC Tetramer Core Facility, National Institute of Allergy and Infectious Diseases (Atlanta, GA). MHC I-Kb/SIINFEKL tetramer complexes conjugated with streptavidin–phycoerythrin (PE) (SIINFEKL-PE) were purchased from Beckman Coulter (Fullerton, CA). Each batch of SIINFEKL tetramer complexes was titrated and normalized with OT-I splenic T-cells before use in experiments.
Antibodies anti-CD8a (clone 53-6.7; conjugated to PE, FITC, or APC), anti-interferon-γ (IFNγ) (clone 2572311; conjugated to PE), and anti-Vα2 (clone B20.1; conjugated to FITC or APC) were purchased from PharMingen (San Diego, CA). Antibodies purified from hybridoma lines producing anti-CD11b/Mac-1 (clone M1/70; rat IgG2b), anti-CD45R/B220 (clone RA3-6B2; rat IgG2a), anti-CD11a/CD18 [leukocyte function-associated antigen 1 (LFA-1); clone 2D7], anti-CD49d/CD29 [very late antigen-4 (VLA-4); clone PS/2], anti-CD62L (clone Mel 14), and anti-CD154 (CD40L; clone MR-1) (American Type Culture Collection, Rockville, MD) were conjugated with FITC or cyanine 5 as described previously (Fabry et al., 1992). An isotype control antibody, rat IgG isotype (clone 554685; conjugated to PE), was also purchased from PharMingen and used for background compensation control in cytofluorimetry and immunohistochemistry.
OVA (albumin from chicken egg white; grade 98%) and pigeon cytochrome c (PCC) proteins were purchased from Sigma (St. Louis, MO). OVA and PCC preparations were tested for endotoxin content and showed no B-cell-activating capacity for naive splenocytes isolated from C57BL/6 mice (data not shown).
Immunization with OVA protein antigen and application of anti-CD154 antibodies. For IC antigen delivery, mice were anesthetized by intraperitoneal injection of a ketamine (90 mg/kg)–xylazine (10 mg/kg) mixture. OVA (60 μg/20 μl sterile PBS) or an equal volume of PBS was injected into the right frontal lobe with an insulin syringe attached to a penetrating depth controller. The injection was restricted to the ventral–posterior region of the frontal lobe and a penetrating depth of 1.5 mm from the surface of the brain. Solution was injected slowly, after which the syringe was held in place for 1 additional minute to reduce back filling of injected solution. In this study, a single IC-OVA immunization was sufficient to induce a significant OVA-specific immune response in the CNS and was used for most experiments. To dissect the effect of a small traumatic injury and an irrelevant protein antigen on the recruitment of OVA-specific CD8+ T-cells into the brain, a vehicle (PBS) or an irrelevant protein antigen, PCC, was administrated. These control IC immunizations usually do not lead to a detectable OVA-specific immune response. To boost and maximize the magnitude of an OVA-specific immune response, IC immunizations were repeated 14 d after the primary injection in these experiments.
For subcutaneous (SC) antigen delivery, 100 μg of OVA diluted in 50 μl of PBS was emulsified thoroughly with an equal volume of Complete Freund's Adjuvant (CFA) (Sigma). Experimental animals were injected subcutaneously into the subscapular region with the emulsified OVA (100 μg/100 μl), and SC-OVA immunization was repeated 14 d after the primary injection to boost the OVA-specific immune response.
For antibody treatment, mice received a single intraperitoneal injection of purified anti-CD154 monoclonal antibody (CD40L clone MR-1; 200 μg/200 μl of PBS) immediately after aseptic cerebral injury (ACI).
Induction of ACI. ACI, a well characterized experimental model, was induced after the standard procedure (Swartz et al., 2001). Briefly, C57BL/6 mice were deeply anesthetized and placed on a stereotaxic instrument (Stoelting, Wood Dale, IL). The skull was exposed by a 1- to 2-cm sagittal incision in the skin, and a liquid nitrogen-chilled steel rod (3 mm in diameter), centered ∼2 mm posterior to bregma and 1.2 mm lateral to midline on the left side, was held against the exposed intact skull for 6 s. The incision was stitched, and the mice were monitored until they awakened.
Peripheral irradiation of experimental animals. To test whether CNS persisting antigen-specific CD8+ T-cells are recruited continuously from the periphery, mice received peripheral irradiation 1 week after IC-OVA immunization. Lead shielding over the head, not including the cervical area, was used to protect lymphocytes in the brain while the mice were irradiated at 900 rad with an x-ray machine. One week later, mice were killed, and lymphocytes were isolated.
To generate antigen-presenting cells for in vitro antigen presentation assays, isolated splenic lymphocytes from naive C57BL/6 mice (1 × 107 cells per milliliter) were irradiated at 2000 rad with a Mark 1 Shepherd irradiator (J. L. Shepherd and Associates, San Fernando, CA).
Lymphocyte isolation. Seven days after the final immunization, or 2 d after ACI, experimental mice and age-matched naive mice were deeply anesthetized and perfused transcardially with PBS. Spleens, cervical lymph nodes (CLNs), and brains were dissected, weighed, and transferred into cold HBSS (Cellgro, Herndon, VA). Spleen and CLN tissues were processed to single-cell suspensions by standard methods (Qing et al., 2000). Brain tissue was minced with scissors and processed to single-cell suspensions with a Medimachine (Becton Dickinson, San Jose, CA). Lymphocytes from brain were isolated from the interface of a 50:30% Percoll (Amersham Biosciences, Piscataway, NY) gradient as described previously (Sewell et al., 2004). For some mice, antigen was injected into one hemisphere, and ACI was induced in the contralateral hemisphere. After these procedures, brains were dissected along the midline, two to three hemispheres with the same treatment were pooled, and lymphocytes were isolated from each group of hemispheres as described above.
Blood contamination of brain lymphocyte preparations was analyzed with proflavin staining (Rodriguez-Peralta, 1968; Brabb et al., 2000). In these experiments, mice received intravenous administration of proflavin hydrochloride (20 μg/ml, 200 μl per mouse) (Sigma) and were perfused with PBS 10 min later. More than 85% of lymphocytes isolated from the brain were negative for proflavin (data not shown), indicating a very low level of contamination by blood lymphocytes.
The isolated lymphocytes were washed with cold HBSS twice and resuspended in staining buffer (HBSS containing 5% fetal bovine serum and 0.1% NaN3). For each brain sample, the total number of isolated cells per gram of tissue was calculated. The phenotype of T-cells was examined by pooling lymphocytes isolated from five identically treated mice and evenly separating these cells into three or four groups for flow cytofluorimetry analysis.
Carboxyfluorescein diacetate succinimidyl ester labeling and adoptive transfer of lymphocytes. Carboxyfluorescein diacetate succinimidyl ester (CFSE) passively diffuses into cells. It is colorless and nonfluorescent until the acetate groups are removed by intracellular esterases to yield highly fluorescent carboxyfluorescein succinimidyl ester. The succinimidyl ester group reacts with intracellular amines, forming fluorescent conjugates that are retained in the cytoplasm; therefore, when cells divide, CFSE labeling is distributed equally between two daughter cells, which are half as fluorescent as the parents. As a result, each successive generation in a population of proliferating cells is marked by a halving of fluorescent intensity that is readily detected by a flow cytometer (Lyons, 1999). Single-cell suspensions from spleens and brains of C57BL/6 or OT-I mice (2 × 107cells per milliliter) were incubated with 2.5 μm CFSE in HBSS for 10 min at 37°C. Fetal bovine serum was added to a concentration of 20% to arrest CFSE staining. Cells washed in PBS were processed for flow cytometry analysis, in vitro rechallenge, or adoptive transfer. For adoptive transfer, 1 × 107 cells in 200 μl of PBS were injected into the tail vein of each mouse 24 h before antigen microinjection into the CNS.
Analysis of proliferation of brain lymphocytes in vitro. Proliferation of lymphocytes isolated from brains of IC-OVA-immunized or naive C57BL/6 mice was assessed. Cells from two to four mice treated identically were pooled, labeled with CFSE, placed into 96-well culture plates (1 × 105 CFSE-stained cells per well in 200 μl of medium), and cocultured with antigen-presenting cells (4 × 105 per well; irradiated naive C57BL/6 spleen cells) in the presence or absence of 5 μg/ml OVA. Three days later, cells were harvested, stained, and processed for flow cytofluorimetry analysis.
Cytofluorimetry analysis of cells isolated from brains, CLNs, and spleens. Cells (1 × 106 isolated from spleen or CLN or ∼5 × 104 from brain) were incubated with monoclonal antibodies, anti-CD11b/Mac-1 (FITC), -CD45R/B220 (FITC), -CD8a (PE or APC), -Vα2 (FITC or APC), and SIINFEKL tetramer (PE or APC) for 30–60 min on ice or at room temperature. Unlabeled antibody recognizing receptor II/III that binds the crystallizable fraction (Fc) of Ig molecule (clone 2.4G2) was used to block binding to Fc receptors.
Lymphocytes were analyzed by four-color flow cytometry (FACScalibur instrument; Becton Dickinson) for expression of intracellular proteins and cell-surface markers. Fluorescence-activated cell sorter data were analyzed with CellQuest (Becton Dickinson) or FlowJo (Tree Star, Ashland, OR). For each sample, cells were gated on the basis of positive and negative controls, and the percentage and the absolute number of CD8+ T-cells and antigen-specific CD8+ T-cells within the lymphocyte population were calculated.
Immunohistochemistry analysis and confocal microscopy. Mice under deep anesthesia were perfused with sterile PBS followed by 2% paraformaldehyde in PBS. Brains were postfixed in 2% paraformaldehyde in PBS and sectioned at 50 μm intervals with a vibratome. Free-floating sections were collected and costained with antibodies to CD8 and SIINFEKL tetramers (60 min on ice), according to the protocol provided by the MHC Tetramer Core Facility, National Institute of Allergy and Infectious Diseases. CD8+ T-cells and SIINFEKL+ cells were detected immunocytochemically in a series of sections, and their distribution was examined and imaged with a Bio-Rad MRC 1000 confocal laser scanning microscope (Bio-Rad, Hercules, CA) at 100–200× magnification. Digital data were exported into Adobe Photoshop (version 5.0) for further analysis and presentation. To visualize individually labeled cells and obtain semiquantitative data regarding the number of labeled cells among different brain regions, images (96 × 96 μm) were taken from the center of the IC-OVA injection, an anatomically correspondent area in the contralateral cerebral cortex, or the border region of the ACI on two series of sections of each brain (two mice in each group), under 1000× magnification. CD8+ and SIINFEKL+ single- or double-positive cells on each image were counted, and the group mean number of cells per square millimeter was calculated.
Statistical data analysis. Data were analyzed statistically with two-tailed t test. All values were expressed as mean ±SE.
Results
Intracerebral antigen delivery is required for antigen-specific CD8+ T-cell recruitment into the brain
We have demonstrated previously that antigen-specific CD8+ T-cells accumulate in the CNS and persist in the brain up to 42 d after IC-OVA microinjection (Ling et al., 2003). To dissect the effect of IC antigen injection and peripheral activation of T-cells on the recruitment of these cells into the brain, experimental animals were separated into four groups and received SC-OVA, IC-OVA, IC-PBS, or IC-PCC immunizations. Because a secondary immunization (with the same antigen) might achieve a higher magnitude of immune responses, mice in these experiments received the secondary immunization 14 d after the primary injection to boost OVA-specific immune response. Seven days after the last immunization, the percentage of peripheral SIINFEKL+ cells within the CD8+ T-cell population increased in both the spleens and CLNs of mice that received IC-OVA or SC-OVA immunizations, but not in mice given IC-PBS or IC-PCC injections (data not shown). In the brain, the percentage of CD8+ T-cells that were OVA specific (Fig. 1, top panels, dots in the top left) and the absolute number of SIINFEKL+ CD8+ T-cells (Fig. 1, bottom table) significantly increased only after IC-OVA microinjection (Fig. 1, IC-OVA in bottom table and top panel). Subcutaneous antigen deliveries (SC-OVA) or IC-PBS injections did not increase the antigen-specific T-cell frequency or absolute number in the brain (Fig. 1). In parallel, when mice received IC-PCC, an irrelevant protein antigen, the number of CD8+ T-cells (Fig. 1, dots in top left quadrant of each plot) increased significantly in the brain (more than 10-fold higher than that with IC-PBS), but without any detectable change in OVA-specific cells (SIINFEKL+).
Figure 1.

Intracerebral immunization is necessary to induce the accumulation of protein antigen-specific CD8+ T-cells in brain. C57BL/6 mice received an IC or SC injection of OVA, PBS, or PCC on days 0 and 14 and were perfused on day 21. Brain lymphocytes were isolated and analyzed according to Materials and Methods. The dot plots are gated on CD45R/B220– and CD11b/Mac-1– viable lymphocytes. The value in each plot is the percentage of SIINFEKL+ cells within the CD8+ T-cell population. Data presented are representative analyses of two separate experiments, each with eight mice. The group mean number of SIINFEKL+ CD8+, CD45R/B220–, and CD11b/Mac-1– T-cells per gram of brain tissue is also listed in the bottom table. *p < 0.001 in a t test.
Altogether these data indicate that both SC- and IC-OVA immunization can induce an antigen-specific immune response in peripheral lymphoid organs, but newly expanded and activated CD8+ T-cells do not migrate into the brain parenchyma without the presence of their specific (cognate) antigen in the brain. These data also suggest that local cognate antigen is necessary for the recruitment of antigen-specific CD8+ T-cells into the brain parenchyma, and peripheral activation itself, or a small-needle traumatic injury induced by PBS injection, is not sufficient to induce an accumulation of CD8+ T-cells in the brain. An IC immunization with PCC did not increase any recruitment of OVA-specific CD8+ T-cells, further confirming that IC T-cell recruitment was antigen specific.
Antigen-specific CD8+ T-cells enter the CNS after proliferation cycles in peripheral lymphoid organs
Antigen-specific CD8+ T-cells were recruited into the brain after IC protein injection, but it was not clear whether these antigen-specific T-cells expanded in the peripheral lymphoid organs in response to the presentation of antigen drained from the brain before their migration into the CNS or proliferated in the brain tissue in response to local antigen presentation. To address this concern, lymphocytes were isolated from OT-I mice and labeled with CFSE. OT-I mice are transgenic for a T-cell receptor recognizing OVA257–264 (SIINFEKL) peptide bound to H2-Kb (Kelly et al., 1993; Hogquist et al., 1994). In these mice, therefore, the number of OVA-specific CD8+ T-cells is >20 times higher than that in wild-type mice. CFSE+ OT-I cells were injected into the tail vein of C57BL/6 mice, and protein antigen (OVA) was injected intracerebrally 24 h later. Proliferation and localization of CFSE+ OVA-specific CD8+ T-cells were analyzed by cytofluorimetry, as shown in Figure 2. One day after IC-OVA injection, adoptively transferred (CFSE+) OVA-specific (SIINFEKL+) CD8+ T-cells appeared in the CLN and the spleen; however, there were no detectable CFSE-labeled cells in the brain at the same time. By 3 d after IC-OVA injection, adoptively transferred OT-I T-cells that had undergone multiple proliferation cycles were detected in higher numbers in the CLNs and with lower frequency in the spleen. At the same time, OT-I cells also appeared in the brain, but only cells that had already proliferated were found. Interestingly, 4 d later (day 7), all OT-I cells recovered from the brain showed an additional one to two cycles of proliferation (Fig. 2).
Figure 2.

Antigen-specific T-cells accumulate in the brain after completing multiple proliferation cycles in the periphery. C57BL/6 mice received an IC-OVA immunization 24 h after intravenous transfer of 1 × 107 spleen cells isolated from naive OT-I mice and stained with CFSE as described in Materials and Methods. Lymphocytes were isolated from spleens, CLNs, and brains 1, 3, or 7 d after IC-OVA immunization and stained with Kb/SIINFEKL tetramers and antibodies to CD8. The histograms are gated on viable CD8+CFSE+SIINFEKL+ cells (adoptively transferred OT-I cells). Data presented are representative analyses from 12 mice in two independent experiments.a
These data indicate that antigen-specific T-cells accumulate in the brain after peripheral expansion, and these cells also may be capable of further proliferation in situ in the brain tissue presumably in response to cognate antigen presentation by resident antigen-presenting cells.
Antigen-specific CD8+ T-cells preferentially localize to sites of cognate antigen deposition in the brain
We have demonstrated that OVA-specific CD8+ T-cells were recruited into the brain parenchyma after IC-OVA immunization. To define the localization pattern of these infiltrated CD8+ T-cells in the brain, we analyzed the distribution of both OVA-specific and nonspecific CD8+ T-cells in brain sections 7 d after a single IC-OVA immunization. SIINFEKL tetramer reagent and anti-CD8 antibody were used in this confocal microscopy study to identify individual OVA-specific and nonspecific CD8+ T-cells and to demonstrate the distribution of these cells in the brain (Fig. 3A–D). We also quantitatively analyzed the number of antigen-specific CD8+ T-cells in each hemisphere with cytofluorimetry (Fig. 3F). Cells isolated from brain hemispheres containing antigen injection sites were compared with those isolated from the noninjected contralateral cerebral cortical area (Fig. 3E). In brains of nonimmunized C57BL/6 mice, OVA-specific CD8+ T-cells were undetectable by both cytofluorimetry analysis (Ling et al., 2003) and immunocytochemical–confocal analysis (data not shown). Antigen-specific CD8+ T-cells densely accumulated at the OVA microinjection site and were rarely detected in the contralateral brain hemisphere (Fig. 3A,B). In parallel with the visible localization of antigen-specific T-cells at the injection site, there was a significant increase in the percentage of both CD8+ and tetramer+ cells in the injected hemisphere as compared with the control brain hemisphere (Fig. 3F). In the cortical area contralateral to the antigen injection site, SIINFEKL tetramer+ cells were not detected, although a small number of nontetramer-specific CD8+ T-cells were scattered in the tissue (Fig. 3A,C). For further comparison, we counted and calculated the mean number of labeled cells per square millimeter (Fig. 3A–D, number at bottom left) in the center of the injection site and in an anatomically similar area of the contralateral cerebral cortex. These digital data, derived from selective and relatively small brain areas (4 × 96 × 96 μm), provided only semiquantitative information and did not represent the total number of lymphocytes in the brain hemisphere. Even with these restrictions, our data indicated a significant difference in the number of CD8+ and SIINFEKL+ cells between the antigen-containing and antigen-free regions in the brain. There was an increase in the number of CD8+ T-cells of >20-fold and an increase in the number of antigen-specific (SIINFEKL+) CD8 T-cells of >700-fold in the antigen-containing site.
Figure 3.
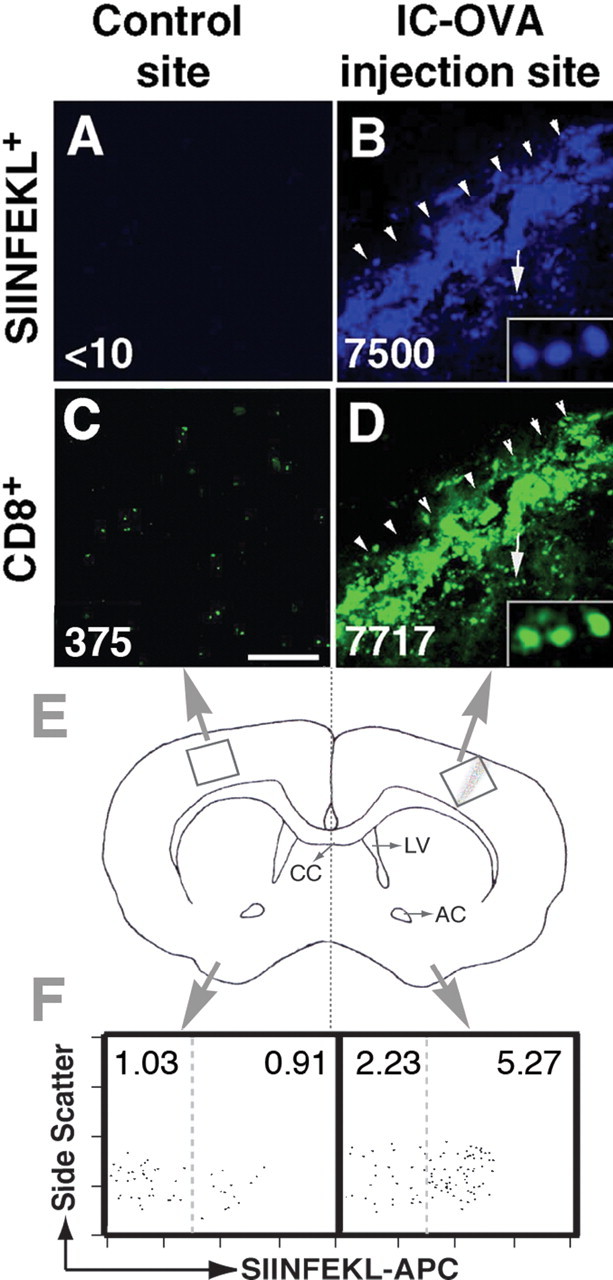
Preferential localization of antigen-specific (MHC I-Kb/SIINFEKL-tetramer+) CD8+ T-cells in the brain parenchyma after a single IC-OVA injection. C57BL/6 mice were perfused 7 d after IC-OVA immunization. Free-floating brain sections and brain lymphocyte suspensions were prepared as described in Materials and Methods. A–D, Confocal images show the specific localization of SIINFEKL+ (blue) and CD8+ T-cells (green) in the brain. The value in each image is the mean number of labeled cells per square millimeter counted at high magnification (1000×) in a square region (96 × 96 μm) in the center of the injection track or contralateral tissue section. B, D, Insets are enlargements of cells indicated by arrows. OVA injection track (B, D) is indicated by arrowheads. E, Schematic diagram illustrates brain regions analyzed. F, Biased accumulation of OVA-specific CD8+ T-cells between two cerebral hemispheres. Brains were dissected from midline (E, dotted line), and lymphocytes were isolated from each half of the brain. The dot plots are gated on CD8+, CD45R/B220–, and CD11b/Mac-1– viable lymphocytes. Values in each square are percentages of SIINFEKL negative (left) and positive (right) cells per total lymphocytes. Data are representative analyses for each group with at least six mice in two independent experiments. CC, Corpus callosum; AC, anterior commissure; LV, lateral ventricle. Scale bar, 250 μm.
Together, these immunocytochemical and cytofluorimetric analyses indicate that antigen-specific CD8+ T-cells preferentially localize to cognate antigen-containing sites in the brain and do not distribute throughout the tissue, likely because of antigen retention by local antigen-presenting cells after IC injection
The persistence of antigen-specific CD8+ T-cells in the CNS is not dependent on their continuous replenishment from the periphery and is not reduced by blocking CD154
We have shown that, after IC-OVA immunization, OVA-specific CD8+ T-cells expanded in the periphery, and newly proliferated cells were recruited into the brain parenchyma. Next we asked whether these cells were recruited continuously from the periphery or persisted in the brain. Experimental animals received peripheral irradiation at a dosage sufficient to abolish cell proliferation in peripheral lymphoid organs, including the CLNs, 7 d after a single IC-OVA immunization, when recruitment of OVA-specific CD8+ T-cells to the brain had reached its peak level. One week after irradiation, the percentage of antigen-specific cells within the CD8+ T-cell population and the mean number of lymphocytes isolated from each spleen and brain were measured and compared with nonirradiated mice given the same immunization. Figure 4 shows that the peripheral irradiation did not reduce either the percentage of CD8+ T-cells that were OVA-specific (Fig. 4, left cytofluorimetry panels) or the total number of brain lymphocytes (Fig. 4, left histogram). Although the percentage of antigen-specific cells CD8+ T-cells in the spleen did not change significantly (Fig. 4, right cytofluorimetry panels) (from 0.3 to 0.7%), the total number of lymphocytes in the spleen was significantly reduced after irradiation (Fig. 4, right histogram), indicating increased cell death and abolished cell proliferation as a result of irradiation. These results indicate that once recruited into the brain, OVA protein-specific CD8+ T-cells can persist in situ for an extended period of time without continual replenishment from peripheral lymphoid organs.
Figure 4.
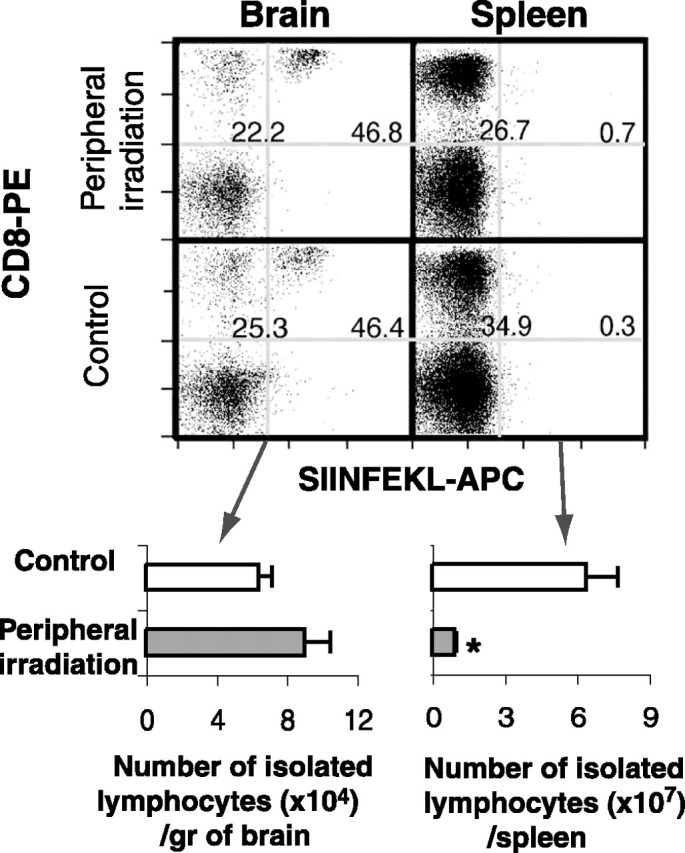
Antigen-specific CD8+ T-cells persist in the brain without replenishment from the periphery. C57BL/6 mice received peripheral irradiation 7 d after IC-OVA immunization and were perfused 1 week later, as described in Materials and Methods. Lymphocytes isolated from brains and spleens were analyzed by flow cytometry. The dot plots are gated on CD45R/B220–, CD11b/Mac-1– viable lymphocytes. The numbers in each plot represent the mean percentage of CD8+ cells within the B220, Mac-1-negative lymphocyte population (left) and the mean percentage of Kb/SIINFEKL+ cells within the CD8+ T-cell population (right), derived from six mice in two separate experiments. Histograms at the bottom show quantitative analyses of the total number of brain lymphocytes (left) and spleen lymphocytes (right) after peripheral irradiation. Bars represent the mean + SE number of CD8+ T-cells derived from six mice. *p < 0.01 in a t test.
It has been suggested that CD40–CD154-mediated interactions are crucial for antigen-specific T-cell activation and in developing T-cell-mediated inflammation in the CNS (Darabi et al., 2004). To directly assess the role of CD154-mediated interactions in the persistence of CD8+ T-cells in brain, we inhibited CD40–CD154 interactions by intraperitoneal injection of anti-CD154 antibodies 7 d after IC-OVA, when the recruitment of OVA-specific CD8+ T-cells has saturated. It has been known that intraperitoneally injected anti-CD154 can access the CNS and inhibit microglia-induced T-cell activation (Tan et al., 2002). Two days after anti-CD154 antibody treatment, CD8+ SIINFEKL+ T-cells in the brain hemisphere containing cognate antigen were analyzed quantitatively. A single treatment significantly reduced both the percentage and the absolute number of antigen-specific CD8+ T-cells in the brain (Fig. 5).
Figure 5.
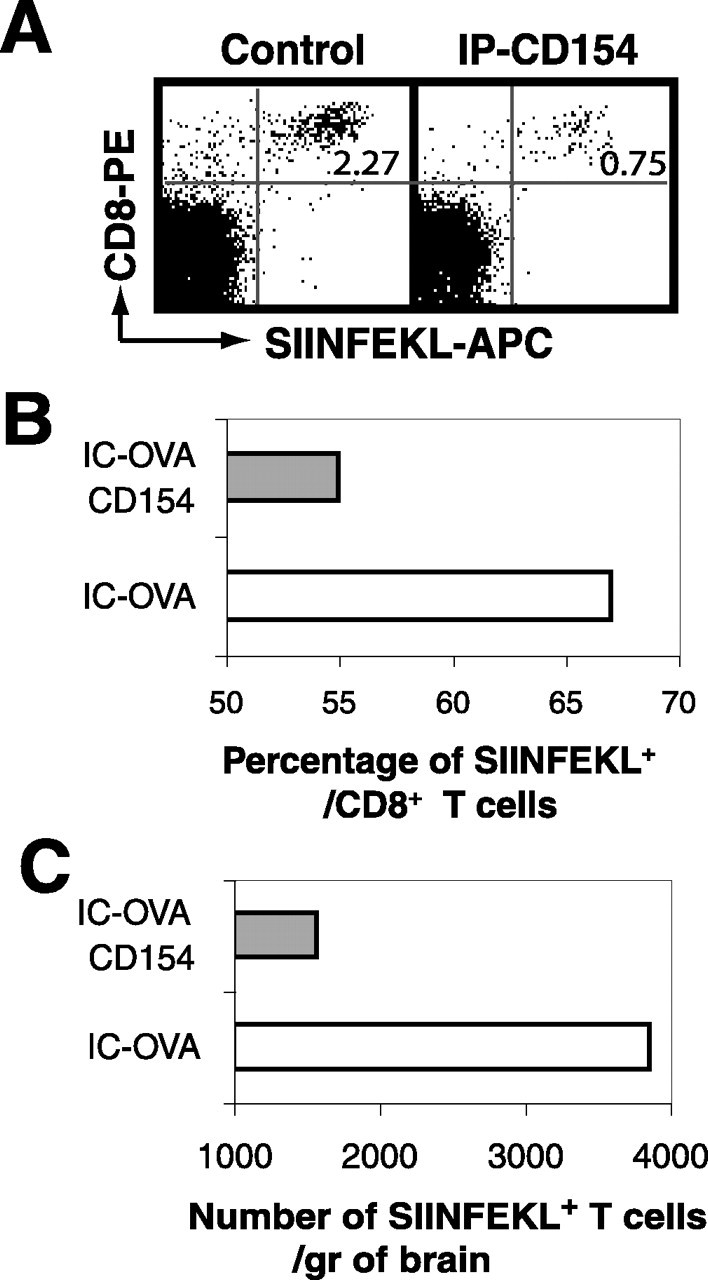
Involvement of CD154 in the persistence of antigen-specific T-cells in the brain parenchyma. C57BL/6 mice received IC-OVA immunization and a single intraperitoneal treatment of anti-CD154 or isotype control antibody 7 d later, as described in Materials and Methods. Two days after anti-CD154 or control treatments, lymphocytes were isolated from the hemisphere that received IC-OVA immunization and pooled for each group. A, Dot plots are gated on viable lymphocytes and show that a single treatment of anti-CD154 antibodies significantly reduces the percentage and absolute number of OVA-specific (SIINFEKL+)/CD8+ cells. The number in each plot is the percentage of SIINFEKL+ CD8+ cells. B, C, Quantitative analysis of the percentage of SIINFEKL+ cells within the CD8+ T-cell population (B) and the total number of SIINFEKL+ CD8+ T-cells (C) in each side of the cerebral hemisphere after anti-CD154 treatment. Data presented are derived from 6 mice and comparable with the result derived from 10 mice with the same treatment, but with a different reagent to detect OVA-specific CD8+ T-cells in two other independent experiments.
Altogether, these data indicate that the persistence of antigen-specific CD8+ T-cells in the antigen-containing site in the CNS is regulated by interactions mediated via CD154 molecules, in addition to the presence of cognate antigens.
Antigen-specific CD8+ T-cells express an activated phenotype in the brain and respond to antigenic recall stimulation by proliferation
It has been suggested that only peripherally activated T-cells are capable of migrating into the CNS; however, this statement has come under recent scrutiny, because the migration of nonactivated T-cells into the brain has also been demonstrated (Brabb et al., 2000). It has also been reported that antigen-specific T-cells may become anergic after being recruited into the CNS, which is an inhibitory environment for lymphocyte activation (Matsumoto et al., 1993; Irani et al., 1997; Brabb et al., 2000). To directly assess the activation status of infiltrated cells in our model, CD8+ T-cells isolated from the brains of OVA-microinjected animals were analyzed for the expression of the surface T-cell activation markers CD62L, CD11a/CD18 (LFA-1), and CD49d/CD29 (VLA-4). Seven days after IC-OVA immunization, only ∼1% of CD8+ T-cells in the spleen were OVA specific (SIINFEKL+), whereas >60% of brain isolated CD8+ T-cells bound SIINFEKL tetramers, indicating a significant accumulation of antigen-specific T-cells in the brain (Fig. 6A) in the absence of a global increase of OVA-specific CD8+ T-cells. In the spleen, most CD8+ cells showed a CD62Lhigh, an LFA-1low, and a VLA-4low naive phenotype (Fig. 6A, left panels). In contrast, most brain CD8+ T-cells, including SIINFEKL– cells, expressed a CD62Llow-, an LFA-1high-, and a VLA-4high-activated phenotype (Fig. 6A, right panels). These data demonstrate that in this experimental system, most of the brain-accumulating antigen-specific CD8+ T-cells carried an activated phenotype (91.5% are CD62Llow, 97% are LFA-1high, and 88% are VLA-4high).
Figure 6.
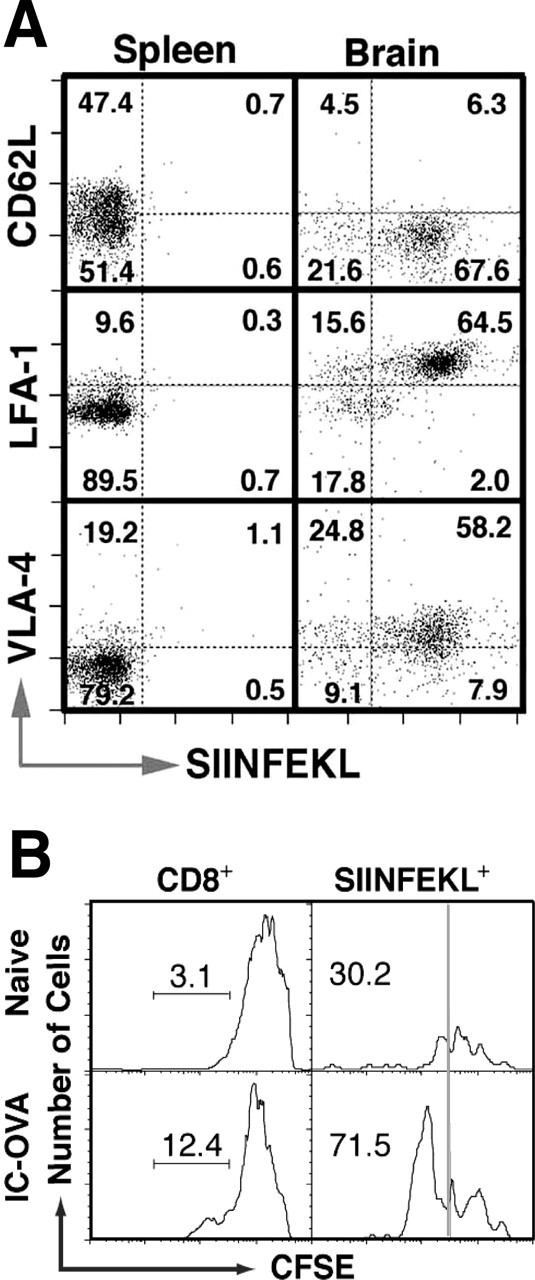
Brain OVA-specific CD8+ T-cells express an activated phenotype and respond to recall antigen stimulation by proliferation. A, Phenotype of OVA-specific (SIINFEKL+) CD8+ T-cells isolated from spleens (left) and brains (right) of C57BL/6 mice 7 d after IC-OVA immunization. Dot plots are gated on viable CD8+ T-cells. For each plot, the quadrant percentage is indicated. B, Proliferation of brain OVA-specific CD8+ T-cells in response to OVA stimulation in vitro. Lymphocytes were isolated from brains of C57BL/6 mice with or without IC-OVA immunization, stained with CFSE, and incubated with OVA for 3 d, as described in Materials and Methods. Histograms are gated on viable CD8+ cells (left panels) or SIINFEKL+ cells (right panels). The number in each plot is the percentage of proliferating cells within an indicated population.
To study whether antigen-specific CD8+ T-cells could respond to secondary antigenic stimulation, we isolated these cells from the brain 7 d after a single IC-OVA microinjection and restimulated them in a recall response with OVA in the presence of irradiated antigen-presenting cells. Figure 6B shows that brain-originated antigen-specific T-cells were capable of further proliferation in vitro, suggesting that these cells were functionally responsive; however, when intracellular IFNγ staining was performed, <2% of the brain CD8+ and SIINFEKL+ cells produced IFNγ in response to stimulation (data not shown).
These data indicate that OVA-specific CD8+ T-cells that accumulated in the brain after IC-OVA injection have an activated phenotype and are capable of proliferation in vitro after contact with their specific antigen, but produce little if any IFNγ.
Antigen-specific CD8+ T-cells also accumulate at sterile traumatic injury sites in the brain
In the previous experiment, we have shown that antigen-specific CD8+ T-cells preferentially migrate into the cognate antigen-containing injection site in the brain. To dissect the effect of sterile trauma in the absence of antigen presentation versus presented cognate antigen in CD8+ T-cell localization in the brain, ACI was induced on the contralateral hemisphere 7 d after OVA microinjection, when the brain recruitment of OVA-specific CD8+ T-cells had reached saturation (Ling et al., 2003). As described previously, ACI resulted in an inflammatory site in the brain where chemokines, cytokines, and complement components were upregulated (Swartz et al., 2001; Fee et al., 2004). Two days after ACI induction, the distribution of antigen-specific CD8+ T-cells in both hemispheres was analyzed in a manner similar to that described in Figure 3. In brains of mice with IC-OVA and ACI, increased numbers of CD8+ T-cells were seen in both the IC-OVA injection site and the ACI site (Fig. 7A), where no accumulation was observed in uninjected control brains (data not shown), although the density of CD8+ T-cells was relatively lower in the ACI site than in the IC-OVA injection site. Similar to the mice that received IC-OVA only, in the injected hemisphere, CD8+ T-cells preferentially accumulate in the injection site and were mostly SIINFEKL+ (Fig. 7A, top right panel). Fewer CD8+ T-cells in the ACI site were SIINFEKL+ (Fig. 7A, top left panel), suggesting that OVA-specific CD8+ T-cells were also capable of localizing to areas of inflammation in the absence of cognate antigen presentation in the brain. The mean number of labeled cells per square millimeter (the number in each image) was again calculated from limited, selected, and anatomically similar areas (4 × 96 × 96 μm) (Fig. 7, central area of black bars in the schematic diagram) to show a significant effect of IC-OVA and ACI on T-cell accumulation. Compared with mice that received IC-OVA only (Fig. 3, left panel), ACI resulted in a significant increase of CD8+ T-cells and OVA-specific (SIINFEKL+) T-cells in the hemisphere without IC-OVA (Fig. 7C). Cytofluorimetry analyses further indicated that both OVA-specific and nonspecific CD8+ T-cells were able to access a small sterile trauma inflammatory site in the brain, although more OVA-specific CD8+ T-cells remained at the antigen site (Fig. 7B). These data indicate that although OVA-specific CD8+ T-cells preferentially accumulate at antigen presentation sites, they also localize to sterile injury sites in the absence of in situ cognate antigen.
Figure 7.
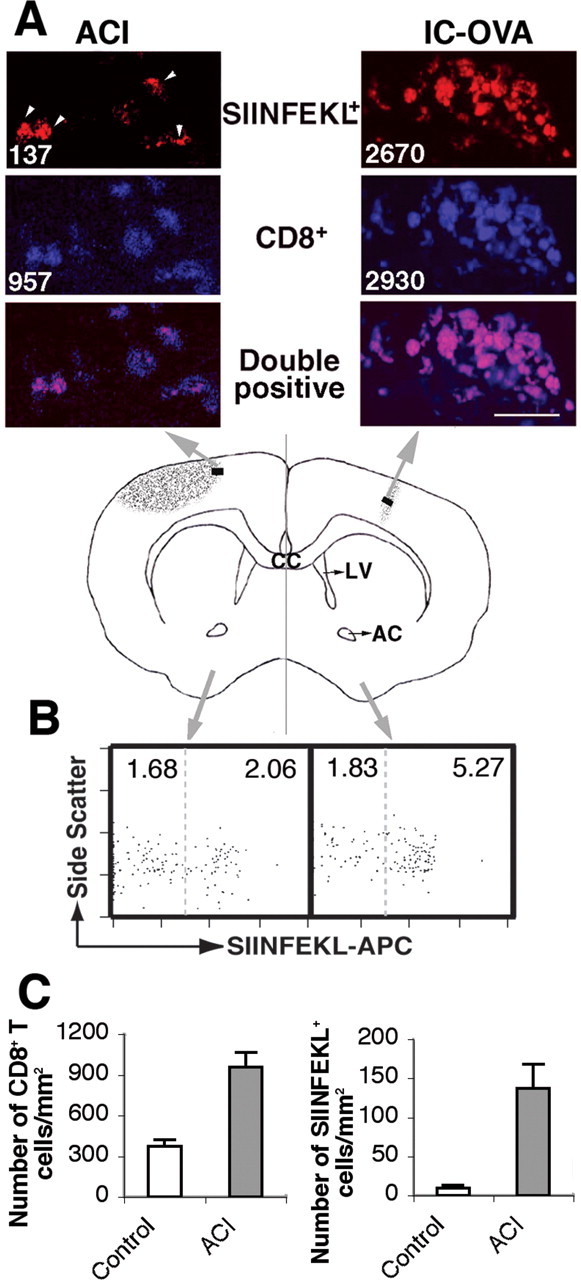
Antigen-specific CD8+ T-cells migrate to sterile traumatic injury sites in the brain. C57BL/6 mice received IC-OVA immunization on the right side of the cerebral cortex and an ACI on the left side 7 d later. Mice were perfused 2 d after ACI, and free-floating sections and lymphocyte suspensions were prepared from the brains, as described in Materials and Methods. A, Confocal images of brain sections show OVA-specific (SIINFEKL+; red) and CD8+ (blue) T-cells in the brain parenchyma. CD8+ T-cells, most of which are also SIINFEKL+, densely accumulate in the IC-OVA injection site (right panels). Comparatively fewer CD8+ T-cells are found in the ACI injury site (left panels), and only a portion of them are SIINFEKL+ (indicated by arrowheads). The value in each image is the number of labeled cells per square millimeter derived from the brain region (black rectangles) indicated in the schematic diagram of the middle panel. B, Flow cytometry analyses of brain lymphocytes. Lymphocytes were isolated from each half-brain and stained with Kb/SIINFEKL tetramers and antibodies to CD11b/Mac-1, CD45R/B220, and CD8. The plots are gated on CD8+, CD45R/B220–, and CD11b/Mac-1– viable lymphocytes. Values in each square are percentages of SIINFEKL negative (left) and positive (right) cells per total lymphocytes. C, Effect of ACI on the infiltration of CD8+ T-cells in the hemisphere contralateral to IC-OVA immunization. Left panel, CD8+ T-cells; right panel, CD8+SIINFEKL+T-cells. Data presented are representative for each group, with six mice in two independent experiments. CC, Corpus callosum; AC, anterior commissure; LV, lateral ventricle. Scale bar, 25 μm.
The accumulation of antigen-specific CD8+ T-cells at sterile trauma sites cannot be suppressed with anti-CD154 antibody injection
We have shown in Figure 5 that the persistence of antigen-specific CD8+ T-cells in the antigen-containing site was suppressed by anti-CD154 antibody treatment. To investigate the involvement of CD154-mediated interactions in the accumulation of antigen-specific CD8 T-cells in traumatic injury sites, ACI was induced on the contralateral hemisphere 7 d after OVA microinjection, and the accumulation of antigen-specific CD8+ T-cells was analyzed after a single anti-CD154 antibody treatment. Surprisingly, neither the percentage nor the absolute number of antigen-specific CD8+ T-cells at the ACI site was significantly affected by this treatment (Fig. 8). These data suggest that the accumulation of antigen-specific CD8+ T-cells in cognate antigen-expressing or trauma-induced inflammatory sites is regulated by different mechanisms: one involves CD154 molecular interactions, whereas the other does not.
Figure 8.
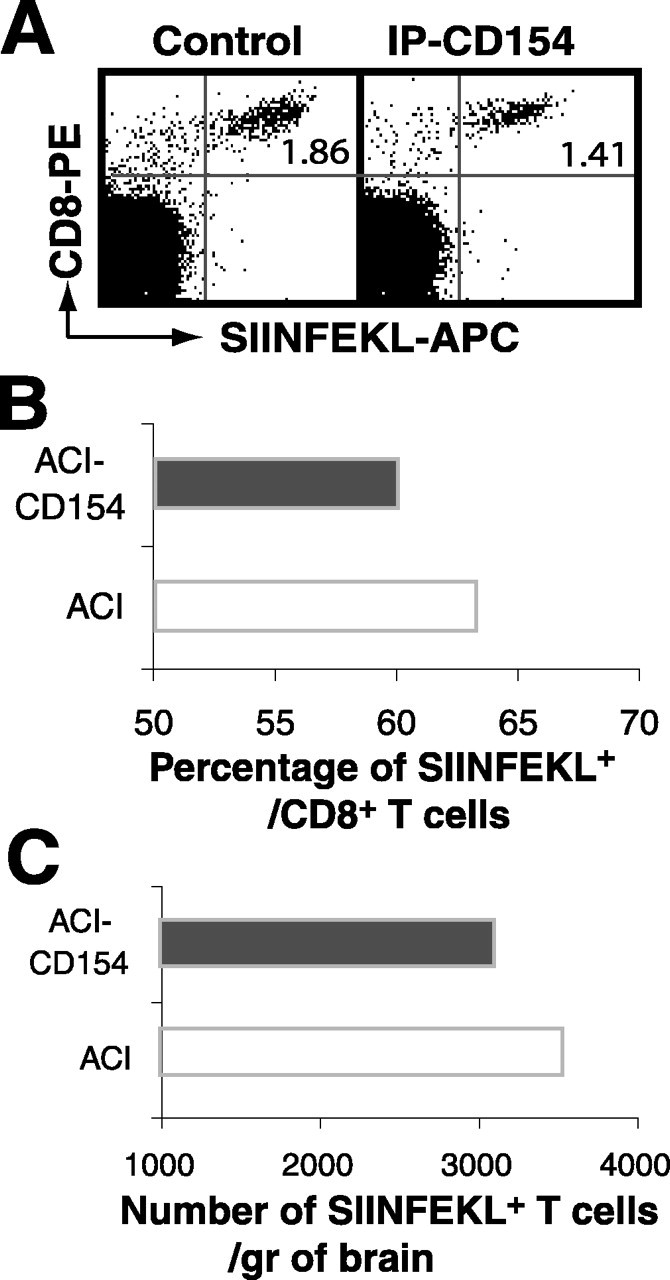
Anti-CD154 antibody treatment does not significantly modify the accumulation of antigen-specific T-cells in traumatic injury sites. C57BL/6 mice received IC-OVA immunization on the right side of the cerebral cortex and an ACI on the left side 7 d later. Immediately after ACI, mice also received a single intraperitoneal treatment of anti-CD154 or isotype control antibodies as described in Materials and Methods. Two days after anti-CD154 or control treatments, lymphocytes were isolated from the hemisphere with ACI, pooled for each group, and analyzed. A, Dot plots are gated on viable lymphocytes, showing that a single treatment of anti-CD154 antibodies significantly reduces neither the percentage nor the absolute number of OVA-specific (SIINFEKL+)/CD8+ cells in the hemisphere with ACI. The number in each plot is the quadrant percentage for SIINFEKL+CD8+ cells. B, C, Quantitative analysis of the percentage of SIINFEKL+ cells within the CD8+ T-cell population (B) and the total number of SIINFEKL+ CD8+ T-cells (C) in the cerebral hemisphere after anti-CD154 treatment. Data presented are derived from six mice and are comparable with results derived from four mice with the same treatment, but with a different reagent to detect OVA-specific CD8+ T-cells in another independent experiment.
Discussion
Our study was designed to evaluate the contribution of cognate antigen and traumatic injury to the accumulation of antigen-specific CD8+ T-cells in the CNS under noninfectious conditions. We immunized mice with OVA, a soluble protein antigen. OVA is capable of eliciting a strong immune response, including the clonal expansion of OVA-specific CD8+ and CD4+ T-cells, through various routes of immunization; therefore, it has been widely used in studies of mechanisms that regulate immune responses (Kelly et al., 1993; Hogquist et al., 1994). We show here that after IC-OVA immunization, the localized retention of cognate antigen (IC-injected OVA) directs the recruitment and preferential localization of OVA-specific CD8+ T-cells to the CNS. Small sterile traumatic injury itself does not induce a detectable recruitment of CD8+ T-cells in the brain; however, preactivated antigen-specific CD8+ T-cells can localize to sites of traumatic injury in the brain.
Previously, we have shown that intracerebrally microinjected OVA protein was retained in CD11b+ cells at injection sites in the brain (Ling et al., 2003). In this study, we demonstrate that antigen-specific CD8+ T-cells preferentially accumulate at the same location. This result indicates that the local persistence of cognate antigen and antigen-presenting cells is critical for T-cell homing to and accumulation in the CNS as well as to other nonlymphoid organs (Calzascia et al., 2005; Obst et al., 2005). Whether CNS-accessing T-cells are mostly activated or naive has been the focus of multiple investigations. Our data demonstrate that regardless of their specificity, most of the brain-infiltrating antigen-specific CD8+ T-cells are activated, based on the expression of VLA-4high, LFA-1high, and CD62Llow (Fig. 5); however, systemic activation of antigen-specific T-cells leads to their accumulation in the brain only when their cognate antigen is present. IC immunization is required for recruitment of protein antigen-specific CD8+ T-cells to the brain parenchyma. It has been suggested from studying other nonlymphoid tissues that the route of immunization is crucial for the preferential homing of T-cells (Azukizawa et al., 2003; Itano et al., 2003; Mullins et al., 2003). In agreement, our data also suggest that the route of immunization is an important factor in regulating T-cell migration into the brain.
After IC immunization, antigen-specific CD8+ T-cells access the brain after multiple proliferation cycles in the periphery (Fig. 2) in response to drainage of brain protein antigen (Ling et al., 2003) and to migration of antigen-pulsed dendritic cells from the brain to the CLNs (Karman et al., 2004b). After migrating into the CNS, these antigen-specific CD8+ T-cells may also proliferate in situ (Fig. 2). Antigen-specific CD8+ T-cells can persist in the CNS for a long period of time without inducing any detectable inflammation (Ling et al., 2003) (data not shown). When these cells are isolated from the brain and rechallenged in vitro by their specific antigen, they are capable of proliferating (Fig. 6B). These results suggest that with optimal stimuli, brain protein antigen-specific CD8+ T-cells could be activated in vivo and participate in the amplification of T-cell-mediated immune responses in the CNS.
In addition to accumulating in the cognate antigen retention site, activated protein antigen-specific CD8+ T-cells can also accumulate at traumatic injury sites. Although their accumulation is much more prominent at cognate antigen-containing sites, a significantly increased number of antigen-specific T-cells can be observed at trauma sites. These antigen-specific T-cells have most likely migrated from cognate antigen-expressing sites. This conclusion is based on the fact that ACI was induced 7 d after antigen microinjection into the brain, and, at this time, the frequency of antigen-specific CD8+ T-cells returns to the preimmunization levels in peripheral lymphoid organs but remains constantly high in the brain (Ling et al., 2003). These antigen-specific T-cells persist in the brain without continuous replenishment from the periphery and preferentially accumulate in cognate antigen retention sites. The factors that are possibly involved in the migration of activated CD8+ T-cells from an antigen site to trauma sites in the brain are presently unknown. It is generally accepted that accumulation and localization of antigen-specific T-cells to antigen-containing sites are promoted by locally activated antigen-presenting cells. In agreement with this, we have shown that OVA-specific CD8+ T-cells preferentially accumulate in the IC-OVA injection site (Figs. 1, 2), where OVA has been processed and internalized by local antigen-presenting cells (Ling et al., 2003). Because proteolyzed OVA (indicating antigen presentation) is restricted mainly to the injection site and not detectable in the contralateral cortex where ACI is induced (Ling et al., 2003), it is unlikely that the initial migration of antigen-specific CD8+ T-cells to trauma sites in the brain is supported by antigen-presentation processes. Consistent with this notion, the accumulation and persistence of antigen-specific T-cells at trauma sites were not inhibited by blocking the CD154 costimulatory molecule (Fig. 8). CD40–CD154 interactions have been proposed to be critical in T-cell-mediated inflammation in the CNS (Darabi et al., 2004). CD40 is a transmembrane glycoprotein expressed by a large variety of antigen-presenting cells, including dendritic cells, macrophages, and microglia (Tan et al., 1999; Quezada et al., 2004). Its ligand, CD154 (CD40L), is an integral membrane protein expressed on activated, but not resting, T-cells (Bourgeois et al., 2002). During binding antigen, APCs upregulate CD40 expression and consequently present antigen to antigen-specific T-cells. Engagement of CD40–CD154 between APCs and activated antigen-specific T-cells induces the upregulation of costimulatory molecules and triggers cognate T-cells to expand clonally and to differentiate into effector cells. Likewise, the interruption of CD40–CD154 engagement may result in inadequate antigen presentation or T-cell tolerance (Quezada et al., 2004). Our data indicate that the persistence of antigen-specific CD8+ T-cells in an antigen-containing site in the CNS is regulated by interactions mediated via CD154 molecules, but the migration of brain antigen-specific CD8+ T-cells into trauma sites is independent of CD154-mediated interactions. This observation suggests that anti-CD40–CD154 blocking approaches proposed for T-cell-mediated CNS inflammatory diseases should be reevaluated when T-cell migration into trauma sites would need to be inhibited.
The function of protein antigen-specific T-cells at traumatic injury sites in the brain is somewhat controversial. Evidence indicates that injury-directed migration of T-cells would be beneficial in the chronic phase of traumatic injury healing processes in the CNS (Serpe et al., 1999, 2000, 2003; Hauben et al., 2000; Moalem et al., 2000; Schwartz, 2001; Hofstetter et al., 2003). It has been shown that after peripheral myelin basic protein immunization, both myelin basic protein-specific and nonspecific T-cells infiltrate the CNS and preferentially migrate to the site of traumatic injury. These T-cells produce multiple neurotrophic factors and dramatically reduce the loss of neurons around the injury sites (Hammarberg et al., 2000). After facial nerve transection, CNS antigen-specific CD4+ T-cells are recruited to facial nucleus and promote facial motoneuron survival, but non-CNS antigen-specific CD4+ T-cells fail to provide any support (Byram et al., 2004), suggesting that the protective function of T-cells may be antigen specific in the CNS.
In addition to their neuroprotective role, activated T-cells can also potentially exert neurodegenerative actions, as demonstrated by the demyelination in multiple sclerosis (a CNS autoimmune disease) (Carson, 2002; Yan et al., 2003; Nitsch et al., 2004; Aktas et al., 2005) and by the increased damage during the acute phases of traumatic brain injury (Fee et al., 2003; Hofstetter et al., 2003). Altogether these studies suggest a “double-edged sword” effect of antigen-specific T-cells in the CNS. The accumulation of protein antigen-specific CD8+ T-cells in traumatic injury sites may contribute to the focal appearance of lesions in the CNS in T-cell-mediated inflammation. The initial step in developing these lesions may be the induction of chemokines and proinflammatory mediators produced at trauma sites by endothelial cells of the blood–brain barrier and by infiltrating mononuclear cells (Whitney et al., 1999; Liu et al., 2001; Hulshof et al., 2002; Sorensen et al., 2002). The production of proinflammatory cytokines (such as interleukin-12-p35 and interleukin-6), chemokines [such as RANTES (regulated upon activation, normal T-cell expressed and secreted), Eotaxin, MIF (macrophage migration inhibitory factor), and MCP-1 (monocyte chemoattractant protein 1)], and complement activation products (such as C3 and C5) has been clearly demonstrated at brain trauma sites (Swartz et al., 2001; Fee et al., 2003, 2004). Many of these mediators may potentially affect T-cell migration. Undoubtedly, injuries in the CNS can trigger an inflammatory process involving the infiltration of leukocytes to the parenchyma. In various experimental models, circulating leukocytes adhere to the vascular wall through adhesion molecules, particularly vascular cell adhesion molecule 1 (VCAM-1); however, blockage with anti-VCAM-1 antibody did not influence cellular infiltration at traumatic injury sites in the brain (Justicia et al., 2005). This latter finding supports our hypothesis that leukocyte infiltration at traumatic injury sites is less influenced by adhesion molecules. Because of this unregulated infiltration, these cells might behave differently from those leukocytes that are more dependent on adhesion molecules for infiltration.
The accumulation of T-cells in the brain may contribute to the induction of T-cell-mediated inflammatory diseases in the CNS. Understanding the mechanisms that regulate such accumulation will help us to control these diseases. For example, it has been proposed that there is an early low-grade phase of MS lesion formation when the recirculation of lymphocytes is moderately enhanced, probably because of a low-grade vessel injury. This early low-grade infiltration profoundly influences the late phase of the disease and leads to an extensive immunopathology that is focused on the target organ, the CNS (Traugott, 1989). Factors leading to localized vessel injuries and early low-grade accumulation of T-cells in MS are not well understood. The result of this study provides information about possible causes of the early low-grade T-cell infiltration; thus it may accelerate the development of new therapeutic treatment to terminate or reduce the later phase of MS. In conclusion, our data indicate that protein antigen-specific CD8+ T-cells localize to cognate antigen-expressing sites and persist for an extended period of time without inducing further inflammation in the CNS. These antigen-specific CD8 T-cells can respond to a sterile injury in the CNS by migrating to trauma sites. Their accumulation at a trauma site in the brain may contribute to the induction of T-cell-mediated inflammatory diseases in the CNS.
Footnotes
This work was supported by National Institutes of Health Grant RO1-NS 37570-01A2 to Z.F. We thank Khen Macvilay for careful cytofluorimetry data acquisition, Satoshi Kinoshita for expert histopathology service, and Emily Reinke, Jozsef Karman, and Dr. Laura Hogan for their critical review of this manuscript and valuable comments.
Correspondence should be addressed to Dr. Zsuzsa Fabry, Department of Pathology, University of Wisconsin, 1300 University Avenue, 6130 Medical Sciences Center, Madison, WI 53706. E-mail: zfabry@facstaff.wisc.edu.
DOI:10.1523/JNEUROSCI.3502-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260731-11$15.00/0
References
- Aktas O, Smorodchenko A, Brocke S, Infante-Duarte C, Topphoff US, Vogt J, Prozorovski T, Meier S, Osmanova V, Pohl E, Bechmann I, Nitsch R, Zipp F (2005) Neuronal damage in autoimmune neuroinflammation mediated by the death ligand TRAIL. Neuron 46: 421–432. [DOI] [PubMed] [Google Scholar]
- Arand M, Melzner H, Kinzl L, Bruckner UB, Gebhard F (2001) Early inflammatory mediator response following isolated traumatic brain injury and other major trauma in humans. Langenbecks Arch Surg 386: 241–248. [DOI] [PubMed] [Google Scholar]
- Azukizawa H, Kosaka H, Sano S, Heath WR, Takahashi I, Gao XH, Sumikawa Y, Okabe M, Yoshikawa K, Itami S (2003) Induction of T-cell-mediated skin disease specific for antigen transgenically expressed in keratinocytes. Eur J Immunol 33: 1879–1888. [DOI] [PubMed] [Google Scholar]
- Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S, Ravid R, Rajewsky K (2000) Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med 192: 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booss J, Esiri MM, Tourtellotte WW, Mason DY (1983) Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci 62: 219–232. [DOI] [PubMed] [Google Scholar]
- Bourgeois C, Rocha B, Tanchot C (2002) A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science 297: 2060–2063. [DOI] [PubMed] [Google Scholar]
- Brabb T, von Dassow P, Ordonez N, Schnabel B, Duke B, Goverman J (2000) In situ tolerance within the central nervous system as a mechanism for preventing autoimmunity. J Exp Med 192: 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byram SC, Carson MJ, DeBoy CA, Serpe CJ, Sanders VM, Jones KJ (2004) CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J Neurosci 24: 4333–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzascia T, Masson F, Di Berardino-Besson W, Contassot E, Wilmotte R, Aurrand-Lions M, Ruegg C, Dietrich PY, Walker PR (2005) Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity 22: 175–184. [DOI] [PubMed] [Google Scholar]
- Carson MJ (2002) Microglia as liaisons between the immune and central nervous systems: functional implications for multiple sclerosis. Glia 40: 218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserr HF, DePasquale M, Harling-Berg CJ, Park JT, Knopf PM (1992) Afferent and efferent arms of the humoral immune response to CSF-administered albumins in a rat model with normal blood-brain barrier permeability. J Neuroimmunol 41: 195–202. [DOI] [PubMed] [Google Scholar]
- Darabi K, Karulin AY, Boehm BO, Hofstetter HH, Fabry Z, LaManna JC, Chavez JC, Tary-Lehmann M, Lehmann PV (2004) The third signal in T cell-mediated autoimmune disease? J Immunol 173: 92–99. [DOI] [PubMed] [Google Scholar]
- Fabry Z, Waldschmidt MM, Hendrickson D, Keiner J, Love-Homan L, Takei F, Hart MN (1992) Adhesion molecules on murine brain microvascular endothelial cells: expression and regulation of ICAM-1 and Lgp 55. J Neuroimmunol 36: 1–11. [DOI] [PubMed] [Google Scholar]
- Fee D, Crumbaugh A, Jacques T, Herdrich B, Sewell D, Auerbach D, Piaskowski S, Hart MN, Sandor M, Fabry Z (2003) Activated/effector CD4+ T cells exacerbate acute damage in the central nervous system following traumatic injury. J Neuroimmunol 136: 54–66. [DOI] [PubMed] [Google Scholar]
- Fee DB, Sewell DL, Andresen K, Jacques TJ, Piaskowski S, Barger BA, Hart MN, Fabry Z (2004) Traumatic brain injury increases TGF beta RII expression on endothelial cells. Brain Res 1012: 52–59. [DOI] [PubMed] [Google Scholar]
- Fontana A, Frei K, Bodmer S, Hofer E (1987) Immune-mediated encephalitis: on the role of antigen-presenting cells in brain tissue. Immunol Rev 100: 185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay FW, Drye TJ, Dick GW, Esiri MM (1997) The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis: identification and characterization of the primary demyelinating lesion. Brain 120: 1461–1483. [DOI] [PubMed] [Google Scholar]
- Hammarberg H, Lidman O, Lundberg C, Eltayeb SY, Gielen AW, Muhallab S, Svenningsson A, Linda H, van Der Meide PH, Cullheim S, Olsson T, Piehl F (2000) Neuroprotection by encephalomyelitis: rescue of mechanically injured neurons and neurotrophin production by CNS-infiltrating T and natural killer cells. J Neurosci 20: 5283–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauben E, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, Akselrod S, Neeman M, Cohen IR, Schwartz M (2000) Autoimmune T cells as potential neuroprotective therapy for spinal cord injury. Lancet 355: 286–287. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Morimoto C, Burks JS, Kerr C, Hauser SL (1988) Dual-label immunocytochemistry of the active multiple sclerosis lesion: major histocompatibility complex and activation antigens. Ann Neurol 24: 523–531. [DOI] [PubMed] [Google Scholar]
- Hensler T, Sauerland S, Bouillon B, Raum M, Rixen D, Helling HJ, Andermahr J, Neugebauer EA (2002) Association between injury pattern of patients with multiple injuries and circulating levels of soluble tumor necrosis factor receptors, interleukin-6 and interleukin-10, and polymorphonuclear neutrophil elastase. J Trauma 52: 962–970. [DOI] [PubMed] [Google Scholar]
- Hickey WF (1991) Migration of hematogenous cells through the bloodbrain barrier and the initiation of CNS inflammation. Brain Pathol 1: 97–105. [DOI] [PubMed] [Google Scholar]
- Hofstetter HH, Sewell DL, Liu F, Sandor M, Forsthuber T, Lehmann PV, Fabry Z (2003) Autoreactive T cells promote post-traumatic healing in the central nervous system. J Neuroimmunol 134: 25–34. [DOI] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR (1994) T cell receptor antagonist peptides induce positive selection. Cell 76: 17–27. [DOI] [PubMed] [Google Scholar]
- Hulshof S, Montagne L, De Groot CJ, Van Der Valk P (2002) Cellular localization and expression patterns of interleukin-10, interleukin-4, and their receptors in multiple sclerosis lesions. Glia 38: 24–35. [DOI] [PubMed] [Google Scholar]
- Ingulli E, Ulman DR, Lucido MM, Jenkins MK (2002) In situ analysis reveals physical interactions between CD11b+ dendritic cells and antigen-specific CD4 T cells after subcutaneous injection of antigen. J Immunol 169: 2247–2252. [DOI] [PubMed] [Google Scholar]
- Irani DN, Lin KI, Griffin DE (1997) Regulation of brain-derived T cells during acute central nervous system inflammation. J Immunol 158: 2318–2326. [PubMed] [Google Scholar]
- Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, Jenkins MK (2003) Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity 19: 47–57. [DOI] [PubMed] [Google Scholar]
- Justicia C, Martin A, Rojas S, Gironella M, Cervera A, Panes J, Chamorro A, Planas AM (2005) Anti-VCAM-1 antibodies did not protect against ischemic damage either in rats or in mice. J Cereb Blood Flow Metab, in press. [DOI] [PubMed]
- Karman J, Ling C, Sandor M, Fabry Z (2004a) Dendritic cells in the initiation of immune responses against central nervous system-derived antigens. Immunol Lett 92: 107–115. [DOI] [PubMed] [Google Scholar]
- Karman J, Ling C, Sandor M, Fabry Z (2004b) Initiation of immune responses in brain is promoted by local dendritic cells. J Immunol 173: 2353–2361. [DOI] [PubMed] [Google Scholar]
- Kelly JM, Sterry SJ, Cose S, Turner SJ, Fecondo J, Rodda S, Fink PJ, Carbone FR (1993) Identification of conserved T cell receptor CDR3 residues contacting known exposed peptide side chains from a major histocompatibility complex class I-bound determinant. Eur J Immunol 23: 3318–3326. [DOI] [PubMed] [Google Scholar]
- Lenzlinger PM, Morganti-Kossmann MC, Laurer HL, McIntosh TK (2001) The duality of the inflammatory response to traumatic brain injury. Mol Neurobiol 24: 169–181. [DOI] [PubMed] [Google Scholar]
- Ling C, Sandor M, Fabry Z (2003) In situ processing and distribution of intracerebrally injected OVA in the CNS. J Neuroimmunol 141: 90–98. [DOI] [PubMed] [Google Scholar]
- Liu JS, Zhao ML, Brosnan CF, Lee SC (2001) Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol 158: 2057–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y, Zou L, Liu H, Lu H, Yuan X, Robertson CS, Yang K (2003) Altered expression of randomly selected genes in mouse hippocampus after traumatic brain injury. J Neurosci Res 71: 710–720. [DOI] [PubMed] [Google Scholar]
- Lyons AB (1999) Divided we stand: tracking cell proliferation with carboxyfluorescein diacetate succinimidyl ester. Immunol Cell Biol 77: 509–515. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Hanawa H, Tsuchida M, Abo T (1993) In situ inactivation of infiltrating T cells in the central nervous system with autoimmune encephalomyelitis: the role of astrocytes. Immunology 79: 381–390. [PMC free article] [PubMed] [Google Scholar]
- McGavern DB, Truong P (2004) Rebuilding an immune-mediated central nervous system disease: weighing the pathogenicity of antigen-specific versus bystander T cells. J Immunol 173: 4779–4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGavern DB, Homann D, Oldstone MB (2002) T cells in the central nervous system: the delicate balance between viral clearance and disease. J Infect Dis 186 [Suppl 2]: S145–S151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moalem G, Yoles E, Leibowitz-Amit R, Muller-Gilor S, Mor F, Cohen IR, Schwartz M (2000) Autoimmune T cells retard the loss of function in injured rat optic nerves. J Neuroimmunol 106: 189–197. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T (2001) Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock 16: 165–177. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T (2002) Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care 8: 101–105. [DOI] [PubMed] [Google Scholar]
- Mullins DW, Sheasley SL, Ream RM, Bullock TN, Fu YX, Engelhard VH (2003) Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J Exp Med 198: 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsch R, Pohl EE, Smorodchenko A, Infante-Duarte C, Aktas O, Zipp F (2004) Direct impact of T cells on neurons revealed by two-photon microscopy in living brain tissue. J Neurosci 24: 2458–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obst R, van Santen HM, Mathis D, Benoist C (2005) Antigen persistence is required throughout the expansion phase of a CD4+ T cell response. J Exp Med 201: 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing Z, Sewell D, Sandor M, Fabry Z (2000) Antigen-specific T cell trafficking into the central nervous system. J Neuroimmunol 105: 169–178. [DOI] [PubMed] [Google Scholar]
- Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ (2004) CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol 22: 307–328. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Kivisakk P, Kidd G (2003) Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol 3: 569–581. [DOI] [PubMed] [Google Scholar]
- Ray SK, Dixon CE, Banik NL (2002) Molecular mechanisms in the pathogenesis of traumatic brain injury. Histol Histopathol 17: 1137–1152. [DOI] [PubMed] [Google Scholar]
- Reinhardt RL, Bullard DC, Weaver CT, Jenkins MK (2003) Preferential accumulation of antigen-specific effector CD4 T cells at an antigen injection site involves CD62E-dependent migration but not local proliferation. J Exp Med 197: 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Peralta LA (1968) Hematic and fluid barriers of the retina and vitreous body. J Comp Neurol 132: 109–124. [DOI] [PubMed] [Google Scholar]
- Schwartz M (2001) Harnessing the immune system for neuroprotection: therapeutic vaccines for acute and chronic neurodegenerative disorders. Cell Mol Neurobiol 21: 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe CJ, Kohm AP, Huppenbauer CB, Sanders VM, Jones KJ (1999) Exacerbation of facial motoneuron loss after facial nerve transection in severe combined immunodeficient (scid) mice. J Neurosci 19: RC7(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe CJ, Sanders VM, Jones KJ (2000) Kinetics of facial motoneuron loss following facial nerve transection in severe combined immunodeficient mice. J Neurosci Res 62: 273–278. [DOI] [PubMed] [Google Scholar]
- Serpe CJ, Coers S, Sanders VM, Jones KJ (2003) CD4+ T, but not CD8+ or B, lymphocytes mediate facial motoneuron survival after facial nerve transection. Brain Behav Immunol 17: 393–402. [DOI] [PubMed] [Google Scholar]
- Sewell DL, Nacewicz B, Liu F, Macvilay S, Erdei A, Lambris JD, Sandor M, Fabry Z (2004) Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol 155: 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen TL, Trebst C, Kivisakk P, Klaege KL, Majmudar A, Ravid R, Lassmann H, Olsen DB, Strieter RM, Ransohoff RM, Sellebjerg F (2002) Multiple sclerosis: a study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J Neuroimmunol 127: 59–68. [DOI] [PubMed] [Google Scholar]
- Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, Raine CS (2001) Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol 166: 7579–7587. [DOI] [PubMed] [Google Scholar]
- Swartz KR, Liu F, Sewell D, Schochet T, Campbell I, Sandor M, Fabry Z (2001) Interleukin-6 promotes post-traumatic healing in the central nervous system. Brain Res 896: 86–95. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M (1999) Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science 286: 2352–2355. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Mullan M (2002) CD40-CD40L interaction in Alzheimer's disease. Curr Opin Pharmacol 2: 445–451. [DOI] [PubMed] [Google Scholar]
- Traugott U (1989) Detailed analysis of early immunopathologic events during lesion formation in acute experimental autoimmune encephalomyelitis. Cell Immunol 119: 114–129. [DOI] [PubMed] [Google Scholar]
- Vass K, Lassmann H, Wekerle H, Wisniewski HM (1986) The distribution of Ia antigen in the lesions of rat acute experimental allergic encephalomyelitis. Acta Neuropathol (Berl) 70: 149–160. [DOI] [PubMed] [Google Scholar]
- Whitney LW, Becker KG, Tresser NJ, Caballero-Ramos CI, Munson PJ, Prabhu VV, Trent JM, McFarland HF, Biddison WE (1999) Analysis of gene expression in multiple sclerosis lesions using cDNA microarrays. Ann Neurol 46: 425–428. [DOI] [PubMed] [Google Scholar]
- Williams KC, Hickey WF (1995) Traffic of hematogenous cells through the central nervous system. Curr Top Microbiol Immunol 202: 221–245. [DOI] [PubMed] [Google Scholar]
- Woodroofe MN, Bellamy AS, Feldmann M, Davison AN, Cuzner ML (1986) Immunocytochemical characterisation of the immune reaction in the central nervous system in multiple sclerosis: possible role for microglia in lesion growth. J Neurol Sci 74: 135–152. [DOI] [PubMed] [Google Scholar]
- Xiao BG, Link H (1999) Antigen-specific T cells in autoimmune diseases with a focus on multiple sclerosis and experimental allergic encephalomyelitis. Cell Mol Life Sci 56: 5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SS, Wu ZY, Zhang HP, Furtado G, Chen X, Yan SF, Schmidt AM, Brown C, Stern A, LaFaille J, Chess L, Stern DM, Jiang H (2003) Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat Med 9: 287–293. [DOI] [PubMed] [Google Scholar]