

Abstract
Spinal and bulbar muscular atrophy (SBMA) is a hereditary neurodegenerative disease caused by an expansion of a trinucleotide CAG repeat encoding the polyglutamine tract in the androgen receptor (AR) gene. To elucidate the pathogenesis of polyglutamine-mediated motor neuron dysfunction, we investigated histopathological and biological alterations in a transgenic mouse model of SBMA carrying human pathogenic AR. In affected mice, neurofilaments and synaptophysin accumulated at the distal motor axon. A similar intramuscular accumulation of neurofilament was detected in the skeletal muscle of SBMA patients. Fluoro-gold labeling and sciatic nerve ligation demonstrated an impaired retrograde axonal transport in the transgenic mice. The mRNA level of dynactin 1, an axon motor for retrograde transport, was significantly reduced in the SBMA mice resulting from pathogenic AR-induced transcriptional dysregulation. These pathological events were observed before the onset of neurological symptoms, but were reversed by castration, which prevents nuclear accumulation of pathogenic AR. Overexpression of dynactin 1 mitigated neuronal toxicity of the pathogenic AR in a cell culture model of SBMA. These observations indicate that polyglutamine-dependent transcriptional dysregulation of dynactin 1 plays a crucial role in the reversible neuronal dysfunction in the early stage of SBMA.
Keywords: polyglutamine, spinal and bulbar muscular atrophy, androgen, neurofilament, axonal transport, retrograde, dynactin
Introduction
Spinal and bulbar muscular atrophy (SBMA), or Kennedy's disease, is a hereditary neurodegenerative disease resulting from a loss of bulbar and spinal motor neurons (Kennedy et al., 1968; Sobue et al., 1989). Patients present with muscle atrophy and weakness of proximal limbs associated with bulbar palsy, tongue atrophy and contraction fasciculation (Katsuno et al., 2006). The disease affects exclusively adult males, whereas females carrying the mutant androgen receptor (AR) are seldom symptomatic (Schmidt et al., 2002). The molecular basis of SBMA is an expansion of a trinucleotide CAG repeat, which encodes the polyglutamine tract in the first exon of the AR gene (La Spada et al., 1991). This type of mutation has also been found to cause a variety of neurodegenerative disorders, termed polyglutamine diseases, such as Huntington's disease (HD), several forms of spinocerebellar ataxia, and dentatorubral pallidoluysian atrophy (Gatchel and Zoghbi, 2005). Although expression of the causative gene in each of these diseases is ubiquitous, selective neuronal cell death is observed in disease-specific areas of the CNS, suggesting a common molecular basis for these polyglutamine diseases.
Nuclear accumulation of pathogenic protein containing elongated polyglutamine is a crucial step in the pathophysiology of these diseases, providing an important therapeutic target (Adachi et al., 2005; Banno et al., 2006). The aberrant polyglutamine protein has a propensity to form aggregates in the nucleus and inhibits the function of transcriptional factors and coactivators, resulting in transcriptional perturbation (Cha, 2000; Gatchel and Zoghbi, 2005). In support of this hypothesis, altered expression of a variety of genes has been demonstrated in transgenic mouse models of polyglutamine diseases (Sugars and Rubinsztein, 2003). Although polyglutamine-induced transcriptional dysregulation is likely to be central to the pathogenesis of polyglutamine diseases, it has yet to be elucidated which genes are responsible for the selective neurodegeneration (Gatchel and Zoghbi, 2005).
No treatments have been established for polyglutamine diseases, but the androgen blockade therapy, surgical or medical castration, has shown striking therapeutic effects in the SBMA transgenic mouse model (Katsuno et al., 2002, 2003; Chevalier-Larsen et al., 2004). Androgen deprivation strongly inhibits the ligand-dependent nuclear accumulation of pathogenic AR protein, resulting in a striking improvement in neurological and histopathological findings of male mice.
In the present study, we investigated the molecular pathophysiology of motor neuron dysfunction in a transgenic mouse model of SBMA. Polyglutamine-induced transcriptional dysregulation of the dynactin p150 subunit (dynactin 1), an axonal motor-associated protein, resulted in perturbation of retrograde axonal transport in spinal motor neurons in the early stage of the disease. These processes were reversed by castration, which inhibits nuclear accumulation of pathogenic AR. A defect in axonal trafficking of neurofilaments and synaptic vesicles, the potential molecular basis for the reversible pathogenesis, appears to contribute to the initiation of symptoms, and may account for the selective degeneration of motor neurons in SBMA.
Materials and Methods
Generation and maintenance of transgenic mouse.
AR-24Q and AR-97Q mice were generated as described previously (Katsuno et al., 2002). Briefly, the full-length human AR fragment harboring 24 or 97 CAGs was subcloned into the HindIII site of the pCAGGS vector (Niwa et al., 1991) and microinjected into BDF1-fertilized eggs. Five founders with AR-97Q were obtained. These mouse lines were maintained by backcrossing them to C57BL/6J mice. All symptomatic lines (2–6, 4–6, and 7–8) were examined in the present study. All animal experiments were approved by the Animal Care Committee of the Nagoya University Graduate School of Medicine. Mice were given sterile water ad libitum. In the experiments where it was called for, sodium butyrate [a histone deacetylase (HDAC) inhibitor] was administered at a concentration of 4 g/L in distilled water from 5 weeks of age until the end of the analysis, as described previously (Minamiyama et al., 2004).
Neurological testing and castration after onset.
Mice were subjected to the Rotarod task (Economex Rotarod; Colombus Instruments, Columbus, OH), and cage activity was measured (AB system; Neuroscience, Tokyo, Japan) as described previously (Katsuno et al., 2002). Gait stride was measured in 50 cm of footsteps, and the maximum value was recorded for each mouse. The onset of motor impairment was determined using weekly rotarod task analyses. Male AR-97Q mice were castrated or sham-operated via the abdominal route under ketamine–xylazine anesthesia (50 mg/kg ketamine and 10 mg/kg xylazine, i.p.) within 1 week after the onset of rotarod impairment.
Immunohistochemistry and immunofluorescent analysis.
Ten-micrometer-thick sections were prepared from paraffin-embedded tissues, and immunohistochemistry was performed as described previously (Katsuno et al., 2002). Formalin-fixed tail samples were washed with 70% ethanol and decalcified with 7% formic acid–70% ethanol for 7 d before embedding in paraffin. Sections to be immunostained for dynactin 1, dynein intermediate chain, dynein heavy chain, and dynamitin were first microwaved for 20 min in 50 mm citrate buffer, pH 6.0. Sections to be immunostained for polyglutamine (1C2 antibody) were treated with formic acid for 5 min at room temperature. The following primary antibodies were used: anti-dynactin 1 (p150glued, 1:250; BD Transduction, San Diego, CA), anti-dynein intermediate chain (1:500; Millipore, Temecula, CA), anti-dynein heavy chain (1:100; Sigma-Aldrich, St. Louis, MO), anti-dynamitin (1:1000; BD Transduction), anti-polyglutamine, 1C2 (1:10,000; Millipore), antiphosphorylated high molecular weight neurofilament (NF-H) (SMI31, 1:1000; Sternberger Monoclonals, Lutherville, MD), anti-nonphosphorylated NF-H (SMI32, 1:5000; Sternberger Monoclonals), and anti-synaptophysin (1:10,000; Dako, Glostrup, Denmark).
For immunofluorescent analysis of skeletal muscle, mice were deeply anesthetized with ketamine–xylazine and perfused with PBS followed by 4% paraformaldehyde fixative in phosphate buffer, pH 7.4. Gastrocnemius muscles were dissected free, frozen quickly by immersion in cooled acetone and powdered CO2. Longitudinal, 30 μm, cryostat sections were placed on a silane-coated slide in a drop of 3% disodium EDTA, air dried at room temperature, and fixed in methanol/acetone (50:50 v/v). After blocking with PBS containing 5% goat serum and 1% BSA for 30 min at room temperature, sections were incubated with 5 μg/ml Oregon green-conjugated α-bungarotoxin (Invitrogen, Eugene, OR) for 60 min at room temperature. Sections were incubated with antiphosphorylated NF-H (SMI31, 1:5000; Sternberger Monoclonals), anti-synaptophysin (1:50,000; Dako), or anti-Rab3A (1:5000; BD Transduction) antibodies at 4°C overnight, and then with Alexa-546-conjugated goat anti-mouse IgG (1:1000; Invitrogen). Sections were examined with an IX71 inverted microscope (Olympus, Tokyo, Japan). For double staining of the skeletal muscle, paraffin-embedded sections were treated with TNB blocking buffer (PerkinElmer, Boston, MA) and incubated with anti-AR antibody (N-20, 1:500; Santa Cruz Biotechnology, Santa Cruz, CA) together with antiphospho-NF-H.
For immunostaining of human tissues, autopsy specimens of lumbar spinal cord and intercostal muscle obtained from a genetically diagnosed SBMA patient (78-year-old male) and those from a neurologically normal patient (75 years old) were used. The collection of tissues and their use for this study were approved by the Ethics Committee of Nagoya University Graduate School of Medicine. Spinal cord sections at 10 μm were incubated with anti-dynactin 1 antibody (p150glued, 1:250; BD Transduction). Thirty-micrometer-thick cryostat sections of intercostal muscle were incubated with 150 μg/ml Alexa-488-conjugated α-bungarotoxin (Invitrogen) and then with antiphosphorylated NF-H (SMI31, 1:200; Sternberger Monoclonals).
Retrograde Fluoro-gold neurotracer labeling.
For labeling neurons with intramuscular injection of tracer, mice were anesthetized with ketamine–xylazine, and a small incision was made in the skin of the left calf to expose the gastrocnemius muscle. A total volume of 4.5 μl of 2.5% Fluoro-gold solution (Biotium, Hayward, CA) in PBS was injected in three different parts of the muscle (proximal, middle, and distal) using a 10 μl Hamilton syringe. For labeling by the nerve stump method, the sciatic nerve was exposed and transected at mid-thigh level. A small polyethylene tube containing 2.5% Fluoro-gold solution was applied to the proximal stump of the cut sciatic nerve, and sealed with Vaseline to prevent leakage. Mice were anesthetized 44 h after Fluoro-gold administration with ketamine–xylazine and perfused with PBS followed by 4% paraformaldehyde in phosphate buffer, pH 7.4. Spinal cords were removed and postfixed with 4% paraformaldehyde in phosphate buffer for 2 h, floated in 10 and 15% sucrose for 4 h each and in 20% sucrose overnight. The samples were sectioned longitudinally on a cryostat at 30 μm and mounted on silane-coated slides. The number of Fluoro-gold labeled motor neurons was counted in serial spinal cord sections with an IX71 inverted microscope (Olympus) using a wide-band UV filter. Some specimens were immunostained for dynactin immediately after the number of Fluoro-gold-labeled motor neurons was counted.
Western blot analysis.
SH-SY5Y cells were lysed in CellLytic lysis buffer (Sigma-Aldrich) containing a protease inhibitor mixture (Roche, Mannheim, Germany) 2 d after transfection. Mice were killed under ketamine–xylazine anesthesia. Their tissues were snap-frozen with powdered CO2 in acetone and homogenized in 50 mm Tris, pH 8.0, 150 mm NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, and 1 mm 2-mercaptoethanol containing 1 mm PMSF and 6 μg/ml aprotinine and then centrifuged at 2500 × g for 15 min at 4°C. The supernatant fractions were separated on 5–20% SDS-PAGE gels (10 μg protein for the nerve roots or 40 μg for the spinal cord, per lane) and then transferred to Hybond-P membranes (Amersham Pharmacia Biotech, Buckinghamshire, UK), using 25 mm Tris, 192 mm glycine, 0.1% SDS, and 10% methanol as transfer buffer. Immunoblotting was performed using the following primary antibodies: anti-dynactin 1 (p150glued, 1:250; BD Transduction), anti-dynein intermediate chain (1:1000; Millipore), anti-dynein heavy chain (1:200; Sigma-Aldrich), anti-dynamitin (1:250; BD Transduction), anti-α-tubulin (1:5000; Sigma-Aldrich), antiphosphorylated NF-H (SMI31, 1:100,000; Sternberger Monoclonals), and anti-nonphosphorylated NF-H (SMI32, 1:1000; Sternberger Monoclonals). The immunoblots were digitalized (LAS-3000 imaging system; Fujifilm, Tokyo, Japan), signal intensities of three independent blots were quantified with Image Gauge software version 4.22 (Fujifilm), and the means ± SD were expressed in arbitrary units.
Ligation of mouse sciatic nerve.
Under anesthesia with ketamine–xylazine, the skin of the right lower limb was incised. The right sciatic nerve was exposed and ligated at mid-thigh level using surgical thread. For immunofluorescent analysis, operated mice were decapitated under deep anesthesia with ketamine–xylazine 8 h after ligation and perfused with 4% paraformaldehyde fixative in phosphate buffer, pH 7.4. The right sciatic nerve segment, including at least 5 mm both proximal and distal to the ligated site, was removed. The nonligated, left sciatic nerve was also taken out in the same manner as the right nerve. The removed nerves were placed into fixative for 4 h, transferred consecutively to 10, 15, and 20% sucrose in 0.01 m PBS, pH 7.4, for 4 h each at 4°C, mounted in Tissue-Tek OCT compound (Sakura, Tokyo, Japan), and frozen with powdered CO2 in acetone. Ten-micrometer-thick cryostat sections were prepared from the frozen tissues, blocked with normal goat serum (1:20), incubated with anti-synaptophysin (1:50,000; Dako) at 4°C overnight, and then with Alexa-546-conjugated goat anti-mouse IgG (1:1000; Invitrogen). Immunofluorescent images were recorded with an IX71 inverted microscope (Olympus), and the signal intensities were quantified using Image Gauge software, version 4.22 (Fujifilm) and expressed in arbitrary units.
For immunoblotting of axonal proteins, the sciatic nerve segments 1 mm both proximal and distal to the ligated site were removed without paraformaldehyde fixation, and frozen in with powdered CO2 in acetone. Protein extraction and Western blotting were performed as described above.
In situ hybridization. Formalin-fixed, paraffin-embedded 6-μm-thick sections of the spinal cord were deparaffinized, treated with proteinase K, and processed for in situ hybridization using an ISHR kit (Nippon Gene, Tokyo, Japan) according to the manufacturer's instructions. Dynactin 1 cDNA was obtained from spinal cords of wild-type mice. The primers, 5′-AGATGGTGGAGATGCTGACC-3′ and 5′-GAGCCTTGGTCTCAGCAAAC-3′, were phosphorylated with T4polynucleotide kinase (Stratagene Cloning Systems, La Jolla, CA). The cDNA was inserted into the pSPT 19 vector (Roche). Dioxigenin-labeled cRNA antisense and sense probes, 380 bp long, were generated from this plasmid using T7 and SP6 polymerase (Roche), respectively. Spinal cord sections were hybridized for 16 h at 42°C washed in formamide–4× SSC (50:50 v/v) at the same temperature, treated with RNase A at 37°C, and washed again in 0.1× SSC at 42°C. The signals were detected immunologically with alkaline phosphatase-conjugated anti-dioxigenin antibody and incubated with NBT/BCIP (Roche) for 16 h at 42°C. Slices were counterstained with methyl green. To quantify the intensity of the signals in the cell bodies of spinal motor neurons, three nonconsecutive sections from a wild-type littermate and those of a transgenic mouse from lines 7–8 or 4–6 were analyzed using the NIH Image program (version 1.62). Sections adjacent to those used for in situ hybridization were processed for immunohistochemistry using anti-polyglutamine antibody as described above.
Quantitative real-time PCR.
Dynactin 1 mRNA levels were determined by real-time PCR as described before (Ishigaki et al., 2002; Ando et al., 2003). Briefly, total RNA (5 μg each) from AR-97Q and wild-type spinal cord were reverse transcribed into first-strand cDNA using SuperScript II reverse transcriptase (Invitrogen). Real-time PCR was performed in a total volume of 50 μl, containing 25 μl of 2× QuantiTect SYBR Green PCR Master Mix and 0.4 μm of each primer (Qiagen, Valencia, CA), and the product was detected by the iCycler system (Bio-Rad Laboratories, Hercules, CA). The reaction conditions were 95°C for 15 min and then 45 cycles of 15 s at 95°C followed by 60 s at 55°C. For an internal standard control, the expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was simultaneously quantified. The following primers used were 5′-CTCAGAGGAGCCCAGATGA-3′ and 5′-GCTGGTCTTGCGGTACAGT-3′ for dynactin 1, 5′-GAGAGCATGGAGCTGGTGTA-3′ and 5′-CCAACCACGAAGTTGTTGAC-3′ for dynein intermediate chain, 5′-TACCAGGTGGGAGTGCATTA-3′ and 5′-CAGTCACTATGCCCATGACC-3′ for dynein heavy chain, 5′-ACAAGCGTGGAACACATCAT-3′ and 5′-TCTTTCCAATGCGATCTGAG-3′ for dynamitin, and 5′-CCTGGAGAAACCTGCCAAGTAT-3′ and 5′-TGAAGTCGCAGGAGACAACCT-3′ for GAPDH. The threshold cycle of each gene was determined as the number of PCR cycles at which the increase in reporter fluorescence was 10 times the baseline signal. The weight of the gene contained in each sample was equal to the log of the starting quantity and the standardized expression level in each mouse was equal to the weight ratio of each gene to that of GAPDH.
For the real-time PCR with mRNA extracted from SH-SY5Y cells, the following primers were used: 5′-CTTGGAAGCGATGAATGAGA-3′ and 5′-TAGTCTGCAACGTCTCCTG-3′ for dynactin 1, and 5′-AGCCTCAAGATCATCAGCAAT-3′ and 5′-GGACTGTGGTCATGAGTCCTT-3′ for GAPDH.
Plasmid vectors and cell culture.
Human AR cDNAs containing 24 or 97 CAG repeats were subcloned into pcDNA3.1 (Invitrogen) as described previously (Kobayashi et al., 2000). Human dynactin 1 cDNA was also subcloned into pcDNA3.1 (Invitrogen). The human neuroblastoma cells (SH-SY5Y, #CRL-2266; American Type Culture Collection, Manassas, VA) were plated in 6-well dishes in 2 ml of DMEM/F12 containing 10% fetal bovine serum with penicillin and streptomycin, and each dish was transfected with 2 μg of the vector containing AR24, AR97, or mock and with 2 μg of the vector containing dynactin 1 or mock using Opti-MEM (Invitrogen) and Lipofectamine 2000 (Invitrogen) and then differentiated in differentiation medium (DMEM/F12 supplemented with 5% fetal calf serum and 10 μm retinoic acid) for 2 d. Two days after transfection, cells were stained with propidium iodide (Invitrogen, Eugene, OR) and mounted in Gelvatol. Quantitative analyses were made from triplicate determinations. Duplicate slides were graded blindly in two independent trials as described previously (Katsuno et al., 2005).
Statistical analyses.
We analyzed data using the Kaplan-Meier and log-rank test for survival rate, ANOVA with post hoc test (Dunnett) for multiple comparisons, and an unpaired t test from Statview software version 5 (Hulinks, Tokyo, Japan).
Results
Accumulation of axonal proteins in distal motor axons of SBMA mouse
To clarify the molecular basis of neuronal dysfunction in SBMA, we analyzed histopathological alterations in the spinal cords of transgenic mice carrying full-length human AR with 97 CAGs (AR-97Q mice) (Katsuno et al., 2002, 2003). We first focused on the expression and phosphorylation level of NF-H because affected mice demonstrate axonal atrophy in the ventral nerve root (Katsuno et al., 2002). Although it has been widely accepted that NF-H phosphorylation is a crucial factor determining axon caliber, neither the amounts nor the phosphorylation levels of NF-H in spinal cord or ventral root were decreased in male AR-97Q mice compared with wild-type littermates (supplemental Fig. 1A–C, available at www.jneurosci.org as supplemental material). The distribution of NF-H in the anterior horn of AR-97Q mice was also indistinguishable from that of wild-type or AR-24Q mice bearing human AR with a normal polyglutamine length (Fig. 1 A). However, AR-97Q mice demonstrated a striking accumulation of both phosphorylated and nonphosphorylated NF-H in skeletal muscle, a phenomenon not observed in AR-24Q or wild-types (Fig. 1 A). Although motor neurons originating in the anterior horn are always affected in SBMA, because the primary motor neurons projecting their axons to the anterior horn are not affected, no accumulation is seen in this region. The damage to motor neurons originating within the anterior horn results in accumulation of NFs in the skeletal muscle, instead of the spinal cord. A similar accumulation of the middle molecular weight NF was also observed (data not shown). To clarify whether this phenomenon is specific to neurofilaments, we performed immunohistochemistry on both spinal cord and muscle with an antibody against synaptophysin, a transmembrane glycoprotein of synaptic vesicles that is also retrogradely transported in axons (Li et al., 1995). In AR-97Q mice, synaptophysin accumulated among the muscle fibers in a pattern similar to that of NF-H, whereas no such accumulation was observed in unaffected mice (Fig. 1 B).
Figure 1.

Accumulation of neurofilament and synaptophysin in the distal end of motor axons. A, Immunohistochemistry of skeletal muscle and spinal cord from AR-97Q (4–6), AR-24Q, and wild-type mice (12 weeks) using an antibody for phosphorylated or nonphosphorylated NF-H. B, Immunohistochemistry for synaptophysin shows findings parallel to those of neurofilament. C, Age-dependent change in antiphosphorylated NF-H immunohistochemistry in skeletal muscle of SBMA mice. D, Immunofluorescence of mouse skeletal muscle using α-bungarotoxin (green) in combination with antiphospho-NF-H antibody (red). Phosphorylated NF-H accumulates in the distal end of motor axons in AR-97 mice (7–8, 12 weeks). E, Antiphospho-NF-H immunofluorescence with α-bungarotoxin staining in skeletal muscle from a human SBMA patient showing similar neurofilament accumulation. F, Double-labeling of skeletal muscle from an AR-97Q mouse (4–6, 12 weeks) using antiphospho-NF-H antibody (green) and anti-AR (red) shows that accumulated NF-H does not colocalize with AR. Scale bars, 100 μm.
We then investigated the time course of abnormally accumulated NF in skeletal muscle. Because the onset of motor dysfunction occurs at 9–10 weeks in AR-97Q mice, NF pathology before and after the onset was examined. Anti-NF immunostaining demonstrated that intramuscular NF accumulation was detectable as early as 7 weeks before the onset of muscle weakness in this mouse model, and aggravated thereafter (Fig. 1 C). These observations suggest that intramuscular accumulation of NF plays a role in the motor neuron dysfunction in this mouse model of SBMA.
To confirm the distribution of NF-H and synaptophysin in skeletal muscle, we examined the localization of these proteins in relation to the neuromuscular junction. Immunohistochemistry using α-bungarotoxin to mark the junctions, and fluorescent-labeled antibodies showed that both NF-H and synaptophysin accumulated in the most distal motor axon adjacent to neuromuscular junctions (Fig. 1 D). A similar intramuscular accumulation of neurofilament was detected in the skeletal muscle of SBMA patients (Fig. 1 E). Although pathogenic AR accumulated in the nuclei of skeletal muscle in the AR-97Q mice, the accumulation of NF-H did not colocalize with AR (Fig. 1 F). Moreover, immunoprecipitation demonstrated no interaction between AR and NF-H (data not shown). These findings exclude the possibility that pathogenic AR directly interrupts the axonal trafficking.
Retrograde axonal transport is disrupted in SBMA mouse
To elucidate the molecular basis of the abnormal distribution of NF and synaptophysin, we studied axonal transport in this mouse model of SBMA. Axonal components undergo anterograde and/or retrograde axonal transport. Proteins including NF and synaptophysin are bidirectionally transported, whereas some components such as Rab3A, a small GTP binding protein, are transported only anterogradely (Li et al., 1995; Roy et al., 2000). The distribution of Rab3A in skeletal muscle of SBMA mice was equivalent to that of wild-type mice, whereas synaptophysin and neurofilaments accumulated in the most distal motor axons of the SBMA mice only (Figs. 1 D, 2 A).
Figure 2.

Perturbation of retrograde axonal transport in SBMA mice. A, Immunofluorescence of mouse skeletal muscle using α-bungarotoxin (green) labeling the endplate together with anti-synaptophysin antibody (red) or anti-Rab3A antibody (red). Accumulation of Rab3A is not detected in wild-type or AR-97Q mice (7–8, 12 weeks). B, Immunohistochemistry for synaptophysin in the sciatic nerve 8 h after ligation and representative quantification of immunoreactivity. Accumulation of synaptophysin immunoreactivity is decreased on the distal side (arrows) of the ligation site (arrowhead) in preonset (7 weeks) and advanced stage (13 weeks) AR-97Q mice. C, Immunoblots of the sciatic nerve segments on both proximal and distal sides of the ligation. The total amount of proteins extracted from the contralateral nonligated sciatic nerve was analyzed as a control. D, E, Retrograde labeling of lumbar motor neurons of AR-97Q (7–8), AR-24Q, or wild-type mice (12 weeks) by Fluoro-gold injection into the gastrocnemius muscle (D) and the number of labeled neurons (E) (n = 5 for each group). F, The number of motor neurons labeled by Fluoro-gold using the sciatic nerve stump method (n = 5 for each group). Scale bars: A, B, D, 100 μm. Error bars indicate SD.
To further examine the nature of the axonal transport anomaly in SBMA mice, the sciatic nerve was ligated at mid-thigh level. Because the transport rate of NF is slower than other axonal components, we analyzed the transport of synaptophysin and Rab3A in this ligation study (Fig. 2 B,C). In wild-type mice, synaptophysin accumulated predominantly on the proximal side of the ligation, but also on the distal side. Although synaptophysin and Rab3A accumulations proximal to the site of ligation were notable in both preonset and advanced stages of AR-97Q mice, their accumulation on the distal side was decreased before the onset of symptoms and was progressively inhibited. These findings suggest that disrupted retrograde axonal transport gives rise to the accumulation of axonal proteins in the distal motor axon terminals of SBMA mice before the onset of motor impairment.
To confirm this hypothesis, we analyzed retrograde neuronal labeling with the fluorescent tracer Fluoro-gold after its injection into the mouse calf muscle. The number of Fluoro-gold-labeled spinal motor neurons was significantly less in affected AR-97Q mice compared with AR-24Q or wild-type mice (Fig. 2 D,E). To exclude the possibility that synaptic pathology contributed to diminished uptake of the tracer, we also examined Fluoro-gold labeling using direct application of the tracer into the sciatic nerve stump (Sagot et al., 1998). Again, AR-97Q mice showed fewer motor neurons labeled by Fluoro-gold applied directly to the proximal stump of the sciatic nerve than did the AR-24Q mice (Fig. 2 F), suggesting that neither synaptic retraction nor disconnection is the basis for disruption of axonal transport. Furthermore, it should be noted that the decrease in the number of labeled neurons preceded the onset of motor symptoms in both of these experiments. These observations suggest that the disruption of retrograde transport plays an early role in the pathogenesis of motor neuron degeneration in SBMA.
Transcriptional dysregulation of dynactin 1 in SBMA
Retrograde axonal transport is microtubule-dependent and is regulated by the axon motor protein dynein and its associated protein complex, dynactin. To elucidate the molecular mechanism compromising retrograde axonal transport in SBMA mice, we examined the levels of various dynein and dynactin protein subunits. Immunohistochemistry of spinal cord sections demonstrated that the spinal motor neurons from AR-97Q mice had lower levels of dynactin 1, the largest subunit of dynactin, than did those from either wild-type or AR-24Q mice (Fig. 3 A). In the ventral root, significantly decreased levels of dynactin 1 were apparent before the onset of motor symptoms (Fig. 3 B). Although the level of dynein heavy chain was diminished in the advanced disease stage in SBMA mice, this phenomenon was not observed before the onset of symptoms (Fig. 3 B). No alterations were observed in the levels of dynein intermediate chain or dynamitin, the p50 subunit of dynactin, throughout the disease course (Fig. 3 A,B). To confirm the role of dynactin 1 in the pathogenesis of human SBMA, we also examined the protein level in autopsy specimens. As observed in the mouse model, the protein level of dynactin 1 was decreased in the anterior horn cells and in the ventral roots of SBMA patients (Fig. 3 C).
Figure 3.

Decreased levels of dynactin 1 in SBMA. A, Immunohistochemistry for motor proteins regulating retrograde axonal transport, dynactin 1, dynein intermediate chain (IC), dynein heavy chain (HC), and dynamitin in the spinal cord from AR-97Q (4–6), AR-24Q, and wild-type mice (12 weeks). Dynactin 1 is markedly diminished in the motor neurons of AR-97Q mice. B, Western blot analysis for motor proteins in the ventral spinal root from presymptomatic or advanced AR-97Q mice (4–6) compared with those from AR-24Q and wild-type mice. C, Dynactin 1 immunohistochemistry in the anterior horn and the ventral root of an SBMA patient and a normal subject. D, Anti-dynactin 1 immunohistochemistry in various affected (spinal cord and brainstem) and nonaffected (hippocampus and visual cortex) tissues from wild-type and AR-97Q mice. Data from AR-97Q mice are compared with immunohistochemistry using the anti-polyglutamine antibody, 1C2. Scale bars: A, 100 μm; C, D, 50 mm. Error bars indicate SD.
To examine the cell specificity of reduced dynactin 1 levels we compared anti-dynactin 1 immunohistochemistry with that of anti-polyglutamine using the 1C2 antibody in various tissues from wild-type and AR-97Q mice (Fig, 3 D). The immunoreactivity of dynactin 1 was markedly diminished in 1C2-positive tissues, but not in those lacking nuclear polyglutamine staining. This observation suggests that the reduction in dynactin 1 is relevant to the polyglutamine-mediated neuropathology. In addition, to investigate whether reduced levels of dynactin 1 were correlated with defective retrograde axonal transport, we analyzed anti-dynactin 1 immunohistochemistry in spinal cord sections labeled by Fluoro-gold (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The levels of dynactin 1 were decreased in the spinal motor neurons of AR-97Q mice concomitantly with decreased intensities of Fluoro-gold labeling. Together, these data strongly suggest that depletion of dynactin 1 is responsible for the disruption of retrograde axonal transport in SBMA.
To clarify the pathological mechanism responsible for reducing the levels of dynactin 1 protein in SBMA, mRNA levels were determined by in situ hybridization in AR-97Q and wild-type mice. Although dynactin 1 mRNA was expressed in virtually all motor neurons in the anterior horn, the expression was markedly repressed in AR-97Q mice (Fig. 4 A). Moreover, the levels of dynactin 1 mRNA were significantly lower in those motor neurons demonstrating nuclear accumulation of pathogenic AR compared with those without 1C2 nuclear staining (Fig. 4 B). Real-time quantitative PCR also demonstrated a significant decrease in dynactin 1 mRNA levels in the spinal cords of AR-97Q mice at all disease stages compared with those of wild-types (Fig. 4 C). The level of dynein heavy chain mRNA was decreased in the advanced stage, but not in the preonset period. The levels of dynein intermediate chain mRNA and dynamitin mRNA were not altered either before or after the onset of motor symptoms.
Figure 4.
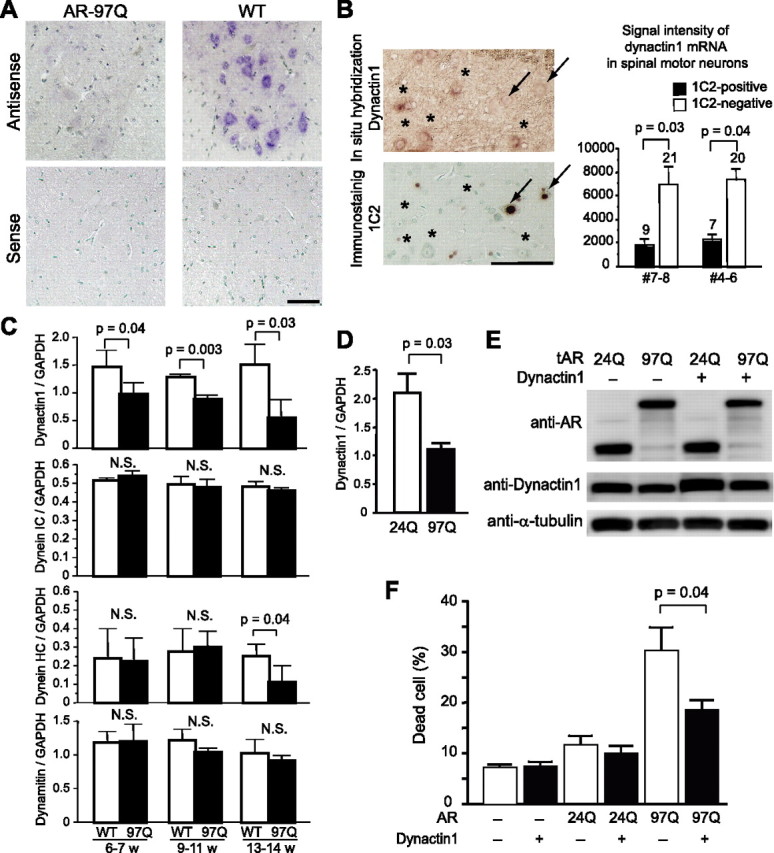
Transcriptional dysregulation of dynactin 1 in spinal motor neurons of SBMA mouse and effects of dynactin 1 overexpression. A, In situ hybridization of dynactin 1 mRNA in the anterior horn of wild-type and AR-97 (4–6, 9 weeks) transgenic mice. Note the marked decrease in dynactin 1 mRNA levels in the spinal motor neurons of AR-97Q compared with those in wild-type mice. B, In situ hybridization of dynactin 1 in the anterior horn. The adjacent sections were processed for anti-polyglutamine using the 1C2 antibody and the signals were quantified in representative AR-97Q mice (7–8, 9 weeks; 4–6, 10 weeks). Dynactin 1 mRNA expression is markedly decreased in the motor neurons demonstrating nuclear accumulation of pathogenic AR (arrows), but not in those lacking clear nuclear staining with anti-polyglutamine antibody (asterisks). The number above each bar indicates cell count. C, The mRNA levels of dynactin 1 and other motor proteins in the spinal cords of wild-type and AR-97Q mice (7–8, 13 weeks) (n = 4 for each group) demonstrated by real-time, RT-PCR. Data shown are ratios of the various mRNA levels to GAPDH mRNA levels. D, The mRNA levels of dynactin 1 in SH-SY5Y cells expressing either AR-24Q or AR-97Q (n = 4). E, Immunoblots of SH-SY5Y cells expressing either AR-24Q or AR-97Q with or without overexpression of exogenous dynactin 1. F, Frequency of cell death detected by propidium iodide staining. Dynactin 1 overexpression significantly reduced cell death in the cells bearing AR with elongated polyglutamine. Scale bars: A, B, 100 μm. Error bars indicate SD (n = 6 for each group). IC, Intermediate chain; HC, heavy chain.
To investigate the role that diminished levels of dynactin 1 play in neurodegeneration in SBMA, we tested whether overexpression of this protein suppressed the cellular toxicity usually observed in the presence of expanded polyglutamine. In SH-SY5Y cells bearing truncated AR containing an expanded polyglutamine, the level of dynactin 1 was decreased both in mRNA and in protein (Fig. 4 D,E). In this cellular model of SBMA, overexpression of dynactin 1 alleviated cell death exerted by pathogenic AR (Fig. 4 E).
In SBMA mice, the level of dynactin 1 protein in spinal motor neurons was restored by oral administration of sodium butyrate, an HDAC inhibitor that increases the level of histone acetylation leading to promotion of gene transcription (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) (Minamiyama et al., 2004). Sodium butyrate-mediated upregulation of dynactin 1 also eventually alleviated the neurofilament accumulation in skeletal muscle (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), although this treatment had no influence on the subcellular distribution of pathogenic AR protein (Minamiyama et al., 2004). These observations indicate that nuclear accumulation of aberrant AR in the nuclei of motor neurons leads to a decrease at the transcription level of dynactin 1, resulting in perturbation of retrograde axonal transport and subsequent motor neuron dysfunction.
Castration reverses symptoms and pathology of SBMA mouse
To examine the reversibility of the phenotypes resulting from polyglutamine-induced neuronal dysfunction, we investigated the effect of castration on early symptomatic SBMA mice. Male AR-97Q mice (7–8 and 4–6) demonstrate a rapid aggravation of neuromuscular phenotypes and usually succumb 3–4 weeks after the onset of motor impairment. The motor-impaired phenotype of the SBMA mouse is dependent on circulating testosterone levels, and we reported previously that castration during the presymptomatic period (4 weeks), to eliminate testosterone, drastically prevents the development of neurological symptoms such as weakness, amyotrophy, and shortened life span (Katsuno et al., 2002). In the present study, we castrated male AR-97Q mice within 1 week after the onset of rotarod task impairment. Castration reversed motor dysfunction in AR-97Q mice, even though it was performed after the onset of symptoms (Fig. 5 A). Most mice showed a reduction in daily activity and body weight loss at the onset of rotarod task defect; these symptoms were also reversed by castration. In accordance with these observations, postonset castration significantly prolonged the life span of the male AR-97Q mice. We confirmed the reversal of motor symptoms by analyzing gait strides in a series of mouse footsteps (Fig. 5 B).
Figure 5.

Symptomatic and histopathological reversibility of the SBMA phenotype in AR-97Q mice. A, Castration of early symptomatic AR-97Q mice within 1 week after symptomatic manifestation resulted in significant improvement of the symptomatic phenotypes: rotarod task (7–8), cage activity (4–6), body weight (4–6), and survival rate (4–6). There are significant differences in all parameters between the sham-operated (n = 10) and castrated (n = 10) male AR-97Q mice (p < 0.0001, p < 0.0001, p = 0.0001, and p = 0.0006, respectively). B, Representative footprints of an individual AR-97Q mouse (2–6) at the early onset of motor symptoms and after he had been castrated within 1 week after the onset of rotarod impairment, compared with those of a wild-type mouse. Quantification of the gait stride data (n = 4). C, Nuclear accumulation of pathogenic AR with expanded polyglutamine in the tail muscle of one individual male AR-97Q mouse (4–6). D, Castration after motor impairment onset significantly reduced the number of nuclei stained by an anti-polyglutamine antibody, 1C2 (n = 4). Scale bar: C, 100 μm. Error bars indicate SD.
To confirm the rescue effects of castration on histopathology, we investigated the nuclear accumulation of pathogenic AR in the skeletal muscle of tail sections sampled over time from the same mouse. Although the number of nuclei positively stained with 1C2 continued to increase for 2 weeks after the castration, at 4 weeks there was a significant decrease in expanded polyglutamine AR-positive nuclei (Fig. 5 C,D). This time course corresponds approximately to the that of the symptomatic improvements, suggesting that nuclear accumulation of pathologic AR contributes to neuronal dysfunction and consequent symptomatic manifestation in SBMA mice.
Castration reverses dynactin 1 expression and restores retrograde axonal transport
It is important to determine whether disrupted retrograde axonal transport resulting from transcriptional dysregulation of dynactin 1, contributes to the reversible motor neuronal dysfunction in the early disease stage of SBMA mice. We therefore investigated axonal transport and the level of dynactin 1 expression in transgenic mice within 1 week after the onset of rotarod task impairment. In this early stage of the disease, the mice already demonstrated a reduction in the number of spinal motor neurons labeled by Fluoro-gold (Fig. 6 A). Castration of symptomatic AR-97Q mice restored Fluoro-gold staining in the spinal motor neurons to a similar level as seen in wild-types (compare Figs. 2 D, 6 A). Castration after the onset of muscle weakness reduced the intramuscular accumulation of neurofilaments and synaptophysin in AR-97Q mice (Fig. 6 B,C). Immunohistochemistry of spinal cord showed that postsymptomatic castration also eliminated nuclear accumulation of pathogenic AR as detected by the 1C2 antibody, and restored anti-dynactin 1 immunoreactivity in motor neurons (Fig. 6 D). Immunoblotting demonstrated that the level of dynactin 1 protein, but not that of dynein heavy chain, was decreased in the ventral root of AR-97Q mice in the early symptomatic stage (Fig. 6 E). Castration after the onset of motor impairment restored dynactin 1 to its normal levels in the ventral root, whereas it had no effect on dynactin 1 expression in wild-type mice (Fig. 6 E). These observations indicate that the castration-mediated restoration of dynactin 1 expression improves retrograde axonal transport and contributes to the reversal of neuromuscular phenotypes in SBMA mice at an early stage of the disease process.
Figure 6.

Hormonal intervention restores expression level of dynactin 1 and improves axonal transport. A, Fluoro-gold labeling of spinal cord from early symptomatic (7–8; 9–11 weeks) and castrated (7–8; 13–16 weeks) male AR-97Q mice (n = 5 for each group). B, Immunohistochemistry of skeletal muscle for NF-H and synaptophysin. C, Immunohistochemistry for phosphorylated NF-H in the tail muscle of an individual male AR-97Q mouse (4–6). Castration after onset of symptoms depletes NF-H accumulation in the skeletal muscle. D, Immunohistochemistry of the spinal cords of early symptomatic (4–6; 11 weeks) and castrated (4–6; 15 weeks) male AR-97Q mice using anti-dynactin 1 and 1C2. Castration eliminated nuclear accumulation of expanded polyglutamine AR. E, Immunoblots of ventral roots from early symptomatic (4–6; 11 weeks) and castrated (4–6; 15 weeks) AR-97Q mice together with that from wild-type littermates (15 weeks) using antibodies against dynactin 1, dynein heavy chain (HC), and α-tubulin. Scale bars: A–D, 100 μm. Error bars indicate SD (n = 3 for each group).
Discussion
Reversibility of neuronal dysfunction in SBMA
The fundamental pathological feature of polyglutamine diseases is the loss of neurons in selected regions of the CNS. Neuronal cell death, however, is often undetectable in mildly affected HD patients despite the presence of definite clinical features (Vonsattel et al., 1985). The early HD symptoms may thus result from functional alterations within neurons rather than cell death (Walker et al., 1984). In mouse models of polyglutamine diseases, it has been postulated that neuronal dysfunction, without cell loss, is sufficient to cause neurological symptoms (Mangiarini et al., 1996; Clark et al., 1997). These observations indicate that the pathogenesis of polyglutamine diseases is potentially reversible at an early stage. This hypothesis is supported by the observation that arrest of gene expression after the onset of symptoms reverses behavioral and neuropathological abnormalities in conditional mouse models of polyglutamine diseases (Yamamoto et al., 2000; Zu et al., 2004). The present study supports this hypothesis in that castration after the onset of motor deficit reverses behavioral and histopathological abnormalities by preventing nuclear accumulation of the pathogenic AR protein. These findings imply that cellular protective responses successfully abrogate the toxicity of polyglutamine-containing pathogenic protein, unless it perpetually accumulates in the nucleus.
Protein quality control systems, including molecular chaperones, the ubiquitin-proteasome system, and autophagy have been shown to reduce polyglutamine toxicity in various animal models of polyglutamine diseases (Adachi et al., 2003; Ravikumar et al., 2004; Katsuno et al., 2005; Waza et al., 2005). It is thus logical that inhibition of AR translocation into the nucleus restores the protein degradation machinery, such as ubiquitin-proteasome system, leading to the reduction in the amount of aggregates as well as the improvement of neuronal dysfunction in the SBMA mice (Waza et al., 2005).
Defective retrograde axonal transport in SBMA
The SBMA mice we examined demonstrated impairment of retrograde axonal transport, resulting in the accumulation of neurofilaments and synaptophysin in the distal motor axon. Many proteins required for neuronal survival are synthesized within neuronal perikarya and are transported along the axon toward the synaptic terminals (Shea, 2000). A bidirectional delivery system consisting of anterograde and retrograde transport enables the recycling of cytoskeletons and synaptic vesicle-associated proteins. A histopathological hallmark of amyotrophic lateral sclerosis (ALS) is the accumulation of neurofilaments in cell bodies and proximal axons of affected motor neurons, presumably caused by compromised anterograde axonal transport; nevertheless, this finding has not been observed in SBMA (Sobue et al., 1990; Julien 2001). Transgenic SBMA mice demonstrate marked neurofilament storage in the distal motor axons, but not in the proximal axons or cell bodies. Neurofilament accumulation at motor endplates has also been reported in a transgenic mouse model of spinal muscular atrophy, another lower motor neuron disease (Cifuentes-Diaz et al., 2002). Axonal transport of NF depends on the dynein/dynactin system, disruption of which results in accumulation of neurofilaments at the distal axon in both cultured cells and transgenic mice (LaMonte et al., 2002; He et al., 2005). When combined, these findings indicate that the accumulation of axonal components in distal motor axons appears to be a substantial pathology associated with degeneration of lower motor neurons.
In the present study, synaptophysin showed an accumulation pattern similar to that of neurofilaments, whereas the distribution of Rab3A, another synaptic vesicle-associated protein, was not altered in this mouse model. Crush injury experiments have shown that although both proteins are delivered from cell bodies into axons, of the two only synaptophysin undergoes retrograde transport (Li et al., 1995, 2000). In addition, Fluoro-gold labeling experiments clearly demonstrated the disruption of retrograde, but not anterograde axonal transport in the spinal motor neurons of SBMA mice before the onset of muscle weakness. Together, the pathogenesis of motor neuronal dysfunction in SBMA is likely to be based on the perturbation of retrograde axonal transport, and not on an excessive transport of total axonal proteins.
Axonal transport impairment has been implicated in the pathogenesis of HD and SBMA (Gunawardena et al., 2003; Szebenyi et al., 2003). Although axonal inclusion interferes with axonal transport in a cell model of SBMA (Piccioni et al., 2002), AR containing expanded polyglutamine may also inhibit anterograde and/or retrograde axonal transport without visible aggregate formation (Szebenyi et al., 2003; Morfini et al., 2006). Accumulation of neurofilaments at nerve terminals has also been documented in a mouse model of HD (Ribchester et al., 2004). In our SBMA mice, pathogenic AR did not colocalize with accumulated neurofilament, nor did it form axonal inclusions. More intriguingly, sodium butyrate-mediated gene upregulation attenuated the accumulation of neurofilaments, but did not alter the intracellular distribution of AR. These observations suggest that the defective retrograde axonal transport in SBMA mice does not result from the direct interaction between aberrant AR and axonal components, but rather from a secondary mechanism resulting from expanded polyglutamine.
Dynactin in motor neuron disease
The present study indicates that a decrease in the level of dynactin 1, the p150 subunit of dynactin, in affected neurons is a fundamental early event in the pathogenesis of SBMA. Dynactin is a multiprotein complex regulating dynein, a microtubule-dependent molecular motor for retrograde axonal transport. A mutation in DCTN1, the gene encoding dynactin 1, has been identified in a family with an autosomal dominant form of lower motor neuron disease and in another with ALS (Puls et al., 2003; Münch et al., 2005). A gene expression analysis of sporadic ALS patients revealed a significant decrease in dynactin 1 mRNA (Jiang et al., 2005). Overexpression of dynamitin dissociates the dynactin complex, resulting in late-onset motor neuron degeneration in a transgenic mouse model of motor neuron disease (LaMonte et al., 2002). These observations specifically link an impaired dynactin function to the pathogenesis of motor neuron diseases.
The pathological alteration in individual polyglutamine diseases is limited to distinct subsets of neurons, suggesting that the causative protein context influences the distribution of lesions. Motor neurons are selectively affected in SBMA, although pathogenic ARs are expressed in a wide range of neuronal and non-neuronal tissues (Doyu et al., 1994). A decreased level of dynactin 1 may contribute to this pathological selectivity, because a mutation in the DCTN1 gene causes a lower motor neuron disease resembling SBMA (Puls et al., 2003, 2005).
Link between altered transcription and neuronal dysfunction
Numerous studies have shown that nuclear accumulation of pathogenic polyglutamine-proteins is essential for neurodegeneration, although cytoplasmic events may also contribute to the pathogenesis (Gatchel and Zoghbi, 2005). Polyglutamine aggregation sequesters a variety of fundamental cellular factors including heat shock proteins and proteasomal components as well as transcriptional factors and coactivators. cAMP response element-binding protein-binding protein (CBP), a transcriptional coactivator, colocalizes with intranuclear inclusions in SBMA patients as well as in transgenic SBMA mice (McCampbell et al., 2000; Nucifora et al., 2001). In addition to its sequestration in inclusion bodies, the histone acetyltransferase activity of CBP is also inhibited by soluble polyglutamine-protein (Steffan et al., 2001). This theory suggests that HDAC inhibitors, which upregulate transcription through acetylation of nuclear histone, may open new avenues in the development of therapeutics. In a fly model of HD, the HDAC inhibitors, sodium butyrate and suberoylanilide hydroxamic acid, increased histone acetylation, leading to the mitigation of neurodegeneration (Steffan et al., 2001). These compounds also improve motor dysfunction in mouse models of HD and SBMA (Hockly et al., 2003; Minamiyama et al., 2004).
In the present study, a reduction in the level of dynactin 1 protein is ascribed to polyglutamine-mediated transcriptional dysregulation, because the mRNA level of this protein is decreased in expanded polyglutamine AR-positive spinal motor neurons. It should be noted that this diminution was significant in the neurons demonstrating nuclear accumulation of pathogenic AR, implying that polyglutamine-induced transcriptional perturbation underlies this pathological process. This hypothesis is confirmed by the observation that administration of sodium butyrate, an HDAC inhibitor, restores dynactin 1 expression, resulting in elimination of neurofilament accumulation at distal motor axons. Although, because of the nonspecific nature of sodium butyrate, we cannot at this time rule out the possibility that expression of some other protein was also elevated, leading to the elimination of neurofilament accumulation.
Given that the expression of other axon motor proteins regulating retrograde axonal transport, such as dynein intermediate chain, dynein heavy chain and dynamitin are not altered before the onset of symptoms, the reduction in dynactin 1 appears to instigate the neurodegeneration in SBMA. In addition to our study, the selective perturbation of certain subsets of gene transcription has been demonstrated in other animal models of polyglutamine diseases (Sugars and Rubinsztein 2003; Sopher et al., 2004), although the precise mechanism has yet to be elucidated.
In summary, the present study demonstrates that the pathogenesis of SBMA is a reversible dysfunction of motor neurons that occurs in the early stages of the disease. Polyglutamine-induced transcriptional alteration of dynactin 1 appears to disrupt retrograde axonal transport, contributing to the early reversible neuronal dysfunction. These observations suggest that transcriptional alteration and subsequent involvement of retrograde axonal transport are substantial therapeutic targets for SBMA.
Footnotes
This work was supported by a Center of Excellence grant from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and by grants from the Ministry of Health, Labor and Welfare, Japan. We have no financial conflict of interest that might be construed to influence the results or interpretation of this manuscript. We thank Jun-ichi Miyazaki for kindly providing the pCAGGS vector.
References
- Adachi H, Katsuno M, Minamiyama M, Sang C, Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu M, Sobue G. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci. 2003;23:2203–2211. doi: 10.1523/JNEUROSCI.23-06-02203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Yoshida M, Hashizume Y, Sobue G. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain. 2005;128:659–670. doi: 10.1093/brain/awh381. [DOI] [PubMed] [Google Scholar]
- Ando Y, Liang Y, Ishigaki S, Niwa J, Jiang Y, Kobayashi Y, Yamamoto M, Doyu M, Sobue G. Caspase-1 and -3 mRNAs are differentially upregulated in motor neurons and glial cells in mutant SOD1 transgenic mouse spinal cord: a study using laser microdissection and real-time RT-PCR. Neurochem Res. 2003;28:839–846. doi: 10.1023/a:1023258923002. [DOI] [PubMed] [Google Scholar]
- Banno H, Adachi H, Katsuno M, Suzuki K, Atsuta N, Watanabe H, Tanaka F, Doyu M, Sobue G. Mutant androgen receptor accumulation in spinal and bulbar muscular atrophy scrotal skin: a pathogenic marker. Ann Neurol. 2006;59:520–526. doi: 10.1002/ana.20735. [DOI] [PubMed] [Google Scholar]
- Cha JH. Transcriptional dysregulation in Huntington's disease. Trends Neurosci. 2000;23:387–392. doi: 10.1016/s0166-2236(00)01609-x. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen ES, O'Brien CJ, Wang H, Jenkins SC, Holder L, Lieberman AP, Merry DE. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci. 2004;24:4778–4786. doi: 10.1523/JNEUROSCI.0808-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Nicole S, Velasco ME, Borra-Cebrian C, Panozzo C, Frugier T, Millet G, Roblot N, Joshi V, Melki J. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet. 2002;11:1439–1447. doi: 10.1093/hmg/11.12.1439. [DOI] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, Orr HT. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyu M, Sobue G, Kimata K, Yamamoto K, Mitsuma T. Androgen receptor mRNA with increased size of tandem CAG repeat is widely expressed in the neural and nonneural tissues of X-linked recessive bulbospinal neuronopathy. J Neurol Sci. 1994;127:43–47. doi: 10.1016/0022-510x(94)90133-3. [DOI] [PubMed] [Google Scholar]
- Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanism and principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyglutamine proteins in Drosophila . Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- He Y, Francis F, Myers KA, Yu W, Black MM, Baas PW. Role of cytoplasmic dynein in the axonal transport of microtubules and neurofilaments. J Cell Biol. 2005;168:697–703. doi: 10.1083/jcb.200407191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki S, Liang Y, Yamamoto M, Niwa J, Ando Y, Yoshihara T, Takeuchi H, Doyu M, Sobue G. X-linked inhibitor of apoptosis protein is involved in mutant SOD1-mediated neuronal degeneration. J Neurochem. 2002;82:576–584. doi: 10.1046/j.1471-4159.2002.00998.x. [DOI] [PubMed] [Google Scholar]
- Jiang YM, Yamamoto M, Kobayashi Y, Yoshihara T, Liang Y, Terao S, Takeuchi H, Ishigaki S, Katsuno M, Adachi H, Niwa J, Tanaka F, Doyu M, Yoshida M, Hashizume Y, Sobue G. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann Neurol. 2005;57:236–251. doi: 10.1002/ana.20379. [DOI] [PubMed] [Google Scholar]
- Julien JP. Amyotrophic lateral sclerosis: unfolding the toxicity of the misfolded. Cell. 2001;104:581–591. doi: 10.1016/s0092-8674(01)00244-6. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Doyu M, Minamiyama M, Sang C, Kobayashi Y, Inukai A, Sobue G. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med. 2003;9:768–773. doi: 10.1038/nm878. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci USA. 2005;102:16801–16806. doi: 10.1073/pnas.0506249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Waza M, Banno H, Suzuki K, Tanaka F, Doyu M, Sobue G. Pathogenesis, animal models and therapeutics in spinal and bulbar muscular atrophy (SBMA) Exp Neurol. 2006;200:8–18. doi: 10.1016/j.expneurol.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968;18:671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem. 2000;275:8772–8778. doi: 10.1074/jbc.275.12.8772. [DOI] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- Li JY, Jahn R, Dahlstrom A. Rab3a, a small GTP-binding protein, undergoes fast anterograde transport but not retrograde transport in neurons. Eur J Cell Biol. 1995;67:297–307. [PubMed] [Google Scholar]
- Li JY, Pfister KK, Brady ST, Dahlstrom A. Cytoplasmic dynein conversion at a crush injury in rat peripheral axons. J Neurosci Res. 2000;61:151–161. doi: 10.1002/1097-4547(20000715)61:2<151::AID-JNR6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Sobue G. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2004;13:1183–1192. doi: 10.1093/hmg/ddh131. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci. 2006;9:907–916. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- Münch C, Rosenbohm A, Sperfeld AD, Uttner I, Reske S, Krause BJ, Sedlmeier R, Meyer T, Hanemann CO, Stumm G, Ludolph AC. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol. 2005;58:777–780. doi: 10.1002/ana.20631. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- Piccioni F, Pinton P, Simeoni S, Pozzi P, Fascio U, Vismara G, Martini L, Rizzuto R, Poletti A. Androgen receptor with elongated polyglutamine tract forms aggregates that alter axonal trafficking and mitochondrial distribution in motor neuronal processes. FASEB J. 2002;16:1418–1420. doi: 10.1096/fj.01-1035fje. [DOI] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Puls I, Oh SJ, Sumner CJ, Wallace KE, Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer A, Powers R, Finnegan K, Holzbaur EL, Fischbeck KH, Ludlow CL. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Ribchester RR, Thomson D, Wood NI, Hinks T, Gillingwater TH, Wishart TM, Court FA, Morton AJ. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington's disease mutation. Eur J Neurosci. 2004;20:3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x. [DOI] [PubMed] [Google Scholar]
- Roy S, Coffee P, Smith G, Liem RK, Brady ST, Black MM. Neurofilaments are transported rapidly but intermittently in axons: implications for slow axonal transport. J Neurosci. 2000;20:6849–6861. doi: 10.1523/JNEUROSCI.20-18-06849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagot Y, Rosse T, Vejsada R, Perrelet D, Kato AC. Differential effects of neurotrophic factors on motoneuron retrograde labeling in a murine model of motoneuron disease. J Neurosci. 1998;18:1132–1141. doi: 10.1523/JNEUROSCI.18-03-01132.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt BJ, Greenberg CR, Allingham-Hawkins DJ, Spriggs EL. Expression of X-linked bulbospinal muscular atrophy (Kennedy disease) in two homozygous women. Neurology. 2002;59:770–772. doi: 10.1212/wnl.59.5.770. [DOI] [PubMed] [Google Scholar]
- Shea TB. Microtubule motors, phosphorylation and axonal transport of neurofilaments. J Neurocytol. 2000;29:873–887. doi: 10.1023/a:1010951626090. [DOI] [PubMed] [Google Scholar]
- Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A. X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain. 1989;112:209–232. doi: 10.1093/brain/112.1.209. [DOI] [PubMed] [Google Scholar]
- Sobue G, Hashizume Y, Yasuda T, Mukai E, Kumagai T, Mitsuma T, Trojanowski JQ. Phosphorylated high molecular weight neurofilament protein in lower motor neurons in amyotrophic lateral sclerosis and other neurodegenerative diseases involving ventral horn cells. Acta Neuropathol (Berl) 1990;79:402–408. doi: 10.1007/BF00308716. [DOI] [PubMed] [Google Scholar]
- Sopher BL, Thomas PS, Jr, LaFevre-Bernt MA, Holm IE, Wilke SA, Ware CB, Jin LW, Libby RT, Ellerby LM, La Spada AR. Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron. 2004;41:687–699. doi: 10.1016/s0896-6273(04)00082-0. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila . Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Walker FO, Young AB, Penney JB, Dovorini-Zis K, Shoulson I. Benzodiazepine and GABA receptors in early Huntington's disease. Neurology. 1984;34:1237–1240. doi: 10.1212/wnl.34.9.1237. [DOI] [PubMed] [Google Scholar]
- Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, Orr HT. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci. 2004;24:8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]