

Abstract
The M-current (IM), comprised of Kv7 channels, is a voltage-activated K+ conductance that plays a key role in the control of cell excitability. In hippocampal principal cells, IM controls action potential (AP) accommodation and contributes to the medium-duration afterhyperpolarization, but the role of IM in control of interneuron excitability remains unclear. Here, we investigated IM in hippocampal stratum oriens (SO) interneurons, both from wild-type and transgenic mice in which green fluorescent protein (GFP) was expressed in somatostatin-containing interneurons. Somatodendritic expression of Kv7.2 or Kv7.3 subunits was colocalized in a subset of GFP+ SO interneurons, corresponding to oriens-lacunosum moleculare (O-LM) cells. Under voltage clamp (VC) conditions at −30 mV, the Kv7 channel antagonists linopirdine/XE-991 abolished the IM amplitude present during relaxation from −30 to −50 mV and reduced the holding current (Ihold). In addition, 0.5 mm tetraethylammonium reduced IM, suggesting that IM was composed of Kv7.2-containing channels. In contrast, the Kv7 channel opener retigabine increased IM amplitude and Ihold. When strongly depolarized in VC, the linopirdine-sensitive outward current activated rapidly and comprised up to 20% of the total current. In current-clamp recordings from GFP+ SO cells, linopirdine induced depolarization and increased AP frequency, whereas retigabine induced hyperpolarization and arrested firing. In multicompartment O-LM interneuron models that incorporated IM, somatodendritic placement of Kv7 channels best reproduced experimentally measured IM. The models suggest that Kv3- and Kv7-mediated channels both rapidly activate during single APs; however, Kv3 channels control rapid repolarization of the AP, whereas Kv7 channels primarily control the interspike interval.
Keywords: antiepileptic, action potential, hippocampus, interneuron, M-current, neuromodulation, potassium channel
Introduction
The M-current (IM) is a K+ conductance known to operate at subthreshold voltages and is a common target of neuromodulation throughout the CNS (Jentsch, 2000; Cooper and Jan, 2003). In CA1 pyramidal cells, IM is inhibited by muscarinic acetylcholine receptor (mAChR) activation (Halliwell and Adams, 1982), controls action potential (AP) frequency (Gu et al., 2005), and generates a medium afterhyperpolarization (mAHP) after single or multiple APs (Gu et al., 2005) that prevents the development of a postspike afterdepolarization (Yue and Yaari, 2004). In these cells, IM is probably composed of Kv7.2 and Kv7.3 channel subunits (Wang et al., 1998; Shah et al., 2002), the expression of which is critical in controlling cell and network excitability (Peters et al., 2005).
Hippocampal interneurons are also highly sensitive to neuromodulation (Parra et al., 1998), which may be important in promoting coherent oscillations and synchronization in the hippocampus (Toth et al., 1997; Buzsaki, 2002; Cobb and Davies, 2005; Lawrence et al., 2006b). Although interneurons are strongly activated by acetylcholine (McQuiston and Madison, 1999; Lawrence et al., 2006a) and possess numerous K+ conductances (Zhang and McBain, 1995a, b; Martina et al., 1998, 2000; Lien et al., 2002; Lien and Jonas, 2003), the question of whether interneurons possess IM has received little attention. Recently, we demonstrated that oriens-lacunosum moleculare (O-LM) cells generate a large mAChR-dependent afterdepolarization that is partially dependent on IM inhibition (Lawrence et al., 2006a), but the nature of how IM itself impacts excitability in stratum oriens (SO) interneurons has not yet been explored in detail. Moreover, the cellular localization of Kv7 channels in neurons has recently become a point of interest. Although early immunocytochemical evidence indicated that Kv7 subunits were localized somatodendritically on hippocampal pyramidal cells (Cooper et al., 2000; Shah et al., 2002) and interneurons (Cooper et al., 2001), an additional population of Kv7 channels has been more recognized recently on the axon initial segment and nodes of Ranvier of CNS neurons (Devaux et al., 2004; Chung et al., 2006; Pan et al., 2006). Kv7 channel antagonists modulate axonal excitability (Devaux et al., 2004), raising uncertainty about whether axonal or dendritic Kv7 channels mediate IM-dependent changes in cellular excitability originally described in hippocampal pyramidal neurons (Yue and Yaari, 2004; Gu et al., 2005) and interneurons (Lawrence et al., 2006a).
In this study, we identified Kv7 channels in CA1 SO interneurons through both immunocytochemical and electrophysiological means. We built realistic multicompartment interneuron models and explored how the cellular localization of Kv7 channels influenced the somatic detection of IM measured experimentally. We found that both experimentally measured IM and its influence on cellular excitability could be best reproduced by somatodendritic placement of Kv7 channels. Using our model, we found that IM is strongly recruited by APs and is a major dendritic conductance controlling the interspike interval (ISI) in hippocampal interneurons.
Materials and Methods
Immunocytochemistry.
All experiments were conducted in accordance with animal protocols approved by the National Institutes of Health. To permit quantification of Kv7.2 (KCNQ2) and Kv7.3 (KCNQ3) subunits in anatomically identified interneuronal populations, a mouse strain was chosen in which green fluorescent protein (GFP) is expressed in a population of inhibitory neurons [the GFP-expressing inhibitory neurons (GIN) line of transgenic mice (Oliva et al., 2000)]. In the CA1 hippocampus, GIN mice label a population of somatostatin-positive (SOM+) interneurons that project axonally to stratum lacunosum moleculare (SLM) (Oliva et al., 2000). GFP+ SO interneurons are solely comprised of the well characterized O-LM cells, whereas GFP+ cells in stratum radiatum (SR) form a relatively newly discovered population of SOM+ neurons that also project to SLM. Male mice of both GIN and wild-type (B6/C57J; Charles River Laboratories, Wilmington, MA) animals, 3–6 months of age, were perfused using an ice-cold 0.1 m phosphate buffer solution containing 4% paraformaldehyde and 15% picric acid, adjusted to pH 7.4 with NaOH. Briefly, the brains were fixed overnight, cryoprotected in 30% sucrose for several days, sliced to 30 μm thickness using a freezing microtome, and kept at 4°C for up to 3 weeks until used. Free-floating sections were incubated with primary antibodies for 12–48 h in a 0.1 m PBS containing 0.1% Triton X-100 and 4% BSA (Sigma-Aldrich, St. Louis, MO). Rabbit-KCNQ2 (1:3000) and rabbit-KCNQ3 (1:2000) primary antibodies were used (Chemicon, Temecula, CA), comparable with that originally described by Yus-Najera et al. (2003). In slices containing GFP+ interneurons, the goat anti-rabbit secondary antibody Alexa Fluoro 633 (1:1000 dilution; Invitrogen, Carlsbad, CA) was chosen to ensure sufficient separation from the GFP wavelength. After incubation of the secondary antibody for 60 min at room temperature, processed slices were mounted onto Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA), allowed to dry, immersed in Mowiol (Calbiochem, La Jolla, CA), coverslipped, and dried overnight. Confocal microscopy was performed using either Leica (Exton, PA) TCS or Zeiss (Thornwood, NY) LSM 510 2-Photon Meta confocal microscopes. For the laminar quantification of Kv7.2+ or Kv7.3+ cells within GFP+ interneuronal populations, cell counts, detected by eye, were based on a total of four brains, 20 slices per brain, and 40 images (both hemispheres) of CA1 hippocampus. Images of CA1 hippocampus were loaded into the confocal module of Neurolucida (Autoneuron v.6.0; MicroBrightField, Williston, VT) and quantified in contour mode. GFP/488 and Kv7/633 channels were viewed independently and together to ensure unambiguous colocalization of Kv7 subunits in GFP+ populations. As controls, sections omitting the primary antibody, or omitting both primary and secondary antibodies, were processed in parallel and imaged at the same confocal settings (i.e., gain and pinhole size).
Electrophysiology.
Transverse hippocampal slices (300 μm) were cut from 14- to 21-d-old mice from 129J1(50%) × CF1(50%), C57/B6, or FVB/N backgrounds. The dissection solution consisted of the following (in mm): 130 NaCl, 3.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 10 glucose, 7 MgCl2, 0.5 CaCl2, saturated with 95% O2/5% CO2, pH 7.4. In more recent experiments, a partial sucrose cutting ringer was used, consisting of the following (in mm): 80 NaCl, 24 NaHCO3, 25 glucose, 75 sucrose, 2.5 KCl, 1.25 NaH2PO4, 4 MgCl2, 0.5 CaCl2, 3 Na-pyruvate, 1 ascorbate, saturated with 95% O2/5% CO2, pH 7.4. Experiments were conducted using an upright Zeiss microscope and submerged slice chamber (Infrapatch; Luigs and Neumann, Ratingen, Germany). The speed of solution exchange was ∼1 min, as estimated by a single exponential fit to the mean change in Ihold at −30 mV by 10 μm linopirdine (see Fig. 2I). Loose patch and whole-cell patch-clamp recordings were made from visually identified interneurons on the SO/alveus border of the CA1 using an Axoclamp 200B or Multiclamp 700A amplifier (Molecular Devices, Union City, CA), filtered at 3 kHz (Bessel filter; Frequency Devices, Haverhill, MA), and digitized at 10–40 kHz (Digidata 1320A and pClamp Software; Molecular Devices). Loose-patch recordings were chosen over tight seal recordings for their noninvasiveness and excellent signal-to-noise ratio (Nunemaker et al., 2003; Pouille and Scanziani, 2004). A 3–5 MΩ electrode was placed on the soma, and negative suction was applied to the pipette until a low resistance seal of ∼7–20 MΩ was achieved. Action currents were recorded in voltage clamp (VC) mode with no voltage applied (V = 0). Stability of the loose-patch recording was monitored via a −1 mV, 100 ms duration step at 30 s intervals. Unless otherwise stated, for whole-cell recordings, the extracellular solution (ECS) contained the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, and 20 glucose. dl-APV (100 μm), DNQX (25 μm), and gabazine (5 μm) were added to the ECS to block NMDA, AMPA, kainate, and GABAA receptors, respectively. Except in loose-patch and current-clamp (CC) recordings (see Fig. 7), tetrodotoxin (1 μm) was added and CsCl (5 mm) was replaced with equimolar NaCl in VC experiments to block Na+ channels and the hyperpolarization-activated current Ih, respectively. For VC recordings in which strong depolarizing voltage steps were introduced, CaCl2 was lowered to 0–0.5 mm and MgCl2 elevated to 2–4 mm to reduce the activation of a slow Ca2+-activated K+ conductance (Lawrence et al., 2006a). In some experiments, 50 μm CdCl2 or 2 mm EGTA was added to further reduce the influence of IAHP. Recording pipettes were filled with the following (in mm): 135 K-gluconate, 10 HEPES, 0.1 EGTA, 2 Na2ATP, 0.3 Na2GTP, 20 KCl, pH 7.3. In more recent experiments, to take additional precautions against the possibility of IM rundown (Suh and Hille, 2002), an intracellular solution was used containing the following (in mm): 110 K-gluconate, 10 HEPES, 0.1 EGTA, 4 Mg-ATP, 0.3 Na2GTP, 10 Na-phosphocreatine, pH 7.2. Biocytin (0.2%) was added for post hoc morphological processing of each recorded cell. Linopirdine and XE-991 (XE) were purchased from Tocris Cookson (Ellisville, MO); all other drugs were purchased from Sigma-Aldrich. Voltage measurements were not corrected for a calculated liquid junction potential of −13.8 mV but were accounted for in multicompartment models.
Figure 2.

Detection of IM in SO interneurons using linopirdine, XE-991, and retigabine. In VC, a step was applied from −30 to −50 mV at 15 s intervals. After a 2 min baseline period, a 4 min application of 10 μm linopirdine (A, B), 4 μm XE-991 (C, D), or 10 μm retigabine (E, F) was applied to the bath. Traces enumerated in A, C, and E are the average of three traces within a 45 s window as indicated by the bars in B, D, and F, respectively. Traces were filtered at 500 Hz for display. Control traces (gray) are overlaid for comparison with drug conditions (black). IM amplitudes (G) and Ihold (H) at −30 mV for mean data (columns) and individual cells (points) for linopirdine (n = 19), XE-991 (XE; n = 11), and retigabine (ret; n = 15) conditions are shown. Each condition in G and H was paired with its initial control value obtained in a 45 s window before the onset of application of the drug. GFP+ SO cells, anatomically identified O-LM cells, and unidentified SO cells are indicated by black symbols, gray symbols, and open symbols, respectively. I, Time course of changes in Ihold during application of linopirdine (circles), XE-991 (squares), or retigabine (triangles), normalized to the initial Ihold before drug application.
Figure 7.
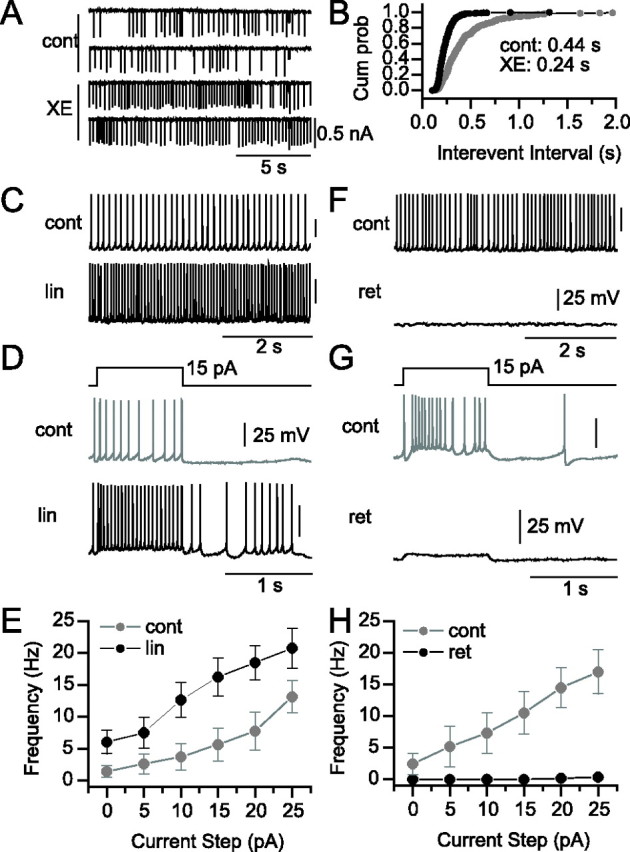
Effects of IM antagonists on AP firing pattern. Loose-patch recording (A) and corresponding cumulative probability plot (Cum prob; B) of action current intervals before (cont) or after (XE) application of 8 μm XE-991. Square current steps near the end of traces in A indicate −1 mV, 100 ms duration seal tests. Spontaneous AP firing (C) and response (D) to a 1 s, 15 pA current step from the resting potential under control conditions (cont) (gray) and in the presence of 20 μm linopirdine (lin) (black). E, Plot of current step amplitude versus AP frequency across a 0–25 pA range for control (gray) and 20 μm linopirdine (black) conditions (p < 0.05; n = 8). Spontaneous AP firing (F) and response (G) to a 1 s, 15 pA current step from the resting potential under control conditions (cont) (gray) and in the presence of 10 μm retigabine (ret) (black). H, Plot of current step amplitude versus AP frequency across a 0–25 pA range for control (cont) (gray) and 10 μm retigabine (ret) (black) conditions (p < 0.05; n = 7).
Electrophysiological analysis.
Current-clamp analysis and leak subtraction was performed off-line using software written in Axograph 4.7 (Molecular Devices) by the authors. For the analysis of loose-patch recordings, spontaneous action currents were detected with a sliding template (derived from raw traces) using a detection threshold of three times the SD of the noise (Clements and Bekkers, 1997). For current-clamp analysis, APs were detected using a detection threshold of 5 V/s. For leak subtraction, a −5 mV seal test from −60 mV preceded each depolarizing stimulus. The steady-state current, derived from the single exponential fit to the average of 18–24 seal tests, was scaled to the voltage waveforms and subtracted from the average current trace. Outward currents generated by depolarizing voltage steps were the average of three separate trials. Traces obtained in control conditions and in the presence of IM antagonists were both leak subtracted before obtaining XE-991 or linopirdine-sensitive components via digital subtraction.
For outward current traces measured in control and in the presence of IM antagonists, deactivation was sufficiently described by three exponentials (see Fig. 6A), as judged by a more than twofold reduction in χ2 compared with a two exponential fit (Origin; Microcal, Northampton, MA). To overcome the poor signal-to-noise ratio in the XE-991 or linopirdine-subtracted traces, fitted exponentials curves obtained in control and drug condition were subtracted from each other. This yielded a curve that described the XE-991 or linopirdine-sensitive component well. Data are presented as means ± SEM.
Figure 6.
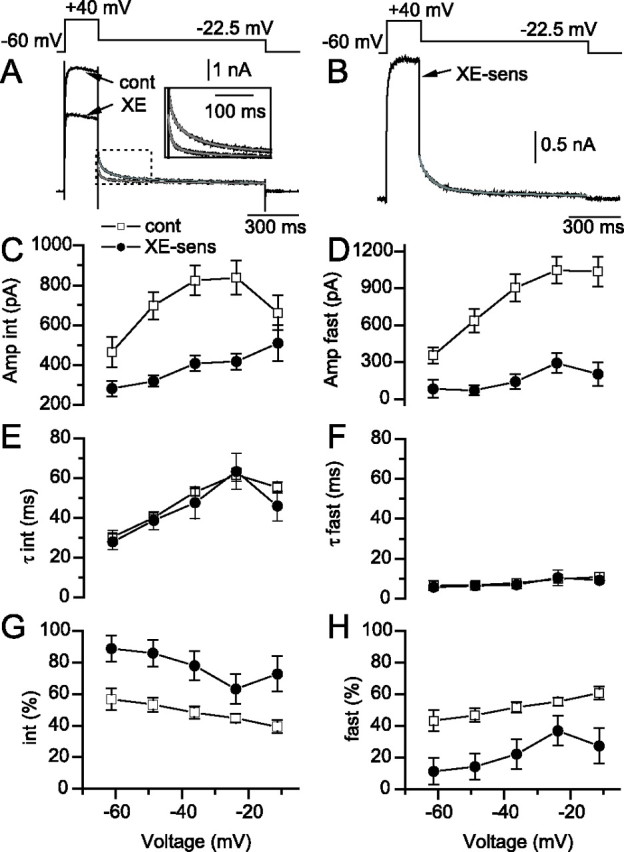
IM underlies an intermediate component of outward current deactivation. A, Triple exponential fits (gray) to the deactivation of the outward current after deactivation of −22.5 mV in control (cont) or in the presence of 4 μm XE-991 (XE). Inset, Expanded region showing that the trace obtained in the presence of XE-991 deactivates faster than the trace in control conditions. B, XE-991-senstive (XE-sens) trace (black) and fit (gray) obtained via digital subtraction of the traces and fits in A. Amplitudes (C, D), time constants (E, F), and percentages (G, H) of each component, plotted against the test potential for control (open squares) and XE-991-sensitive traces (filled circles). Percentages in G and H are calculated from the combined amplitude (amp) of fast and intermediate (int) components.
Transfection of human embryonic kidney 293 cells with Kv3.1/Kv3.2 plasmids.
Human embryonic kidney 293 (HEK293) cells were cultured in DMEM + 10% FBS (Invitrogen). Transfections were performed using FuGENE6 Transfection Reagent (Roche Applied Science), according to the specifications of the manufacturer. cDNA clones expressing Kv3.1b or Kv3.2a were cotransfected with pIRES2-EGFP (Clontech, Cambridge, UK) at a ratio of 4:1. A total amount of 0.5 μg of DNA was used to transfect 5e4 HEK293 in 1 ml of suspension. After transfection, cells were plated on poly-d-lysine-coated 18 mm glass coverslips in 12-well plates. Outward currents were generated from isolated GFP+ HEK293 cells 24–48 h after transfection. Data acquisition was made on two channels. On one channel, recording stability was monitored via the peaks of the capacitance transients; on the second channel, leak subtraction was performed with an on-line P/4 protocol using pClamp software.
Neurolucida reconstructions.
In a subset of electrophysiological recordings, slices were fixed in 4% paraformaldehyde (>24 h) in 0.1 m phosphate buffer. Slices were immersed in PBS and resectioned into 70 μm sections on a freezing microtome. Biocytin staining was revealed using an avidin-biotin-peroxidase reaction (ABC Standard Kit; Vector Laboratories, Burlingame, CA). Slices were mounted on gelatin-coated glass slides, dehydrated, and coverslipped with Permount. Digital reconstruction of interneuron morphologies was performed with a Zeiss bright-field light microscope driven by Neurolucida software (MicroBrightField, Colchester, VT).
Multicompartment modeling of O-LM interneurons.
Vector coordinate files of two O-LM cells (see Fig. 4, cells 1 and 2) were imported from Neurolucida to the shareware program cvapp (www.compneuro.org/CDROM/docs/cvapp.html), which enabled the user to test electrical connectivity between soma and all processes. The reconstructions were then imported into NEURON (http://www.neuron.yale.edu).
Figure 4.

Anatomical identification of IM-containing SO interneurons. Neurolucida reconstructions of 4 O-LM cells (cells 1–3, 5) and one putative basket cell (cell 4) are shown. Strata are labeled stratum oriens (o), pyramidale (p), radiatum (r), and lacunosum moleculare (lm). Cells 1 and 2 were used in subsequent simulations. Arrows denote where the axon emerged from the dendrite (cells 1 and 2) or cell body (cells 3–5).
The passive properties of a cell included the membrane capacitance, C, the axial resistivity, Ra, the leak channel conductance, ḡL, and the reversal potential of the leak channel, EL. For the multicompartment models here, an additional potassium leak channel was incorporated with conductance, ḡKL, and reversal potential, EK. Passive properties (Ra, ḡL, EL, and ḡKL) were optimized uniformly throughout the somatic, dendritic, and axonal tree of the full morphology models to match experimentally measured current traces using a voltage step protocol from −60 to −65 mV for each particular cell (see Fig. 8F). The membrane capacitance, C, was fixed at 0.9 μF/cm2 (Gentet et al., 2000).
Figure 8.

Somatodendritic localization of Kv7 channels best reproduced IM-mediated outward currents. A–D, In a multicompartment O-LM interneuron model, IM was localized to soma only (A), soma and primary dendrites (B), soma and all dendrites (C), or axon only (D). Compartmental regions lacking (black) and containing (red) IM are indicated. Insets, IM-mediated outward currents generated by a family of 400 ms somatic voltage step commands from −60 to +35 mV in −15 mV increments. A fixed gM of 0.75 pS/μm2 was used in all simulations. E, IM-mediated current, measured at end of voltage step, versus command voltage for the four IM distributions shown in A–D. F, In the actual electrophysiological recording from the cell that was modeled, stepping from −60 to −65 mV generates a capacitive transient, which enables an estimate of the surface area that is voltage clamped (38.0 pF). Assuming a specific capacitance of 0.9 μF/cm2, somatic VC control is estimated to correspond to the surface area colored in blue. prox dend, Proximal dendrite.
Although little is known about the properties of channels in interneuronal axons, the axonal surface area makes up a large fraction of the total surface area of the interneuron. To limit errors produced by choosing arbitrary parameters for the large surface area of the axonal membrane, and to speed computation, the axonal morphology was reduced to the point at which it enters the SLM region of the hippocampus. Because the axonal morphology was reduced, the passive properties were reoptimized in the reduced axon segments while keeping the somatic and dendritic passive properties fixed to the previously determined values (see Table A1 in Appendix for values, available at www.jneurosci.org as supplemental material). This provided a correction for reducing the surface area. These reduced morphologies were used for all simulations.
Input resistance (Rm) was measured experimentally from each cell using a seal test protocol from −60 to −65 mV. The steady-state current difference before and after the voltage step was used in combination with the known voltage difference to calculate Rm of each cell. The experimentally measured values for Rm were 349 MΩ for cell 1 and 506 MΩ for cell 2. With the optimized passive properties, as described above, the model interneurons have a Rm of 318 MΩ for cell 1 and 524 MΩ for cell 2. Membrane time constants (τm) measured for O-LM interneurons range in value from 33 to 66 ms (Gloveli et al., 2005). For the model interneurons, τm values were 49 ms for cell 1 and 45 ms for cell 2, which fall within this experimentally measured range.
A kinetic model for IM was designed from values obtained from experiments. The model was based on three main experimental parameters. First, activation time constants were obtained by fitting single exponentials to linopirdine-sensitive current traces. Second, the deactivation kinetics of IM, with special attention given to the slow relaxation time constant of deactivation from −30 to −50 mV, was modeled based on experimental data. Finally, the experimental somatic current voltage (I–V) relationship of the IM-mediated component was used to estimate the density and voltage dependence of gM in the models. Tail currents were not consistently observed experimentally, resulting from possible space clamp and leak subtraction errors associated with the whole-cell recording configuration. However, the model explicitly incorporated the series resistance (Rs) and accounted for space-clamp errors related to the dendritic geometry of the neuron. This allowed V1/2 and k values to be adjusted in the model until the experimentally derived relationship between the command voltage and the somatically generated current was reproduced. The optimal values for V1/2 and k were −27 mV and 7, respectively, in agreement with values reported in the literature (Jow and Wang, 2000; Prole et al., 2003). To simulate the effect of XE991, the conductance gM was set to 0. The effect of the drug retigabine was simulated by shifting the V1/2 of the steady-state activation curve from −27 to −60 mV (Wickenden et al., 2000) while keeping the conductance value the same as in the control cases.
The equations describing dynamics are summarized briefly here. First, the discrete version of the cable equation, an approximation to the original partial differential cable equation, describes the evolution of voltage in each compartment k, Vk as follows:
 |
where C = 0.9 μF/cm2 for all compartments. The sum is taken over all compartments n that are connected to compartment k. γn,k is the internal conductance (reciprocal of axial resistivity; 1/Ra) between the respective compartments. Iionic,k is the transmembrane ionic current for compartment k. Inward currents, which depolarize the membrane, are by convention, negative. For simplicity, the compartment subscript k will be dropped from hereafter.
The term Iionic encompasses various ionic currents that were chosen from literature and experiments performed specifically on this interneuron subtype (Migliore et al., 1995; Zhang and McBain, 1995a, b; Maccaferri and McBain, 1996; Destexhe et al., 1998; Martina et al., 2000; Lien et al., 2002; Saraga et al., 2003). These currents include the traditional Hodgkin–Huxley sodium current, INa, two (fast and slow) delayed rectifier K+ currents, IKdrf and IKdrs, the transient K+ current, IA, the L-type calcium current, ICaL, the T-type calcium current, ICaT, the calcium-activated K+ current, IK(Ca), the hyperpolarization-activated cation current, Ih, and the muscarinic K+ current, IM. In each compartment and at each time, the current through a channel is proportional to its maximal conductance, ḡ, multiplied by its respective Hodgkin–Huxley gating variables, which include activation variables, m, and an inactivation variable, h, where applicable. Some channels do not inactivate and therefore the inactivation variable, h, is not included for these currents. m and h gating variables are representatives of the activation and inactivation variables for the particular ionic current and are not to be considered the same for the different currents.
The sum of the intrinsic membrane terms that, in each compartment, contribute to Iionic can be written as follows:
 |
where the reversal potential for sodium, ENa = 50 mV, for potassium, EK = −95 mV, and for the hyperpolarization-activated cation channel, Eh = −32.9 mV. When referring to conductance (g) throughout the text, this conductance is equal to the conductance density, ḡ, multiplied by the activation variable, m. See Table A2 and A3 (available at www.jneurosci.org as supplemental material) for conductance values, distributions, and kinetic equations.
The somatodendritic distribution of Ih is not known for interneurons. Because in hippocampal pyramidal neurons, Ih is distributed in both the soma and dendrites (Magee, 1998), we compared both somatic only and somatic-dendritic distributions of Ih. The density of h-channels was adjusted in the two cases so as to produce the same “sag” when a hyperpolarizing step current of 100 pA was applied to the soma (soma only, 5.00 × 10−5 S/cm2; somatodendritic, 1.56 × 10−6 S/cm2). Neither the spontaneous frequency (2.6 vs 2.3 Hz) nor the response of the cell to 100 pA of tonic depolarizing current injected into the soma (9.4 vs 9.2 Hz) differed substantially between soma only and somatodendritic conditions, respectively. Because results were very similar for both distributions of Ih, all subsequent simulations were performed with only somatic Ih.
All VC simulations, excluding the −30 to −50 mV protocol, were done with only IM and passive properties included in the models. All ion channels described above were included for the CC simulations. For the −30 to −50 mV VC simulations, the conductance values for INa, IK(Ca), ICaT, ICaL, and Ih were set to zero, simulating the experimental effects of TTX, CdCl2, and 5 mm CsCl.
Results
Laminar and cellular distribution of Kv7 channels in hippocampal interneurons
Although Kv7 channels have been detected in the somata of SO CA1 parvalbumin (PV)-positive interneurons (Cooper et al., 2001), probably corresponding to a subpopulation of basket cells (Somogyi and Klausberger, 2005), an additional interneuronal population of PV-negative Kv7+ interneurons was found in SO that has not been identified either neurochemically or morphologically. To determine whether Kv7 subunits were present in anatomically identified interneuron populations, we used the so-called GIN mice, in which GFP is expressed predominantly in somatostatin-positive interneurons (Oliva et al., 2000). In these mice, GFP+ SO cells in CA1 hippocampus are comprised entirely of O-LM cells (Oliva et al., 2000), an abundant and well characterized SO cell type (Maccaferri and Lacaille, 2003; Maccaferri, 2005). Although GFP+ cells are present in other hippocampal layers, these cells represent SOM+ interneurons, the axons of which also ramify exclusively in SLM (Oliva et al., 2000).
Kv7.2 immunoreactivity was strong in the somata of pyramidal cells but also in interneurons in SO (Fig. 1B). A subset of GFP+ SO cells expressed Kv7.2+ (Fig. 1A,B, arrows). In addition, Kv7.2 expression was detected in the dendrites of GFP+ SO cells (Fig. 1C,D, filled arrows). Similar labeling of Kv7.3 in both soma and dendrites was detected in GFP+ SO cells (Fig. 1E–H). Quantification of Kv7 subunits in GFP+ cells revealed that as a population, 39.7 ± 6.3% (150 of 377) of GFP+ SO cells were Kv7.2 positive. However, GFP+ SR/SLM interneurons did not express Kv7.2 to the same extent (16.8 ± 7.3%, 32 of 191 cells; p < 0.05; n = 4). For Kv7.3 subunits, a similar distribution was observed: Kv7.3 labeling was detected in 34.3 ± 4.0% (143 of 417) of GFP+ SO cells but only 16.14 ± 4.1% (36 of 223) of GFP+ SR/SLM cells (p < 0.01; n = 4). The expression of Kv7.2 and Kv7.3 subunits to similar extents within SO and SR/SLM GFP+ cell populations are consistent with the idea that Kv7.2/Kv7.3 heteromers form a common channel composition in the hippocampus (Cooper et al., 2000; Hadley et al., 2003; Delmas and Brown, 2005). Axonal labeling of Kv7.2 and Kv7.3 was essentially absent in SLM, where the axons of GFP+ cells form a dense plexus (Fig. S1, available at www.jneurosci.org as supplemental material). In summary, immunocytochemical labeling of Kv7.2 and Kv7.3 subunits was localized to somatodendritic regions of CA1 SO interneurons and is more abundantly expressed in GFP+ SO cells (most likely O-LM cells) than in other GFP+ cell populations.
Figure 1.
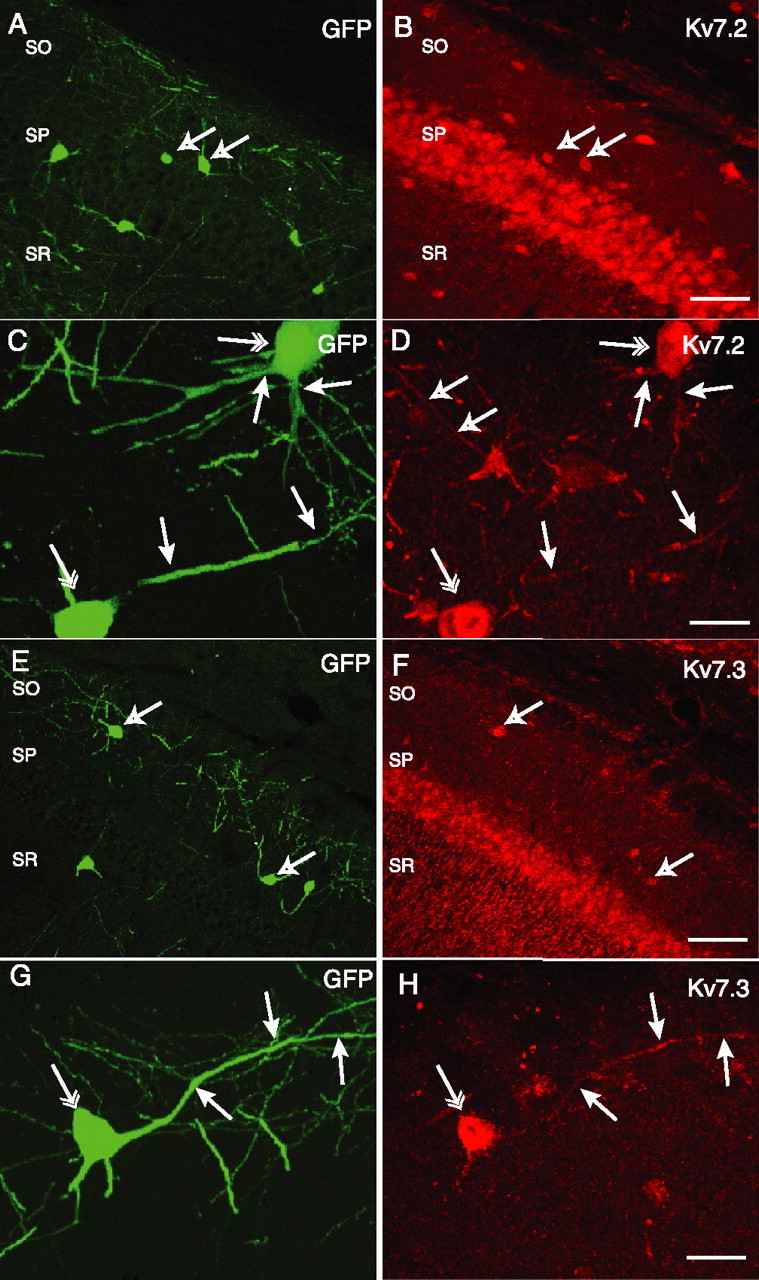
Somatodendritic expression of Kv7.2 (A–D) and Kv7.3 (E–H) in GFP+ hippocampal interneurons. Confocal images of CA1 hippocampal slices from GIN transgenic mice, which express GFP in somatostatin-positive CA1 hippocampal interneurons in SO, SP, and stratum radiatum (SR), are shown. GFP expression (A) and Kv7.2+ immunoreactivity (B) reveal colocalization of Kv7.2 in a subpopulation of GFP+ cells (arrows). GFP+ cells in SO exhibit Kv7.2 immunoreactivity on somata (C, double arrows) and dendrites (C, D, single filled arrows). Open arrows in D indicate a Kv7.2-immunoreactive dendrite from a GFP-negative interneuron. GFP+ interneurons (E) also colocalize with Kv7.3 immunoreactivity (F, arrows). In a GFP+ SO interneuron (G), somatic (H, double arrow) and dendritic (H, single arrows) Kv7.3 immunoreactivity was also apparent. Scale bars: B, F, 100 μm; D, H, 25 μm.
Electrophysiological detection of Kv7 channels in SO interneurons
Using a protocol that reveals IM through its distinct voltage dependence and time course of deactivation from other K+ channels (Lamas et al., 1997; Schnee and Brown, 1998), we determined whether IM could be revealed in SO interneuron populations. In this protocol, the cell was held at −30 mV; 1 s long, −20 mV steps were introduced every 15 s (Fig. 2). Under control conditions, a step to −50 mV forced deactivation of a slowly relaxing outward current that decayed with a time constant of ∼106 ms (Fig. 2A), consistent with the presence of IM. The amplitude of the single exponential fit, in this case 20 pA, was used as an index of how much IM was present. After repolarization to −30 mV, IM was observed to reactivate, although this observation was not always in a symmetrical manner because of the reactivation of one or more additional conductances present. After obtaining a 2–3 min baseline, the Kv7 antagonist linopirdine (10 μm) was added to the bath, causing a complete loss of measurable IM over the course of 4 min (Fig. 2A), accompanied by a reduction in Ihold from 221 to 86 pA (Fig. 2B). As demonstrated in Figure 2, C and D, a similar time course and extent of block was observed using the Kv7 antagonist XE-991 (4 μm), which abolished the 21 pA IM-mediated current detected in control conditions and reduced Ihold from 160 to 21 pA at −30 mV. In contrast, the Kv7 channel opener retigabine had the reverse effect, enhancing the amplitude of the IM-mediated relaxation from 18 to 32 pA (Fig. 2E), which was accompanied by an increase in Ihold from 167 to 355 pA (Fig. 2F).
Distributions of IM amplitude and drug-induced changes in Ihold at −30 mV for individual cells and the mean are plotted in Figure 2G–I. In linopirdine experiments, a measurement of IM amplitude could be obtained in 19 of 27 (70.4%) SO interneurons; the average IM amplitude was 11 ± 1.2 pA, which decayed with an average time constant of 193 ± 57 ms. After 4 min application of 10 μm linopirdine at −30 mV, IM amplitude was abolished to 0.1 ± 0.1 pA (Fig. 2G) (n = 19; p = 4 × 10−8), and Ihold was reduced from 122 ± 11 to 71 ± 9 pA (Fig. 2H) (n = 19; p = 7 × 10−6). The initial IM amplitude and linopirdine-induced change in Ihold were correlated (p = 0.013). In eight cells, IM was not detected; however, linopirdine produced a smaller but significant decrease in Ihold at −30 mV (−17.5 ± 3.8 pA; p = 0.002). Similarly, in experiments in which XE-991 was used, IM was detected in 11 of 17 (64.7%) SO cells (mean, 10.5 ± 1.7 pA). In the presence of XE-991, IM was completely abolished (Fig. 2G) (p = 0.0001; n = 11), and the mean Ihold at −30 mV decreased from 146 ± 16 to 82 ± 16 pA (Fig. 2H) (p = 0.0002; n = 11). A significant correlation was also seen between IM amplitude and the shift in Ihold induced by XE-991 (p = 0.0008; n = 11). Retigabine increased the mean IM amplitude from 12.1 ± 2.6 to 21.0 ± 3.1 pA (Fig. 2G) (p = 0.002; n = 15) and the mean Ihold at −30 mV from 128 ± 16 to 227 ± 32 pA (Fig. 2H) (p = 0.0001; n = 15). The initial IM amplitude and the magnitude of the increase in Ihold at −30 mV by retigabine were correlated (p = 0.016; n = 15). Figure 2I plots the mean Ihold at −30 mV, normalized to the initial Ihold, for each of the three drug conditions. Note that linopirdine and XE-991 overlap well in both magnitude and time course of inhibition of Ihold at −30 mV. In summary, using three different modulators of Kv7 channels, these data reveal the presence of IM in SO interneurons.
Tetraethylammonium sensitivity of interneuronal IM
In recombinant and native systems, Kv7.2-containing subunits are selectively blocked by submillimolar concentrations of tetraethylammonium (TEA) (Hadley et al., 2000, 2003; Shah et al., 2002). To determine whether Kv7.2-containing channels contributed to IM in SO interneurons, 0.5 mm TEA was applied to SO interneurons held at −30 mV (Fig. 3). For these experiments, SO recordings were limited to those in which IM amplitude was detected (Fig. 3A). A 4 min application of 0.5 mm TEA blocked the IM amplitude from 26.9 ± 6.9 to 13.1 ± 4.4 pA (p = 0.002; n = 10) (Fig. 3B) and reduced Ihold at −30 mV (from 160.3 ± 39.0 to 132.2 ± 37.4 pA; p = 2.6 × 10−5; n = 10) (Fig. 3C). We then asked whether 4 μm XE-991 could occlude the effects of TEA on IM amplitude and Ihold. Application of 4 μm XE-991 inhibited IM (38.6 ± 6.9 to 6.2 ± 1.1 pA) and reduced Ihold at −30 mV (246.3 ± 26.2 to 192.5 ± 29.4 pA; p = 0.0006), but subsequent application of 0.5 mm TEA plus 4 μm XE-991 did not produce a further reduction in IM amplitude (5.8 ± 1.6 to 5.7 ± 2.0 pA; p = 0.97; n = 7) (Fig. 3B) or Ihold (188.9 ± 41.2 to 185.5 ± 35.6 pA; p = 0.86; n = 7) (Fig. 3C). These findings suggest that a substantial fraction of IM is carried by TEA-sensitive, Kv7.2-containing subunits.
Figure 3.

TEA sensitivity of interneuron IM. TEA (0.5 mm) was bath applied to SO interneurons or after inhibition of IM with 4 μm XE-991 (XE). A, Current response to a −20 mV step from −30 mV under control conditions (cont) and after a 4 min application of 0.5 mm TEA. Gray lines in A indicate single exponential fits from which IM amplitude was obtained. Plots of IM amplitude (B) and Ihold (C) at −30 mV for each condition (n = 10) are shown. Filled symbols indicate recordings from morphologically identified O-LM cells.
Morphology of IM-containing SO interneurons
In 48 cells from wild-type animals in which sufficient biocytin filling of the axon permitted identification of the cell type, 43 IM-containing cells were found to be O-LM cells (89.6%), consistent with O-LM cells being a commonly encountered cell type in SO (Maccaferri, 2005). Other IM-containing cells included four cells in which the axon terminated primarily in the pyramidal cell layer (8.3%), which most likely correspond to basket cells (Fig. 4, cell 4), and one cell in which the axon ramified primarily in SO (2.1%). Representative Neurolucida reconstructions of five IM-containing cells are shown in Figure 4. As previously described in rats (Martina et al., 2000), O-LM cell morphologies featured cells in which the axon originated either from a proximal dendrite (cells 1, 2) or the soma (cells 3, 5).
In a subset of recordings, we recorded from 27 GFP+ SO cells. In several cells in which sufficient axonal labeling allowed the unambiguous identification of the cell type, GFP+ SO cells were found to project to LM, as reported previously (Oliva et al., 2000). Consistent with the heterogeneity in immunocytochemical detection of Kv7.2/Kv7.3 in GFP+ cell populations (Fig. 1), the magnitude of IM (Fig. 2G, black symbols) and change in Ihold (Fig. 2H, black symbols) fell across a 2–20 pA range in recorded GFP+ SO cells. In addition, in 4 of 17 (23.5%) GFP+ SO cells, an IM amplitude was not detected (data not shown).
IM-mediated outward currents in SO interneurons
After sustained depolarization, hippocampal CA1 SO interneurons produce large outward K+ currents (Zhang and McBain, 1995b; Martina et al., 1998; Lien et al., 2002). In addition to a transient IA-type conductance, the outward conductance is comprised of a sustained component thought to be carried by Kv3-like channels and to a smaller extent by Ca2+-sensitive conductances (Zhang and McBain, 1995b; Lien et al., 2002). Importantly, in previous studies, Kv3-like outward currents in SO interneurons were isolated based on their TEA sensitivity (Zhang and McBain, 1995b; Martina et al., 1998; Lien et al., 2002), which we now show will also block interneuronal IM in these neurons (Fig. 3). Moreover, the same protocols used to activate the sustained outward current would be expected to activate IM (Martina et al., 1998; Lien et al., 2002).
To address whether an IM-like conductance was present in the sustained outward current of hippocampal CA1 SO interneurons, we introduced a large depolarizing voltage step from −60 mV and determined to what extent IM antagonists blocked the outward current. As illustrated in Figure 5A (inset), a +100 mV voltage step command from −60 mV generated an outward current of 7.8 nA amplitude, which was reduced to 6.8 nA after incubation of the IM antagonist XE-991 (8 μm). Digital subtraction of the control current from the current obtained in the presence of XE-991 yielded a ∼1 nA IM-mediated current that was 13.6% of the total outward current. As revealed by application of XE-991 or linopirdine, plots of individual and pooled isolated IM outward current amplitudes (Fig. 5A) and the fractional contribution of IM to the total outward current (Fig. 5B) are shown. With increasing concentration of XE-991, increasingly larger XE-991-sensitive components of the outward current were revealed, as well as the fraction of cells exhibiting IM-mediated currents. Linopirdine (10 μm) also revealed large IM-mediated outward currents (1590 ± 374 pA) in six of six cells (p = 0.008, compared with vehicle), comprising 24.2 ± 5.5% of the total outward current.
Figure 5.
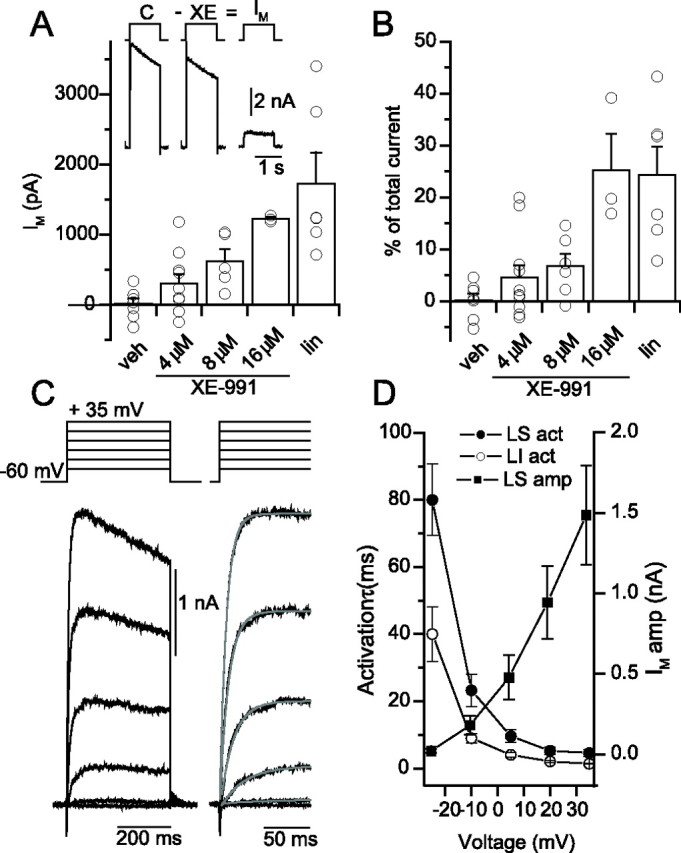
XE-991- and linopiridine-sensitive components of outward currents in SO interneurons. A, Inset, Digital subtraction of the outward current generated by a +100 mV voltage step command from −60 mV in control (C) from that generated after 10 min incubation in 8 μm XE-991 (XE) yields the isolated outward current mediated by IM. Dose dependence of IM antagonist effect, plotted as IM amplitude (A) and as a percentage (B) of the total outward current for vehicle (veh), 4–16 μm XE-991, and 10 μm linopirdine (lin) conditions. Recording stability was first ascertained in the absence of antagonists (vehicle). Recordings in which ΔRs was <3% after 10 min of whole-cell recording generated subtracted currents that were typically ±300 pA (mean 43 ± 86 pA; n = 7), a negligible amount (0.2 ± 1.3%) of the total outward current (7.5 ± 0.7 nA). Experiments with IM antagonists used the same criteria for recording stability and time point in the recording (at 10 min after application of antagonists). The IM-mediated current was significantly outside of the distribution of the vehicle for 4 of 11 cells in 4 μm XE-991 (733 ± 162 pA; p = 0.01), four of six cells in 8 μm XE-991 (742 ± 164 pA; p = 0.01), three of three cells in 16 μm XE-991 (1227 ± 24 pA; p = 4 × 10−6), and six of six cells in 10 μm linopirdine (1590 ± 374 pA; p = 0.008). C, Linopirdine-sensitive outward currents generated in response to a family of voltage steps from −60 mV to +35 mV. Right, Expanded region showing single exponential fits (gray) to the rise of the outward current. D, Time constant of activation of the linopirdine-sensitive component (LS act; filled circles), the linopirdine-insensitive component (LI act; open circles), and the amplitude of the linopirdine-sensitive component (LS amp; filled squares) plotted versus command voltage for a population of six SO cells.
In a separate set of experiments, we examined the time course of activation of the IM-mediated current by applying a series of increasingly more depolarizing voltage steps in the absence and presence of 10 μm linopirdine (Fig. 5C). After delivering a voltage step from −60 to −25 mV, the isolated IM activated with an average time constant of 80.0 ± 10.7 ms, consistent with the relatively slow time course of activation of the IM-mediated current (Selyanko and Brown, 1999; Shah et al., 2002). However, with increasingly stronger levels of depolarization, the time course of IM activation decreased (Fig. 5D, filled circles) as the amplitude of the IM outward current increased (Fig. 5D, filled squares). However, the time course of activation of the linopirdine-insensitive component of the outward current (Fig. 5D, open circles) was faster at test voltages between −25 and +35 mV (p < 0.05; n = 6), indicating that linopirdine-sensitive and -insensitive components of the outward current were kinetically distinct from one another.
In addition, we examined how IM antagonists altered deactivation of the outward current (Fig. 6). In control conditions, depolarization to a command potential of +40 mV generated a 6.4 nA outward current, which, after stepping back to −22.5 mV, generated a complex time course of deactivation that was well described by three exponentials (Fig. 6A, gray). Digital subtraction of the exponential curves obtained in control and in the presence of XE-991 described the deactivation of the IM-mediated component well (Fig. 6B). Inspection of the amplitudes (Fig. 6C,D), time constants (Fig. 6E,F), and percentages of each component (Fig. 6G,H) revealed that the XE-991-sensitive current is predominantly composed of an intermediate time constant of deactivation (Fig. 6G). The fast component (Fig. 6D,F), which probably reflects deactivation of Kv3 channels, was relatively insensitive to XE-991 (Fig. 6H). Similar results were obtained using 10 μm linopirdine (n = 8; data not shown). Although dendritic filtering will influence the kinetics of deactivation of IM, these results nevertheless show that IM mediates a substantial fraction of the intermediate component present in the deactivation of the outward current.
Although we show that the outward currents isolated by Kv7 antagonists can be distinguished from the faster (presumably delayed rectifier) component of the outward current based on both their activation (Fig. 5D) and deactivation (Fig. 6C–H) time courses, the observation that the linopirdine-sensitive component activates more rapidly at depolarized potentials (Fig. 5D) than appreciated in recombinant Kv7 channels (Wang et al., 1998) raised the possibility that delayed rectifiers may have some affinity for Kv7 antagonists. Slow-delayed rectifier channels in CA1 pyramidal cells, which are primarily composed of Kv2 channels (Martina et al., 1998), have no appreciable affinity for linopirdine at 10 μm (Schnee and Brown, 1998). However, whether fast delayed rectifiers, which are primarily composed of Kv3 channels in SO interneurons (Martina et al., 1998), have any affinity for Kv7 antagonists is not known.
We tested the effects of linopirdine or XE-991 on recombinant Kv3 channels expressed in HEK293 cells. As shown in Fig. S2 (available at www.jneurosci.org as supplemental material), neither linopirdine nor XE-991 at 20 μm substantially altered the amplitude of Kv3.1- or Kv3.2-mediated outward currents. In addition, we performed a subset of recordings from SO interneurons in the continuous presence of l mm 4-AP, which blocks the majority of Kv3-mediated current (Fig. S2, available at www.jneurosci.org as supplemental material) (Lien et al., 2002). Under these conditions, 10 μm linopirdine attenuated IM amplitude after stepping from −30 to −50 mV (10.9 ± 2.3 to 0.3 ± 0.3 pA; p = 0.01) and induced changes in Ihold at −30 mV (102 ± 16 to 68 ± 16 pA; p = 0.04; n = 5), similar to that seen in the absence of 4-AP. After strong depolarization to +20 mV, rapidly activating (τ = 4.5 ± 1.8 ms), linopirdine-sensitive outward currents (1024 ± 327 pA; n = 5) were observed in the presence of 1 mm 4-AP. Therefore, we conclude that linopirdine- and XE-991-sensitive components of the outward current are indeed carried by Kv7 channels, and that the magnitude and kinetics of the IM-mediated outward current are unlikely to be substantially contaminated by a Kv3-mediated outward current component.
Effects of IM modulators on AP firing pattern
As an initial assessment of the physiological consequences of IM, we took advantage of the observation that many SO interneurons fire spontaneously (McBain et al., 1994; Zhang and McBain, 1995a). Spontaneous firing was monitored as action currents in loose-patch configuration, a noninvasive approach that yields excellent signal-to-noise (Fig. 7A). After bath application of 8 μm XE-991, the mean time interval between APs decreased from 0.44 to 0.24 s (Fig. 7B), corresponding to an 82% increase in AP frequency. In a population of nine cells [three cells using XE-991 (8 μm) and six cells using linopirdine (20 μm)], AP frequency increased by 90.9% (p = 0.039), suggesting that Kv7 channels influence spontaneous firing frequency in SO interneurons.
Whole-cell current-clamp recordings were then performed in GFP+ SO cells (Fig. 7C–E). Consistent with loose-patch recordings, 20 μm linopirdine increased the average firing frequency from 1.0 ± 0.7 to 4.5 ± 1.5 Hz (Fig. 7C) (n = 11; p = 0.015), which was accompanied by a depolarization from −57.7 ± 1.5 to −53.1 ± 1.8 mV (n = 11; p = 0.005). In response to 1 s current steps between 5 and 25 pA, linopirdine induced an increase in firing frequency at each current step tested (Fig. 7D,E) (n = 8; p < 0.05). When 15 and 20 pA current step amplitudes were introduced, linopirdine induced robust increases in firing frequency by 10.6 ± 2.6 Hz (n = 8; p = 0.005) and 10.8 ± 2.7 Hz (n = 8; p = 0.005), respectively. However, at 25 pA, the linopirdine-induced increase in frequency was smaller (7.6 ± 3.2 Hz; n = 8; p = 0.045), suggesting that linopirdine effects narrowed as the cell approached maximum firing frequency. We saw no indication that linopirdine altered AP repolarization, providing additional confirmation that linopirdine does not block Kv3 channels. Finally, as an independent means of assessing the effect of IM on interneuronal excitability, we applied the Kv7 channel opener retigabine (10 μm). In seven of seven neurons, retigabine caused a cessation of spontaneous firing (Fig. 7F), hyperpolarized the cells from −53.6 ± 2.7 to −58.8 ± 1.4 mV (n = 7; p = 0.027), and greatly shunted depolarization-induced firing (Fig. 7G,H).
Therefore, IM appeared to primarily control the ISI and was particularly effective in influencing firing frequency in response to small depolarizing current steps near the resting potential.
Somatodendritic localization of Kv7 channels best reproduced IM-mediated outward currents
To understand how the presence and localization of IM influenced interneuron properties, we turned to multicompartment models of interneurons. Because O-LM cells were the most abundant IM-containing SO cell type, we chose to model two O-LM cells (Fig. 4, cells 1–2) in which IM had been pharmacologically detected. In construction of the model, Kv7 channels were localized to different cellular regions: soma only, soma and all dendrites, soma and primary dendrites, and axon only (Fig. 8A–D). Primary dendrites are defined here as dendrites that originate from the soma and extend to the first branch point.
For each IM distribution, the conductance density was adjusted to generate ∼1 nA of outward current after voltage command to +40 from −60 mV, a value close to that measured in our whole-cell recordings. The density obtained was different for each of the four IM distributions explored (Table 1). Values ranged from 0.12 pS/μm2 for the somatodendritic distribution to 6.40 pS/μm2 for the soma only distribution. Interestingly, our predicted density for the soma only distribution of IM (5.65–6.4 pS/μm2) is very similar to the density used by Hu et al. (2002) in which they included only somatic IM (6.7 pS/μm2). For the distribution in which IM was limited to the axon only, conductance levels required to generate a ∼1 nA somatic current were ∼280–1200 pS/μm2, ∼50–190 times larger than the density needed in the soma alone and 1600–2300 times larger than needed for somatodendritic distributions. Moreover, the high axonal Kv7 channel density made IM the dominant repolarizing conductance shaping AP half-width, which greatly abbreviated the AP amplitude, ceased spontaneous firing, and, unlike that measured experimentally (Martina et al., 2000), no longer permitted AP initiation in the axon-bearing dendrite. When IM density was set to 0.75 pS/μm2 for the four distributions, somatodendritic IM was the largest (Fig. 8A–D, insets), with the other distributions generating less somatic current (Fig. 8E).
Table 1.
Conductance densities for different IM distributions
| IM distribution | Conductance density (pS/μm2) |
Conductance density (pS/μm2) |
|---|---|---|
| Cell 1 | Cell 2 | |
| Soma | 6.40 | 5.65 |
| Soma and all dendrites | 0.75 | 0.12 |
| Soma and primary dendrites | 3.00 | 2.57 |
| Axon | 1200 | 280 |
| Calculation from experiments | 1.95 | 2.29 |
An independent estimate of the IM density was obtained using experimental measurements of the capacitance transient, which can be used to determine the approximate surface area that is sampled by the somatic recording electrode. In stepping from −60 to −65 mV under whole-cell VC conditions, the capacitance transient was generated in the same cells that were modeled (Fig. 8F, inset) (cell 1, 38.0 pF; cell 2, 32.4 pF). Assuming a specific membrane capacitance of 0.9 μF/cm2 and EK of −95 mV, the density required to generate 1 nA of outward current is as follows:
 |
 |
 |
A similar calculation for cell 2 yields a density of 2.29 pS/μm2. These density values lie between those predicted for IM distributed somatodendritically and IM distributed in the soma and primary dendrites only (Table 1). This calculated distribution also corresponds well with the immunocytochemical localization of Kv7.2 and 7.3 subunits to the soma and dendrites (Fig. 1).
Simulation of IM inhibition and enhancement in O-LM interneuron models
Having estimated IM density and compartmental localization to somatodendritic regions (IM distributed in the soma and all dendrites was used for all subsequent simulations), we then proceeded to determine the influence of IM on firing properties in O-LM interneuron models using VC and CC protocols similar to the experimental conditions in Figures 2 and 7. Making gM equal to zero, which simulated the XE-991 case (“XE”), increased spontaneous firing (Fig. 9A2) compared with control conditions (Fig. 9A1). Simulating the action of the Kv7 channel opener retigabine [“ret” (Wickenden et al., 2000)] arrested spontaneous firing (Fig. 9A3), similar to experimental observations. In response to a 1 s long, 15 pA current step, the XE condition increased AP frequency without changing AP half-width (Fig. 9B2,E), whereas the ret condition shunted AP firing (Fig. 9B3,E) compared with control conditions (Fig. 9B1,E). Interestingly, after introduction of a 5 Hz sinusoidal current, the model exhibited an increase in AP reliability (Fig. 9C1,C2) compared with that seen in O-LM cells after mAChR activation (Lawrence et al., 2006). After simulating the effects of XE-991 (Fig. 9D, red) or retigabine (Fig. 9D, blue) in VC, the model also reproduced the shifts in Ihold and changes IM amplitude apparent (Fig. 9D). Finally, as observed in experimental conditions, the effects of IM inhibition or enhancement on firing diminished as the model approached maximal firing frequencies (Fig. 9F), suggesting that IM is most effective in modulating firing frequency at low or moderate current steps. Thus, inhibition or enhancement of somatodendritic IM in O-LM cell models reproduces salient aspects of IM-related effects observed experimentally.
Figure 9.

Simulation of IM inhibition and augmentation in O-LM interneuron model. Simulations were performed on cell 1 with a somatodendritic IM distribution. A, Spontaneous firing simulating in control (A1), XE-991 (A2), and retigabine (A3) conditions. B, Introduction of 1 s long, 15 pA step into the soma for control (B1), XE (B2), and retigabine (B3). C, Sinusoidal current (20 pA) at 5 Hz introduced into the soma for control (C1), XE (C2), and retigabine (C3). D, In VC, a step was applied from −30 to −50 mV for control (gray), XE (red), and retigabine (blue). E, AP frequency in response to 0–25 pA current pulses from the resting potential for control (cont; gray), XE (red), and retigabine (ret; blue) conditions. F, Plot of AP frequency, as a percentage of control frequency, for XE (red) and retigabine (blue) conditions across a wide range of current steps. Note that the effects of XE and retigabine on firing frequency diminish with increasing current step amplitude. The dotted box indicates the range of current step values shared with E.
The role of gM in shaping interneuronal AHP and interspike interval
Under what conditions can interneuronal gM be activated? In hippocampal pyramidal cells, after eliciting multiple APs, IM mediates a mAHP (Storm, 1990; Gu et al., 2005). Interestingly, IM mediates the mAHP even after a single AP (Gu et al., 2005), which was only reproduced when IM was modeled to have rapid activation kinetics at voltages more positive than −20 mV (Gu et al., 2005). Having experimentally confirmed that IM activates rapidly in SO interneurons (Fig. 5D), we investigated conditions under which gM is activated in the O-LM model (Fig. 10). After activation by a single AP (Fig. 10A), gM reached maximal activation (∼40%) during AP repolarization (Fig. 10Aii). Despite this strong activation, IM plays a negligible role in AP repolarization, because gKdrf and gK(Ca) are the dominant K+ conductances (Fig. 10Aiii), with IM contributing only 6% to the total active K+ conductance during repolarization (Fig. 10Aiv). As gKdrf and gK(Ca) rapidly decay after the AP, gM remains active for several hundreds of milliseconds (Fig. 10Aiii) so that gM becomes the major conductance during the ISI (Fig. 10Aiv). These observations explain not only how gM can influence the AHP and spontaneous firing but also why gM contributes so little to shaping the AP waveform (Fig. 10Bi-iii). The model also accounts, in part, for how gM can influence the ISI yet, paradoxically, have little effect on the amplitude of the AHP after a 1 s train of APs (Fig. 10C,D) (Lawrence et al., 2006a). When gM is inhibited, the accompanying increase in AP frequency (Fig. 10Cii) results in an enhanced accumulation of gK(Ca) (Fig. 10Dii,iii), which can obscure a reduction in AHP mediated by gM inhibition (Fig. 10Di). Finally, the model is also consistent with published work (Lawrence et al., 2006a), indicating that linopirdine does not change the first spike latency when the cell is depolarized from −60 mV. At this voltage, IM is active by only 0.12%, contributing very little to Rm or τm. Overall, gM appears to be strongly recruited by APs but does not contribute to shaping excitability until the ISI, when faster deactivating conductances that shape the AP waveform no longer dominate.
Figure 10.

The role of gM in shaping interneuronal AHP and ISI. Model 1 was used with gM distributed somatodendritically at a density of 0.75 pS/μm2. A, Introduction of a 100 μs, 1 nA current into the soma from −60 mV elicits an AP (i) followed by an AHP (black) that is reduced in magnitude and duration when gM is 0 (blue trace). Plotted on the same time scale are gM open probability (ii), conductances gM (iii; black), gKdrf (red), and gK(Ca) (green), and the fractional contribution of each conductance (iv) as a percentage of the total active K+ conductance (all K+ conductances except leak conductances). B, Spontaneous firing conditions with the same parameters monitored as in A. C, Voltage responses after introduction of a 100 pA, 1 s current step from −60 mV into the soma in the presence (i, black) or absence (ii, blue) of gM. D, In an expanded region during the posttrain AHP (C, dashed box), voltage responses (i), gK(Ca) (ii), and the fractional contribution of gK(Ca) (iii) are shown in the presence (black) or absence (blue) of gM. To suppress spontaneous firing, holding currents of −15 or −15.3 pA were used when gM was present or absent, respectively. APs shown in i are truncated for display.
Discussion
Interneuronal Kv7 channels are localized somatodendritically
In this paper, we present immunocytochemical, electrophysiological, and computational evidence that converges on the conclusion that a population of Kv7 channels is localized somatodendritically in O-LM cells. As evidenced by Kv7.2 immunocytochemistry and susceptibility to block by 0.5 mm TEA, the molecular identity of IM on SO interneurons is most likely to be Kv7.2/Kv7.3 heteromers (Shah et al., 2002). Although we do not know to what extent Kv7.5 may be expressed in SO interneurons, strong Kv7.5 expression is expected to confer a lower degree of TEA sensitivity (Schroeder et al., 2000). Although both immunocytochemical and electrophysiological data indicate considerable heterogeneity in the expression of Kv7 channels, the relationship between immunocytochemical detection of Kv7 subunits and IM amplitude is likely to be complex, involving not only practical limits in the detection threshold for each these parameters but probably also other parameters such as channel open probability, PIP2 (phosphatidylinositol 4,5-bisphosphate) level, dendritic geometry, and the level of tonic mAChR-mediated inhibition. However, as implied by the observation that retigabine had a detectable effect in all recordings, it is possible that all SO interneurons express Kv7 channels to some extent.
To explore how the localization of Kv7 channels influenced the detection of somatically recorded IM, we used realistic multicompartment models in which Kv7 channels were localized to different regions of the soma, dendrites, or axon. Our results indicate that IM characteristics obtained with whole-cell recordings point to a somatodendritic localization. Although we cannot rule out that interneuron axons also possess Kv7 channels, our results argue that axonal channels are unlikely to fully account for IM measured somatically because of the poor space clamp on the axon and the unrealistically high axonal densities required to generate the magnitude of IM seen experimentally. Indeed, in comparing the capacitance of the cell estimated experimentally at the soma with the total capacitance in the O-LM model, it is likely that voltage control is limited to a ∼100 μm region surrounding the soma (Fig. 8F), which corresponds well with the cellular location where immunoreactivity for Kv7 channels is strongest (Fig. 1). Finally, modulating somatodendritic IM in the model (Fig. 9) recapitulates the effects of linopirdine and retigabine on AP firing measured experimentally (Fig. 7). Thus, our electrophysiological results are most likely explained by somatodendritically localized Kv7 channels.
Kv7 channels are specialized to control ISI in SO interneurons
In hippocampal pyramidal cells, IM is well known for its role in spike frequency accommodation and has therefore been associated with neurons that exhibit strong spike frequency accommodation. Given that O-LM interneurons exhibit only modest spike frequency accommodation (Lien et al., 2002; Lawrence et al., 2006a), the discovery of IM in O-LM cells may be somewhat surprising. Moreover, the overlap in TEA sensitivity between IM and Kv3-like delayed rectifier channels may be an additional reason that it has not been appreciated in nucleated patches from SO cells before this study (Lien et al., 2002). Interestingly, in five of six nucleated patches from SO cells, we did detect a change in Ihold at −30 mV (−1.8 ± 0.3 pA; n = 5; p = 0.008) after 3 min wash-in of 4 μm XE-991, a value comparable with the magnitude of change in Ihold predicted in a nucleated patch model using our obtained estimate of IM density (data not shown). What function might IM have in O-LM cells, and how does IM differ from that of other K+ currents in O-LM cells? In this paper, we show that although Kv3 and Kv7 channels are both activated by similar depolarizing stimuli, the two channel types play fundamentally different roles in the regulation of interneuronal excitability because of their different time courses of activation (Fig. 5D) and deactivation (Figs. 2, 6G,H). Kv3 channels are strongly activated by the AP, comprising the dominant conductance mediating repolarization of the AP. The rapid activation and deactivation kinetics of Kv3 channels enables rapid and precise repolarization of the AP and endows interneurons with the capacity to fire APs at high frequencies (Atzori et al., 2000; Rudy and McBain, 2001; Lien et al., 2002); indeed, introducing a Kv3-like conductance into a pyramidal cell transforms it to a fast spiking phenotype (Lien and Jonas, 2003). Neuromodulation of Kv3 channels changes spike width (Atzori et al., 2000), which may influence downstream synaptic transmission by impacting calcium entry during the spike (Goldberg et al., 2005). Kv3 channels also ensure that sodium channels undergo sufficient recovery from inactivation, thus setting the upper limit on firing frequency (Atzori et al., 2000; Rudy and McBain, 2001). In contrast, Kv7 channels activate during the AP but do not influence spike width or AP repolarization, primarily because they comprise a minor conductance during AP repolarization. However, IM becomes a dominant conductance relative to Kv3-mediated conductances during the ISI. The ability of IM to influence AP frequency without influencing AP half-width may be a mechanism by which SO interneurons can tune ISI while preserving the fidelity of calcium entry that might occur during the AP. IM inhibition also has consequences on other conductances, but it is interesting that cholinergic signaling mechanisms in O-LM cells appear to have evolved to repress a Ca2+-dependent K+ conductance after mAChR activation (Lawrence et al., 2006a) that would otherwise probably be enhanced by IM inhibition (Fig. 10D).
Interneuronal IM may also be important in the broader context of cellular and network oscillations (Cooper et al., 2001). In CA1 pyramidal cells, IM and Ih mediate a subthreshold theta resonance (Hu et al., 2002). A theta resonance has been identified in SO interneurons that is similar to that of pyramidal cells (Pike et al., 2000). The potential contribution of IM and Ih to theta resonance, coupled to their depolarized resting potential relative to pyramidal cells (Gloveli et al., 2005; Lawrence et al., 2006a), tendency to fire spontaneously (McBain et al., 1994; Maccaferri and McBain, 1996), relatively high Rm (Gloveli et al., 2005; Lawrence et al., 2006a), intrinsic spike frequency preference (Pike et al., 2000; Lawrence et al., 2006a), and susceptibility to entrainment by GABAergic afferents of the medial septum (Toth et al., 1997), not only put additional constraints on the range of ISIs available to SO interneurons but also offers mechanisms by which ISI can be tuned through extrinsic neuromodulation. Interestingly, mAChR modulation in O-LM cells, which is associated with IM inhibition (Lawrence et al., 2006a), appears to amplify theta rhythmicity by enhancing AP reliability to theta frequency oscillatory input (Lawrence et al., 2006b), a finding that is reproduced in our O-LM model (Fig. 9C). Thus, neuromodulation of somatodendritic IM and Ih in SO interneurons may be mechanisms by which ISI can be tuned to alter the degree of engagement of SO interneurons in network activity.
Pathophysiological implications of IM in hippocampal interneurons
Recently, the Kv7 channels have been shown to factor prominently into inherited human epilepsies. Benign familial neonatal convulsions, a type of epilepsy involving genetic mutations of Kv7.2/Kv7.3 channels, result in epileptic activity early in development (Jentsch, 2000). Linopirdine also induces epileptic activity in hippocampal slices in 1- to 2-week-old animals (Pena and Alavez-Perez, 2006). In experimental models in which Kv7.2 channels are globally suppressed, CA1 pyramidal cells exhibit hyperexcitability and reduced IM (Peters et al., 2005; Otto et al., 2006). However, an interneuronal component of reduced expression of somatodendritic Kv7.2 channels to changes in network excitability has not been addressed. At first glance, increased release of GABA resulting from reduced expression of interneuron Kv7.2 channels might be expected to ameliorate or reduce seizure susceptibility. However, GABA is excitatory early in development; seizure susceptibility is actually increased by GABA and reduced by GABAA antagonists (Khazipov et al., 2004). Thus, it is possible that suppression of IM in interneurons could exacerbate epileptogenic activity through enhanced GABAergic transmission (Cossart et al., 2005).
Conversely, IM channel openers such as retigabine are steadily gaining recognition for their advantageous antiepileptic properties (Rogawski, 2006). Although retigabine has been described as a “GABA enhancer” (Czuczwar and Patsalos, 2001) for its action at postsynaptic GABAA receptors (Otto et al., 2002), our observation that retigabine effectively arrests spontaneous firing of interneurons (Fig. 7) predicts that retigabine may impair AP driven GABAergic transmission to some extent. Although it is unexpected that the actions of an antiepileptic agent would include a reduction in GABAergic transmission, reducing excitability of both pyramidal cells and interneurons may be more effective in disrupting pathological network synchrony than reducing excitability of pyramidal cells alone. Thus, the antiepileptic action of retigabine might be reinterpreted as antimuscarinic in nature, effectively counteracting the action of acetylcholine in promoting rhythmicity of the hippocampal network via modulation of IM in pyramidal cells and interneurons (Cobb and Davies, 2005; Lawrence et al., 2006b).
In summary, our immunocytochemical, electrophysiological, and modeling evidence converges on the conclusion that most IM detected somatically in SO interneurons is attributable to somatodendritically activated IM. The activation of somatodendritic IM has a major influence on the control of ISI. Modulation of IM has potent effects on interneuronal excitability, a novel means of influencing the excitability of the hippocampal network.
Footnotes
This work was supported by National Institute of Child Health and Human Development intramural funding to C.J.M., Science and Engineering Research Canada [Natural Sciences and Engineering Research Council of Canada (NSERC)] funding to F.K.S., and an NSERC Postgraduate and Ontario Graduate Scholarship to F.S. J.F.C. and J.M.S. are Howard Hughes Medical Institute–National Institutes of Health Research Scholars. We thank Dr. Zachary Grinspan for preliminary experiments and analysis code, Brian Jeffries for biocytin immunocytochemistry and Neurolucida reconstructions, Dr. Melanie Tallent for immunochemistry protocols, Xioqing Yuan for expert technical assistance with Kv3.1/Kv3.2 transfections, Dr. Bernardo Rudy for Kv3.1/Kv3.2 plasmids, Dr. Michael Rogawski for the gift of retigabine, and Dr. Dax Hoffman for critically reading a previous version of this manuscript.
References
- Atzori M, Lau D, Tansey EP, Chow A, Ozaita A, Rudy B, McBain CJ. H2 histamine receptor-phosphorylation of Kv3.2 modulates interneuron fast spiking. Nat Neurosci. 2000;3:791–798. doi: 10.1038/77693. [DOI] [PubMed] [Google Scholar]
- Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Jan YN, Jan LY. Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proc Natl Acad Sci USA. 2006;103:8870–8875. doi: 10.1073/pnas.0603376103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophys J. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Davies CH. Cholinergic modulation of hippocampal cells and circuits. J Physiol (Lond) 2005;562:81–88. doi: 10.1113/jphysiol.2004.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EC, Jan LY. M-channels: neurological diseases, neuromodulation, and drug development. Arch Neurol. 2003;60:496–500. doi: 10.1001/archneur.60.4.496. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Aldape KD, Abosch A, Barbaro NM, Berger MS, Peacock WS, Jan YN, Jan LY. Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci USA. 2000;97:4914–4919. doi: 10.1073/pnas.090092797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EC, Harrington E, Jan YN, Jan LY. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci. 2001;21:9529–9540. doi: 10.1523/JNEUROSCI.21-24-09529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Bernard C, Ben-Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28:108–115. doi: 10.1016/j.tins.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Czuczwar SJ, Patsalos PN. The new generation of GABA enhancers. Potential in the treatment of epilepsy. CNS Drugs. 2001;15:339–350. doi: 10.2165/00023210-200115050-00001. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Neubig M, Ulrich D, Huguenard J. Dendritic low-threshold calcium currents in thalamic relay cells. J Neurosci. 1998;18:3574–3588. doi: 10.1523/JNEUROSCI.18-10-03574.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci. 2004;24:1236–1244. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentet LJ, Stuart GJ, Clements JD. Direct measurement of specific membrane capacitance in neurons. Biophys J. 2000;79:314–320. doi: 10.1016/S0006-3495(00)76293-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloveli T, Dugladze T, Saha S, Monyer H, Heinemann U, Traub RD, Whittington MA, Buhl EH. Differential involvement of oriens/pyramidale interneurones in hippocampal network oscillations in vitro. J Physiol (Lond) 2005;562:131–147. doi: 10.1113/jphysiol.2004.073007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg EM, Watanabe S, Chang SY, Joho RH, Huang ZJ, Leonard CS, Rudy B. Specific functions of synaptically localized potassium channels in synaptic transmission at the neocortical GABAergic fast-spiking cell synapse. J Neurosci. 2005;25:5230–5235. doi: 10.1523/JNEUROSCI.0722-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol (Lond) 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA. Differential tetraethylammonium sensitivity of KCNQ1–4 potassium channels. Br J Pharmacol. 2000;129:413–415. doi: 10.1038/sj.bjp.0703086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, Brown DA. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci. 2003;23:5012–5019. doi: 10.1523/JNEUROSCI.23-12-05012.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J Physiol (Lond) 2002;545:783–805. doi: 10.1113/jphysiol.2002.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Jow F, Wang K. Cloning and functional expression of rKCNQ2 K(+) channel from rat brain. Brain Res Mol Brain Res. 2000;80:269–278. doi: 10.1016/s0169-328x(00)00146-7. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Khalilov I, Tyzio R, Morozova E, Ben-Ari Y, Holmes GL. Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur J Neurosci. 2004;19:590–600. doi: 10.1111/j.0953-816x.2003.03152.x. [DOI] [PubMed] [Google Scholar]
- Lamas JA, Selyanko AA, Brown DA. Effects of a cognition-enhancer, linopirdine (DuP 996), on M-type potassium currents (IK(M)) and some other voltage- and ligand-gated membrane currents in rat sympathetic neurons. Eur J Neurosci. 1997;9:605–616. doi: 10.1111/j.1460-9568.1997.tb01637.x. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Statland JM, Grinspan ZM, McBain CJ. Cell type-specific dependence of muscarinic signalling in mouse hippocampal stratum oriens interneurones. J Physiol (Lond) 2006a;570:595–610. doi: 10.1113/jphysiol.2005.100875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JJ, Grinspan ZM, Statland JM, McBain CJ. Muscarinic receptor activation tunes mouse stratum oriens interneurons to amplify spike reliability. J Physiol (Lond) 2006b;571:555–562. doi: 10.1113/jphysiol.2005.103218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien CC, Jonas P. Kv3 potassium conductance is necessary and kinetically optimized for high-frequency action potential generation in hippocampal interneurons. J Neurosci. 2003;23:2058–2068. doi: 10.1523/JNEUROSCI.23-06-02058.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien CC, Martina M, Schultz JH, Ehmke H, Jonas P. Gating, modulation and subunit composition of voltage-gated K(+) channels in dendritic inhibitory interneurones of rat hippocampus. J Physiol (Lond) 2002;538:405–419. doi: 10.1113/jphysiol.2001.013066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G. Stratum oriens horizontal interneurone diversity and hippocampal network dynamics. J Physiol (Lond) 2005;562:73–80. doi: 10.1113/jphysiol.2004.077081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G, Lacaille JC. Interneuron diversity series: hippocampal interneuron classifications–making things as simple as possible, not simpler. Trends Neurosci. 2003;26:564–571. doi: 10.1016/j.tins.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, McBain CJ. The hyperpolarization-activated current (Ih) and its contribution to pacemaker activity in rat CA1 hippocampal stratum oriens-alveus interneurones. J Physiol (Lond) 1996;497:119–130. doi: 10.1113/jphysiol.1996.sp021754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina M, Schultz JH, Ehmke H, Monyer H, Jonas P. Functional and molecular differences between voltage-gated K+ channels of fast-spiking interneurons and pyramidal neurons of rat hippocampus. J Neurosci. 1998;18:8111–8125. doi: 10.1523/JNEUROSCI.18-20-08111.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina M, Vida I, Jonas P. Distal initiation and active propagation of action potentials in interneuron dendrites. Science. 2000;287:295–300. doi: 10.1126/science.287.5451.295. [DOI] [PubMed] [Google Scholar]
- McBain CJ, DiChiara TJ, Kauer JA. Activation of metabotropic glutamate receptors differentially affects two classes of hippocampal interneurons and potentiates excitatory synaptic transmission. J Neurosci. 1994;14:4433–4445. doi: 10.1523/JNEUROSCI.14-07-04433.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuiston AR, Madison DV. Muscarinic receptor activity induces an afterdepolarization in a subpopulation of hippocampal CA1 interneurons. J Neurosci. 1999;19:5703–5710. doi: 10.1523/JNEUROSCI.19-14-05703.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore M, Cook EP, Jaffe DB, Turner DA, Johnston D. Computer simulations of morphologically reconstructed CA3 hippocampal neurons. J Neurophysiol. 1995;73:1157–1168. doi: 10.1152/jn.1995.73.3.1157. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, DeFazio RA, Moenter SM. A targeted extracellular approach for recording long-term firing patterns of excitable cells: a practical guide. Biol Proced Online. 2003;5:53–62. doi: 10.1251/bpo46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva AA, Jr, Jiang M, Lam T, Smith KL, Swann JW. Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in GABAergic interneurons. J Neurosci. 2000;20:3354–3368. doi: 10.1523/JNEUROSCI.20-09-03354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto JF, Kimball MM, Wilcox KS. Effects of the anticonvulsant retigabine on cultured cortical neurons: changes in electroresponsive properties and synaptic transmission. Mol Pharmacol. 2002;61:921–927. doi: 10.1124/mol.61.4.921. [DOI] [PubMed] [Google Scholar]
- Otto JF, Yang Y, Frankel WN, White HS, Wilcox KS. A spontaneous mutation involving Kcnq2 (Kv7.2) reduces M-current density and spike frequency adaptation in mouse CA1 neurons. J Neurosci. 2006;26:2053–2059. doi: 10.1523/JNEUROSCI.1575-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul J-Y, Cranstoun SD, Bennett V, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra P, Gulyas AI, Miles R. How many subtypes of inhibitory cells in the hippocampus? Neuron. 1998;20:983–993. doi: 10.1016/s0896-6273(00)80479-1. [DOI] [PubMed] [Google Scholar]
- Pena F, Alavez-Perez N. Epileptiform activity induced by pharmacologic reduction of M-current in the developing hippocampus in vitro. Epilepsia. 2006;47:47–54. doi: 10.1111/j.1528-1167.2006.00369.x. [DOI] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Pike FG, Goddard RS, Suckling JM, Ganter P, Kasthuri N, Paulsen O. Distinct frequency preferences of different types of rat hippocampal neurones in response to oscillatory input currents. J Physiol (Lond) 2000;529:205–213. doi: 10.1111/j.1469-7793.2000.00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Routing of spike series by dynamic circuits in the hippocampus. Nature. 2004;429:717–723. doi: 10.1038/nature02615. [DOI] [PubMed] [Google Scholar]
- Prole DL, Lima PA, Marrion NV. Mechanisms underlying modulation of neuronal KCNQ2/KCNQ3 potassium channels by extracellular protons. J Gen Physiol. 2003;122:775–793. doi: 10.1085/jgp.200308897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA. Diverse mechanisms of antiepileptic drugs in the development pipeline. Epilepsy Res. 2006;69:273–294. doi: 10.1016/j.eplepsyres.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–526. doi: 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Saraga F, Wu CP, Zhang L, Skinner FK. Active dendrites and spike propagation in multi-compartment models of oriens-lacunosum/moleculare hippocampal interneurons. J Physiol (Lond) 2003;552:673–689. doi: 10.1113/jphysiol.2003.046177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnee ME, Brown BS. Selectivity of linopirdine (DuP 996), a neuro transmitter release enhancer, in blocking voltage-dependent and calcium-activated potassium currents in hippocampal neurons. J Pharmacol Exp Ther. 1998;286:709–717. [PubMed] [Google Scholar]
- Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Brown DA. M-channel gating and simulation. Biophys J. 1999;77:701–713. doi: 10.1016/S0006-3495(99)76925-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P. Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol (Lond) 2002;544:29–37. doi: 10.1113/jphysiol.2002.028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol (Lond) 2005;562:9–26. doi: 10.1113/jphysiol.2004.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35:507–520. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- Toth K, Freund TF, Miles R. Disinhibition of rat hippocampal pyramidal cells by GABAergic afferents from the septum. J Physiol (Lond) 1997;500:463–474. doi: 10.1113/jphysiol.1997.sp022033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Yu W, Zou A, Jegla T, Wagoner PK. Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol Pharmacol. 2000;58:591–600. doi: 10.1124/mol.58.3.591. [DOI] [PubMed] [Google Scholar]
- Yue C, Yaari Y. KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yus-Najera E, Munoz A, Salvador N, Jensen BS, Rasmussen HB, Defelipe J, Villarroel A. Localization of KCNQ5 in the normal and epileptic human temporal neocortex and hippocampal formation. Neuroscience. 2003;120:353–364. doi: 10.1016/s0306-4522(03)00321-x. [DOI] [PubMed] [Google Scholar]
- Zhang L, McBain CJ. Potassium conductances underlying repolarization and after-hyperpolarization in rat CA1 hippocampal interneurones. J Physiol (Lond) 1995a;488:661–672. doi: 10.1113/jphysiol.1995.sp020998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, McBain CJ. Voltage-gated potassium currents in stratum oriens-alveus inhibitory neurones of the rat CA1 hippocampus. J Physiol (Lond) 1995b;488:647–660. doi: 10.1113/jphysiol.1995.sp020997. [DOI] [PMC free article] [PubMed] [Google Scholar]