

Abstract
Maintenance of mitochondrial DNA (mtDNA) depends on nuclear-encoded proteins such as mtDNA polymerase (POLG), whose mutations are involved in the diseases caused by mtDNA defects including mutation and deletion. The defects in mtDNA and in intracellular Ca2+ ([Ca2+]i) homeostasis have been reported in bipolar disorder (BD). To understand the relevance of the mtDNA defects to BD, we studied transgenic (Tg) mice in which mutant POLG (mutPOLG) was expressed specifically in neurons. mtDNA defects were accumulated in the brains of mutPOLG Tg mice in an age-dependent manner and the mutant mice showed BD-like behavior. However, the molecular and cellular basis for the abnormalities has not been clarified. In this study, we investigated Ca2+ regulation by isolated mitochondria and [Ca2+]i dynamics in the neurons of mutPOLG Tg mice. Mitochondria from the mutant mice sequestered Ca2+ more rapidly, whereas Ca2+ retention capacity and membrane potential, a driving force of Ca2+ uptake, of mitochondria were unaffected. To elucidate the molecular mechanism of the altered Ca2+ uptake, we performed DNA microarray analysis and found that the expression of cyclophilin D (CyP-D), a component of the permeability transition pore, was downregulated in the brains of mutPOLG Tg mice. Cyclosporin A, an inhibitor of CyP-D, mimicked the enhanced Ca2+ uptake in mutant mice. Furthermore, G-protein-coupled receptor-mediated [Ca2+]i increase was attenuated in hippocampal neurons of the mutant mice. These findings suggest that mtDNA defects lead to enhancement of Ca2+ uptake rate via CyP-D downregulation and alter [Ca2+]i dynamics, which may be involved in the pathogenesis of BD.
Keywords: bipolar disorder, calcium, GPCR, mitochondria, patch clamp, cyclophilin D
Introduction
Defects in mitochondrial DNA (mtDNA) have been implicated in the pathophysiology of various neuropsychiatric diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD) (Schapira, 1999), and bipolar disorder (BD) (Kato and Kato, 2000), in addition of hereditary mitochondrial diseases (MDs). Among MDs, chronic progressive external ophthalmoplegia (CPEO) is known to closely relate to mtDNA defects produced by mutations of nuclear genes (Kaukonen et al., 2000; Spelbrink et al., 2001; Van Goethem et al., 2001). Because CPEO was recently redefined as a multisystem disease including CNS involvement (Suomalainen et al., 1992; Luoma et al., 2004), we suspected that mtDNA defects were relevant to the pathogenic mechanisms in mood disorders. For mtDNA defects, causal mutations in the following three genes have been identified to date: mtDNA polymerase (POLG), adenine nucleotide translocator 1 (ANT1/Slc25a4), and mtDNA helicase (PEO1/Twinkle). We developed transgenic (Tg) mice expressing neuron-specific mutant POLG (mutPOLG) and have shown that mtDNA defects age-dependently increased in the forebrains of mutPOLG Tg mice. These animals exhibited several BD-like behavioral phenotypes (Kasahara et al., 2006). This implicates that the accumulation of mtDNA defects leads to the BD-like behavior of mutPOLG Tg mice, whereas the underlying molecular and cellular mechanisms are not clarified.
Mitochondria participate in regulation of Ca2+ homeostasis by sequestering and releasing Ca2+ in cooperation with endoplasmic reticulum (ER) (Missiaen et al., 1991; Pozzan and Rizzuto, 2000). The mitochondrial Ca2+ uptake is involved principally in inositol 1,4,5-triphosphate (IP3)-mediated Ca2+ mobilization from the internal stores, which localize to the ER (Malli et al., 2003; Rizzuto et al., 2004). Ca2+ overload to mitochondria provokes opening of the permeability transition pore (PTP), disturbing the intracellular Ca2+ ([Ca2+]i) homeostasis by Ca2+ flux through the PTP (Ichas et al., 1997; Bianchi et al., 2004). Perturbation of [Ca2+]i homeostasis by mitochondrial Ca2+ dysregulation may be involved in pathogenesis of neurodegenerative diseases (Ghosh et al., 1999). For example, isolated mitochondria from brains of mice model for HD were prone to opening of the PTP and showed a slow rate of Ca2+ uptake (Panov et al., 2002), implying that mitochondrial Ca2+ dysregulation was associated with the pathophysiology of HD. In BD, altered [Ca2+]i increase, which is dependent on both G-protein-coupled receptor (GPCR) and IP3, has been found in blood cells (Dubovsky et al., 1989; Kusumi et al., 1994; Okamoto et al., 1995; Kato et al., 2003) and in neuronal cells (Hahn et al., 2005) of patients. These observations brought us a hypothesis that [Ca2+]i homeostasis would be disturbed in the neurons of the mutant mice and that accumulation of mtDNA defects might be responsible for the altered [Ca2+]i dynamics. In this study, we characterized Ca2+ retention capacity (CRC) and membrane potential of isolated mitochondria from the brains of Tg mice and investigated GPCR-mediated Ca2+ dynamics in CA1 pyramidal neurons of mutPOLG Tg mice by combining a whole-cell recording with a fluorometric Ca2+ imaging in the hippocampal slice preparation.
Materials and Methods
Chemicals and drugs.
All fluorescent dyes were purchased from Invitrogen (Grand Island, NY): 10-nonyl acridine orange (NAO), 5,5′,6,6′-tetracloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1), 1,1′,3,3,3′,3′-hexamethylindodicarbocyanine iodide [DiIC1(5)], Calcium Green-5N (CaG-5N), bis-fura-2, and fura-6F. Percoll solution was purchased from GE Healthcare Bio-Science (Piscataway, NJ). Cyclosporin A (CsA), carbonyl cyanide 4-(trifluoromethoxy)phenyl-hydrazone (FCCP), atropine sulfate, carbamylcholine chloride [carbachol (CCh)], low molecular weight heparin (molecular weight, 3000 Da), and cyclopiazonic acid (CPA) were purchased from Sigma (St. Louis, MO). Amino-phosphonopentanoic acid (APV), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), (S)-3,5-dihydroxyphenylglycine (DHPG), and α-methyl-4-carboxyphenylglycine (MCPG) were obtained from Tocris (Ellisville, MO). Chelex 100 resin was obtained from Bio-Rad (Richmond, CA).
Animals.
In the mutPOLG Tg mice, mutant mtDNA polymerase lacking proofreading activity because of a D181A substitution was attached to the promoter of calmodulin kinase II α. The mutPOLG Tg mice were always used as heterozygotes. Male mutant mice were used for mating to avoid possible transmission of mtDNA mutations from the maternal side. The genotyping was performed as described previously (Kasahara et al., 2006). Controls were wild-type littermates whenever possible. All experimental procedures involving animal preparation were approved by the Wako Animal Experiment Committee, RIKEN.
Isolation of mitochondria.
Mitochondria of the mutPOLG Tg mice and the littermates (70–75 weeks old) were isolated using a discontinuous Percoll gradient developed by Sims (1990). Briefly, a mouse was decapitated and the brain was transferred to an ice-cold isolation buffer [(in mm): 320 sucrose, 1 EGTA, and 10 MOPS (4-morpholinepropanesulfonic acid), pH 7.4]. The forebrain tissue was dissected and homogenized in 12% Percoll with a Dounce homogenizer. The homogenate was layered on a discontinuous gradient of 26 and 40% Percoll in the isolation buffer and centrifuged in a Beckman Coulter (Fullerton, CA) JA 25.15 rotor at 30,700 × g for 5 min at 4°C, followed by two washing steps in the isolation buffer at 16,700 and 7300 × g, respectively. Bovine serum albumin (final, 0.1%) was added at the last washing step. After the final sedimentation, the mitochondrial pellet was suspended in 0.3 ml of a Chelex-treated Ca2+-free buffer containing the following (in mm): 210 sucrose, 20 KCl, 3 glycylglycine, and 1 KH2PO4, pH 7.2. The suspension was divided into aliquots for measurements of CRC that were immediately performed after the isolation. Mitochondrial protein concentration was determined by the Bradford protein assay (usually 0.2–0.3 mg/ml). There was no difference in the averaged mitochondrial yield between genotypes.
Mitochondrial Ca2+ retention capacity and Ca2+ removal.
The mitochondrial pellet was resuspended in 150 μl of Ca2+-free buffer, and the suspension was added to 450 μl of buffer containing 20 mm glutamate plus 2 mm malate, or 10 mm succinate: the total solution was 0.6 ml in volume. Extramitochondrial free Ca2+ concentration ([Ca2+]exm) was monitored with 25 nm Calcium Green-5N [excitation (Ex), 488 nm; emission (Em), 530 nm] in a stirred cuvette at 30°C in a spectrofluorometer (F2500; Hitachi, Tokyo, Japan). The time resolution of the [Ca2+]exm measurement was set at 0.5 s. To evaluate the CRC, 1.5 μl of Ca2+ solution was added at 1 min intervals; the stock solutions of 0.625, 1.250, and 3.125 mm CaCl2 gave final concentrations of 1.6, 3.1, and 7.8 μm, respectively, which approximately corresponded to 25, 50, and 100 nmol of Ca2+ per milligram of mitochondrial protein. When mitochondria were incubated in the buffer containing succinate, 3.1 μm Ca2+ solution was repeatedly added after four additions of 1.6 μm. However, when mitochondria were incubated in the buffer containing glutamate plus malate, 7.8 μm Ca2+ solution was repeatedly added after four additions of 1.6 μm Ca2+ solutions. To obtain representative parameters of Ca2+ uptake by mitochondria, the recovery phase of [Ca2+]exm transients was normalized by the maximal peak amplitude and best fitted with the biexponential function. The percentage of change in fluorescence was calculated by ΔF/Fmax × 100%, where ΔF = F − F0 and Fmax = Fpeak − F0. F is the fluorescence intensity at any time point after the addition of Ca2+, and F0 is baseline fluorescence obtained from the average of 20 data points before the addition of Ca2+. The recovery time was measured from the peak to 37% (decay time) of peak [Ca2+]exm transients using KyPlot software (version 4.0; Kyens, Tokyo, Japan).
Mitochondrial membrane potential.
We estimated mitochondrial membrane potential (ΔΨm) using JC-1 or DiIC1(5) probe dye, of which the fluorescent intensity was analyzed by a fluorescence-activated cell sorter (Epics Elite ESP; Beckman Coulter). Flow cytometric analysis is useful to analyze mitochondrial function (Mattiasson et al., 2003), because a signal from each mitochondrion can be detected. Heteroplasmic mutations in mtDNA biochemically diversify individual mitochondrion as a result of the levels of mutations. Therefore, the population comparison is appropriate for evaluating the difference between groups. Mitochondria isolated from the brains of mutPOLG Tg or wild-type mice were suspended in an analysis buffer composed of 210 mm sucrose, 20 mm KCl, 3 mm glycylglycine, 1 mm KH2PO4, and 0.5 mm MgCl2, pH 7.2, containing additive components of 10 mm succinate plus 0.1 μg/ml rotenone or 20 mm glutamate plus 2 mm malate. The mitochondrial suspension (7.5–12.5 μg of mitochondrial protein in 200 μl) was stained by 5 μg/ml JC-1 or 10 nm DiIC1(5) at 37°C for 10 min in darkness. No stained samples were incubated in the buffer without a probe. Some samples were combined with 6.25 μm Ca2+ or 1 μm FCCP, a proton ionophore, before incubation at 37°C. The mitochondrial particles were gated based on light scattering properties in the side-scattering (SSC) and forward-scattering (FSC) modes, and 10,000 events per sample were collected. ΔΨm was assessed using a fluorescent potentiometric dye, JC-1, which displayed a green fluorescence of monomers (Ex, 490 nm; Em, 530 nm) and a red fluorescence of the dimer (Ex, 490 nm; Em, 590 nm). When dye was incorporated in mitochondria that retain a high membrane potential, JC-1 forms aggregates to exhibit the red fluorescence. Energized mitochondrial particle showed high emission intensity at both the wavelengths. The number of depolarized mitochondrial particles, defined as a ratio of red to green of <0.3, was counted. FCCP was applied as a negative control experiment to determine depolarized mitochondria. DiIC1(5) was also used to detect ΔΨm changes (Ex, 635 nm; Em, 660 nm) by combining with 100 nm NAO, which selectively stains mitochondria (Ex, 488 nm; Em, 525 nm). ΔΨm was measured in NAO-positive particles that were also selected from background based on light-scattering properties (SSC and FSC). In every experiment, 98% of the events were NAO positive.
RNA sampling and DNA microarray analysis.
Male mutPOLG Tg and their wild-type littermates were killed at the age of 25–34 weeks old. The bilateral frontal cortices and hippocampi were rapidly dissected, and total RNA samples were extracted from the brains of six mutPOLG Tg or wild-type littermates using Trizol reagent (Invitrogen). The microarray analysis was conducted using RNA samples derived from five pairs of animals on the basis of the quality of mRNA. Five micrograms of total RNA of each sample was reverse-transcribed into cDNA, and biotinylated cRNA was synthesized from the cDNA by in vitro transcription. DNA microarray experiments were performed using MG_430A GeneChips (Affymetrix, Santa Clara, CA), which contained 42,153 probe sets. The hybridization signal on the chip was scanned by a GeneArray scanner and processed by GeneSuite software (Affymetrix). The raw data were initially analyzed using MAS5 (Affymetrix), and then imported into GeneSpring 6.1 software (Silicon Genetics, Redwood, CA). The fluorescence intensity of each spot on the microarray was divided by its median value using GeneSpring 6.1 software and normalized. The probe sets for mitochondria-related genes were chosen either from the NetAffx database on the distributor's web site with the keywords “mitochondria” or “mitochondrion” or from the gene list of “mitochondria” in the GeneSpring software, which classified probe sets based on the information from Gene Ontology. Only the probe sets that were called as being “present” in more than one-half of the samples were analyzed, and 1221 probe sets were identified. For statistical analysis, a two-tailed paired t test was performed between the mutPOLG Tg mice and their littermates, and a value of p < 0.05 was considered statistically significant.
Real-time quantitative PCR.
To quantify mRNA levels of several interesting genes, we performed real-time quantitative PCR analysis (TaqMan technology) using commercially available probe–primer sets (Applied Biosystems, Foster City, CA) as described previously (Kakiuchi et al., 2003). The relative levels of each mRNA were calculated by 2−CT (CT standing for the cycle number at which the signal reached the threshold) and normalized to the corresponding β-actin mRNA level. Each CT value used for these calculations was the mean of four values obtained when each reaction was performed in quadruplicate.
Electrophysiology and intracellular Ca2+ imaging.
Fluorometric measurement of intracellular Ca2+ ([Ca2+]i) was performed according to the method of Nakamura et al. (1999) with slight modification. Transverse hippocampal slices (300 μm thick) were prepared from age-matched 5- to 7-week-old mutPOLG Tg and wild-type mice as described previously (Kubota et al., 2001). Each mouse was anesthetized under ether vapor anesthesia and quickly decapitated. The brain was chilled in ice-cold cutting solution, which is a modified artificial CSF (aCSF) containing the following (in mm): 120 choline-Cl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2.5 MgCl2, 0.5 CaCl2, and 20 glucose. Slices were made in the cutting solution using a tissue slicer (VT1000S; Leica, Wetzlar, Germany) and transferred to aCSF, which was composed of the following (in mm): 124 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. The slice was mounted on a recording chamber on an upright microscope (BX50WI; Olympus, Tokyo, Japan) and superfused with the aCSF during experiments at a rate of 1.5 ml/min. The cutting solution and aCSF had an osmolarity of 280 mOsm and a final pH of 7.4 after it was saturated with 95% O2 and 5% CO2. The recording chamber composition was changed by 90% in 3 min. The whole-cell recordings were made in CA1 pyramidal neurons using patch pipettes pulled from 1.5 mm outer diameter, thick-walled borosilicate glass tubing (GC-150 F-10; Harvard, Kent, UK) using an electrode puller (P-97; Sutter Instruments, Novato, CA). The patch pipette contained an internal solution of the following (in mm): 140 K-gluconate, 4 NaCl, 4 Mg-ATP, 0.3 Na-GTP, and 10 HEPES, pH 7.4 with KOH; and the osmolarity was adjusted to 280 mOsm with sucrose. This solution was supplemented with 300 μm of a calcium indicator, bis-fura-2, in the experiments of imaging for action potential (AP)-induced [Ca2+]i transient. In the experiments to see the enhancing effects of DHPG on AP-induced [Ca2+]i transient, a low-affinity Ca2+ indicator, fura-6F, was used. A tight seal on CA1 pyramidal cell soma was made under visual control using a 40× water immersion lens and video-enhanced differential interference contrast (DIC) optics, and the electrical recordings were performed in current-clamp mode at room temperature. The backpropagating APs were induced by depolarizing pulses (1 ms duration), which were evoked from the patch electrode (resistance, 3–5 MΩ) with a patch-clamp amplifier (Axoclamp 2B; Molecular Devices, Union City, CA). The signals were sampled at 100 μs intervals and digitized at 10 kHz (Digidata 1322A; Molecular Devices). Holding potentials, data acquisition, and analysis were controlled by pCLAMP 7.0 software (Molecular Devices). Time-lapse [Ca2+]i imaging was acquired at 30 ms intervals for 6.4 s using a CCD camera (C4742-95; Hamamatsu Photonics, Hamamatsu, Japan). The fluorescence images were analyzed with AQUACOSMOS system (version 2.0; Hamamatsu Photonics). The regions of interests (ROIs) (3 × 3 pixels) were put on the cell body and dendrites of each neuron. Relative [Ca2+]i levels associated with electrical activities were measured with single-wavelength imaging (Ex, 380 nm; Em, 530 nm) and were expressed as fluorescence change [−ΔF/F = (Fi − Fb)/(Fb − Fa)], where Fi is the average fluorescence of the ROI in each image, Fb is the baseline fluorescence averaged over at least 10 images before stimulation, and Fa is the autofluorescence. Normalized in this way, F is the fluorescence intensity when the cell was rest, and ΔF is the change in fluorescence during activity. In some cases, background intensity was defined as fluorescence in each ROI that was located outside the neuron of interest. Determination of absolute Ca2+ concentration by a ratiometric measurement cannot catch up with rapid [Ca2+]i transient in neurons, which makes it difficult to compare the [Ca2+]i levels between Tg mice and wild-type mice. Thus, we used the ratio of ΔF/F after DHPG application to the averaged ΔF/F before drug application. [Ca2+]i change was assessed by measuring peak amplitude and area under the curve (AUC) for each set of responses, to reflect actions on both the amplitude and the duration of the [Ca2+]i response. To detect the effect of a metabotropic glutamate receptor (mGluR) agonist, 20 μm CNQX and 50 μm APV were added to block ionotropic glutamate receptors in some experiments (see Figs. 4, 5).
Figure 4.

Effect of various pharmacological agents on GPCR-mediated [Ca2+]i increase. A, Effect of mGluR agonist or antagonist on AP-induced [Ca2+]i increase at the soma of hippocampal pyramidal neuron filled with the low-affinity indicator fura-6F (Kd, 6 μm) in wild-type mice. Time-lapse imaging of fluorescence changes (top) and voltage responses induced by APs at 30 Hz (bottom) were taken before (−DHPG) and during (+DHPG) bath application of 30 μm DHPG, a group I mGluR agonist. DHPG-induced [Ca2+]i increase in the presence of 1 mm MCPG, a mGluR antagonist (MCPG+DHPG), after washing out of MCPG (+DHPG), and without the presence of any agents (wash). B, Effect of a cholinergic agonist, CCh, on AP-induced [Ca2+]i increase. Time-lapse imaging of fluorescence changes (top) and voltage responses induced by APs at 30 Hz (bottom) were taken before (−CCh) and during (+CCh) bath application of 30 μm CCh. After application of a muscarinic receptor antagonist, atropine (1 μm), for 5 min, CCh was applied (atropine+CCh). C, Dependence of DHPG-induced [Ca2+]i increases on intracellular Ca2+ stores. Time-lapse imaging of fluorescence changes (top) and voltage responses induced by APs at 30 Hz (bottom) were taken before (−DHPG) and during (+DHPG) bath application of 30 μm DHPG. An endoplasmic reticulum Ca2+/ATPase blocker, CPA, is known to cause depletion of the stores. After application of 20 μm CPA for 7.5 min, DHPG was applied (CPA+DHPG). D, Dependence of DHPG-induced [Ca2+]i increases on IP3 receptor activation. Fluorescence changes (top) were taken before (−DHPG) and during (+DHPG) bath application of 30 μm DHPG. Recordings were made with a pipette containing solution with an impermeable IP3 receptor blocker, 2 mg/ml heparin (right), or no additives (left). Voltage responses induced by APs at 30 Hz at the middle are shown on an expanded timescale at the bottom. The arrows indicate GPCR-mediated membrane depolarization during AP firing. A set of measurements was done using slices derived from an animal. The perfusate contained 20 μm CNQX and 50 μm APV in all of the experiments.
Figure 5.
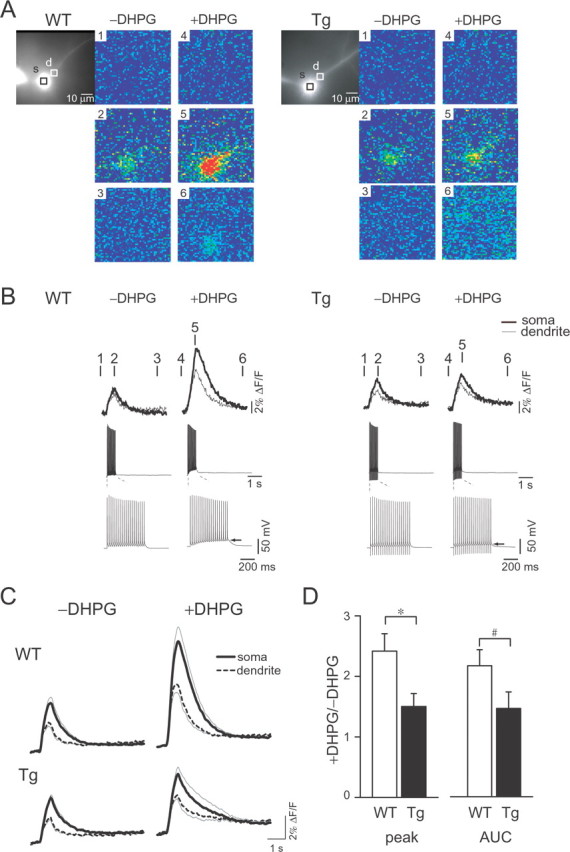
GPCR-mediated [Ca2+]i increase in the neurons of wild-type and Tg mice. A, Hippocampal pyramidal neurons were filled with the low-affinity indicator fura-6F (Kd, 6 μm). DIC images of the neurons are shown with regions of interest located at the soma (black rectangles) and the dendrites (white rectangles) in wild-type (WT) and mutPOLG Tg (Tg) mice, respectively. Pseudocolor images of spatiotemporal [Ca2+]i distribution reflect the basal level of [Ca2+]i before AP firing (panels 1 and 4), the peak of [Ca2+]i increase during AP firing (panels 2 and 5), and the recovered level of [Ca2+]i after AP firing (panels 3 and 6), respectively. Images taken before (−DHPG) and during (+DHPG) bath application of 30 μm DHPG are shown. B, [Ca2+]i increases at the soma (thick lines) and the dendrites (thin lines) were superimposed for the wild-type (WT) and mutPOLG Tg (Tg) mice, respectively. [Ca2+]i increase was induced by APs and observed by time-lapse imaging of fluorescence changes (top). The distinct time points (1–6) over traces correspond to the [Ca2+]i images of panels 1–6 in A. Traces of membrane potential at the middle are shown on an expanded timescale at the bottom. The arrows indicate GPCR-mediated membrane depolarization during AP firing. C, Averaged traces of [Ca2+]i increases were given at the soma (thick lines) and the dendrite (broken lines) in wild-type (WT) and mutPOLG Tg (Tg) mice, respectively. The AP-induced [Ca2+]i increases before (−DHPG) and during (+DHPG) bath application of DHPG are shown. Data at each point are expressed as mean ± SEM, which is indicated by the gray line. D, Statistical analysis of DHPG-induced [Ca2+]i changes. The amplitude of peak (peak) and AUC of fluorescence changes at the soma are compared with the wild-type (WT) and mutPOLG Tg (Tg) mice (n = 11 and 15, respectively). Data were given as the ratio of during application of DHPG to before application of drugs. [Ca2+]i increase during application of DHPG was attenuated in the neurons of mutPOLG Tg mice (peak, *p < 0.05; AUC, #p < 0.1). Data are expressed as mean ± SEM.
Statistics.
Statistical calculation was performed using KyPlot software (version 4.0; Kyens) or SPSS software (version 10.0; SPSS, Chicago, IL). Results were presented as mean ± SEM and were evaluated using a paired or a two-sample Student's t test when appropriate. Paired t test was used to test for difference in the presence or absence of drug. Interaction between CsA treatment and Ca2+ addition in the experiment of isolated mitochondria was statistically evaluated by two-way repeated-measures ANOVA with the factors of Ca2+ addition and CsA treatment. Degree of freedom for repeated-measures ANOVA was adjusted for sphericity by Greenhouse–Geisser method. Post hoc analysis of the difference between recovery time with or without CsA was conducted by the Mann–Whitney U test. Values of p < 0.05 were considered statistically significant.
Results
Ca2+ retention capacity and Ca2+ uptake kinetics in isolated mitochondria
Mitochondrial Ca2+ uptake through Ca2+ uniporter is associated with the activity of the mitochondrial respiratory chain, which consists of a set of five complexes (I to V). None of the proteins of complex II are encoded by mtDNA (Larsson and Clayton, 1995). To assess the impact of accumulated mtDNA defects on mitochondrial Ca2+ transport, we performed two experiments; in one experiment, we used the substrate for complex I (glutamate plus malate), and the substrate for complex II (succinate) was used in another experiment. We added aliquots of Ca2+ solution to mitochondrial suspension and determined the CRC by monitoring [Ca2+]exm. In the experiment using a buffer containing glutamate plus malate, four additions of 1.6 μm final concentration of Ca2+ (short arrows) induced small [Ca2+]exm transients, and 7.8 μm final concentration of Ca2+ (long arrows) induced larger changes (Fig. 1A). The [Ca2+]exm increased at first and was completely restored to the basal level by mitochondrial uptake. At the point marked by asterisks (Fig. 1A), PTP opened and Ca2+ was released from the matrix to the outside of mitochondria. Finally, a similar number of additions was required to open the PTP in mitochondria from wild-type and mutPOLG Tg mice. The point of PTP opening was defined according to the previous studies (Ichas et al., 1997; Fontaine et al., 1998). Next, we measured the CRC of mitochondria incubated in a buffer containing succinate, in which the mitochondria are energized through complex II components independent of mtDNA involvement. By additions of 7.8 μm Ca2+ in the buffer containing succinate, PTP opening was induced more readily than was done in the buffer containing glutamate plus malate (data not shown). Thus, we added final 1.6 and 3.1 μm Ca2+ solution to measure the CRC. Four additions of 1.6 μm final concentration of Ca2+ (short arrows) and several additions of 3.1 μm final concentration of Ca2+ (long arrows) induced [Ca2+]exm transients in a dose-dependent manner (Fig. 1C). There was no clear distinction in the CRC of mitochondria between mutPOLG Tg mice and wild-type mice (Fig. 1E, left, n = 6, p = 0.81; right, n = 6, p = 0.85, Student's t test), whereas we noticed a difference in [Ca2+]exm kinetics in response to the first addition of 7.8 μm Ca2+ solution. The traces of [Ca2+]exm shown in Figure 1B were obtained from six independent experiments in the substrate for complex I and superimposed. In mitochondria of mutPOLG Tg mice, the [Ca2+]exm were rapidly restored back toward the basal level, and the recovery time was significantly shorter than in mitochondria from wild-type mice (Fig. 1F, left, n = 6, p < 0.05, Student's t test). No significant difference was observed between genotypes in the buffer containing the substrate for complex II (Fig. 1D,F, right, n = 6, p = 0.27, Student's t test).
Figure 1.
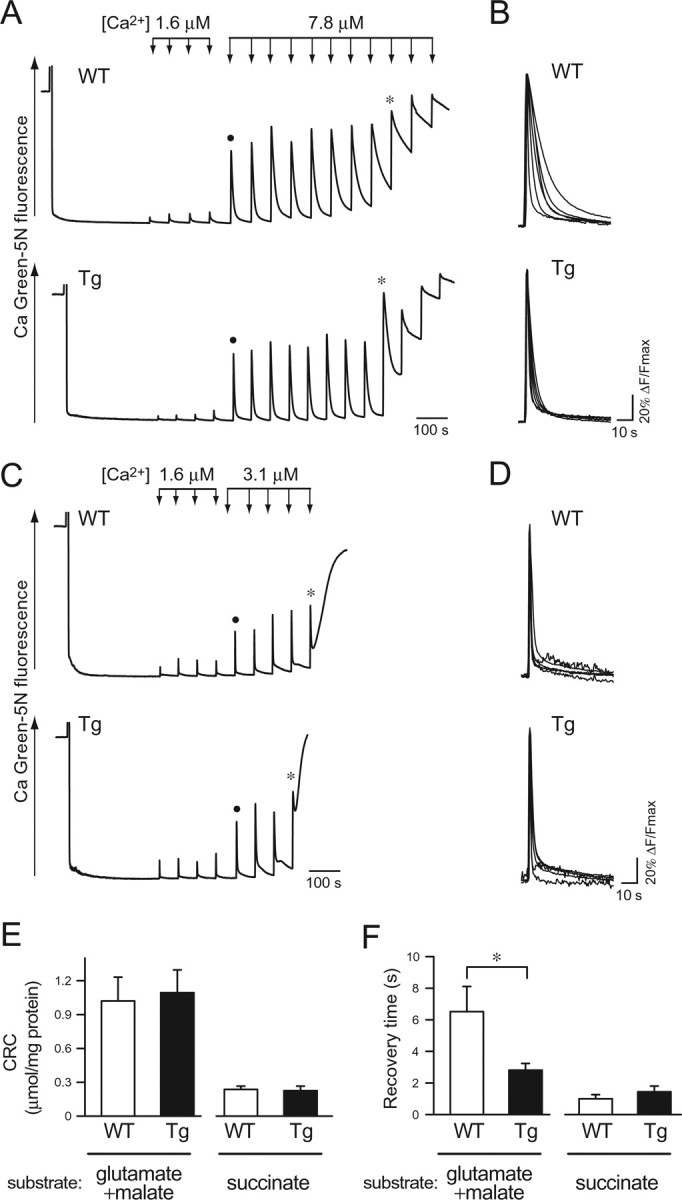
Mitochondrial CRC and uptake kinetics. A, CRC of mitochondria from wild-type (WT) and mutPOLG Tg (Tg) mice in a buffer containing 20 mm glutamate plus 2 mm malate, which is a substrate for complex I. Changes in extramitochondrial Ca2+ concentration ([Ca2+]exm) were monitored by a fluorescent Ca2+-sensitive dye, Calcium Green-5N. Ca2+ solution was applied by four additions at a final concentration of 1.6 μm (short arrows) and subsequent additions at a final concentration of 7.8 μm (long arrows) at 60 s intervals. An asterisk on the trace indicates the point at which Ca2+-overloaded mitochondria released Ca2+. A closed circle indicates the trace that was used for the analysis in B. The representative data of six experiments are shown. B, Kinetics of [Ca2+]exm responses in a buffer containing 20 mm glutamate plus 2 mm malate. The fluorescence changes in response to the first additions of 7.8 μm Ca2+ in six independent experiments were superimposed on an expanded timescale after normalization by maximal peak amplitude. C, CRC of mitochondria from wild-type (WT) and mutPOLG Tg (Tg) mice in a buffer containing 10 mm succinate, a substrate for complex II. Ca2+ solution was applied by four additions of 1.6 μm Ca2+ (short arrows) and subsequent additions of 3.1 μm Ca2+ (long arrows) at 60 s intervals. An asterisk on the trace indicates the point at which Ca2+-overloaded mitochondria released Ca2+. A closed circle indicates the trace that was used for the analysis in D. The representative data of seven independent experiments are shown. D, Kinetics of [Ca2+]exm responses in the buffer containing 10 mm succinate. The fluorescence changes in response to the first additions of 3.1 μm Ca2+ in six independent experiments were superimposed on an expanded timescale after normalization by maximal peak amplitude. E, Statistical analysis of results from the CRC assays in the buffer containing glutamate plus malate or succinate. Total amount of Ca2+ (micromoles of Ca2+ per milligram of protein) necessary to induce Ca2+ release was compared between mitochondria from wild-type (WT) and mutPOLG Tg (Tg) mice (n = 6). Data are expressed as mean ± SEM. F, Statistical analysis of the recovery times for Ca2+ uptake in two different conditions. Mitochondria from mutPOLG Tg (Tg) mice sequestered Ca2+ more rapidly than did those from wild-type (WT) mice (n = 6; *p < 0.05) when a CRC assay was performed in the buffer containing glutamate plus malate. Data are expressed as mean ± SEM.
Mitochondrial membrane potential in isolated mitochondria
As a mechanism underlying the enhanced Ca2+ uptake of mitochondria from mutPOLG Tg mice, it was assumed that a higher ΔΨm across the mitochondrial inner membrane would provide a higher driving force for the Ca2+ uptake. We stained mitochondria with a ΔΨm-sensitive dye, JC-1, including the substrate for complex I and assessed ΔΨm (Fig. 2A); a depolarized mitochondria particle was defined on the basis of a ratio of the red-to-green signals (R/G). The mitochondria of mutant mice did not display higher R/G than those from wild-type mice under the steady-state condition (Fig. 2B, control, p = 0.64, Student's t test). A treatment with Ca2+ corresponding to the level of one addition in the above CRC assay (100 nmol of Ca2+ per milligram of mitochondrial protein) decreased the fluorescence intensity of the red signal of JC-1, indicating that mitochondria were depolarized in both genotypes (Fig. 2A, +Ca2+). The addition of Ca2+ resulted in a clear shift in the peak of histogram to a lower R/G value (Fig. 2B, +Ca2+). A remarkable loss of fluorescence was induced after exposure to the protonophore FCCP, indicating the ΔΨm collapse (Fig. 2C, +FCCP). There were no differences between genotypes in ΔΨm or ΔΨm response under any of the conditions (Fig. 2C, control, p = 0.64; +FCCP, p = 0.94; +Ca2+, p = 0.48, Student's t test). These observations were confirmed when samples were incubated with the complex II substrate (data not shown). However, the aggregate formation of JC-1 may provide a higher estimate in a population of polarized mitochondria, which interfere with detection of the difference in genotypes. Therefore, we verified our result using another ΔΨm-dependent dye, DiIC1(5), that exhibits a 630 nm excitability of emission spectrum, which is also used with a mitochondria-targeted dye, NAO. By the double staining with NAO and DiIC1(5), mitochondria particles were identified. The NAO-labeled particles showing higher intensity of DiIC1(5) signal were determined to be more “energized” mitochondria incubated in the complex I substrate (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). No significant differences were detected in the mean intensities between genotypes under any of the conditions (steady state, p = 0.66; FCCP, p = 0.10; Ca2+, p = 0.21, Student's t test). Neither was there any difference in ΔΨm response between genotypes. These results were confirmed by a measurement taken in the buffer containing the complex II substrate (data not shown).
Figure 2.

Mitochondrial membrane potential. A, Measurement of mitochondrial membrane potential (ΔΨm) in isolated mitochondria using a ΔΨm-sensitive dye, JC-1. Isolated brain mitochondria from wild-type (WT) and mutPOLG Tg (Tg) mice were subjected to flow-cytometric analysis in a buffer containing 20 mm glutamate plus 2 mm malate. A potentiometric dye, JC-1, accumulates in mitochondria. The emission fluorescence exhibited green and red, which were plotted on the x- and y-axes, respectively. Each point indicates fluorescence intensity from each mitochondrion under steady-state condition (control) and treatment with 1 μm FCCP (+FCCP) or 6.25 μm Ca2+ (+Ca2+). A negative shift in red signals along the y-axis indicated a loss of ΔΨm. B, Frequency distributions of red-to-green fluorescence ratio (R/G) of JC-1. Depolarized mitochondria particles were counted by defining that the R/G was <0.3 (a gray broken line indicates the threshold). A loss of ΔΨm was evaluated by decreased R/G. Ca2+ exposure increased the population of depolarized mitochondria from wild-type (WT) and mutPOLG Tg (Tg) mice. C, Quantifications of depolarized mitochondria. The population of depolarized mitochondria was averaged and compared with wild-type (WT) and mutPOLG Tg (Tg) under steady-state condition (control) and treatment with Ca2+ (+Ca2+) or FCCP (+FCCP). There were no significant differences between genotypes (n = 6) in ΔΨm or ΔΨm response under any of the conditions. Data are expressed as mean ± SEM.
Differentially expressed genes in brains of mutPOLG Tg mice
Our findings were not attributable to respiratory failure but caused by some unknown mechanisms related to Ca2+ transport. To clarify the molecular basis of the abnormality in mitochondria of mutPOLG Tg mice, we performed DNA microarray analysis of the brains from male mutPOLG Tg and wild-type littermates of 34–35 weeks of age (n = 5 for each genotype). The expression of four genes related to mitochondria was significantly affected commonly in the two regions of the brain in mutPOLG Tg mice; two genes were downregulated and the other two genes were upregulated (Table 1). We performed quantitative real-time RT-PCR to validate the microarray results and confirmed the downregulation of Ppif and Glud1 genes in the hippocampal tissues of mutPOLG Tg mice compared with wild-type mice (Table 1) (n = 6 for each genotype). The level of Ppif [cyclophilin D (CyP-D)] and Glud1 (glutamate dehydrogenase) genes were significantly decreased in mutPOLG Tg mice. Because the Ppif gene encodes CyP-D, which is a critical component of PTP and regulates Ca2+ sensitivity to PTP opening, we considered that the downregulated expression of the Ppif gene would be relevant to the enhanced Ca2+ uptake in mutPOLG Tg mice. To determine whether several genes encoding proteins required for the opening of PTP was entirely suppressed as well as the Ppif gene, we monitored mRNA levels of the genes encoding the other proteins constituting the PTP components [e.g., adenine nucleotide translocator (ANT1/Slc25a4), voltage-dependent anion channel (Vdac1), hexokinase (Hk1), creatine kinase (Ckmt1), and peripheral benzodiazepine receptor (Bzrp)]. The relative amounts of mRNAs for these genes were not significantly changed in the brains of mutPOLG Tg mice (data not shown). Our experimental and analytical procedures were validated by a higher expression level of the mutPolg gene in mutant mice than wild-type mice. The mutPolg gene expression is likely to be engaged in upregulation of mtDNA topoisomerase I gene (Top1mt) expression. Because Top1mt protein has been known to be involved in the mtDNA replication (Moraes, 2001; Zhang et al., 2001), this upregulation was thought to be the influence of the transgene, mutPolg.
Table 1.
Differentially expressed genes in the mutPOLG Tg mice brains
| Gene |
DNA microarray |
RT-PCR |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Hippocampus |
Frontal cortex |
Hippocampus |
|||||||
| Symbol | Description | Accession no. | Affymetrix ID | Fold changea | p value* | Fold changea | p value* | Fold changea | p value* |
| Ppif | Cyclophilin D | NM_134084 | 1416940_at | 0.91 | 0.038 | 0.93 | 0.008 | 0.84 | 0.021 |
| Glud1 | Glutamate dehydrogenase 1 | BI329832 | 1416209_at | 0.93 | 0.049 | 0.97 | 0.040 | 0.84 | 0.020 |
| Top1mt | mtDNA topoisomerase 1 | AF362952 | 1460370_at | 1.12 | 0.005 | 1.05 | 0.002 | Not tested | |
| Polg | mtDNA polymerase | BG064799 | 1423272_at | 1.49 | 0.045 | 1.32 | 0.007 | Not tested | |
aFold changes were given as ratio of signals of mutPOLG Tg mice relative to those of wild-type mice.
*Student's t test for paired comparison (two-tailed).
Effect of the cyclophilin D inhibitor on rate of Ca2+ uptake in isolated mitochondria
We speculated that the decrease of CyP-D would be responsible for the abnormal Ca2+ uptake in mitochondria from mutPOLG Tg mice. A CyP-D inhibitor, CsA, prevents PTP from opening at the adenine nucleotide translocator (ANT)-binding site of CyP-D (Hansson et al., 2003). So, we tested whether CyP-D inhibition would enhance Ca2+ uptake in mitochondria from wild-type mice (n = 6). PTP opening occurred after six additions of 7.8 μm Ca2+ in the absence of CsA (Fig. 3A, −CsA). In the presence of 2 μm CsA, over 20 additions of 7.8 μm Ca2+ were required to open the PTP (Fig. 3A, +CsA). This indicated that CsA inhibited PTP opening and significantly increased the CRC (Fig. 3A, inset) (p < 0.05, paired t test). We then measured the recovery time of [Ca2+]exm to evaluate the rate of Ca2+ uptake. Shortening of the recovery time by CsA became evident after repeated addition of Ca2+ (Fig. 3B, a–c, +CsA and −CsA). This finding was confirmed in five independent experiments. The recovery time was almost unchanged in the presence of CsA even when Ca2+ solution was added repeatedly (Fig. 3C, +CsA), which was similarly observed in mutPOLG Tg mice (Fig. 3, compare C, D).
Figure 3.

Effect of the cyclophilin D inhibitor, cyclosporin A, on Ca2+ uptake in mitochondria. A, CRC was measured in the absence (−CsA) or presence (+CsA) of 1 μm CsA in a buffer containing glutamate plus malate. Changes in [Ca2+]exm were monitored by the fluorescent Ca2+-sensitive dye, Calcium Green-5N. Ca2+ solution was subsequently applied at a final concentration of 7.8 μm for untreated mitochondria (−CsA) or for CsA-treated mitochondria (+CsA) at 60 s intervals. The traces marked by a–c were used for analysis in B. Inset, The CRC was determined from three independent experiments (n = 6; *p < 0.05). Data are expressed as mean ± SEM. B, Kinetics of [Ca2+]exm changes. Representative traces of [Ca2+]exm marked by a–c in A were expanded. The traces were normalized by maximal peak amplitude and compared between the absence (−CsA) and presence (+CsA) of CsA. After the repeated addition of Ca2+, the recovery time became shorter in the presence of CsA than in the absence of CsA. As shown in C, CsA apparently decreased the recovery time of Ca2+. C, Changes in recovery time with sequential additions of Ca2+, plotted by the amount of Ca2+ added in the absence (−CsA) or presence (+CsA) of CsA. Each circle corresponds to a Ca2+ addition, and each trace of [Ca2+]exm is from one experiment. Two-way repeated-measures ANOVA in the first four additions revealed a significant interaction between the number of Ca2+ addition and CsA treatment (F = 4.78; df = 1.08; p < 0.05, repeated two-way ANOVA, Greenhouse–Geisser correction). Although there was no statistically significant difference of recovery time after the first addition of Ca2+ (p = 0.35, Mann–Whitney U test), recovery time in the presence of CsA (+CsA) became shorter than that in the absence of CsA at the fourth addition of Ca2+ (p < 0.05, Mann–Whitney U test). D, Changes in recovery time with the subsequent additions of Ca2+. Each circle corresponds to a Ca2+ addition and each trace of [Ca2+]exm is from one experiment using mitochondria from wild-type (WT) and mutPOLG Tg (Tg) mice in the absence of CsA.
Electrical membrane properties and action potential-induced Ca2+ transient in hippocampal neurons
To examine the effect of enhanced Ca2+ uptake of isolated mitochondria of mutPOLG Tg mice on the [Ca2+]i dynamics, hippocampal slice preparation was used. Whole-cell recordings were made from the soma of pyramidal neurons in area CA1. Basic neuronal excitability of hippocampal pyramidal neurons of mutPOLG Tg mice was similar to that of wild-type mice (see supplemental Table 1, available at www.jneurosci.org as supplemental material) (n = 33 for each genotype). Because mitochondria participate in regulating [Ca2+]i according to the level of [Ca2+]i change induced by a varying frequency of AP generation (David et al., 1998; Peng, 1998), we elicited a repetitive AP in pyramidal neurons of the mutant mice and compared the induced [Ca2+]i changes with those in wild-type mice (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). The magnitude of [Ca2+]i transient was measured using two parameters, the amplitude or the time of peak, and revealed no significant difference between genotypes (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material).
Involvement of Ca2+ store in GPCR-mediated [Ca2+]i increase in hippocampal neurons
Because IP3-mediated Ca2+ response was reported to be enhanced in platelets from BD patients (Dubovsky et al., 1989; Yamawaki et al., 1996), we had hypothesized that reduced mitochondrial Ca2+ uptake may result in increased cytosolic [Ca2+]i response in BD (Kato and Kato 2000). Contrary to our expectation, however, mitochondrial Ca2+ uptake was enhanced in mutant mice. To test how the enhanced Ca2+ uptake in mitochondria affects cytosolic [Ca2+]i response of mutPOLG Tg mice, we studied the agonist-stimulated [Ca2+]i response in hippocampal neurons. Although it is difficult to assess this impact quantitatively in neurons, previous studies showed that GPCR activation paired by backpropagating APs could induce Ca2+ mobilization from internal stores (Nakamura et al., 1999). To apply this experimental paradigm for investigating the molecular basis of altered Ca2+ signaling in the neurons of Tg mice, we first pharmacologically characterized this response. After group I mGluR agonist, DHPG (30 μm), was applied, all of the neurons tested were slightly depolarized within the first few minutes as described previously (Rae et al., 2000). In the presence of DHPG, the membrane potential gradually depolarized during the AP firing (Figs. 4D, 5B, bottom, +DHPG, indicated by arrows), which was often followed by a spontaneous bursting discharge and excessive [Ca2+]i increase in some neurons. Because this Ca2+ change hampered an evaluation of IP3-mediated Ca2+ mobilization, these cells were excluded from the analysis. When DHPG was applied, the same number of APs (30 Hz) as before elicited a larger [Ca2+]i increase (+DHPG) compared with before addition of DHPG (−DHPG) in hippocampal CA1 pyramidal neurons of wild-type mice (Fig. 4A). DHPG binds to mGluR, stimulates Gq protein, and produces IP3 to activate IP3 receptor (IP3R), releasing Ca2+ from internal store in neurons (Berridge, 1998). The mGluR receptor antagonist, MCPG (1 mm), significantly inhibited this enhancement (26.6 ± 12.4%; n = 4; p < 0.01, paired t test) (Fig. 4A, MCPG+DHPG). The enhanced[Ca2+]i increase was restored by washing out of MCPG (+DHPG), supporting the role of mGluR in this reaction as suggested by previous studies (Nakamura et al., 2000; Rae et al., 2000). Application of a cholinergic receptor agonist, 30 or 300 μm CCh, also caused a similar enhancement (Fig. 4B), which was abolished by a muscarinic receptor antagonist, atropine (1 μm) (25.9 ± 2.3%; n = 3; p < 0.01, paired t test). Two agonists acting on different receptors but affecting the common intracellular signaling pathway, Gq–PLCβ (phospholipase Cβ)–IP3 (Shirasaki et al., 1994; Power and Sah, 2002), showed similar Ca2+ enhancement, suggesting the role of this pathway in this enhancement. To further verify the source of [Ca2+]i, the effect of an ER Ca2+/ATPase blocker, CPA, was examined. DHPG-induced [Ca2+]i increase was attenuated in the presence of CPA (Fig. 4C). An IP3R blocker, heparin (2 mg/ml), also abolished the enhancement of DHPG- or CCh-induced [Ca2+]i increase (Fig. 4D or data not shown). These results further confirmed that the enhancement of [Ca2+]i increase by GPCR stimulation is mediated by Ca2+ release from the ER through IP3R.
Effect of GPCR agonist on AP-induced [Ca2+]i transient in hippocampal neurons of wild-type and mutPOLG Tg mice
[Ca2+]i transient induced by a train of APs was not different in both genotypes. Additionally, DHPG effect on AP-induced [Ca2+]i increase was observed in neurons of mutPOLG Tg mice. In the absence of DHPG, a train of 20 APs similarly induced cytosolic and dendritic [Ca2+]i transients both in the neurons of wild-type and mutPOLG Tg mice (Fig. 5A,B, −DHPG). The magnitude of membrane depolarization after DHPG application did not differ between genotypes (−3.5 ± 1.4 mV in neurons of wild-type mice and −3.5 ± 1.7 mV in neurons of mutPOLG Tg mice; p = 0.40, Student's t test). The different population of cells showing the spontaneous bursting in the presence of DHPG did not found between wild-type (4 of 19 cells) and mutPOLG Tg mice (8 of 19 cells) (p = 0.14, Fisher's exact test). In the presence of DHPG, [Ca2+]i increase was enhanced in neurons of both genotypes (Fig. 5C, +DHPG). The impact of DHPG was estimated by a change in the peak amplitude or the AUC of the [Ca2+]i increase at the soma. Ratio of the [Ca2+]i increase after DHPG application to that before the application was significantly smaller in mutPOLG Tg mice (n = 11) compared with that in wild-type mice (n = 15) (Fig. 5D, peak, *p < 0.05; AUC, #p < 0.1, Student's t test). The modulatory effect of DHPG on AP-induced [Ca2+]i increase was less in the neurons of mutPOLG Tg mice than that of wild-type mice.
Discussion
We investigated Ca2+ dynamics in mutPOLG Tg mice harboring neuron-specific defects in mtDNA age dependently. We proposed that the mutant mice would be a model of BD, because it not only fulfills construct validity based on mitochondrial dysfunction hypothesis of BD but also fulfills face and predictive validities: its BD-like phenotypes such as periodic activity change were improved by a mood stabilizer. The present study demonstrated that the rate of Ca2+ uptake was enhanced in isolated mitochondria from the mutant mice and GPCR-mediated [Ca2+]i increase was attenuated in the hippocampal neurons of the mutPOLG Tg mice. These findings might shed light on the pathophysiology of BD.
Molecular basis of enhanced rate of Ca2+ uptake in mitochondria from mutPOLG Tg mice
The recovery time of [Ca2+]exm in isolated mitochondria from mutPOLG Tg mice was shorter than that in wild-type mice (Fig. 1B), although mitochondrial CRC was not altered (Fig. 1A,E). This result could be explained by two functional states of PTP, persistent and brief openings (Ichas and Mazat, 1998). Persistent PTP opening with a large conductance is thought to associate with a disruption of mitochondrial outer membrane and a subsequent release of cytochrome c to the cytosol (Weiss et al., 2003), and translocation of cytochrome c is often accompanied with apoptosis and a loss of ΔΨm (Schild et al., 2001). In contrast, brief PTP opening with a small conductance is induced by binding of CyP-D in the matrix to ANT in the inner membrane of mitochondria and enables Ca2+ to be released. A role of CyP-D in brief PTP opening has been well established by forming the pore that associates with the conformational change of inner membrane (Crompton, 1999).
In this study, we found downregulation of Ppif encoding CyP-D in the forebrains of mutPOLG Tg mice (Table 1). Moreover, application of CyP-D inhibitor, CsA, facilitated Ca2+ uptake (Fig. 3B) and mimicked the altered Ca2+ uptake kinetics in mitochondria from mutPOLG Tg mice (Fig. 3C,D) in addition to marked increase of CRC (Fig. 3A) as shown in a previous study (Fontaine et al., 1998). Thus, we can speculate that downregulation of CyP-D inhibited the brief PTP opening, which resulted in the enhanced mitochondrial Ca2+ uptake in mutPOLG Tg mice. This finding is consistent with the reports that CsA facilitates mitochondrial Ca2+ sequestration in brain mitochondria (Levy et al., 2003) or chromaffin cells (Montero et al., 2001). Mitochondria from CyP-D-deficient mice caused accumulation of a larger amount of Ca2+ (Baines et al., 2005; Basso et al., 2005; Nakagawa et al., 2005). The downregulation of CyP-D, however, may be a consequence of the pathological Ca2+ homeostasis caused by the accumulation of mtDNA defects. Additional studies will be required for elucidating the causal relationship.
Relationship between enhanced Ca2+ uptake rate in isolated mitochondria and attenuated DHPG-induced Ca2+ increase in hippocampal neurons of mutPOLG Tg mice
Regarding synaptic Ca2+ transients, mitochondrial Ca2+ uptake is known to contribute to Ca2+ clearance at the presynaptic terminals (Billups and Forsythe, 2002) and postsynaptic sites (Pivovarova et al., 2002). The enhanced rate of Ca2+ uptake in mitochondria of the mutPOLG Tg mice should result in attenuated Ca2+ signaling in the neurons. As we expected, GPCR-mediated [Ca2+]i increase, which is primarily coupled to the IP3 mobilization, was influenced in mutant mice (Fig. 5). Previous studies demonstrated that GPCR-mediated [Ca2+]i increase was not affected by a ryanodine receptor (RyR) inhibitor and was triggered by photolysis of caged IP3 (Nakamura et al., 1999; Yamamoto et al., 2000). The present result that IP3-mediated [Ca2+]i increase was sensitive to the store depletion and IP3R blockade (Fig. 4C,D) is consistent with those previous findings. However, [Ca2+]i transient by AP-induced depolarization was not altered in neurons of mutPOLG Tg mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). One possible explanation for the lack of influence on this component is that [Ca2+]i increase by GPCR activation is much larger than that by AP-induced depolarization. When [Ca2+]i reaches 1–2.5 μm, mitochondria exert their function for Ca2+ clearance in synaptic terminals (Billups and Forsythe, 2002; Kim et al., 2005). In pyramidal neurons, [Ca2+]i level induced by depolarization is ∼1 μm, whereas if the depolarization is evoked by paired with an activation of GPCR, peak [Ca2+]i level augments to ∼3 μm (Nakamura et al., 1999). The Ca2+ concentration applied to mitochondrial suspension in this study (3.1 or 7.8 μm) may be comparable with the latter situation. Close contact of mitochondria with ER at microdomain may cause even higher Ca2+ level, which may further enhance mitochondrial Ca2+ uptake (Rizzuto et al., 1998).
It is reasonable to attribute the attenuation of agonist-induced [Ca2+]i enhancement to accelerated mitochondrial Ca2+ uptake based on previous observations (Hajnóczky et al., 1999; Pivovarova et al., 2004). It should be noted, however, that a possible role of adaptive changes secondary to mitochondrial dysfunction cannot be ruled out. For example, reduced Ca2+ levels in ER or a promoted Ca2+ extrusion through the plasma membrane, a decrease in Ca2+ influx from plasma membrane can also explain the present findings. Ca2+-induced Ca2+ release mediated by RyR or IP3R may also be involved.
Additionally, adenine nucleotides including cAMP (Snyder and Supattapone, 1989; Shimizu et al., 1993; Giovannucci et al., 2000), interactions with other proteins (Gee et al., 2003; Tozzi et al., 2003; Wanaverbecq et al., 2003), or phosphorylation by various kinases (Wagner et al., 2003; Skeberdis et al., 2006) can modulate IP3R function. Because both serotonin- and thrombin-induced Ca2+ mobilizations in platelets are reportedly enhanced in BD patients (Yamawaki et al., 1998), we anticipated that altered mitochondrial Ca2+ regulation may enhance agonist-induced [Ca2+]i increase in neurons of mutPOLG Tg mice. Although our results are in the opposite direction to the finding in platelets from patients (Fig. 5), they are compatible with a recent study demonstrating that GPCR-mediated [Ca2+]i increase was diminished in the olfactory neurons from BD patients (Hahn et al., 2005).
Possible role of mitochondrial Ca2+ transport in cell death and/or respiratory failure
The enhanced Ca2+ uptake in mitochondria from mutPOLG Tg mice was seen in the buffer containing a substrate for complex I (Fig. 1B,F), but not in the buffer containing a substrate for complex II (Fig. 1D,F). Many of the complex I subunits are encoded by mtDNA, whereas all of the complex II subunits are by nuclear DNA. This fact supports the association between the mtDNA defects and the enhanced mitochondrial Ca2+ uptake. Similar discrepancy between substrates has been discussed in studies using cybrids from patients with mtDNA mutations (Moudy et al., 1995; Brini et al., 1999) and those from patients with AD or PD (Sheehan et al., 1997a,b). A slower rate of Ca2+ uptake in these cybrids was improved by a treatment with succinate, which is the substrate for complex II. This is in accord with our finding that the rate of Ca2+ uptake was not different between genotypes when we used the buffer containing succinate.
Unlike mitochondria of the model mice for HD, we found no difference between genotypes in the ΔΨm as measured by flow cytometric analysis (Fig. 2; supplemental Fig. 1, available at www.jneurosci.org as supplemental material), and the CRC was not altered in mitochondria of the mutPOLG Tg mice. Preliminary observation showed no different histological features and no marked cell death in the brains of mutPOLG Tg mice. It would be interesting to study how amounts of mutations could enhance Ca2+ uptake rate in mitochondria from mutPOLG Tg mice without causing energy dissipation. Another group reported that transgenic mice expressing heart-specific mutPOLG suffered from a cardiomyopathy attributable to an accumulation of mtDNA defects (Zhang et al., 2000). Nevertheless, neither an abnormality of mitochondrial respiration nor a loss of ATP could be detected in the hearts of these mice (Mott et al., 2001; Zhang et al., 2003). More studies are still needed to elucidate the mechanisms by which mtDNA defects alter mitochondrial Ca2+ uptake. It will be interesting to see the cellular phenotypes in different lines of mice with accumulation of mtDNA defects, such as knock-in mice of POLG mutation (Trifunovic et al., 2004; Kujoth et al., 2005) and mutant mice with defect in Twinkle (Tyynismaa et al., 2005).
Relationship of altered Ca2+ dynamics in behavioral phenotypes of mutPOLG Tg mice
The mutPOLG Tg mice apparently exhibited a different pattern in wheel-running activity from wild-type mice, but were not impaired in a comprehensive series of learning and memory tests (Kasahara et al., 2006). In this study, it was difficult to provide a precise explanation with respect to the cause of behavioral phenotypes in mutPOLG Tg mice. However, the mtDNA defects in neurons might underlie BD-like behavioral phenotypes of mutPOLG Tg mice through the attenuation of GPCR-mediated [Ca2+]i increase and the subsequent intracellular events.
Footnotes
This work was supported by grants to the Laboratory for Molecular Dynamics of Mental Disorders, RIKEN Brain Science Institute (BSI), a grant-in-aid from the Japanese Ministry of Health and Labor, and grants-in-aid from the Japanese Ministry of Education, Culture, Sports, Science and Technology. We thank staff members in the Division of Animal Experiments and Common Instrumentations (Research Resources Center, RIKEN BSI) for technical assistance and members of our laboratory for useful discussion. We are indebted to the Research Resources Center for flow cytometry analysis and Dr. A. Takashima (Laboratory for Alzheimer's Disease, RIKEN BSI) for use of the Beckman high-performance centrifuge. We also thank Dr. A.V. Panov (Emory University School of Medicine, Atlanta, GA) for helpful suggestions. This paper is dedicated to our coauthor, Dr. Takeshi Nakamura, who unexpectedly passed away while this paper was being prepared. He was a devoted and insightful scientist, and his contribution to this project is deeply appreciated.
References
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bianchi K, Rimessi A, Prandini A, Szabadkai G, Rizzuto R. Calcium and mitochondria: mechanisms and functions of a troubled relationship. Biochim Biophys Acta. 2004;1742:119–131. doi: 10.1016/j.bbamcr.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2002;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M, Pinton P, King MP, Davidson M, Schon EA, Rizzuto R. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat Med. 1999;5:951–954. doi: 10.1038/11396. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett JN, Barrett EF. Evidence that mitochondria buffer physiological Ca2+ loads in lizard motor nerve terminals. J Physiol (Lond) 1998;509:59–65. doi: 10.1111/j.1469-7793.1998.059bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubovsky SL, Christiano J, Daniell LC, Franks RD, Murphy J, Adler L, Baker N, Harris RA. Increased platelet intracellular calcium concentration in patients with bipolar affective disorders. Arch Gen Psychiatry. 1989;46:632–638. doi: 10.1001/archpsyc.1989.01810070058010. [DOI] [PubMed] [Google Scholar]
- Fontaine E, Ichas F, Bernardi P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J Biol Chem. 1998;273:25734–25740. doi: 10.1074/jbc.273.40.25734. [DOI] [PubMed] [Google Scholar]
- Gee CE, Benquet P, Gerber U. Group I metabotropic glutamate receptors activate a calcium-sensitive transient receptor potential-like conductance in rat hippocampus. J Physiol (Lond) 2003;546:655–664. doi: 10.1113/jphysiol.2002.032961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh SS, Swerdlow RH, Miller SW, Sheeman B, Parker WD, Jr, Davis RE. Use of cytoplasmic hybrid cell lines for elucidating the role of mitochondrial dysfunction in Alzheimer's disease and Parkinson's disease. Ann NY Acad Sci. 1999;893:176–191. doi: 10.1111/j.1749-6632.1999.tb07825.x. [DOI] [PubMed] [Google Scholar]
- Giovannucci DR, Groblewski GE, Sneyd J, Yule DI. Targeted phosphorylation of inositol 1,4,5-trisphosphate receptors selectively inhibits localized Ca2+ release and shapes oscillatory Ca2+ signals. J Biol Chem. 2000;275:33704–33711. doi: 10.1074/jbc.M004278200. [DOI] [PubMed] [Google Scholar]
- Hahn CG, Gomez G, Restrepo D, Friedman E, Josiassen R, Pribitkin EA, Lowry LD, Gallop RJ, Rawson NE. Aberrant intracellular calcium signaling in olfactory neurons from patients with bipolar disorder. Am J Psychiatry. 2005;162:616–618. doi: 10.1176/appi.ajp.162.3.616. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Hager R, Thomas AP. Mitochondria suppress local feedback activation of inositol 1,4, 5-trisphosphate receptors by Ca2+ J Biol Chem. 1999;274:14157–14162. doi: 10.1074/jbc.274.20.14157. [DOI] [PubMed] [Google Scholar]
- Hansson MJ, Persson T, Friberg H, Keep MF, Rees A, Wieloch T, Elmer E. Powerful cyclosporin inhibition of calcium-induced permeability transition in brain mitochondria. Brain Res. 2003;960:99–111. doi: 10.1016/s0006-8993(02)03798-8. [DOI] [PubMed] [Google Scholar]
- Ichas F, Mazat JP. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim Biophys Acta. 1998;1366:33–50. doi: 10.1016/s0005-2728(98)00119-4. [DOI] [PubMed] [Google Scholar]
- Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Kakiuchi C, Iwamoto K, Ishiwata M, Bundo M, Kasahara T, Kusumi I, Tsujita T, Okazaki Y, Nanko S, Kunugi H, Sasaki T, Kato T. Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat Genet. 2003;35:171–175. doi: 10.1038/ng1235. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Kubota M, Miyauchi T, Noda Y, Mouri A, Nabeshima T, Kato T. Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Mol Psychiatry. 2006;11:577–593. doi: 10.1038/sj.mp.4001824. [DOI] [PubMed] [Google Scholar]
- Kato T, Kato N. Mitochondrial dysfunction in bipolar disorder. Bipolar Disord. 2000;2:180–190. doi: 10.1034/j.1399-5618.2000.020305.x. [DOI] [PubMed] [Google Scholar]
- Kato T, Ishiwata M, Mori K, Washizuka S, Tajima O, Akiyama T, Kato N. Mechanisms of altered Ca2+ signalling in transformed lymphoblastoid cells from patients with bipolar disorder. Int J Neuropsychopharmacol. 2003;6:379–389. doi: 10.1017/S1461145703003717. [DOI] [PubMed] [Google Scholar]
- Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, Keranen S, Peltonen L, Suomalainen A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- Kim MH, Korogod N, Schneggenburger R, Ho WK, Lee SH. Interplay between Na+/Ca2+ exchangers and mitochondria in Ca2+ clearance at the calyx of Held. J Neurosci. 2005;25:6057–6065. doi: 10.1523/JNEUROSCI.0454-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota M, Murakoshi T, Saegusa H, Kazuno A, Zong S, Hu Q, Noda T, Tanabe T. Intact LTP and fear memory but impaired spatial memory in mice lacking CaV2.3 (α1E) channel. Biochem Biophys Res Commun. 2001;282:242–248. doi: 10.1006/bbrc.2001.4572. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Kusumi I, Koyama T, Yamashita I. Serotonin-induced platelet intracellular calcium mobilization in depressed patients. Psychopharmacology. 1994;113:322–327. doi: 10.1007/BF02245204. [DOI] [PubMed] [Google Scholar]
- Larsson NG, Clayton DA. Molecular genetic aspects of human mitochondrial disorders. Annu Rev Genet. 1995;29:151–178. doi: 10.1146/annurev.ge.29.120195.001055. [DOI] [PubMed] [Google Scholar]
- Levy M, Faas GC, Saggau P, Craigen WJ, Sweatt JD. Mitochondrial regulation of synaptic plasticity in the hippocampus. J Biol Chem. 2003;278:17727–17734. doi: 10.1074/jbc.M212878200. [DOI] [PubMed] [Google Scholar]
- Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L, Majamaa K, Somer H, Suomalainen A. Parkinsonism, premature menopause, and mitochondrial DNA polymerase γ mutations: clinical and molecular genetic study. Lancet. 2004;364:875–882. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- Malli R, Frieden M, Osibow K, Graier WF. Mitochondria efficiently buffer subplasmalemmal Ca2+ elevation during agonist stimulation. J Biol Chem. 2003;278:10807–10815. doi: 10.1074/jbc.M212971200. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Friberg H, Hansson M, Elmér E, Wieloch T. Flow cytometric analysis of mitochondria from CA1 and CA3 regions of rat hippocampus reveals differences in permeability transition pore activation. J Neurochem. 2003;87:532–544. doi: 10.1046/j.1471-4159.2003.02026.x. [DOI] [PubMed] [Google Scholar]
- Missiaen L, Taylor CW, Berridge MJ. Spontaneous calcium release from inositol trisphosphate-sensitive calcium stores. Nature. 1991;352:241–244. doi: 10.1038/352241a0. [DOI] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Albillos A, García-Sancho J, Alvarez J. Mitochondrial Ca2+-induced Ca2+ release mediated by the Ca2+ uniporter. Mol Biol Cell. 2001;12:63–71. doi: 10.1091/mbc.12.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes CT. What regulates mitochondrial DNA copy number in animal cells? Trends Genet. 2001;17:199–205. doi: 10.1016/s0168-9525(01)02238-7. [DOI] [PubMed] [Google Scholar]
- Mott JL, Zhang D, Stevens M, Chang S, Denniger G, Zassenhaus HP. Oxidative stress is not an obligate mediator of disease provoked by mitochondrial DNA mutations. Mutat Res. 2001;474:35–45. doi: 10.1016/s0027-5107(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Moudy AM, Handran SD, Goldberg MP, Ruffin N, Karl I, Kranz-Eble P, DeVivo DC, Rothman SM. Abnormal calcium homeostasis and mitochondrial polarization in a human encephalomyopathy. Proc Natl Acad Sci USA. 1995;92:729–733. doi: 10.1073/pnas.92.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara JG, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y, Kagaya A, Shinno H, Motohashi N, Yamawaki S. Serotonin-induced platelet calcium mobilization is enhanced in mania. Life Sci. 1995;56:327–332. doi: 10.1016/0024-3205(94)00955-4. [DOI] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Peng YY. Effects of mitochondrion on calcium transients at intact presynaptic terminals depend on frequency of nerve firing. J Neurophysiol. 1998;80:186–195. doi: 10.1152/jn.1998.80.1.186. [DOI] [PubMed] [Google Scholar]
- Pivovarova NB, Pozzo-Miller LD, Hongpaisan J, Andrews SB. Correlated calcium uptake and release by mitochondria and endoplasmic reticulum of CA3 hippocampal dendrites after afferent synaptic stimulation. J Neurosci. 2002;22:10653–10661. doi: 10.1523/JNEUROSCI.22-24-10653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB. Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J Neurosci. 2004;24:5611–5622. doi: 10.1523/JNEUROSCI.0531-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Sah P. Nuclear calcium signaling evoked by cholinergic stimulation in hippocampal CA1 pyramidal neurons. J Neurosci. 2002;22:3454–3462. doi: 10.1523/JNEUROSCI.22-09-03454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzan T, Rizzuto R. High tide of calcium in mitochondria. Nat Cell Biol. 2000;2:E25–E27. doi: 10.1038/35000095. [DOI] [PubMed] [Google Scholar]
- Rae MG, Martin DJ, Collingridge GL, Irving AJ. Role of Ca2+ stores in metabotropic l-glutamate receptor-mediated supralinear Ca2+ signaling in rat hippocampal neurons. J Neurosci. 2000;20:8628–8636. doi: 10.1523/JNEUROSCI.20-23-08628.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Duchen MR, Pozzan T. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE. 2004 doi: 10.1126/stke.2152004re1. 2004:re1. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Mitochondrial involvement in Parkinson's disease, Huntington's disease, hereditary spastic paraplegia and Friedreich's ataxia. Biochim Biophys Acta. 1999;1410:159–170. doi: 10.1016/s0005-2728(98)00164-9. [DOI] [PubMed] [Google Scholar]
- Schild L, Keilhoff G, Augustin W, Reiser G, Striggow F. Distinct Ca2+ thresholds determine cytochrome c release or permeability transition pore opening in brain mitochondria. FASEB J. 2001;15:565–567. doi: 10.1096/fj.00-0551fje. [DOI] [PubMed] [Google Scholar]
- Sheehan JP, Swerdlow RH, Parker WD, Miller SW, Davis RE, Tuttle JB. Altered calcium homeostasis in cells transformed by mitochondria from individuals with Parkinson's disease. J Neurochem. 1997a;68:1221–1233. doi: 10.1046/j.1471-4159.1997.68031221.x. [DOI] [PubMed] [Google Scholar]
- Sheehan JP, Swerdlow RH, Miller SW, Davis RE, Parks JK, Parker WD, Tuttle JB. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer's disease. J Neurosci. 1997b;17:4612–4622. doi: 10.1523/JNEUROSCI.17-12-04612.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M, Nishida A, Yamawaki S. Forskolin and phorbol myristate acetate inhibit intracellular Ca2+ mobilization induced by amitriptyline and bradykinin in rat frontocortical neurons. J Neurochem. 1993;61:1748–1754. doi: 10.1111/j.1471-4159.1993.tb09812.x. [DOI] [PubMed] [Google Scholar]
- Shirasaki T, Harata N, Akaike N. Metabotropic glutamate response in acutely dissociated hippocampal CA1 pyramidal neurones of the rat. J Physiol (Lond) 1994;475:439–453. doi: 10.1113/jphysiol.1994.sp020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR. Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J Neurochem. 1990;55:698–707. doi: 10.1111/j.1471-4159.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- Snyder SH, Supattapone S. Isolation and functional characterization of an inositol trisphosphate receptor from brain. Cell Calcium. 1989;10:337–342. doi: 10.1016/0143-4160(89)90059-6. [DOI] [PubMed] [Google Scholar]
- Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L, Santoro L, Toscano A, Fabrizi GM, Somer H, Croxen R, Beeson D, Poulton J, Suomalainen A, Jacobs HT, Zeviani M, Larsson C. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- Suomalainen A, Majander A, Haltia M, Somer H, Lonnqvist J, Savontaus ML, Peltonen L. Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J Clin Invest. 1992;90:61–66. doi: 10.1172/JCI115856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozzi A, Bengtson CP, Longone P, Carignani C, Fusco FR, Bernardi G, Mercuri NB. Involvement of transient receptor potential-like channels in responses to mGluR-I activation in midbrain dopamine neurons. Eur J Neurosci. 2003;18:2133–2145. doi: 10.1046/j.1460-9568.2003.02936.x. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, Spelbrink JN, Paetau A, Suomalainen A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci USA. 2005;102:17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goethem G, Dermaut B, Löfgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- Wagner LE II, Li WH, Yule DI. Phosphorylation of type-1 inositol 1,4,5-trisphosphate receptors by cyclic nucleotide-dependent protein kinases: a mutational analysis of the functionally important sites in the S2+ and S2− splice variants. J Biol Chem. 2003;278:45811–45817. doi: 10.1074/jbc.M306270200. [DOI] [PubMed] [Google Scholar]
- Wanaverbecq N, Marsh SJ, Al-Qatari M, Brown DA. The plasma membrane calcium-ATPase as a major mechanism for intracellular calcium regulation in neurones from the rat superior cervical ganglion. J Physiol (Lond) 2003;550:83–101. doi: 10.1113/jphysiol.2002.035782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Hashimoto K, Isomura Y, Shimohama S, Kato N. An IP3-assisted form of Ca2+-induced Ca2+ release in neocortical neurons. NeuroReport. 2000;11:535–539. doi: 10.1097/00001756-200002280-00022. [DOI] [PubMed] [Google Scholar]
- Yamawaki S, Kagaya A, Okamoto Y, Shimizu M, Nishida A, Uchitomi Y. Enhanced calcium response to serotonin in platelets from patients with affective disorders. J Psychiatry Neurosci. 1996;21:321–324. [PMC free article] [PubMed] [Google Scholar]
- Yamawaki S, Kagaya A, Tawara Y, Inagaki M. Intracellular calcium signaling systems in the pathophysiology of affective disorders. Life Sci. 1998;62:1665–1670. doi: 10.1016/s0024-3205(98)00125-8. [DOI] [PubMed] [Google Scholar]
- Zhang D, Mott JL, Chang SW, Denniger G, Feng Z, Zassenhaus HP. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics. 2000;69:151–161. doi: 10.1006/geno.2000.6333. [DOI] [PubMed] [Google Scholar]
- Zhang D, Mott JL, Farrar P, Ryerse JS, Chang SW, Stevens M, Denniger G, Zassenhaus HP. Mitochondrial DNA mutations activate the mitochondrial apoptotic pathway and cause dilated cardiomyopathy. Cardiovasc Res. 2003;57:147–157. doi: 10.1016/s0008-6363(02)00695-8. [DOI] [PubMed] [Google Scholar]
- Zhang H, Barcelo JM, Lee B, Kohlhagen G, Zimonjic DB, Popescu NC, Pommier Y. Human mitochondrial topoisomerase I. Proc Natl Acad Sci USA. 2001;98:10608–10613. doi: 10.1073/pnas.191321998. [DOI] [PMC free article] [PubMed] [Google Scholar]