

SUMMARY
Adipose tissue fibrosis is associated with inflammation and insulin resistance in human obesity. In particular, visceral fat fibrosis is correlated with hyperlipidemia and ectopic fat accumulation. Myocardin-related transcription factor A (MRTFA) is an important coactivator that mediates the transcription of extracellular matrix and other fibrogenic genes. Here, we examine the role of MRTFA in the development of adipose tissue fibrosis and identify a signaling pathway that regulates the fate of vascular progenitors. We demonstrate that obesity induces the formation of Sca1−, Sma+, ITGA5+ fibrogenic progenitor cells (FPCs) in adipose tissue. MRTFA deficiency in mice shifts the fate of perivascular progenitors from FPCs to adipocyte precursor cells and protects against chronic obesity-induced fibrosis and accompanying metabolic dysfunction, without a shift in energy expenditure. Our findings highlight the ITGA5-MRTFA pathway as a potential target to ameliorate obesity-associated metabolic disease.
In Brief
Identification of signaling pathways regulating obesity-induced adipose tissue fibrosis has therapeutic potential. Lin et al. show that myocardin-related transcription factor A (MRTFA) drives the recruitment of fibrogenic progenitor cells (FPCs) at the expense of healthy adipocytes. MRTFA-deficient mice are protected from chronic diet-induced obesity and fibrosis without a change in energy expenditure.
INTRODUCTION
The obesity epidemic is increasing the prevalence of metabolic diseases such as diabetes, cardiovascular disease, and hyperlipidemia. Despite considerable effort, current therapies do not address the underlying cause of metabolic dysfunction. Excessive caloric intake leads to increased triglycerides storage in white adipose tissue (WAT) and fat expansion. In particular, high visceral adiposity is associated with an increased risk for insulin resistance and fatty liver disease (Guglielmi et al., 2015; Lafontan and Berlan, 2003; Wajchenberg, 2000). WAT undergoes dynamic extracellular matrix (ECM) remodeling, expanding and contracting in response to caloric excess and deprivation (Sun et al., 2011). In an obese state of persistent nutrient overload, however, chronic inflammation and tissue injury disrupt remodeling and lead to extracellular matrix dysfunction (Sun et al., 2013). Recent studies of adipose tissue from obese individuals link increased extracellular matrix deposition with insulin resistance (Divoux et al., 2010; Spencer et al., 2011). Fibrosis in visceral fat is negatively associated with serum lipid profiles, while fibrosis in subcutaneous fat is correlated with less weight loss post-bariatric surgery (Bel Lassen et al., 2017; Liu et al., 2016). Thus, the modulation of extracellular matrix remodeling may represent strategies to treat obesity-associated insulin resistance.
Myofibroblasts are considered to be the dominant collagen secreting cells during wound healing and in various types of organ fibrosis (Hinz et al., 2007). Recent data suggest that myofibroblasts originate from perivascular progenitors, which have multipotent capacity to differentiate into chondrocytes, osteoblasts, and adipocytes (Kramann et al., 2015). Furthermore, in human obese adipose tissue, PDGFRα+ adipocyte precursor cells (APCs) exhibit profibrotic myofibroblast markers (Marcelin et al., 2017), suggesting that a shift in progenitor cell differentiation to myofibroblasts rather than adipocytes may occur in fibrosis. Thus, the potential to direct progenitors from a myofibroblast fate and favor adipocyte differentiation is an attractive proposition to oppose fibrosis and enhance hyperplasia. Myocardin-related transcription factor A (MRTFA) is a known regulator of myofibroblast differentiation that drives the expression of cytoskeleton and extracellular matrix genes, including α-smooth muscle actin (Sma) and collagens (Luchsinger et al., 2011; Velasquez et al., 2013). Additionally, TGFβ and mechanical stress have been shown to induce myofibroblast differentiation through the activation of MRTFA (Crider et al., 2011). Studies by others and us have shown that MRTFA inhibition enhances white, beige, and bone marrow adipocyte differentiation (Bian et al., 2016; McDonald et al., 2015; Nobusue et al., 2014). Therefore, we hypothesize that MRTFA deficiency may oppose adipose tissue fibrosis and increase adipocyte hyperplasia.
Our previous studies showed that MRTFA deficiency caused postnatal browning of WAT and prevented short-term (6 weeks) HFD-induced weight gain and glucose intolerance through enhancement of energy expenditure. In the present study, we investigate the role of MRTFA in chronic (20 weeks) HFD-induced metabolic dysfunction and adipose tissue fibrosis. We demonstrate that the loss of MRTFA protects against chronic obesity-induced fibrosis, adipose tissue dysfunction, and metabolic comorbidities, such as hyperlipidemia and insulin resistance, without a change in energy balance. The studies also identify ITGA5 as a surface marker of profibrotic perivascular progenitors. We show that MRTFA directs adipose stromal vascular cell fate toward Sca1−, ITGA5+, Sma+ FPCs rather than Sca1+, CD34+, CD29+, PDGFRα+ APCs. Our findings suggest that integrin interactions communicate extracellular mechanical cues to direct perivascular progenitor cell fate in a MRTFA-dependent manner. Moreover, activation of the MRTFA-integrin axis is an important component of adipose tissue fibrosis under obesogenic conditions. We suggest that inhibition of MRTFA activity protects against adipose dysfunction by reducing obesity-associated fibrosis and inflammation.
RESULTS
MRTFA-Deficient Mice Are Protected from Chronic Obesity-Associated Insulin Resistance and Hepatic Steatosis
Obesity-associated insulin resistance is caused by ectopic fat accumulation in non-adipose tissues (Kim et al., 2007). We previously found that MRTFA-knockout (KO) mice are protected from obesity-associated glucose intolerance following a short-term (6 weeks) HFD (McDonald et al., 2015). The absence of MRTFA led to WAT browning, resulting in an enhancement of energy expenditure and consequently a reduction in adipose expansion and weight gain. Chronic obesity, however, can lead to more drastic perturbation of metabolic homeostasis due to extensive adipose inflammation and fibrosis. To evaluate the role of MRTFA in chronic compared with early-onset obesity-induced maladies, we challenged mice with a HFD for 8 and 20 weeks. MRTFA-KO and their wild-type (WT) littermates gained similar amounts of body weight, even though KO mice were smaller than littermate controls prior to HFD feeding, and this weight difference remained throughout (Figures 1A and S1). The slight stature of MRTFA-KO mice is due to a reduction in both fat and lean mass (Figure S1), which can be attributed to the osteopenia previously described (Bian et al., 2016). HFD feeding induced rapid weight gain in WT and KO mice for 8 weeks, after which mice continued to gain weight slowly until plateauing at 16 weeks. Fat depots were smaller in MRTFA-KO mice at 8 weeks, but no significant difference in size was observed at 20 weeks (Figures 1B and 1C). Interestingly, histological examination of adipose tissue sections at 20 weeks showed decreased cell size in MRTFA-KO mice (Figure 1D). Adipocyte cell size assessment showed smaller adipocytes with a leftward shift of the Gaussian distribution in the KO mice (Figures 1E and 1F). The presence of smaller adipocytes appeared not to result from selective hyperplasia in KO mice, at least when measured at 8 weeks of the diet (Figure S1). Liver weights were similar at 8 weeks, but liver weight in WT mice was increased from chronic HFD, while no change was observed in the liver of KO mice. In concordance with decreased liver size, H&E staining showed significantly reduced lipid accumulation in KO mice (Figures 1D and 1G), suggesting that liver from MRTFA-KO mice is protected from diet-induced steatosis. Moreover, whereas no difference in triglyceride level was observed after 8 weeks of HFD feeding, MRTFA-KO mice showed a significant improvement of the triglyceride profile after 20 weeks under the diet compared with WT mice (Figure 1H). MRTFA-KO mice display reduced glucose and insulin levels at 8 and 20 weeks of the diet (Figures 2A and 2B), but glucose tolerance was only improved by absence of MRTFA at the early stage of obesity (Figures 2C and S2); MRTFA deficiency did, however, protect the mice from chronic (20 weeks) diet-induced insulin resistance (Figure 2D). Despite a decrease in insulin secretion, circulating adiponectin levels remain elevated, promoting insulin sensitivity (Figures 2E and S2). Previous studies showed that the improved glucose tolerance in young MRTFA-KO mice was a result of beige adipocytes in inguinal WAT (ingWAT) and enhanced energy expenditure (McDonald et al., 2015). However, the improvements in insulin sensitivity after 20 weeks of HFD cannot be attributed to these processes, as all the parameters, including VO2, VCO2, heat, and respiratory exchange ratio (RER), indicative of enhanced energy expenditure as well as physical activity are equivalent in KO and WT mice (Figures 2F–2K). There was also no difference in food intake (Figure 2L). It is also relevant that the extensive browning of WAT observed during the initial 8–12 weeks of age in KO mice is not retained following a 20 week HFD, as there is no difference in brown adipose gene expression, including Ucp1, Pgc1α, Cidea, Cox7a1, and Cox8b1 (Figure S2) between WT and KO mice. These data demonstrate that mice lacking MRTFA are healthier even after a chronic HFD preventing ectopic fat accumulation and insulin resistance that cannot be explained by enhanced energy expenditure.
Figure 1. MRTFA-Deficient Mice Are Protected from Chronic Obesity-Induced Adipo-cyte Hypertrophy and Hepatic Steatosis.

(A) Cumulative weight gain of 6-week-old WT and MRTFA-KO male mice fed a diet of 60% kcal from fat (high-fat diet) over 20 weeks (n = 7–8).
(B and C) Weights of epididymal (B) and inguinal (C) fat pads at 8 (n = 6) and 20 (n = 7–8) weeks of high-fat diet.
(D) Representative H&E staining sections of epi-WAT, ingWAT, and liver from WT and MRTFA−/− male mice fed a high-fat diet for 20 weeks (scale bar, 100 μm; n = 7–8 per group).
(E and F) Frequency of distribution of epididymal (E) and inguinal (F) adipocyte cell size from >100 randomly selected adipocytes measured from histological sections of each depot (n = 6). (G) Liver weights at 8 (n = 6) and 20 (n = 6–7) weeks of high-fat diet.
(H) Serum triglycerides of WT and MRTFA-KO mice after 20 weeks of high-fat diet (n = 7–8). Data are presented as mean ± SEM. *p < 0.05.
Figure 2. Chronically Obese MRTFA-KO Mice Exhibit Improved Insulin Sensitivity despite Unchanged Energy Expenditure.

(A and B) Fasting blood glucose (A) and fasting insulin (B) levels of WT and MTRFA-KO mice at 8 (n = 5 or 6) and 20 (n = 7–8) weeks of high-fat diet.
(C and D) Glucose (n = 5–6) (C) and insulin (n = 4–5) (D) tolerance tests were performed after 20 weeks of high-fat diet. GTT, glucose tolerance test; ITT, insulin tolerance test.
(E) Serum adiponectin of WT and MRTFA-KO mice after 20 weeks of high-fat diet (n = 7–8).
(F–L) Whole-body energy expenditure of WT and MTRFA-KO mice fed a high-fat diet for 20 weeks (n = 4). (F) Average day-night oxygen consumption, (E) O2 consumption, (H) CO2 production, (I) heat, (J) respiratory exchange ratio, (K) locomotor activity, and (L) daily food intake.
Data are presented as mean ± SEM. *p < 0.05 and **p < 0.01.
MRTFA-KO Mice Are Resistant to Adipose Tissue Fibrosis and Inflammation
Because MRTFA is a major regulator of fibrosis in various tissues including the skin, lung, heart, and eye (Shiwen et al., 2015; Sisson et al., 2015; Small et al., 2010; Yu-Wai-Man et al., 2014), we investigated whether its deficiency also protects adipose tissue from chronic diet-induced fibrosis and associated inflammation. Collagen fiber accumulation, also referred to as pericellular fibrosis, was visualized using both Masson’s trichrome and picrosirius red staining. After 20 weeks of HFD feeding, we observed extracellular collagen surrounding adipocytes in WT epididymal WAT (epiWAT), while almost no collagen stain is visible in KO epiWAT (Figure 3A). Furthermore, collagen levels measured by hydroxyproline showed a reduction in KO epiWAT (Figure 3B). In agreement with human studies showing a tight association between macrophage infiltration and extracellular matrix accumulation, less F4/80 staining was observed in KO epi-WAT (Figure 3A). In parallel, expression of pro-inflammatory markers including MCP-1, TNFα, and IL-10 was reduced in adipose tissue from MRTFA-KO mice (Figure 3C). Consistent with the reduced fibrogenesis, lower gene expression of fibrotic markers including Sma, Col1a1, Col3a1, Col6a3, and Mmp2 was observed in KO epiWAT at 8 weeks of HFD (Figure 3D). Extending the diet out to 20 weeks resulted in elevated gene expression of Col3a1, Col4a1, Col6a3, Mmp2, and Timp1 in WT mice, whereas the MRTFA-KO mice maintained a lower expression of these profibrotic genes (Figure 3D). On the other hand, no difference in fibrotic and pro-inflammatory gene expression was found in inguinal fat depot at 8 and 20 weeks (Figure S3). Additionally, Sma and Col1 protein levels were significantly diminished in MRTFA-KO mice after 20 weeks of HFD feeding (Figure 3E). Immunofluorescence showed Sma+ cells surrounding dead, perilipin-negative cells, suggesting that myofibroblasts migrate and surround dead cells (Figures 3F and S3). Additionally, using a transgenic GFP reporter of Col1a1 gene transcription, collagen-producing cells were significantly reduced in KO epiWAT (Figure 3G). Relatedly, the decreased fibrosis is accompanied with decreased Tgfβ1 and increased Adipoq expression suggesting that MRTFA deficiency maintains a healthier adipose tissue homeostasis (Figures 3H and 3I). These data demonstrate that the absence of MRTFA protects against chronic diet-induced adipose tissue fibrosis and inflammation.
Figure 3. MRTFA-KO Mice Are Resistant to Chronic Obesity-Induced Adipose Tissue Fibrosis and Inflammation.

(A) Representative images of Masson’s trichrome, picrosirius red, and F4/80 staining (magenta) in epiWAT at 20 weeks of high-fat diet (n = 6). (B) Quantification of collagen accumulation via hydroxyproline content (n = 4).
(C) Relative expression by qRT-PCR of inflammatory genes from epiWAT of WT and MRTFA−/− male mice (n = 6–7).
(D) qRT-PCR relative expression of relevant profibrotic genes involved in myofibroblast differentiation and extracellular matrix production from epiWAT of WT and KO mice fed a high-fat diet for 8 (n = 5–6) and 20 (n = 7–8) weeks.
(E) Representative western blot of Sma and Col1 protein expression from epiWAT of 20 week high-fat diet fed mice (n = 4). (F and G) Immunofluorescence images of perilipin (F), Sma, and GFP (G) in epiWAT (n = 6).
(H and I) Real-time qPCR of Tgfβ1 (H) and Adipoq (I) expression (n = 6–7). Data are presented as mean ± SEM. *p < 0.05 and **p < 0.01.
MRTFA Deficiency Decreases ITGA5+ FPCs and Enhances the APC Population
Obesity-induced adipose fibrosis has been proposed to result from hypoxia-induced expression of extracellular matrix proteins by mature white adipocytes as well as profibrogenic activation of stromal progenitors. To identify which adipose tissue compartment is responsible for the extensive increase in fibrotic gene expression (see Figure 3D), we first analyzed mRNAs from adipocytes versus stromal vascular (SV) cells in WAT of mice fed a low-fat diet (LFD) and a HFD. The data demonstrate that SV cells is the principal contributor to obesity-induced fibrosis (Figure S4). Myofibroblasts have been shown to be principal cells contributing to fibrosis in other tissues, such as skin and lung (Hinz et al., 2007), which are recruited and activated from vascular progenitors in response to profibrogenic factors. TGFβ is one such factor that signals through several pathways, including SMADs, MAPKs, and MRTFA to regulate expression of extracellular matrix and contractile proteins. We postulate that MRTFA participates in adipose fibrosis by regulating key determinants of vascular cell fate to myofibroblasts. To identify these determinants, we first investigated myofibroblast differentiation in WT and MRTFA-KO epididymal stromal vascular (epiSV) cells via longitudinal expression analysis of select fibrogenic genes during TGFβ treatment (Figure 4A). Interestingly, the gene expression patterns varied significantly from each other, likely because of the stimulation of different signaling pathways by the TGFβ receptor as mentioned above. Sma and Itga5 expression was induced at days 4 and 8 by TGFβ in WT cells but reduced in MRTFA-null cells, while Col1a1 and CD9 expression was induced in both WT and KO cells. On the other hand, TGFβ repressed Col6a3 gene expression, which was further reduced in MRTFA-null cells. Importantly, Itga5 and Sma appear to be coordinately regulated by MRTFA and TGFβ and are therefore appropriate candidates for markers of MRTFA-associated stromal vascular cell fate decisions during fibrosis. In addition to myofibroblasts, Sma has been shown to be expressed in vascular cells, which give rise to APCs that are identified as Lin−, CD29+, CD34+, PDGFRα+, Sca1+ cells. Our studies (not shown) have determined that greater than 90% of the APC population express Sca1. To identify fibrogenic progenitors, we sorted Sma+ cells on the basis of mCherry fluorescence from WT and KO mice expressing mCherry under the control of the Sma promoter. Flow cytometry of epiSV cells from chronically obese (20 weeks of HFD) WT and MRTFA-KO mice showed a significant decrease in Sca1−, Sma+ cells in KO epiWAT (Figures 4B, 4C, and S4). Interestingly, MRTFA deficiency led to a corresponding increase in the Sca1+, Sma+ population (Figures 4C and S4). Further analysis demonstrated that this increase was likely due to an enhancement of the APC population (Lin−, CD29+, CD34+, PDGFRα+, Sca1+) in MRTFA-KO epiSV cells (Figures 4D, 4E, and S4). The fact that ITGA5 is coordinately expressed with Sma, we also analyzed the distribution of cells expressing this integrin. Fluorescence-activated cell sorting (FACS) analysis showed that Sca1−, ITGA5-expressing cells were lower in MRTFA-KO mice than in WT mice (Figures 4F, 4G, and S4). Interestingly, feeding mice a 20-week HFD significantly enhanced the population of Sca1−, ITGA5+ cells (Figures 4H and 4I). On the basis of these data, we suggest that Sca1−, Sma+, ITGA5+ cells are profibrotic and may arise from APCs or their progenitors. Furthermore, ITGA5+ cells express PDGFRα, which has been described as a pericyte marker as well as a mediator of tissue fibrosis (Figure S4).
Figure 4. MRTFA Deficiency Decreases ITGA5+ FPCs and Enhances APC Population.

(A) Real-time qPCR of fibrogenic genes of WT and MRTFA epiSV cells treated with TGFβ at days 0, 2, 4, and 8 (n = 3).
(B and C) Flow cytometry of Sca1 and Sma expressing cells from WT and MRTFA-KO mice fed a high-fat diet for 20 weeks. (B) Gating strategy of CD45−, CD31−, Sma+, Sca1+, and Sca1− cells. (C) Frequency of Sca1−, Sma+ and Sca1+, Sma+ cells (n = 4).
(D and E) Flow cytometry of epiSV cells from WT and MRTFA-KO mice fed a high-fat diet for 20 weeks. (D) Gating strategy of APCs, CD45−, CD31−, CD34+, CD29+, Sca1+, PDGFRa+ cells. (E) Frequency of APCs in WT and KO epiWAT (n = 4).
(F and G) Flow cytometry of ITGA5 expression in Sca1−, Sma+ cells from WT and MRTFA-KO mice fed a high-fat diet for 20 weeks. (F) Gating strategy of CD45−, CD31−, Sca1−, Sma+, ITGA5+ cells. (G) Frequency of Sca1−, Sma+, ITGA5+ cells in WT and KO epiWAT (n = 5).
(H and I) Flow cytometry of epiSV cells from mice fed LFD or high-fat diet for 20 weeks. (H) Gating strategy of Sca1−, ITGA5+ and Sca1+, ITGA5+ cells from epiWAT. (I) Frequency of Lin−, Sca1−, ITGA5+ and Lin−, Sca1+, ITGA5+ cells (n = 4).
Sca1− but Not Sca1+ Cells Respond to TGFβ and Undergo Differentiation into Myofibroblasts
To gain insight into the phenotype of Sca1+ APCs versus Sca1− cells, we isolated them from SV cells of mice fed a HFD using magnetic-activated cell sorting (MACS) (Figure 5A) or FACS (Figure 5B) and analyzed expression of potential regulators of vascular cell fate. Genes generally associated with fibrogenesis, including Sma, Itga5, Tgfb, Gli1, Pdgfra, and Pdgfrb, were much more highly expressed in Sca1− cells compared with Sca1+ cells. Interestingly, CD9, a gene recently identified as a marker of FPCs (Marcelin et al., 2017), is equally expressed in Sca1− and Sca1+ cells (Figure 5A). To determine whether absence of MRTFA influences the expression of these genes, we analyzed Sca1− and Sca1+ cells from WT and KO epiWAT. MRTFA deficiency resulted in a significantly lower expression of Itga5 and its ligand fibronectin (Fn1) in Sca1− compared with Sca1+ cells. The other genes, including Sma, Pdgfra, CD9, Gli1, and Pref1, were unaffected by MRTFA deficiency (Figure 5C). To gain further insight into the phenotype of Sca1− and Sca1+ cells, we treated each population with TGFβ and analyzed expression of select target genes (Figures 5D and 5E). The majority of the fibrogenic genes (Itga5, Itgb1, Pdgfra, Sma, and Col6a3,) were unresponsive to TGFβ in Sca1+ cells compared with their dramatic induction in Sca1− cells. CD9 expression did not respond to TGFβ in either Sca1+ or Sca1− cells. Of interest is the observation that TGFβ suppresses expression of Sca1 and CD34 in Sca1+ cells, suggesting that obesity-associated inflammation (elevated TGFβ) might convert adipocyte precursor cells to a profibrogenic lineage.
Figure 5. Sca1− but Not Sca1+ Cells Respond to TGFβ and Undergo Differentiation into Fibrogenic Cells.

(A) Expression of profibrotic markers from MACS-isolated Sca1+ and Sca1− cells of mice fed a HFD for 16 weeks (n = 6).
(B) Expression of profibrotic markers of CD45−, Sca1+, Sma+ and CD45−, Sca1−, Sma+ cells isolated by FACS of SV cells from lean epiWAT of transgenic Sma-mCherry mice (pooled from three animals).
(C) qRT-PCR relative expression of fibrogenic markers in the Sca1+ and Sca1− cells isolated from epiWAT of WT and MRTFA-KO mice fed a HFD for 20 weeks using MACS (n = 4–5).
(D and E) Sca1+ (D) and Sca1− (E) cells isolated from epiWAT as in (C) were treated with or without TGFβ (1 ng/mL) for 8 days. qRT-PCR gene expression analysis of fibrotic markers (n = 4).
Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
DISCUSSION
Currently available remedies for obesity and its associated metabolic diseases, including lifestyle modification, have proved ineffective, highlighting the need for new therapeutic approaches (Fildes et al., 2015). Recent studies linked adipose tissue fibrosis with insulin resistance and hyperlipidemia, suggesting that antifibrotic agents have a strong potential for metabolic disease treatment. Our work leads to an understanding of signaling pathways that affect adipose tissue fibrosis. Here, we report that absence of MRTFA protects against chronic HFD-induced fibrosis, inflammation, insulin insensitivity, and ectopic lipid deposition without a change in energy expenditure. The studies have also identified ITGA5 as a surface marker of FPCs. We demonstrate that obesity induces the recruitment of perivascular, profibrogenic Sma+, ITGA5+, Sca1− cells in adipose tissue, which is attenuated in MRTFA-deficient mice. We propose that ITGA5-MRTFA signaling favors the formation of FPCs rather than APCs in obese adipose tissue. Inhibition of the ITGA5-MRTFA pathway may have potential to treat obesity-associated maladies by opposing myofibroblast differentiation and enhancing adipocyte precursor cell recruitment. Moreover, we demonstrate that myofibroblast recruitment plays a pivotal role in the development of adipose tissue fibrosis upon chronic high fat feeding. Attenuation of this process through suppression of MRTFA signaling prevents extracellular matrix deposition, macrophage infiltration, and inflammation. This conclusion is in accordance with mechanisms regulating fibrosis in other tissues, whereby perivascular progenitors have been shown to differentiate into myofibroblasts and become the major cellular source of fibrogenic proteins (Kramann et al., 2015).
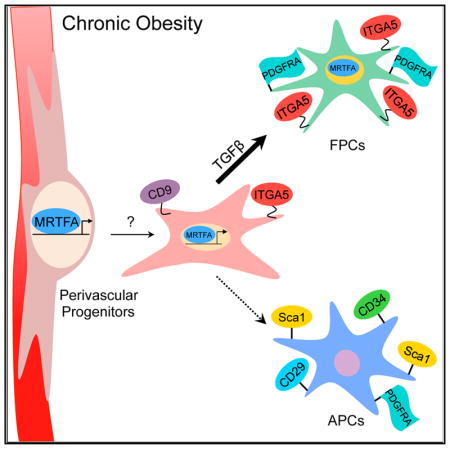
Our observations are also in agreement with recent studies by Clement and colleagues, who demonstrated that obesity induces formation of FPCs from PDGFRα+, CD9+ adipose tissue progenitors (Marcelin et al., 2017). Scherer and colleagues presented a different model for adipose fibrosis in which hypoxic adipocytes in obese adipose tissue secrete collagen VI, which is proteolytically cleaved to produce endotrophin (Khan et al., 2009). They further showed that overexpression of endotrophin in adipocytes stimulates fibrosis and exacerbates inflammation induced by a HFD. Regardless of the cellular origin(s) of fibrogenic proteins, it is likely that macrophage-mediated inflammation and TGFβ secretion are critical components of adipose fibrosis (Vila et al., 2014). Supporting our data that early myofibroblast activation and macrophage infiltration are key in fibrosis progression, time course transcriptional profiling found an early increase of collagen genes upon HFD feeding coincident with increases in expression of proinflammatory cytokines and chemokines (Kwon et al., 2012). Overall, these results suggest that HFD induces macrophage infiltration and secretion of TGFβ, which initiates recruitment of perivascular progenitors. Our present study showed that this process leads to MRTFA-dependent expression of ITGA5 and other adhesion proteins, promoting myofibroblast formation (Figure 6). This model does not exclude the possibility of myofibroblast activation through endotrophin stimulation of macrophages to release TGFβ (Zhao et al., 2016).
Figure 6. A Model Illustrating MRTFA-Associated Regulation of Perivascular Cell Fate.

Under chronic obesity conditions, MRTFA directs differentiation of CD9+ perivascular progenitors to ITGA5+ fibrogenic progenitor cells (FPCs) in response to fibrotic factors (TGFβ) at expense of adipocyte precursor cells.
The vasculature presents a niche of stem cells that possess self-renewal, clonogenic, and multipotent capacities. These perivascular progenitors are recruited in response to various stimuli and differentiate into the desired cell type (i.e., adipocytes and myofibroblasts) (Jiang et al., 2014; Kramann et al., 2015; Lee et al., 2013; Vishvanath et al., 2016). The extracellular matrix, which consists of fibronectin, collagens, and laminins, likely directs cell fate toward myofibroblasts as opposed to adipocytes. It has long been known that fibronectin inhibits adipogenesis via cytoskeletal rearrangements and alterations in cell morphology (Spiegelman and Ginty, 1983). Recent studies have shown that expression of integrins and regulation of adipose-derived stem cell proliferation and differentiation are coordinated (Liu et al., 2005; Morandi et al., 2016). Integrins are transmembrane receptors that sense the mechanical stiffness of the extracellular matrix and can alter cytoskeleton tension, leading to actin rearrangement and cell spreading, which reduces G-actin levels and activates MRTFA-SRF transcriptional activity. Thus, we hypothesize that the recruitment of progenitors from the perivascular niche requires vessel detachment and dynamic integrin signaling that modifies the actin cytoskeleton and cell morphology. Furthermore, as it has been demonstrated that APCs, in response to obesity and TGFβ, have suppressed adipogenesis and instead have a more profibrotic phenotype (Keophiphath et al., 2009; Marcelin et al., 2017), our data suggest that MRTFA is an important regulator that shifts the cell fate of perivascular progenitors between FPCs and APCs. It is also important to mention that the tetraspanin CD9, a recently identified marker of profibrotic progenitors in adipose tissue (Marcelin et al., 2017) can associate with RGD-integrins and regulate adhesion and migration in different cells (Leung et al., 2011; Yu et al., 2017). Therefore, CD9 and ITGA5 (CD49e) may function together to facilitate recruitment of FPCs from the vascular compartment. Unlike ITGA5, CD9 is not regulated by MRTFA, and its expression is suppressed by TGFβ during the differentiation of Sca1− cells to myofibroblasts.
Adipose tissue fibrosis is a progressive disease in which chronic low-grade inflammation stimulates the deposition of extracellular matrix. Treating fibrosis and the persistent inflammation that accompanies it in the obese state is especially challenging and leads to questions regarding the feasibility of preventing disease progression and reversing fibrosis (Henegar et al., 2008). The general approach to develop novel therapeutics for fibrosis has been to target individual receptors and pathways, such as LPA receptors and TGFβ with antagonists or neutralizing antibodies, respectively. However, because the majority of the signaling pathways ultimately activate Rho GTPase and downstream MRTF/SRF activity, therapeutic targeting of this downstream convergence point may have a higher likelihood of being efficacious. For example, the ROCK inhibitors fasudil and Y-27632 have been shown to attenuate pulmonary fibrosis in a bleomycin model (Shimizu et al., 2001; Zhou et al., 2013), and recently, an inhibitor of MRTFA/SRF transcriptional activity, CCG-1423, was identified in a luciferase transactivation screen (Evelyn et al., 2007). Importantly, a lead analog, CCG-203971, has been shown to resolve preexisting pulmonary fibrosis (Sisson et al., 2015). By targeting a downstream convergence point of multiple signaling pathways, MRTFA-ITGA5 inhibitors could represent effective antifibrotic therapeutics. In conclusion, treating adipose tissue fibrosis via the inhibition of MRTFA may be a method to decrease extracellular matrix accumulation as well as enhance progenitor recruitment for formation of healthy adipocytes.
EXPERIMENTAL PROCEDURES
Animals
C57Bl6 mice were purchased from The Jackson Laboratory, and MRTFA+/− mice were obtained from Dr. Eric Olsen (University of Texas Southwestern). Transgenic Sma-mCherry and Col1a1-GFP mice were obtained from Dr. David Rowe at the University of Connecticut. Mice were housed in a temperature-controlled environment with a 12 hr light-dark cycle and ad libitum water and standard chow diet. For diet studies, 6-week-old mice were fed a diet of 60% kcal from fat (HFD) or normal chow (10% kcal from fat) (Research Diets). For EdU studies, mice were fed a HFD for 8 weeks and then injected intraperitoneally (i.p.) with EdU (100 mg/kg/day) every 2 days for 2 weeks before euthanasia. All animal studies were approved by the Boston University School of Medicine Institutional Animal Care and Use Committee.
Body Composition and Indirect Calorimetry Measurements
For indirect calorimetry measurements, animals were individually housed in metabolic chambers maintained at 20°C to 22°C on a 12 hr light-dark cycle. Mice were provided the HFD mentioned above and water ad libitum. Mice were acclimated to Comprehensive Laboratory Animal Monitoring System (CLAMS) cages for 48 hr, and data were recorded every 18 min for a 48 hr period. Metabolic measurements (oxygen consumption, carbon dioxide production, respiratory exchange ratio, and locomotor activity) were obtained continuously using the Comprehensive Laboratory Animal Monitoring System consisting of an open-circuit calorimeter and motion detectors. For food intake measurements, mice were individually housed, and food was weighed each at the same time each day for 4 days. Food intake is expressed as grams of food per 30 g body weight. Body composition was assessed by noninvasive quantitative magnetic resonance spectroscopy (EchoMRI).
Glucose Tolerance Test and Insulin Tolerance Test
Mice were fasted for 6 hr with free access to water. For the glucose tolerance test, glucose was administered intraperitoneally (1 g/kg). For the insulin tolerance test, insulin was administered intraperitoneally (0.5 mU/g). Blood glucose was measured via the tail vein with a glucometer (Contour) just prior to injection and at 15, 30, 60, and 120 min after injection. For glucose-stimulated insulin secretion, blood was collected before glucose injection and 30 min after injection.
Analysis of Blood Parameters
Glucose was measured using a glucose meter. Plasma insulin and adiponectin levels were determined using an ELISA (Millipore). Plasma triglyceride levels were measured using an enzymatic colorimetric kit (Thermo Fisher Scientific).
SVF Cell Culture
Epididymal fat pads were isolated, minced, and digested in collagenase I and collagenase II (Worthington) for 40 min in 37°C. The digest was filtered through a 40 μm cell strainer and centrifuged at 500 × g for 10 min. The stromal vasculature fraction was resuspended in red blood cell lysis solution (0.154 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) for 3 min and then centrifuged at 500 × g for 10 min. SV cells were resuspended and cultured in DMEM containing 10% fetal bovine serum (FBS). For myofibroblast differentiation, cells were stimulated with human recombinant TGFβ1 (1 ng/mL) every 2 days. For adipogenic differentiation, cells were induced with dexamethasone (5 μM), isobutyl-methylxanthine (500 μM), and insulin (4 μg/mL) for 48 hr and then maintained with insulin for 6 days.
FACS Analysis
Freshly isolated epiSV cells were resuspended in FACS buffer (PBS containing 1% BSA). For adipocyte progenitors, anti-Sca1-BV395 (D7), anti-CD140a-PE-Cy7-A (APA5), anti-CD45.2-BV737 (104), anti-CD31 eF450 (390), anti-CD34-eF660 (RAM34), anti-CD29-PE-eF710 (eBioHMb1-1), and anti-Itga5 AF647 (5H10-27) were used. For EdU incorporation, isolated stromal vascular cells were processed with Click-iT Plus EdU Alexa Flour 647 flow cytometry assay kit (Life Technologies), according to the manufacturer’s instructions. Briefly, cells were first stained with cell-surface markers, next cells were fixed and per-meabilized, and last ethynyl moiety of EdU was covalently linked with picolyl azide coupled to Alexa Flour 647. Flow cytometry analysis was performed with LSRII (BD Biosciences) and FACS with Aria (BD Biosciences).
Real-Time PCR
Total RNA was extracted from frozen tissues and cells using TRIzol reagent according to the manufacturer’s instructions. RNA concentrations were determined on NanaDrop spectrophotometer. Total RNA (100 ng to 1 μg) was transcribed to cDNA using Maxima cDNA synthesis (Thermo Fisher Scientific). Quantitative real-time PCR was performed on ABI Via detection system, and relative mRNA levels were calculated using comparative threshold cycle (CT) method. SYBR green primers are listed in Table S1.
Western Blot
Adipose tissue was homogenized in lysis buffer (100 mM Tris HCl, 2.5 mM EDTA, 0.45% NP-40, 0.45% Tween 20) containing protease and phosphatase inhibitor (Thermo Fisher Scientific). Protein concentrations were quantified using BCA protein assay. Equivalent amounts of total protein were separated on SDS polyacrylamide gels and transferred to nitrocellulose membranes. After blocking, immunoblots were incubated with primary antibodies: rabbit anti-collagen I (600-401-103; Rockland), mouse anti-Sma (A2547; Sigma-Aldrich), and rabbit anti-cyclophilin A (07-313; Sigma-Aldrich). After washing, blots were incubated in horseradish peroxidase (HRP)-conjugated secondary antibodies anti-mouse, anti-rabbit, and anti-goat. Proteins were detected using a BioRad imager.
Collagen Content
Hydroxyproline measurement was performed with hydroxyproline colormetric assay (Sigma-Aldrich). According to the manufacturer’s instructions, frozen tissues were weighed and digested in 10N HCl at 100°C for 3 hr at 10 μL of HCl per milligram WAT. Ten microliters of supernatant was transferred to a 96-well plate and dried in a 60°C oven for 2 hr. Chloramine-T was used to oxidize the free hydroxyproline for the production of a pyrrole, and 4-(dimethylamino)benzaldehyde (DMAB) was added for colormetric quantification.
Histology
Tissue was fixed with paraformaldehyde, paraffin embedded, and sectioned (5 μm). Fibrosis was assayed using H&E, Masson’s trichrome (Sigma-Aldrich), and picrosirius red (Polysciences) stain kits, according to manufactures’ instructions. ImageJ software (NIH) was used to measure adipocyte area, which is represented as the average adipocyte area.
Immunohistochemistry
Tissue sections and cells were stained with mouse anti-Sma (A2547; Sigma-Aldrich), rabbit anti-perilipin (9349S; Cell Signaling), chicken anti-GFP (GFP-1020; Aves Labs), and anti-F4/80 conjugated Alexa 647 (BM8; Biolegend) overnight at 4°C. After washing, sections were incubated for 1 hr at room temperature with fluorophore conjugated secondary antibodies: goat anti-rabbit Alexa 488, goat anti-mouse Alexa 568, and donkey anti-chicken Alexa 647 (Invitrogen). After washing, slides were mounted with Prolong Gold with DAPI (Invitrogen).
Statistical Analysis
Data are represented as mean ± SEM. The significance of differences between two groups was determined using Student’s t test. Statistical significance was defined as p < 0.05.
Supplementary Material
Highlights.
Sca1−, PDGFRa+, ITGA5+ FPCs are recruited in adipose tissue fibrosis
MRTFA deficiency protects against obesity-induced fibrosis and metabolic dysfunction
Genetic ablation of MRTFA shifts progenitor cell fate from profibrotic to adipogenic
ITGA5-MRTFA pathway is a possible target to combat obesity-associated metabolic disease
Acknowledgments
This work was supported by NIH grants DK098830 and DK102199. J.Z.L. was supported by National Research Service Award (NRSA) Fellowships (T32 DK007201 and F32 DK109635) and N.R. by a Société Francophone du Diabète (SFD) and an American Heart Association fellowship (17POST33660875). We thank Anna C. Belkina of the Boston University School of Medicine (BUSM) Flow Cytometry Core Facility for advice and assistance. We acknowledge Tom Balon of the BUSM Metabolic Phenotyping/IVIS Core for his support.
Footnotes
Supplemental Information includes four figures and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.04.057.
DECLARATION OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Conceptualization, J.Z.L. and S.R.F.; Methodology, J.Z.L. and S.R.F.; Investigation, J.Z.L. and N.R.; Formal Analysis, J.Z.L.; Writing – Original Draft, J.Z.L.; Writing – Review & Editing, J.Z.L., N.R., and S.R.F.; Funding Acquisition, J.Z.L. and S.R.F.
References
- Bel Lassen P, Charlotte F, Liu Y, Bedossa P, Le Naour G, Tordjman J, Poitou C, Bouillot JL, Genser L, Zucker JD, et al. The FAT score, a fibrosis score of adipose tissue: predicting weight-loss outcome after gastric bypass. J Clin Endocrinol Metab. 2017;102:2443–2453. doi: 10.1210/jc.2017-00138. [DOI] [PubMed] [Google Scholar]
- Bian H, Lin JZ, Li C, Farmer SR. Myocardin-related transcription factor A (MRTFA) regulates the fate of bone marrow mesenchymal stem cells and its absence in mice leads to osteopenia. Mol Metab. 2016;5:970–979. doi: 10.1016/j.molmet.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crider BJ, Risinger GM, Jr, Haaksma CJ, Howard EW, Tomasek JJ. Myocardin-related transcription factors A and B are key regulators of TGF-β1-induced fibroblast to myofibroblast differentiation. J Invest Dermatol. 2011;131:2378–2385. doi: 10.1038/jid.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, Basdevant A, Guerre-Millo M, Poitou C, Zucker JD, et al. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes. 2010;59:2817–2825. doi: 10.2337/db10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evelyn CR, Wade SM, Wang Q, Wu M, Iñiguez-Lluhí JA, Merajver SD, Neubig RR. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther. 2007;6:2249–2260. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- Fildes A, Charlton J, Rudisill C, Littlejohns P, Prevost AT, Gulliford MC. Probability of an obese person attaining normal body weight: cohort study using electronic health records. Am J Public Health. 2015;105:e54–e59. doi: 10.2105/AJPH.2015.302773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmi V, Cardellini M, Cinti F, Corgosinho F, Cardolini I, D’Adamo M, Zingaretti MC, Bellia A, Lauro D, Gentileschi P, et al. Omental adipose tissue fibrosis and insulin resistance in severe obesity. Nutr Diabetes. 2015;5:e175. doi: 10.1038/nutd.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henegar C, Tordjman J, Achard V, Lacasa D, Cremer I, Guerre-Millo M, Poitou C, Basdevant A, Stich V, Viguerie N, et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 2008;9:R14. doi: 10.1186/gb-2008-9-1-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Berry DC, Tang W, Graff JM. Independent stem cell lineages regulate adipose organogenesis and adipose homeostasis. Cell Rep. 2014;9:1007–1022. doi: 10.1016/j.celrep.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keophiphath M, Achard V, Henegar C, Rouault C, Clément K, Lacasa D. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol Endocrinol. 2009;23:11–24. doi: 10.1210/me.2008-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29:1575–1591. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16:51–66. doi: 10.1016/j.stem.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon EY, Shin SK, Cho YY, Jung UJ, Kim E, Park T, Park JH, Yun JW, McGregor RA, Park YB, Choi MS. Time-course micro-arrays reveal early activation of the immune transcriptome and adipokine dys-regulation leads to fibrosis in visceral adipose depots during diet-induced obesity. BMC Genomics. 2012;13:450. doi: 10.1186/1471-2164-13-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontan M, Berlan M. Do regional differences in adipocyte biology provide new pathophysiological insights? Trends Pharmacol Sci. 2003;24:276–283. doi: 10.1016/S0165-6147(03)00132-9. [DOI] [PubMed] [Google Scholar]
- Lee YH, Petkova AP, Granneman JG. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 2013;18:355–367. doi: 10.1016/j.cmet.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KT, Chan KY, Ng PC, Lau TK, Chiu WM, Tsang KS, Li CK, Kong CK, Li K. The tetraspanin CD9 regulates migration, adhesion, and homing of human cord blood CD34+ hematopoietic stem and progenitor cells. Blood. 2011;117:1840–1850. doi: 10.1182/blood-2010-04-281329. [DOI] [PubMed] [Google Scholar]
- Liu J, DeYoung SM, Zhang M, Zhang M, Cheng A, Saltiel AR. Changes in integrin expression during adipocyte differentiation. Cell Metab. 2005;2:165–177. doi: 10.1016/j.cmet.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Liu Y, Aron-Wisnewsky J, Marcelin G, Genser L, Le Naour G, Torcivia A, Bauvois B, Bouchet S, Pelloux V, Sasso M, et al. Accumulation and changes in composition of collagens in subcutaneous adipose tissue after bariatric surgery. J Clin Endocrinol Metab. 2016;101:293–304. doi: 10.1210/jc.2015-3348. [DOI] [PubMed] [Google Scholar]
- Luchsinger LL, Patenaude CA, Smith BD, Layne MD. Myocardin-related transcription factor-A complexes activate type I collagen expression in lung fibroblasts. J Biol Chem. 2011;286:44116–44125. doi: 10.1074/jbc.M111.276931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelin G, Ferreira A, Liu Y, Atlan M, Aron-Wisnewsky J, Pelloux V, Botbol Y, Ambrosini M, Fradet M, Rouault C, et al. A PDGFRα-mediated switch toward CD9high adipocyte progenitors controls obesity-induced adipose tissue fibrosis. Cell Metab. 2017;25:673–685. doi: 10.1016/j.cmet.2017.01.010. [DOI] [PubMed] [Google Scholar]
- McDonald ME, Li C, Bian H, Smith BD, Layne MD, Farmer SR. Myocardin-related transcription factor A regulates conversion of progenitors to beige adipocytes. Cell. 2015;160:105–118. doi: 10.1016/j.cell.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morandi EM, Verstappen R, Zwierzina ME, Geley S, Pierer G, Ploner C. ITGAV and ITGA5 diversely regulate proliferation and adipogenic differentiation of human adipose derived stem cells. Sci Rep. 2016;6:28889. doi: 10.1038/srep28889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobusue H, Onishi N, Shimizu T, Sugihara E, Oki Y, Sumikawa Y, Chiyoda T, Akashi K, Saya H, Kano K. Regulation of MKL1 via actin cytoskeleton dynamics drives adipocyte differentiation. Nat Commun. 2014;5:3368. doi: 10.1038/ncomms4368. [DOI] [PubMed] [Google Scholar]
- Shimizu Y, Dobashi K, Iizuka K, Horie T, Suzuki K, Tukagoshi H, Nakazawa T, Nakazato Y, Mori M. Contribution of small GTPase Rho and its target protein rock in a murine model of lung fibrosis. Am J Respir Crit Care Med. 2001;163:210–217. doi: 10.1164/ajrccm.163.1.2001089. [DOI] [PubMed] [Google Scholar]
- Shiwen X, Stratton R, Nikitorowicz-Buniak J, Ahmed-Abdi B, Ponticos M, Denton C, Abraham D, Takahashi A, Suki B, Layne MD, et al. A role of myocardin related transcription factor-A (MRTF-A) in scleroderma related fibrosis. PLoS ONE. 2015;10:e0126015. doi: 10.1371/journal.pone.0126015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisson TH, Ajayi IO, Subbotina N, Dodi AE, Rodansky ES, Chibucos LN, Kim KK, Keshamouni VG, White ES, Zhou Y, et al. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am J Pathol. 2015;185:969–986. doi: 10.1016/j.ajpath.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K, Olson EN. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer M, Unal R, Zhu B, Rasouli N, McGehee RE, Jr, Peterson CA, Kern PA. Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. J Clin Endocrinol Metab. 2011;96:E1990–E1998. doi: 10.1210/jc.2011-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman BM, Ginty CA. Fibronectin modulation of cell shape and lipogenic gene expression in 3T3-adipocytes. Cell. 1983;35:657–666. doi: 10.1016/0092-8674(83)90098-3. [DOI] [PubMed] [Google Scholar]
- Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Tordjman J, Clément K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013;18:470–477. doi: 10.1016/j.cmet.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasquez LS, Sutherland LB, Liu Z, Grinnell F, Kamm KE, Schneider JW, Olson EN, Small EM. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc Natl Acad Sci U S A. 2013;110:16850–16855. doi: 10.1073/pnas.1316764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila IK, Badin PM, Marques MA, Monbrun L, Lefort C, Mir L, Louche K, Bourlier V, Roussel B, Gui P, et al. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 2014;7:1116–1129. doi: 10.1016/j.celrep.2014.03.062. [DOI] [PubMed] [Google Scholar]
- Vishvanath L, MacPherson KA, Hepler C, Wang QA, Shao M, Spurgin SB, Wang MY, Kusminski CM, Morley TS, Gupta RK. Pdgfrβ+ mural preadipocytes contribute to adipocyte hyperplasia induced by high-fat-diet feeding and prolonged cold exposure in adult mice. Cell Metab. 2016;23:350–359. doi: 10.1016/j.cmet.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev. 2000;21:697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- Yu J, Lee CY, Changou CA, Cedano-Prieto DM, Takada YK, Ta-kada Y. The CD9, CD81, and CD151 EC2 domains bind to the classical RGD-binding site of integrin αvβ3. Biochem J. 2017;474:589–596. doi: 10.1042/BCJ20160998. [DOI] [PubMed] [Google Scholar]
- Yu-Wai-Man C, Treisman R, Bailly M, Khaw PT. The role of the MRTF-A/SRF pathway in ocular fibrosis. Invest Ophthalmol Vis Sci. 2014;55:4560–4567. doi: 10.1167/iovs.14-14692. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Gu X, Zhang N, Kolonin MG, An Z, Sun K. Divergent functions of endotrophin on different cell populations in adipose tissue. Am J Physiol Endocrinol Metab. 2016;311:E952–E963. doi: 10.1152/ajpendo.00314.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H, Jin TH, Desai L, Bernard K, Thannickal VJ. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest. 2013;123:1096–1108. doi: 10.1172/JCI66700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.