

Abstract
A series of (2RS,4R)-2-arylthiazolidine-4-carboxylic acid amide (ATCAA) was synthesized. Antiproliferative activity against melanoma and prostate cancer cells compared with control cells (fibroblast and RH7777, respectively) was evaluated. Compound 3id showed the best selectivity and growth-inhibition activity against three melanoma cell lines (B16-F1, A375, and WM-164). Compounds 15b and 3ac had good selectivity and potency against four prostate cancer cell lines (DU 145, PC-3, LNCaP, and PPC-1). The structure-activity relationship (SAR) of the side chain, the thiazolidine ring, and phenyl substituents is discussed. Cell cycle analysis showed that the percentage of cancer cells undergoing apoptosis (sub-G1 phase) increased after treatment with 1b and 3ad, which also strongly inhibited melanoma colony formation. In vivo studies on nude mice bearing A375 melanoma tumors showed that compound 1b inhibited tumor growth in a dose-dependent manner. At a dose of 10 mg/kg, 1b significantly inhibited melanoma tumor growth and showed higher efficacy than did dacarbazine at 60 mg/kg.
Keywords: Melanoma, Prostate cancer, Thiazolidine, Antiproliferative activity, Structure-activity relationship, Colony formation, In vivo evaluation
1. Introduction
Cancer ranks second in diseases leading to mortality, following only cardiovascular diseases. One-quarter of all deaths in the United States are caused by cancers.1 Out of the many cancer diseases, the incidence of cutaneous malignant melanoma is increasing rapidly throughout the world. Melanoma metastasizing to major organs (stage IV) is virtually incurable. Currently, dacarbazine (DTIC) is the only FDA-approved drug to treat advanced melanoma; it provides complete remission in only 2% of patients.2–4 Prostate cancer is the most common noncutaneous malignancy in men in Western countries. One out of nine men over 65 years of age is diagnosed with prostate cancer in the United States.5 It accounts for one-third of all male cancer diagnoses and 9% of male deaths as a result of cancer.6
In previous contributions from our laboratory, we discovered that, by replacing the glycerol backbone in lysophosphatidic acid (LPA, 1-acyl-sn-glycerol-3-phosphate), the resulting 2-arylthiazoli-dine-4-carboxylic acid amides (ATCAAs) were potent cytotoxic agents for prostate cancer and melanoma.7–12 One of earlier derivatives (2RS,4 R)-2-phenyl-thiazolidine-4-carboxylic acid octadecylamide 1a and (2RS,4 R)-2-(4-acetamidophenyl)-thiazolidine-4-carboxylic acid hexadecylamide 1b (Fig. 1) were sent to the U.S. National Cancer Institute 60 human tumor cell line anticancer drug screen (NCI-60). The NCI-60 screening data indicated that compounds 1a and 1b strongly inhibited the growth of all nine types of cancer cells with GI50 values ranging from 0.12 μM (leukemia, CCRF-CEM cell line) to 10.9 μM (colon cancer, HCC-15 cell line). 1b was very potent against melanoma (GI50 = 0.13−1.48 μM against all eight tumor cell lines) and prostate cancer (GI50= 0.17−0.27 μM against two tumor cell lines). Based on these preliminary cytotoxic screenings, extensive structure-activity relationship (SAR) and in vivo studies were carried out. In this article, we report the synthesis, biological evaluation, and SAR studies of ATCAA analogues based on 1a and 1b for both melanoma and prostate cancer. These analogs were further assessed for their ability to inhibit melanoma colony formation. The in vivo efficacy of compound 1b was tested in a xenograft model using human A375 melanoma tumors. This class of compounds showed potent antitumor activity and selectivity, which could represent the basis for their development into novel chemotherapeutic drugs.
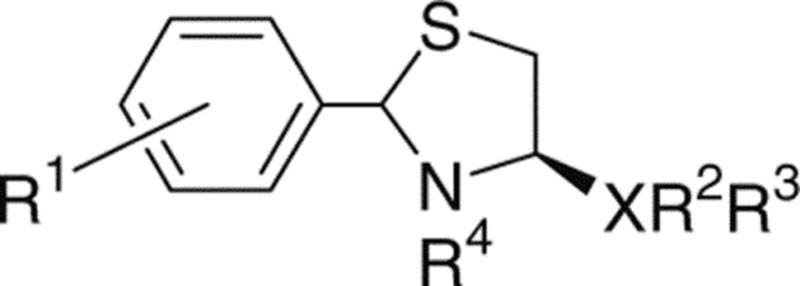
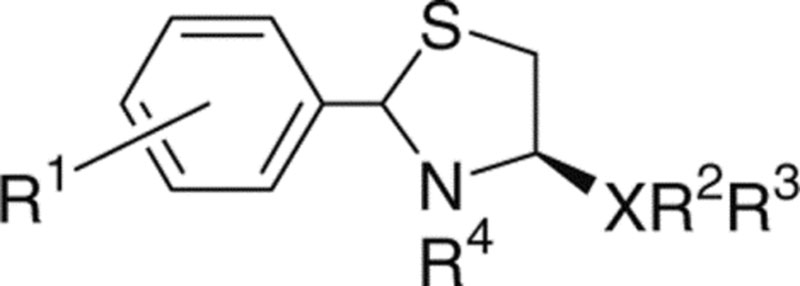
Figure 1.

Structures of LPA and ATCAA.
2. Chemistry
The general synthesis of (2RS,4 R)-2-arylthiazolidine-4-carbox-amides is shown in Scheme 1. L-Cysteine reacted with appropriate benzaldehydes in ethanol and water at ambient temperature to give cyclized (2RS,4 R)-2-arylthiazolidine-4-carboxylic acids (AT-CAs),13 which were converted to the corresponding Boc-protected derivatives 2a–2j. Reaction of 2a–2j with appropriate amines using EDCI/HOBt gave corresponding amides, which were subsequently treated with trifluoroacetic acid (TFA) to form the target compounds 1a, 1b, and 3aa–3jc. Acetylation of the thiazolidine amino group in compound 1b with acetyl chloride gave acylation derivative 4. Ester 5 was obtained by esterifying 2i with DCC/DMAP catalyzed by (1S)-(+)-10-camphorsulfonic acid followed by TFA treatment.14 Reducing amide 1b with LAH or B2H6 did not provide the desired amine 8b; instead, disulfanyl dimer 9 was obtained. 8a and 8b were prepared from (2RS,4R)-2-arylthiazolidine-4-carbal-dehyde 7, which was obtained from reducing Weinreb amide 6.15 The reductive amination of aldehyde 7 with hexadecylamine and NaBH3CN16 followed by Boc deprotection gave amines 8a and 8b.
Scheme 1.

Synthesis of ATCAA. Reagents and conditions: (a) C2H5OH, H2O (70–99%); (b) Boc20, 1 N NaOH, 1,4-dioxane, H2O (89–97%); (c) EDCI, HOBt, Et3N, R2R3NH, CH2Cl2; (d) TFA, CH2Cl2; (e) CH3COCl, pyridine, CH2Cl2 (74%); (f) DCC, DMAP, 0.3 equiv (1S)-(+)-10 camphorsulfonic acid, C16H33OH (94%); (g) EDCI, HOBt, NMM, HNCH3OCH3, CH2Cl2 (71%); (h) LAH, THF (84%); (i) R2NH2, NaBH3CN, HOAc; (j) LAH, THF, 0 °C to rt or B2H6, THF, rt to reflux.
The Fmoc-protected (2RS,4R)-(4-acetylamino-phenyl)-thiazoli-dine-4-carboxylic acid 10 was prepared by reacting L-cysteine with 4-acetylaminobenzaldehyde, followed by reaction with Fmoc-Cl in the presence of triethanolamine (TEA) with a 99% yield. Subsequent EDCI/HOBt coupling reaction yielded carboxamide 11. 4-Aminophenyl intermediate 12 was obtained by acid hydrolysis in methanol. Compound 12 was further reacted with different acid chlorides or sodium cyanate17 to afford derivatives 14a–14c. Compounds 13a–13b and 15a–15c were obtained by deprotection of the Fmoc group from 12 and 14a–14c, respectively (Scheme 2).
Scheme 2.

Reagents: (a) C2H5OH, H2O (78%); (b) Fmoc-Cl, Et3N, CH2Cl2 (99%); (c) EDCI, HOBt, Et3N, R2NH2, CH2Cl2 (71%); (d) CH3COCl, CH3OH (48%); (e) DBU, CH2Cl2 (46–66%) (f) 15a CH3SO2Cl, pyridine (80%); 15b ClCH2COCl, pyridine (51%); 15c NaOCN, HOAc, CH3OH, H2O (88%).
The open-ring compounds 16a–16d were prepared as shown in Scheme 3. Compounds 16a–16c were easily obtained from (R)-2-amino-3-(methylthio)propanoic acid or (R)-3-(benzylthio)-2-(tert-butoxycarbonylamino)propanoic acid. Compound 16d was readily prepared in a five-step procedure from 2-aminoacetic acid at 38.4% overall yield.
Scheme 3.

Reagents: (a) Boc20,1 N NaOH; (b) EDCI, HOBt, C16H33NH2; (c) CF3COOH; (d) N-(4-formyl-phenyl)-acetamide, MeOH, (e) NaBH3CN.
We characterized each compound with nuclear magnetic resonance (NMR), mass spectroscopy, and elemental analysis. Because of the presence of two chiral centers at the thiazolidine ring (C2 and C4 positions, the chirality at C4 is fixed), the 1H NMR spectra of ATCAAs indicated two diastereomers with a ratio of 7:3. The chemical shifts for C2-H, C4-H, and C5-Ha/b on the thiazolidine ring were greatly different between the two diastereomers. For example, in the case of 1b (Fig. 2A), the C2-H signal of the minor isomer appeared downfield (5.58 ppm in CDCl3), while that of the major isomer appeared at 5.32 ppm. The C4-H signal of the major isomer appeared at 4.33 ppm, while the minor isomer appeared at 3.90 ppm. The signals from the two unequivalent protons of C5-Ha and C5-Hb showed four sets of peaks; each set appeared as a doublet of doublet (dd) as a result of geminal coupling and with additional vicinal coupling to C4-H. These peaks were centered at 3.69 ppm, 3.41 ppm, 3.39 ppm, and 3.28 ppm, respectively, with upfield signals partially overlapping with CH2 signals (3.26−3.21 ppm) from the long fatty chain near the carboxylic amide CONHCH2C15H31. The absolute structures of the two diastereomers can be readily assigned using 1D nuclear Overhauser effect (NOE) experiments (Fig. 2). When the peak at 4.33 ppm (C4-H of the major isomer) was irradiated, the peak at 5.32 ppm (C2-H of the major isomer) did not show any detectable NOE (Fig. 2B); when the peak at 3.90 ppm (C4-H of the minor isomer) was irradiated, the peak at 5.58 ppm (C2-H of the minor isomer) showed a strong NOE (Fig. 2C). Because the chirality at C4 is fixed (R-configuration from starting material L-cysteine), the major isomer was clearly (2S,4R), and the minor isomer was (2R, 4R) as indicated in Figure 2. The relative distribution of these two configurations can also be expected based on their molecular structures, because it is sterically more favorable when the two bulky groups are attached to opposite sides of the five-membered ring.
Figure 2.

(A) Expanded 1H NMR spectrum of two diastereomers of 1b (ratio 7:3) in CDCl3; (B) 1D NOE from irradiation of C4-H of the major isomer at 4.33 ppm, no NOE observed at 5.32 ppm; (C) 1D NOE from irradiation of C4-H of minor isomer at 3.90 ppm, shows strong NOE at 5.58 ppm. Thus, protons at C2-H and C4-H are ‘cis-position’ in the minor isomer.
3. Biological results and discussion
3.1. Antiproliferative effects of ATCAA against melanoma and prostate cancer
The diastereomeric mixtures of compounds 3aa–15c were used to evaluate their in vitro inhibitory activity and toxicity against two human melanoma cell lines (A375 and WM-164), one mouse melanoma cell line (B16-F1), four human prostate cancer cell lines (DU 145, PC-3, LNCaP, and PPC-1), fibroblast cells (control cell line for melanoma), and RH7777 (control cell line for prostate cancer). Because preparing pure diastereomers was not easy to achieve, the IC50 values were obtained on diastereomeric mixtures to select the most promising compounds. The standard sulforhodamine B (SRB) assay was used to evaluate the antiproliferative activity of different compounds in melanoma and prostate cancer cells. The results are summarized in Tables 1 and 2. Compounds 1a and 1b were chosen as control compounds for both tumor cell lines in these in vitro studies. Cisplatin (CDDP), DTIC, and Sorafenib (Nexavar, Bay43–9006,18 which had been granted Fast Track designation by FDA to treat advanced melanoma) were used as reference compounds for melanoma. The antiproliferative data showed that all three melanoma cell lines were resistant to cisplatin. DTIC was inactive because of the lack of bioactivation in vitro.19,20 The average IC50 value of Sorafenib on three melanoma cell lines is 5.1 μM, and the selectivity of Sorafenib between fibroblast cells and melanoma cells is threefold. Our purpose in modifying chemical structure was to enhance the selectivity between tumor cells and control cells and to increase or maintain antitumor potency. Thus, we used the average IC50 value of all melanoma and prostate cancer cells for comparison. Selectivity is defined as the ratio of IC50 values in the control cell line and the average in tumor cell lines. From Tables 1 and 2, the most selective compound (3id) had a selectivity of 11.3-fold in melanoma cells against fibroblast cells; compounds 3ac and 15b showed an improved selectivity of 9.1-fold and 9.4-fold for prostate cancer cells, respectively.
Table 1.
Antiproliferative effects of ATCAA in three melanoma cell lines and fibroblast cell line
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | X | R1 | R2 | R3 | R4 | IC50 ± SEM(μM) | |||||
| B16-F1 | A375 | WM-164 | Fibroblast | Aver.a | Ratiob | ||||||
| 3aa | CON | 3,4,5-Trimethoxy | n-C18H37 | H | H | 5.9 ± 0.4 | 4.6 ± 0.2 | 3.0 ±0.1 | 4.7 ± 0.4 | 4.5 | 1.0 |
| 3ab | CON | 3,4,5-Trimethoxy | (Z)-Octadec-8-enyl | H | H | 4.7 ± 0.1 | 2.4 ± 0.1 | 1.3 ±0.1 | 4.8 ± 0.3 | 2.8 | 1.7 |
| 3ac | CON | 3,4,5-Trimethoxy | (E)-Octadec-8-enyl | H | H | 3.2 ± 0.2 | 1.8 ±0.1 | 1.1 ±0.1 | 4.0 ± 0.3 | 2.0 | 2.0 |
| 3ad | CON | 3,4,5-Trimethoxy | n-C16H33 | H | H | 1.6 ± 0.2 | 1.4 ±0.2 | 0.7 ± 0.1 | 2.4 ± 0.4 | 1.2 | 2.0 |
| 3ba | CON | 3,4-Dimethoxy | n-C18H37 | H | H | 14.3 ± 0.5 | 6.9 ± 0.3 | 2.7 ± 0.2 | 20.3 ± 1.1 | 8.0 | 2.5 |
| 3bb | CON | 3,4-Dimethoxy | (Z)-Octadec-8-enyl | H | H | 3.3 ± 0.2 | 1.6 ±0.1 | 1.2 ±0.1 | 18.3 ±0.8 | 2.0 | 9.0 |
| 3bc | CON | 3,4-Dimethoxy | (E)-Octadec-8-enyl | H | H | 3.2 ± 0.3 | 1.4 ±0.1 | 1.1 ±0.1 | 16.3 ±0.7 | 1.9 | 8.6 |
| 3ca | CON | 2-OMe | n-C16H33 | H | H | 4.3 ± 0.2 | 3.0 ± 0.4 | 2.4 ± 0.2 | 7.2 ± 0.3 | 3.2 | 2.2 |
| 3da | CON | 3-OMe | n-C16H33 | H | H | 3.0 ± 0.1 | 1.8 ±0.1 | 1.2 ±0.1 | 2.5 ± 0.2 | 2.0 | 1.3 |
| 3ea | CON | 4-OMe | n-C16H33 | H | H | 2.3 ± 0.3 | 1.5 ±0.1 | 1.0 ±0.1 | 8.1 ±0.5 | 1.6 | 5.1 |
| 3fa | CON | 4-NMe2 | n-C16H33 | H | H | 6.7 ± 0.5 | 1.8 ±0.1 | 1.5 ±0.1 | 21.0±3.1 | 3.3 | 6.3 |
| 3ga | CON | 2-NHAc | n-C16H33 | H | H | 8.0 ± 0.4 | 9.3 ± 0.6 | 3.9 ± 0.5 | 27.8 ±3.6 | 7.1 | 3.9 |
| 3ha | CON | 3-NHAc | n-C16H33 | H | H | 2.2 ± 0.2 | 1.5 ±0.1 | 1.1 ±0.1 | 6.3 ± 0.4 | 1.6 | 3.9 |
| 3ia | CON | 4-NHAc | n-C18H37 | CH3 | H | 18.9 ± 1.3 | 20.6 ±2.1 | 10.7 ±0.9 | >100 | 16.7 | - |
| 31b | CON | 4-NHAc | 1-Adamantanyl | H | H | 96.5 ± 3.6 | 137.5 ±4.2 | 127.5 ±2.8 | >100 | 120.5 | - |
| 3ic | CON | 4-NHAc | 2-Adamantanyl | H | H | 108.2 ± 4.6 | 66.0 ±2.4 | 64.4 ± 3.0 | >100 | 79.5 | - |
| 3id | CON | 4-NHAc | 9H-Fluoren-2-yl | H | H | 3.9 ± 0.3 | 2.1 ±0.1 | 1.7 ±0.1 | 28.9 ±1.0 | 2.6 | 11.3 |
| 3ie | CON | 4-NHAc | Anthracen-2-yl | H | H | 5.6 ± 0.2 | 3.1 ±0.1 | 1.4 ±0.1 | 13.5 ±0.5 | 3.4 | 4.0 |
| 3if | CON | 4-NHAc | 4-Biphenyl | H | H | 5.7 ± 0.3 | 6.2 ± 0.4 | 4.1 ± 0.2 | 33.4 ±3.5 | 5.3 | 6.3 |
| 3ig | CON | 4-NHAc | 2-Benzothiazolyl | H | H | 55.3 ± 2.9 | 45.9 ±3.7 | >100 | >100 | 50.6 | - |
| 3ih | CON | 4-NHAc | (Z)-Hexadec-9-enyl | H | H | 2.1 ±0.1 | 1.9 ±0.1 | 2.0 ±0.1 | 14.0 ± 0.9 | 2.0 | 7.0 |
| 3ii | CON | 4-NHAc | Nonadec-10-ynyl | H | H | 1.6 ±0.1 | 2.7 ± 0.2 | 1.3 ±0.1 | 7.0 ± 0.3 | 1.9 | 3.8 |
| 3jb | CON | H | (Z)-Octadec-8-enyl | H | H | 6.0 ± 0.5 | 4.9 ± 0.7 | 2.4 ± 0.1 | 11.6 ±0.8 | 4.4 | 2.6 |
| 3jc | CON | H | (E)-Octadec-8-enyl | H | H | 10.1 ±0.8 | 5.3 ± 0.4 | 2.4 ± 0.2 | 12.4 ±1.1 | 5.9 | 2.1 |
| 4 | CON | 4-NHAc | n-C16H33 | H | Ac | 15.0 ±0.8 | 12.6 ±0.5 | 20.4 ± 0.8 | 57.7 ±1.3 | 16 | 3.6 |
| 5 | COO | 4-NHAc | n-C16H33 | H | H | 41.9 ±3.1 | 28.0 ±2.1 | 18.5 ±0.4 | >100 | 29.5 | - |
| 8a | CH2N | H | n-C16H33 | H | H | 4.7 ± 0.2 | 13.0 ±0.3 | 4.9 ± 0.1 | 60 ±1.2 | 7.5 | 8.0 |
| 8b | CH2N | 4-NHAc | n-C16H33 | H | H | 11.9 ±0.4 | 14.4 ±0.3 | 3.2 ±0.1 | 84.5 ± 2.5 | 9.8 | 8.6 |
| 15a | CON | 4-NHSO2CH3 | n-C16H33 | H | H | 113.6 ±2.1 | 50.6 ±1.6 | 18.6 ±1.0 | >100 | 60.9 | - |
| 15b | CON | 4-NHCOCH2CI | n-C16H33 | H | H | 93.3 ± 3.6 | 20.4 + 1.3 | 5.7 ± 0.2 | 25.4 ±1.3 | 39.8 | 0.6 |
| 15c | CON | 4-NHCONH2 | n-C16H33 | H | H | 6.5 ± 0.6 | 3.6 ± 0.3 | 1.7 ±0.1 | 7.2 ± 0.4 | 3.9 | 1.8 |
| 1a | CON | H | n-C18H37 | H | H | 15.5 ±0.6 | 15.0 ±0.5 | 4.4 ± 0.2 | 29.8 ±2.1 | 11.6 | 2.6 |
| 1b | CON | 4-NHAc | n-C16H33 | H | H | 2.2 ± 0.3 | 2.1 ± 0.2 | 1.1 ±0.1 | 16.0 ±2.5 | 1.8 | 8.9 |
| DTIC | >100 | >100 | >100 | >100 | - | - | |||||
| CDDP | >100 | >100 | >100 | >100 | - | - | |||||
| Sorafenib | 4.9 ± 0.3 | 5.4 ± 0.5 | 5.0 ± 0.2 | 15.1 ±1.2 | 5.1 | 3.0 | |||||
Average IC50 of three melanoma cells IC50s.
Ratio = (IC50 of fibroblast cell)/(the average IC50 of melanoma cell lines).
Table 2.
Antiproliferative effects of ATCAA in four prostate cancer cell lines and RH7777 cell line
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | X | R1 | R2 | R3 | R4 | IC50 ± SEM (μM) | ||||||
| DU 145 | PC-3 | LNCaP | PPC-1 | RH7777 | Aver.a | Ratiob | ||||||
| 3aa | CON | 3,4,5-Trimethoxy | n-C18H37 | H | H | 4.1 ± 0.5 | 4.9 ± 0.4 | 1.6 ±0.2 | 1.0 ±0.1 | 14.0 ±1.4 | 2.9 | 4.8 |
| 3ab | CON | 3,4,5-Trimethoxy | (Z)-Octadec-8-enyl | H | H | 1.2 ±0.1 | 1.5 ±0.1 | 0.7 ± 0.1 | 0.5 ±0.1 | 8.4 ± 0.8 | 1.0 | 8.6 |
| 3ac | CON | 3,4,5-Trimethoxy | (E)-Octadec-8-enyl | H | H | 0.9 ± 0.1 | 1.2 ±0.1 | 0.6 ± 0.1 | 0.3 ± 0.1 | 6.8 ± 0.7 | 0.8 | 9.1 |
| 3ba | CON | 3,4-Dimethoxy | n-C18H37 | H | H | 5.5 ±1.7 | 4.1 ± 0.5 | 2.4 ± 0.4 | 0.9 ±0.1 | 11.8 ±0.6 | 3.2 | 3.7 |
| 3bb | CON | 3,4-Dimethoxy | (Z)-Octadec-8-enyl | H | H | 2.0 ± 0.2 | 2.6 ± 0.3 | 1.3 ±0.1 | 0.6 ±0.1 | 10.0 ±0.2 | 1.6 | 6.2 |
| 3bc | CON | 3,4-Dimethoxy | (E)-Octadec-8-enyl | H | H | 1.5 ±0.2 | 2.0 ± 0.2 | 0.8 ± 0.1 | 0.3 ± 0.1 | 9.4 ±1.0 | 1.2 | 8.2 |
| 3ca | CON | 2-OMe | n-C16H33 | H | H | 6.5 ± 0.9 | 7.4 ±1.1 | 3.6 ± 0.3 | 1.7 ±0.1 | 17.2 ± 0.3 | 4.8 | 3.6 |
| 3da | CON | 3-OMe | n-C16H33 | H | H | 1.1 ±0.1 | 1.0 ±0.1 | 0.5 ± 0.1 | 0.2 ±0.1 | 3.6 ±0.1 | 0.7 | 5.1 |
| 3ea | CON | 4-OMe | n-C16H33 | H | H | 2.2 ± 0.2 | 2.7 ± 0.3 | 1.1 ±0.1 | 0.4 ± 0.1 | 8.4 ±1.1 | 1.6 | 5.3 |
| 3fa | CON | 4-NMe2 | n-C16H33 | H | H | 3.2 ± 0.3 | 6.0 ± 0.5 | 1.4 ±0.1 | 0.4 ± 0.1 | 10.0 ±1.6 | 2.8 | 3.6 |
| 3ga | CON | 2-NHAc | n-C16H33 | H | H | 4.6 ± 0.5 | 2.7 ± 0.5 | 3.3 ± 0.4 | 1.5 ±0.2 | 13.2 ±1.8 | 3.0 | 4.4 |
| 3ha | CON | 3-NHAc | n-C16H33 | H | H | 2.3 ± 0.2 | 1.6 ±0.2 | 1.4 ±0.2 | 0.5 ± 0.2 | 4.9 ± 0.4 | 1.5 | 3.4 |
| 3ia | CON | 4-NHAc | n-C18H37 | CH3 | H | 3.9 ± 0.4 | 3.7 ± 0.2 | 1.8 ±0.1 | 0.8 ± 0.1 | 9.2 ± 0.9 | 2.6 | 3.6 |
| 3ib | CON | 4-NHAc | 1-Adamantanyl | H | H | >20 | >20 | 5.7 ± 0.9 | >20 | >20 | 5.7 | — |
| 3ic | CON | 4-NHAc | 2-Adamantanyl | H | H | >20 | >20 | 9.7 ±1.0 | >20 | >20 | 9.7 | - |
| 3id | CON | 4-NHAc | 9H-Fluoren-2-yl | H | H | 1.9 ±0.3 | 2.1 ±0.1 | 3.5 ± 0.7 | 1.6 ±0.1 | 5.4 ± 0.7 | 2.3 | 2.4 |
| 3ie | CON | 4-NHAc | Anthracen-2-yl | H | H | 0.9 ± 0.1 | 0.8 ± 0.1 | 1.7 ±0.3 | 0.7 ± 0.1 | 2.6 ± 0.4 | 1.0 | 2.5 |
| 3if | CON | 4-NHAc | 4-Biphenyl | H | H | 5.0 ± 0.5 | 7.6 ± 0.6 | 3.5 ± 0.3 | 2.9 ± 0.1 | 9.5 ± 0.8 | 4.8 | 2.0 |
| 3ig | CON | 4-NHAc | 2-Benzothiazolyl | H | H | >20 | >20 | >20 | >20 | >20 | - | - |
| 3ih | CON | 4-NHAc | (Z)-Hexadec-9-enyl | H | H | 2.4 ± 0.2 | 2.5 ± 0.2 | 1.8 ±0.1 | 1.1 ±0.1 | 7.6 ± 0.8 | 2.0 | 3.9 |
| 3ii | CON | 4-NHAc | Nonadec-10-ynyl | H | H | 1.8 ±0.1 | 2.1 ± 0.6 | 2.0 ± 0.4 | 0.7 ± 0.2 | 6.6±1.3 | 1.7 | 4.0 |
| 3jb | CON | H | (Z)-Octadec-8-enyl | H | H | 1.8 ±0.2 | 2.4 ± 0.2 | 1.6 ±0.1 | 1.0 ±0.1 | 8.3 ± 0.9 | 1.7 | 4.9 |
| 3jc | CON | H | (E)-Octadec-8-enyl | H | H | 1.5 ±0.1 | 2.1 ± 0.2 | 1.4 ±0.1 | 0.6 ±0.1 | 8.9 ± 0.9 | 1.4 | 6.5 |
| 4 | CON | 4-NHAc | n-C16H33 | H | Ac | 14.9 ±2.2 | 9.0 ±1.3 | 10.6 ±0.6 | 6.8 ± 0.8 | 13.0 ±3.0 | 10.3 | 1.3 |
| 5 | COO | 4-NHAc | n-C16H33 | H | H | >20 | >20 | >20 | >20 | >20 | - | - |
| 8a | CH2N | H | n-C16H33 | H | H | 3.1 ±0.3 | 3.4 ± 0.3 | 3.9 ± 0.4 | 2.5 + 0.2 | 6.2 ± 0.4 | 3.2 | 1.9 |
| 8b | CH2N | 4-NHAc | n-C16H33 | H | H | 4.7 ± 0.3 | 4.5 ± 0.5 | 4.7 ± 0.5 | 2.4 ± 0.2 | 7.9 ± 0.6 | 4.1 | 1.9 |
| 13a | CON | 4-NH2 | n-C12H25 | H | H | 2.4 ± 0.1 | 2.9 ± 0.1 | 2.0 ±0.2 | 1.2 ±0.1 | 7.3 ± 0.8 | 2.1 | 3.4 |
| 13b | CON | 4-NH2 | n-C16H33 | H | H | 2.6 ±0.1 | 2.4 ± 0.1 | 1.5 ±0.1 | 0.6 ±0.1 | 9.4 ± 0.1 | 1.8 | 5.3 |
| 15a | CON | 4-NHSO2CH3 | n-C16H33 | H | H | >20 | >20 | 4.8 ±1.0 | 1.1 ±0.1 | >20 | 2.9 | - |
| 15b | CON | 4-NHCOCH2C1 | n-C16H33 | H | H | 0.7 ± 0.2 | 0.9 ± 0.2 | 2.0 ± 0.2 | 0.4 ± 0.1 | 9.4 ± 0.9 | 1.0 | 9.4 |
| 15c | CON | 4-NHCONH2 | n-C16H33 | H | H | 3.2 ± 0.3 | 2.5 ± 0.4 | 1.8 ±0.2 | 0.5 ± 0.2 | 6.7 ± 0.6 | 2.0 | 3.4 |
| 1a | CON | H | n-C18H37 | H | H | 10.8 ±1.8 | 10.0 ±0.7 | 4.2 ± 0.4 | 2.4 ± 0.2 | >20 | 6.9 | - |
| 1b | CON | 4-NHAc | n-C16H33 | H | H | 1.7 ±0.1 | 1.2 ±0.1 | 1.0 ±0.1 | 0.4 ±0.1 | 6.8 ± 0.7 | 1.1 | 6.3 |
Average IC50 of four prostate cells IC50s.
Ratio = (IC50 of RH7777 cell)/(average IC50 of prostate cell lines).
3.2. Effects of different amide chains, C4 chirality, and chain length of ATCAA
ATCAA molecules were designed from LPA structure, which contains a long aliphatic chain. To examine whether the fatty chain played an essential role in cytotoxicity, we explored the possibilities of mimic fatty chain with bulky ring or aromatic systems such as adamantanyl, fluorenyl, and anthrancenyl. Although the anti-proliferative data showed that 1- or 2-adamantanyl amides and 2-benzothiazolylamide (3ib, 3ic, and 3ig) decreased activity against most melanoma and prostate cancer cells, introducing certain aromatic bulky groups such as 9H-fluoren-2-yl (3id), anthrancen-2-yl (3ie), and 4-biphenyl (3if) retained their proliferative activity for both types of cancers. Of particular interest, while introducing 9-fluoren-2-yl (3id) showed a comparable IC50 value of 2.6 μM with 1b (1.8 μM) on melanoma cells, the selectivity of 3id increased to 11.3-fold from 8.9-fold in 1b. This structural modification provided us a new approach to optimize the drug-like properties of ATCAA molecules, in which the hydrophobic fatty lipid chain could be modified with other groups.21
To investigate whether the chiral center at the 4-thiazolidine position had an effect on the potency of ATCAA, the 4S isomer (3ai) was prepared using D-cysteine as starting material. After comparing activity of 4R (3ae) with 4S (3ai) isomers in both prostate cancer cell lines and RH7777 cells (Table 4), no significant stereoselectivity at the thiazolidine 4-position was found. Therefore, changing the chirality at C4 on the thiazolidine ring did not substantially affect either activity or selectivity.
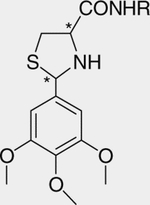
Table 4.
Antiproliferative effects of different chain length amides against prostate cells and R7777 cells
| Compounds | Configuration at 4-position | R | IC50 ± SEM (μM) | ||||
|---|---|---|---|---|---|---|---|
| DU 145 | PC-3 | LNCaP | PPC-1 | RH7777 | |||
 |
3ah (4R) | n-C8H17 | 17.3 ±1.4 | >20 | 2.7 ± 0.7 | 14.0 ±0.5 | >20 |
| 3ag (4R) | n-C10H21 | 4.1 ±0.3 | 4.4 ± 0.3 | 2.2 ± 0.3 | 2.1 ±0.1 | 6.0 ± 0.8 | |
| 3af (4R) | n-C12H25 | 2.8 ±0.1 | 2.9 ±0.1 | 1.4 ±0.1 | 1.0 ±0.1 | 6.5 ± 0.1 | |
| 3ae (4R) | n-C14H29 | 2.2 ±0.1 | 2.6 ±0.1 | 2.0 ± 0.3 | 0.6 ±0.1 | 6.1 ± 0.6 | |
| 3ai (4S) | n-C14H29 | 2.1 ±0.1 | 2.9 ±0.1 | 1.9 ±0.1 | 0.7 ± 0.1 | 5.8 ± 0.8 | |
| 3ad (4R) | n-C16H33 | 1.7 ±0.1 | 1.8 ±0.1 | 0.8 ± 0.1 | 0.4 ±0.1 | 7.2 ± 0.1 | |
| 3aa (4R) | n-C18H37 | 4.1 ±0.7 | 4.9 ± 0.4 | 1.6 ±0.2 | 1.0 ±0.1 | 14.0 ±0.5 | |
| 3ab (4R) | (Z)-Octadec-8-enyl | 1.2 ±0.1 | 1.5 ±0.1 | 0.7 ± 0.1 | 0.5 ± 0.1 | 8.4 ± 0.8 | |
| 3ac (4R) | (E)-Octadec-8-enyl | 0.9 ± 0.1 | 1.2 ±0.1 | 0.6 ± 0.1 | 0.3 ± 0.1 | 6.8 ± 0.7 | |
Short chain lengths with 8–10 carbon atoms in 4-carboxamide fatty chains (3ah, 3ag) displayed low potency. As chain length increased, potency increased, but toxicity also increased as measured on the RH7777 cell line (Fig. 3). Hexadecyl chains (3ad) displayed both the highest potency and selectivity against cancer cells among the examined chains, with an IC50 = 0.4 μM for PPC-1 cells. However, further increasing chain length (18 carbons, 3aa) reduced potency and toxicity. Interestingly, adding either a cis- or transdouble bond in the C18 side chain restored potency dramatically (cis and trans-octadec-8-enyl), demonstrating that both length and composition of the side chain are critical for their activity. trans-Octadec-8-enyl chain (3ac, 3bc, 3jc) showed slightly better activity than did the cis-isomers (3ab, 3bb, 3jb) against prostate cancer cells. Introducing a cis-double bond in the C16 fatty chain (3ih) or an alkyne in the C18 chain (3ii) did not increase overall potency on either cancer cell line. Introducing a branched aliphatic chain (3ia) decreased the potency in both melanoma cells (average IC50 = 16.7 μM, 9.3-fold decrease) and prostate cancer cells (average IC50 = 2.6 μM, 2.4-fold decrease) compared with 1b (average IC50 = 1.8 μM for melanoma, 1.1 μM for prostate cancer).
Figure 3.

Amide carbon chain length relative to IC50 in prostate cancer cell lines (average IC50 of DU 145, PC-3, LNCaP, and PPC-1 cells) and RH7777 cell line.
3.3. Effects of substitutions on 2-phenyl of ATCAA
The antiproliferative effects on both melanoma and prostate cancer indicated that when methoxy, 3,4-dimethoxy, and 3,4,5-tri-methoxy were introduced to 2-phenyl (3aa-3ea), potent activity was preserved or increased compared with that of 1a. For example, (2RS,4R)-2-(3,4,5-trimethoxy-phenyl)thiazolidine-4-carboxylic acid octadecylamide (3aa) (average IC50 = 2.9 μM) was 2.4-fold more potent than was 1a (average IC50 = 6.9 μM) against prostate cancer cells. Subsequently, different groups were introduced to the para-phenyl position. Replacements of 4-NHCOCH3 (1b) with 4-N(CH3)2 (3fa) showed comparable activity on A375 (IC50 = 1.8 vs 2.1 μM) and WM-164 cells (IC50 = 1.5 vs 1.1 μM) but slightly lower selectivity on fibroblast cells (IC50 = 21.0 vs 16.0 μM).
Although both 4-N(CH3)2 (3fa) and 4-NH2 (13b) substituted AT-CAAs were more potent than was 1a against prostate cancer cells (average IC50 = 2.8 μM for 3fa, 1.8 μM for 13b, and 6.9 μM for 1a), they were less potent than was 1b (average IC50 = 1.1 μM and selective ratio = 6.3 for 1b). Methanesulfonamide (15a) or 2-chloroacetamide (15b) were less potent (IC50 = 5.7−113.6 μM) and less toxic (IC50 = 25.4->100 μM) on melanoma cell lines compared with 1b. Introducing a ureido, 4-NHCONH2 (15c) instead of a 4-NHCOCH3 kept partial potency (average IC50 = 3.9 μM) but resulted in loss of selectivity against melanoma cells (1.8-fold for 15c vs 8.9-fold for 1b). A similar trend with 15a and 15c was found in prostate cancer, except that 15b showed improved growth inhibition of prostate cancer cells (average IC50 of 15b is 1.0 μM compared with 6.9 μM for 1a and 1.1 μM for 1b) and a 9.4-fold selectivity against prostate cancer cells.
We also investigated the importance of o-, m-, and p-substitu-tions in the 2-phenyl ring relative to acetylamino and methoxy groups. Interestingly, o-, m-, and p-isomers showed different activities. p-Isomers (3ea, 1b) had the best selectivity and activity for both cancer cell lines. m-Isomers (3da, 3ha) had similar average IC50s against melanoma compared with p-isomers, but their toxicity also increased on both fibroblast (2.5 μM and 6.3 μM) and RH7777 cells (3.6 μM and 4.9 μM). o-MeO and o-NHAc analogues (3ca, 3ga) showed slightly less potency and selectivity on both melanoma and prostate cancer cells.
3.4. Effects of 4-linkage of ATCAA and thiazolidine ring
To investigate the importance of amide linkage on C4 of ATCAA, the carboxamide was replaced with an ester and amine. The isosteric replacement of the amide by an ester resulted in 5 with markedly decreased activity against all cancer cell lines. The amine derivatives 8a and 8b also failed to show any increased potency, although they did show lower toxicity (60–84.5 μM) than did 1a (29.8 μM) and 1b (16 μM) in fibroblast cells.
Thiazolidine-4-carboxylic acid has been reported to be effective for treating advanced cancers.22,23 To examine the importance of a central thiazolidine ring, we synthesized 3-acetyl-thiazolidine compound 4 and a series of thiazolidine ring-opened compounds 16a–16d as shown in Schemes 1 and 3. Antiproliferative activity of 16a–16d against melanoma is summarized in Table 3. When acetyl was introduced to 3-NH of thiazolidine, activity decreased in both cancer cell lines (average IC50 = 16 and 10.3 μM against melanoma and prostate cancer, respectively). Cleavage of both the C-S and C-N bond (16a) led to substantial decrease in both potency (12.9–14.4 μM on melanoma cell lines) and toxicity (61.2 μM on the control cells) compared with 1b and 3ja (0.7–2.1 μM on cancer cells, 2.0–16.0 μM on control cells). Opening the thiazolidine ring from the C-S bond (16b) caused a loss of potency on both cancer and control cells (>100 μM). Cleavage of the C-N bond in the thiazolidine ring (16c) also resulted in decreased activity (12.8–15.1 μM) and no improvement on selectivity compared with 3ja. Removing the C-S bond (16d) reduced the activity of 1b from IC50 = 1.12.1 μM to 10.2–11.9 μM. These results suggested that the presence of the thiazolidine ring is critical for activity. Attempts to reduce the amide with LAH or B2H6 resulted in the C-S bond cleavage in thiazolidine. A disulfanyl dimer (9), which completely lost anticancer activity, was obtained, and this result is consistent with the above finding that the thiazolidine ring played an essential role in ATCAA potency.
Table 3.
Antiproliferative effects of thiazolidine ring-open compounds against two melanoma cell lines and fibroblast cell line
| ID | IC50 ± SEM (μM) | ||
|---|---|---|---|
| A375 | WM-164 | Fibroblast | |
| 16a | 12.9 ± 1.3 | 14.4 ± 0.8 | 61.2 ± 3.6 |
| 16b | 108.5 ± 4.8 | 144.9 ± 5.2 | >100 |
| 16c | 15.1 ± 1.1 | 12.8 ± 2.1 | 16.4 ± 1.2 |
| 16d | 11.9 ± 0.6 | 10.2 ± 0.5 | 24.0 ± 1.3 |
| 3ja | NDa | 0.7 ± 0.1 | 2.2 ± 0.2 |
| 1b | 2.1 ± 0.2 | 1.1 ± 0.1 | 16.0 ± 2.5 |
ND = not determined
3.5. Flow cytometric analysis
Flow cytometric analysis was performed on human melanoma A375 cells and human prostate cancer LNCaP cells to examine the effect of the new synthetics on cell cycle progression. Analysis of the DNA content of control cells (Fig. 4A–D, top graphs) showed typical distribution of peaks corresponding to cells in G1/G0 phase (65.67–66.97%), S-phase (24.16–28.36%), and G2/M-phase (5.97–8.87%). Upon treatment with compounds 3ad and 1b at 1-, 5-, 10-, and 20-μM concentrations for 48 h, flow cytometric analysis showed that these compounds significantly induced apoptosis (sub-G1 phase) in cancer cells in a dose-dependent manner. In both cell lines, the accumulation of cells in sub-G1 phase became apparent at 5 μM (Fig. 4).
Figure 4.

Flow cytometric analysis of selected ATCAA compounds 3ad and 1b. Effect of selected compounds on cell cycle progression of prostate cancer LNCaP and melanoma A375 cells was examined according to the procedure described in Section 5. Sub-G1 phase accumulation was induced by ATCAA compounds.
3.6. Intracellular calcium mobilization assays of ATCAA
The initial design of ATCAA was derived from contriving mimics of lysophosphatidic acid (LPA), a lipid mediator generated via the regulated breakdown of membrane phospholipids that are known to stimulate G-protein-coupled receptors (GPCR) signaling. GPCR activation could be detected through changes in intracellular calcium concentration. To examine whether ATCAA compounds inhibit cancer cell growth via LPA-GPCR pathway, a fluorescence imaging plate reader FLIPR assay (Ca2+ flux response) was conducted to screen 3ad for dose-dependent agonist and antagonist activities on the LPA1-, LPA3-, and LPA5-transfected Chem-1 cells. Oleoyl-LPA and a known LPA antagonist compound Ki1642524 were used as a positive and negative control, respectively. In agonist assay mode, oleoyl-LPA provided EC50 values of 31 nM, 120 nM, and 12 nM for LPA1, LPA3, and LPA5, respectively. When referencing Emax of oleoyl-LPA, 15% activation was considered as an agonist. Compound 3ad did not exhibit any dose-dependent agonist activity on LPA1, LPA3, or LPA5 (Fig. 5). We also tested antagonism of 3ad, and 50% inhibition was considered as an antagonist when referencing EC80. The negative control, Ki16425, had an IC50 value of 46 nM on LPA1. However, 3ad did not exhibit any dose-dependent antagonist activity on the LPA1, LPA3, or LPA5 (Fig. 6). The FLIPR assay results suggested that 3ad did not display cytotoxicity as a result of antagonism or agonism of the LPA receptors. ATCAA compounds exhibit cytotoxicity through other mechanisms.
Figure 5.

FLIPR intracellular calcium mobilization assay in agonist mode. Calcium flux in LPA1-, LPA3-, and LPA5-expressing Chem-1 cell lines induced by oleoyl-LPA. Oleoyl-LPA increased [Ca2+]i in a dose-dependent manner; 3ad and LPA antagonist Ki15425 did not show agonist effect.
Figure 6.

(A) Response of 3ad and oleoyl-LPA on LPA1, LPA3, and LPA5 kinetic data. Based upon these kinetic traces exhibited by 3ad, the relatively small Ca2+ flux is nonLPA receptor mediated. (B) Antagonist data of 3ad, which did not show antagonism on LPA1, LPA3, or LPA5. Ki16425 had an IC50 = 46 nM on LPA1.
3.7. ATCAA inhibits human melanoma colony formation
Cells grown in 96-well plates do not reflect many properties associated with three-dimensional tumors. Human tumor colony formation assay (ex vivo soft agar colony assay) has been suggested as an in vitro method to predict the response of an individual patient’s tumor to chemotherapeutic agents.25,26 As a prelude to in vivo animal experiments, we investigated the ability of 1b and 3ad to inhibit melanoma colony formation by using a well-established method.27–29 We performed studies with A375 human melanoma cells. Compounds 1b and 3ad could effectively inhibit A375 colony formation at 2 μM, the lowest tested concentration (Fig. 7). At a concentration of 20 μM, compound 3ad almost completely inhibited colony formation, while compound 1b inhibited colony formation by 75%. At the highest tested concentration, 100 μM, melanoma colony formation was completely inhibited by both 1b and 3ad.
Figure 7.

Compounds 3ad and 1b inhibit melanoma colony formation.
3.8. Antitumor efficacy of ATCAA on human melanoma cancer A375 xenografted athymic nude mice
We chose human melanoma A375 cells to test efficacy in vivo because ATCAA exhibited potency both in vitro assay and colony formation assay against A375 cells. A375 xenograft tumors also exhibited faster tumor growth rate. While compound 3ad was more potent than was 1b in inhibiting melanoma colony formation, it had much poorer selectivity between melanoma cancer cells and fibroblast cells (2.0-fold for 3ad vs 8.9-fold for 1b) as shown in Table 1. Therefore, we chose 1b for our in vivo studies against A375 melanoma tumors. ATCAA compound 1b was examined for in vivo evaluation and was formulated with 80% Tween 80 and 20% Captex 200 because of its limited water solubility.30 We first determined the maximally tolerated dose (MTD) in ICR mice. These mice are less expensive than nude mice and are commonly used for toxicity testing. The MTD for compound 1b was determined to be 42 mg/kg. Two nontoxic dosages (5 mg/kg and 10 mg/kg) were used. We also included DTIC (60 mg/kg),31 the gold standard for melanoma treatment, as a positive control to assess the efficacy of compound 1b. Briefly, male athymic nude mice were injected subcutaneously with 2.5 × 106 A375 cells. Treatment began on day 7 after tumor inoculation, and 1b was injected once a day. Each group was composed of eight mice. The results are shown in Figure 8. Compound 1b at 5 mg/kg showed moderate melanoma tumor growth inhibition. After 22 days of treatment, the percentage of tumor reduction was 25%. At a higher dose of 10 mg/kg, compound 1b showed significant melanoma tumor growth inhibition with 62% of tumor reduction. Compared with DTIC at a dose of 60 mg/kg, which inhibited tumor growth by 42% at the end of treatment, compound 1b clearly showed superior activity in this human melanoma xenograft model. All mice displayed normal activities, and no significant body weight loss was observed during the experiment.
Figure 8.

Growth of A-375 tumor treated with compound 1b or DTIC. (A) in vivo results of treatment of 1b and DTIC on nude mice bearing A375 tumors compared with control group. (B) Average body weight change in control and treatment groups.
4. Conclusions
We synthesized a series of ATCAA compounds and thiazolidine ring-opened analogues. Chemical modification and structure-activity relationship of ATCAA compounds were investigated with different substituted 2-phenyl, thiazolidine ring, 4-position linkage, and 4-carboxamide groups (Fig. 9) based on biological evaluation against melanoma and prostate cancer cells in vitro. Two compounds, 3id and 3ac, and another, 15b, are promising agents against melanoma and prostate cancer cells, respectively. Compounds 1b and 3ad effectively inhibited A375 melanoma colony formation and induced apoptosis. Compound 1b at 10 mg/kg significantly inhibited melanoma tumor growth in vivo and showed higher efficacy than did DTIC at 60 mg/kg. These compounds did not seem to work via the LPA-GPCR pathway. Additional work is under way to investigate the mechanisms of action of ATCAA and to perform further in vivo studies with other active ATCAA compounds and in prostate cancer xenografted models.
Figure 9.

SAR relationship of ATCAA.
5. Experimental section
5.1. General
All reagents were purchased and used without further purification from Sigma-Aldrich Chemical Co. (St. Louis, MO), Fisher Scientific (Pittsburgh, PA) and AK Scientific, Inc (Mountain View, CA). Moisture-sensitive reactions were conducted in an argon atmosphere. Argon gas was purchased from NexAir Medical Gas, Inc., TN. Routine thin layer chromatography (TLC) was performed on aluminum-backed uniplates (Analtech, Newark, DE). NMR spectra were obtained on a Bruker AX 300 (Billerica, MA) spectrometer. Chemical shifts were reported as parts per million (ppm) relative to TMS in CDCl3 or DMSO-d6. Mass spectral data were collected on a Bruker ESQUIRE electrospray/ion trap instrument in positive and negative ion modes. Elemental analyses were performed by Atlantic Microlab Inc. (Norcross, GA).
5.2. General procedure for preparing (2RS,4R)-2-arylthiazolidine-3, 4-dicarboxylic acid 3-tert-butyl ester 2a-2j
A mixture of L-cysteine (3.16 g, 26.11 mmol) and appropriate aldehyde (26.15 mmol) in ethanol (300 mL) and water (30 mL) was stirred at room temperature for 6–15 h, and the separated solid was collected, washed with diethyl ether, and dried to obtain (2RS,4 R)-2-arylthiazolidine-4-carboxylic acid (ATCA) with yields of 70–99%. At 0 °C, ATCA (5.95 mmol) was dissolved in 1 N NaOH (6 mL) and 1,4-dioxane (15 mL); then di-tert-butyldicarbonate (2.80 g, 12.80 mmol) was added slowly and stirred at room temperature for 1 h. The reaction mixture was concentrated in a vacuum and washed with ethyl acetate (20 mL). The aqueous phase was adjusted to pH 4 by adding 1 N HCl or 5% KHSO4, then extracted with ethyl acetate, dried with magnesium sulfate, filtered, and concentrated in a vacuum to give corresponding compounds 2a–2j as white foam-solids with yields of 89–97%. 2a–2j were used further in the next step of synthesis without purification.
5.3. General procedure for preparing (2RS,4R)-2-arylthiazolidine-4-carboxylic acid amide 1a, 1b, and 3aa-3jc
A mixture of appropriate Boc-protected carboxylic acids (2a–2j, 0.3–0.5 g), EDCI (1.2 equiv), and HOBT (1.05 equiv) in CH2Cl2 (20 mL) was stirred at room temperature for 10 min. To this solution, appropriate amine (1.05 equiv) and Et3N (1.2 equiv) were added, and stirring continued at room temperature for 6–15 h. The reaction mixture was diluted with CH2Cl2 (30 mL) and sequentially washed with water, satd NaHCO3, and brine and dried over MgSO4. The solvent was removed under reduced pressure to yield a crude oil, which was stirred with TFA (0.6–1 mL) in 20 mL CH2Cl2 at room temperature for 1–8 h to cleave the Boc group. The reaction mixture was concentrated, washed with satd NaHCO3, and dried over MgSO4. The solvent was removed to yield a crude solid; ATCAAs 1a–3jc were purified by silica column flash chromatography using hexane/ethyl acetate gradient elution system. Yield was reported as 2-step yield.
5.3.1. (2RS,4R)-2-Phenyl-thiazolidine-4-carboxylic acid octadecylamide (1a)
Yield: 76.1%. 1H NMR (300 MHz, CDCl3) δ 7.63–7.37 (m, 6H), 5.70 (br, 2H), 5.67 and 5.49 (s, s, 1H, 0.3H and 0.7H), 4.90 and 4.63 (br, t, J = 4.5 Hz, 1H), 3.66–3.46 (m, 2H), 3.30–3.26 (m, 2H), 1.54–1.50 (m, 2H), 1.25 (s, 30H), 0.87 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 461.4 [M+H]+. Anal. Calcd for C28H48N2OSCF3COOH: C, 62.69; H, 8.59; N, 4.87. Found: C, 62.94; H, 8.84; N, 4.95.
5.3.2. (2RS,4R)-2-(4-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide (1b)
Yield: 61.3%. 1H NMR (300 MHz, CDCl3) δ 7.54−7.43 (d,d, 4H, J = 8.4 Hz), 7.25 (br, 1H), 5.62 and 5.33 (s, s, 0.3H and 0.7H), 4.36 and 4.05 (br, 1H), 3.69 and 3.43 (dd, m, 2H, J = 4.2 Hz), 3.31−3.24 (m, 2H), 2.19 (s, 3H), 1.53−1.47 (m, 4H), 1.26 (s, 26H), 0.88 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 490.5 [M+H]+, 512.5 [M+Na]+, 488.1 [M−H]−. Anal. Calcd for C28H47N3O2S: C, 68.51; H, 9.61; N, 8.48. Found: C, 68.67; H, 9.67; N, 8.58.
5.3.3. (2RS,4R)-2-(3,4,5-Trimethoxy-phenyl)-thiazolidine-4-carboxylic acid octadecylamide (3aa)
Yield: 65.3%. 1H NMR (300 MHz, CDCl3) δ 7.20 and 6.31 (t, 0.7H, J = 6.6 Hz and br, 0.3H), 6.77 and 6.71 (s, s, 0.7H and 1.3H), 5.55 and 5.28 (s, s, 0.3H and 0.7H), 4.34 and 3.93 (dd, J = 7.8 Hz, 4.2 Hz, m, 1H), 3.88 and 3.87 (s, s, 6H), 3.80 and 3.79 (s, s, 3H), 3.68 and 3.42 (dd, J =11.1 Hz, 4.2 Hz, m, 2H), 3.31–3.19 (m, 2H), 2.52 (br, 1H), 1.50 (m, 2H), 1.25 (s, 30H), 0.87 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 551.5 [M+H]+, 573.5 [M+Na]+, 549.2 [M−H]−. Anal. Calcd for C31H54N2O4S: C, 67.59; H, 9.88; N, 5.09. Found: C, 67.38; H, 9.95; N, 5.09.
5.3.4. (2RS,4R)-2-(3,4,5-Trimethoxy-phenyl)-thiazolidine-4-carboxylic acid octadec-8-cis-enylamide (3ab)
Yield: 58.8%.1H NMR (300 MHz, CDCl3) δ 7.20 and 6.31 (t, 0.7H, J = 6.0 Hz and br, 0.3H), 6.77 and 6.71 (s, s, 0.7H and 1.3H), 5.55 and 5.28 (s, s, 0.3H and 0.7H), 5.37−5.32 (m, 2H), 4.34 and 3.94 (dd, J = 7.2 Hz, 4.2 Hz, m, 1H), 3.88 and 3.87 (s, s, 6H), 3.84 and 3.84 (s, s, 3H), 3.68 and 3.43 (dd, J =11.1 Hz, 4.2 Hz, m, 2H), 3.33−3.21 (m, 2H), 2.01−1.97 (br, 4H), 1.97 (br, 1H), 1.55−1.44 (m, 2H), 1.26 (s, 22H), 0.87 (t, 3H, J =6.9 Hz). MS (ESI) m/z 549.5 [M+H]+, 547.2 [M−H]−. Anal. Calcd for C31H52N2O4S: C, 67.84; H, 9.55; N, 5.10. Found: C, 67.49; H, 9.55; N, 5.12.
5.3.5. (2RS,4R)-2-(3,4,5-Trimethoxy-phenyl)-thiazolidine-4-carboxylic acid octadec-8-trans-enylamide (3ac)
Yield: 42.1%. 1H NMR (300 MHz, CDCl3) δ 7.19 and 6.29 (t, 0.7H, J = 6.3 Hz and br, 0.3H), 6.77 and 6.71 (s, s, 0.7H and 1.3H), 5.55 and 5.28 (s, s, 0.3H and 0.7H), 5.37 (br, 2H), 4.34 and 3.95 (dd, J = 7.8 Hz, 4.2 Hz, m, 1H), 3.88 and 3.87 (s, s, 6H), 3.84 (s, 3H), 3.68 and 3.43 (dd, J =11.1 Hz, 4.2 Hz, m, 2H), 3.31–3.19 (m, 2H), 1.97 (br, 4H), 1.55–1.46 (m, 2H), 1.26 (s, 22H), 0.87 (t, 3H, J = 6.6 Hz). MS (ESI) m/z 583.2 [M+Na]+, 549.5 [M+H]+, 547.2 [M−H]−. Anal. Calcd for C31H52N2O4S: C, 67.84; H, 9.55; N, 5.10. Found: C, 68.10; H, 9.62; N, 5.11.
5.3.6. (2RS,4R)-2-(3,4,5-Trimethoxy-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide (3ad)
Yield: 46.7%. 1H NMR (300 MHz, CDCl3) δ 7.20 and 6.30 (t, 0.7H, J = 6.3 Hz and br, 0.3H), 6.77 and 6.71 (s, s, 0.7H and 1.3H), 5.55 and 5.28 (d, d, 0.3H, J = 10.2 Hz and 0.7H, J = 10.8 Hz), 4.34 and 3.93 (dd, br, m, J = 4.2 Hz, 1H), 3.89 and 3.88 (s, s, 6H), 3.85 and 3.84 (s, s, 3H), 3.68 and 3.45−3.39 (dd, m, 2H), 3.38−3.19 (m, 2H), 2.63 and 2.52 (br, br, 1H), 1.53 (m, 2H), 1.25 (s, 26H), 0.87 (t, 3H, J = 6.3 Hz). MS (ESI) m/z 524.1 [M+H]+, 523.8 [M−H]−. Anal. Calcd for C29H50N2O4S: C, 66.63; H, 9.64; N, 5.36. Found: C, 66.41; H, 9.63; N, 5.30.
5.3.7. (2RS,4R)-2-(3,4,5-Trimethoxy-phenyl)-thiazolidine-4-carboxylic acid tetradecylamide (3ae)
Yield: 51.0%. 1H NMR (300 MHz, CDCl3) δ 7.20 and 6.27 (t, 0.7H, J = 6.3 Hz and br, 0.3H), 6.77 and 6.71 (s, s, 0.7H and 1.3H), 5.56 and 5.28 (d, d, J =10.2 Hz and 0.7H, J =10.5 Hz), 4.34 and 3.93 (dd, J = 4.2 Hz, m, 1H), 3.89 and 3.88 (s, s, 6H), 3.85 and 3.84 (s, s, 3H), 3.68 and 3.45–3.36 (dd, m, 2H), 3.34−3.19 (m, 2H), 2.51 (br, 1h), 1.50 (m, 2H), 1.25 (s, 22H), 0.87 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 495.6 [M+H]+. Anal. Calcd for C27H46N2O4S: C, 65.55; H, 9.37; N, 5.66. Found: C, 65.49; H, 9.41; N, 5.59.
5.3.8. (2RS,4R)-2-(3,4,5-Trimethoxy-phenyl)-thiazolidine-4-carboxylic acid dodecylamide (3af)
Yield: 63.9%. 1H NMR (300 MHz, CDCl3) δ 7.20 and 6.31 (t, br, 0.7H and 0.3H), 6.77 and 6.71 (s, s, 0.7H and 1.3H), 5.55 and 5.29 (d, d, br, 0.3H, J = 7.8 Hz and 0.7H, J = 7.5 Hz), 4.34 and 3.93 (br, m, 1H), 3.89 and 3.88 (s, s, 6H), 3.85 and 3.84 (s, s, 3H), 3.68 and 3.47−3.36 (m, m, 2H), 3.33−3.19 (m, 2H), 2.53 (br, 1H), 1.53 (m, 2H), 1.25 (s, 18H), 0.87 (t, 3H, J =6.9 Hz). MS (ESI) m/z 490.3 [M+Na]+. Anal. Calcd for C25H42N2O4S: C, 64.34; H, 9.07; N, 6.00. Found: C, 64.29; H, 9.06; N, 5.92.
5.3.9. (2RS,4R)-2-(3,4,5-Trimethoxy-phenyl)-thiazolidine-4-carboxylic acid decylamide (3ag)
Yield: 50.0%. 1H NMR (300 MHz, CDCl3) δ 7.21 and 6.32 (br, br, 0.7H and 0.3H), 6.77 and 6.71 (s, s, 0.7H and 1.3H), 5.55 and 5.28 (s, s, 0.3H and 0.7H), 4.34 and 3.94 (dd, J = 7.8 Hz and 4.2 Hz, m, 1H), 3.89 and 3.88 (s, s, 6H), 3.85 and 3.84 (s, s, 3H), 3.68 and 3.45–3.36 (dd, m, 2H), 3.34−3.19 (m, 2H), 1.53 (m, 2H), 1.25 (s, 14H), 0.87 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 461.9 [M+Na]+. Anal. Calcd for C23H38N2O4S: C, 62.98; H, 8.73; N, 6.39. Found: C, 62.98; H, 8.71; N, 6.28.
5.3.10. (2RS,4R)-2-(3,4,5-Trimethoxy-phenyl)-thiazolidine-4-carboxylic acid octylamide (3ah)
Yield: 27.7%. 1H NMR (300 MHz, CDCl3) δ 7.21 and 6.34 (br, br, 0.7H and 0.3H), 6.77 and 6.71 (s, s, 0.7H and 1.3H), 5.55 and 5.28 (s, s, 0.3H and 0.7H), 4.34 and 3.93 (dd, J = 7.8 Hz and 4.2 Hz, m, 1H), 3.89 and 3.88 (s, s, 6H), 3.85 and 3.84 (s, s, 3H), 3.68 and 3.45−3.33 (dd, J = 10.8 Hz and 7.8 Hz, m, 2H), 3.31−3.19 (m, 2H), 2.61 (br, 1H), 1.53 (m, 2H), 1.30−1.26 (br, 30H), 0.87 (t, 3H, J =6.9 Hz). MS (ESI) m/z 411.8 [M+H]+, 433.5 [M+Na]+. Anal. Calcd for C21H34N2O4S: C, 60.11; H, 8.41; N, 6.68. Found: C, 60.36; H, 8.24; N, 6.56.
5.3.11. (2RS,4S)-2-(3,4,5-Trimethoxy-phenyl)-thiazolidine-4-carboxylic acid tetradecylamide (3ai)
Yield: 49.4%. 1H NMR (300 MHz, CDCl3) δ 7.20 and 6.30 (t, br, 0.7H and 0.3H), 6.77 and 6.71 (s, s, 0.7H and 1.3H), 5.56 and 5.28 (s, s, 0.3H and 0.7H), 4.34 and 3.93 (br, m, 1H), 3.89 and 3.88 (s, s, 6H), 3.85 and 3.84 (s, s, 3H), 3.68 and 3.45−3.35 (dd, J = 3.9 Hz, m, 2H), 3.34−3.19 (m, 2H), 2.51 (br, 1H), 1.50 (m, 2H), 1.25 (s, 22H), 0.87 (t, 3H, J = 6.6 Hz). MS (ESI) m/z 495.6 [M+H]+. Anal. Calcd for C27H46N2O4S: C, 65.55; H, 9.37; N, 5.66. Found: C, 65.29; H, 9.42; N, 5.66.
5.3.12. (2RS,4R)-2-(3,4-Dimethoxy-phenyl)-thiazolidine-4-carboxylic acid octadecylamide (3ba)
Yield: 29.0%. 1H NMR (300 MHz, CDCl3) δ 7.51−6.97 (m, 3H), 6.85 (t, 1H), 5.64 and 5.37 (s, s, 1H), 4.69 and 4.51 (dd, br, m, 1H), 3.91, 3.90, 3.89 and 3.88 (s, s, s, s, 6H), 3.64−3.42 (m, 2H), 3.35−3.23 (m, 2H), 2.52 (br, 1H), 1.51–1.43 (m, 2H), 1.25 (s, 30H), 0.87 (t, 3H, J =6.9 Hz). MS (ESI) m/z 521.5 [M+H]+. Anal. Calcd for C30H52N2O3S: C, 69.18; H, 10.06; N, 5.38. Found: C, 69.36; H, 10.01; N, 5.36.
5.3.13. (2RS,4R)-2-(3,4-Dimethoxy-phenyl)-thiazolidine-4-carboxylic acid (Z)-octadec-8-enylamide (3bb)
Yield: 29.1%. 1H NMR (300 MHz, CDCl3) δ 7.27 and 6.38 (br, 0.7H and 0.3H), 7.11−6.80 (m, 3H), 5.58 and 5.31 (s, s, 0.3H and 0.7H), 5.39−5.34 (m, 2H), 4.35 and 4.00 (dd, J = 7.8 Hz and 4.2 Hz, m, 1H), 3.93, 3.92, 3.90 and 3.89 (s, s, s, s, 6H), 3.70 and 3.44 (dd, J =10.8 Hz and 4.2 Hz, m, 2H), 3.34−3.19 (m, 2H), 2.03−1.99 (br, 4H), 1.56–1.44 (m, 2H), 1.28 (s, 22H), 0.89 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 519.3 [M+H]+. Anal. Calcd for C31H50N2O4S: C, 69.45; H, 9.71; N, 5.40. Found: C, 69.17; H, 9.61; N, 5.18.
5.3.14. (2RS,4R)-2-(3,4-Dimethoxy-phenyl)-thiazolidine-4-carboxylic acid (E)-octadec-8-enylamide (3bc)
Yield: 13.3%. 1H NMR (300 MHz, CDCl3) δ 7.26 and 6.33 (br, 1H), 7.09–6.82 (m, 3H), 5.57 and 5.29 (s, s, 0.3H and 0.7H), 5.37 (br, 2h), 4.34 and 3.95 (dd, J = 8.1 Hz and 4.2 Hz, m, 1H), 3.91, 3.90, 3.89 and 3.88 (s, s, s, s, 6H), 3.69 and 3.43 (dd, J = 11.1 Hz and 4.2 Hz, m, 2H), 3.33−3.17 (m, 2H), 1.96 (br, 4H), 1.52−1.43 (m, 2H), 1.26 (s, 22H), 0.87 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 519.5 [M+H]+, 541.3 [M+Na]+, 517.2 [M−H]−. Anal. Calcd for C31H50N2O4S: C, 69.45; H, 9.71; N, 5.40. Found: C, 69.57; H, 9.86; N, 5.20.
5.3.15. (2RS,4R)-2-(2-Methoxy-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide (3ca)
Yield: 38.8%. 1H NMR (300 MHz, CDCl3) δ 7.52−7.27 and 7.04−6.92 (m, 4H), 7.35 (br, 1H), 5.93 and 5.63 (s, s, 1H), 4.37 and 4.23 (dd, J =7.8 Hz and 4.2 Hz, br, 1H), 3.92 and 3.89 (s, s, 3H), 3.67, 3.42−3.35 (dd, m, 2H), 3.33−3.20 (m, 2H), 1.53 (m, 2H), 1.25 (s, 26H), 0.88 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 463.4 [M+H]+. Anal. Calcd for C27H46N2O2S: C, 70.08; H, 10.02; N, 6.05. Found: C, 69.97; H, 9.96; N, 6.02.
5.3.16. (2RS,4R)-2-(3-Methoxy-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide (3da)
Yield: 33.6%. 1H NMR (300 MHz, CDCl3) δ 7.47−6.87 (four doublets, 4H, J = 8.7 Hz), 7.30 (br, 1H), 5.64 and 5.33 (s, s, 1H), 4.36 and 4.25 (dd, J = 4.2 Hz, br, 1H), 3.88 and 3.83 (s, s, 3H), 3.69, 3.46−3.35 (dd, J =10.2 Hz and 4.2 Hz, m, 2H), 3.30−3.24 (m, 2H), 1.53–1.48 (m, 2H), 1.25 (s, 26H), 0.88 (t, 3H, J =7.2 Hz). MS (ESI) m/z 485.5 [M+Na]+. Anal. Calcd for C27H46N2O2S: C, 70.08; H, 10.02; N, 6.05. Found: C, 69.88; H, 9.93; N, 5.97.
5.3.17. (2RS,4R)-2-(4-Methoxy-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide (3ea)
Yield: 28.8%. 1H NMR (300 MHz, CDCl3) δ 7.86–6.88 (m, 4H), 7.36 (br, 1H), 5.69 and 5.32 (s, s, 1H), 4.39 and 4.46 (dd, br, 1H), 3.90 and 3.82 (s, s, 3H), 3.68, 3.50−3.40 (dd, m, 2H), 3.30−3.24 (m, 2H), 1.53−1.45 (m, 2H), 1.26 (s, 26H), 0.88 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 463.1 [M+H]+. Anal. Calcd for C27H46N2O2S: C, 70.08; H, 10.02; N, 6.05. Found: C, 70.13; H, 10.12; N, 6.00.
5.3.18. (2RS,4R)-2-(4-Dimethylamino-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide (3fa)
Yield: 62.5%. 1H NMR (300 MHz, CDCl3) δ 7.33 and 6.70 (dd, J =8.7 Hz, 4H), 7.39 and 6.43 (br, 1H), 5.55 and 5.25 (s, s, 1H), 4.33 and 3.87 (dd, J = 4.2 Hz, t, 1H), 3.69 and 3.45−3.39 (dd, J= 4.2 Hz, m, 2H), 3.32−3.12 (m, 2H), 2.96 and 2.95 (s, s, 3H), 1.55–1.46 (m, 2H), 1.25 (s, 26H), 0.87 (t, 3H). MS (ESI) m/z 476.4 [M+H]+. Anal. Calcd for C28H49N3OS: C, 70.68; H, 10.38; N, 8.83. Found: C, 70.61; H, 10.49; N, 8.70.
5.3.19. (2RS,4R)-2-(2-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide (3ga)
Yield: 52.7%. 1H NMR (300 MHz, CDCl3) δ 8.65 (s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.50 (d, J = 7.8 Hz, 1H), 7.35 (t, J = 7.8 Hz, 1H), 7.16 (t, J = 7.8 Hz, 1H), 6.78 (br, 1H), 5.52 and 5.48 (s, s, 1H), 4.16 (br, 1H), 3.61 and 3.39−3.32 (dd, m, 2H), 3.30–3.20 (m, 2H), 2.90 (br, 1H), 2.25 (s, 3H), 1.54−1.50 (m, 2H), 1.25 (s, 26H), 0.87 (t, 3H, J =6.9 Hz). MS (ESI) m/z 512.3 [M+Na]+, 488.1 [M−h]−. Anal. Calcd for C28H47N3O2S: C, 68.51; H, 9.61; N, 8.48. Found: C, 68.57; H, 9.55; N, 8.47.
5.3.20. (2RS,4R)-2-(3-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide (3ha)
Yield: 70.1%. 1H NMR (300 MHz, CDCl3) δ 7.75 and 7.65 (s, s, 0.7H, 0.3H), 7.51–7.22 (m, 4H), 7.19 and 6.48 (br, 1H), 5.59 and 5.34 (s, s, 0.3H, 0.7H), 4.32 and 3.93 (dd, J = 3.9 Hz, t, J = 6.9 Hz, 0.7H, 0.3H), 3.68 and 3.42−3.34 (dd, J = 3.9 Hz, m, 2H), 3.31–3.16 (m, 2H), 2.60 (br, 1H), 2.17 (s, 3H, J = 6.9 Hz), 1.52 (m, 2H), 1.25 (s, 26H), 0.87 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 512.3 [M+Na]+. Anal. Calcd for C28H47N3O2S: C, 68.51; H, 9.61; N, 8.48. Found: C, 68.54; H, 9.65; N, 8.49.
5.3.21. (2RS,4R)-2-(4-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid (1-methyl-octadecyl)-amide (3ia)
Yield: 68.7%. 1H NMR (300 MHz, CDCl3) δ 9.91 (s, 1H) 7.56 (s, br, 4H), 7.49 (s, br, 1H), 5.77 and 5.76 (s, s, 1H), 4.89 and 4.79 (dd, J = 3.9 Hz, t, br, 0.6H, 0.4H), 3.67 and 3.44 (m, t, 2H), 3.37−3.19 (m, 2H), 3.08 and 2.97 (s, s, 3H), 2.18 (s, 3H), 1.56 (m, 2H), 1.25 (s, 30H), 0.87 (t, 3H, J = 7.2 Hz). MS (ESI) m/z 532.3 [M+Na]+. Anal. Calcd for C31H53N3O2SCF3COOH·H2O: C, 59.70; H, 8.50; N, 6.33. Found: C, 60.15; H, 8.71; N, 6.27.
5.3.22. (2RS,4R)-2-(4-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid adamantan-1-ylamide (3ib)
Yield: 73.0%. 1H NMR (300 MHz, DMSO-d6) δ 10.04 (s, 1H), 7.79 (br, 1H), 7.50 (dd, J = 8.7 Hz, 4H), 5.74 and 5.58 (s, s, 1H), 4.27 and 3.96 (br, 1H), 3.45–3.35 and 3.16−2.99 (m, m, 2H), 2.04 (s, 3H), 1.94 (s, 6H), 1.62 (s, 6H). MS (ESI) m/z 400.3 [M+H]+. Anal. Calcd for C22H29N3O2SCF3COOH: C, 56.13; H, 5.89; N, 8.18. Found: C, 56.39; H, 5.89; N, 8.18.
5.3.23. (2RS,4R)-2-(4-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid adamantan-2-ylamide (3ic)
Yield: 45.3%. 1H NMR (300 MHz, DMSO-d6) δ 9.99 (s, 0.5H), 8.08 and 8.02 (dd, J = 7.2 Hz, 1H), 7.58–7.38 (m, 4H), 5.56 and 5.53 (s, s, 1H), 4.30 and 3.88 (t, J =6.0 Hz, br, 1H), 3.36 and 3.16−2.93 (dd, m, 2H), 2.03 (s, 3H), 1.82–1.49 (m, 15H). MS (ESI) m/z 400.3 [M+H]+, 398.0 [M−H]−. Anal. Calcd for C22H29N3O2S1/2 CF3COOH: C, 60.51; H, 6.51; N, 9.20. Found: C, 60.62; H, 6.83; N, 8.93.
5.3.24. (2RS,4R)-2-(4-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid-(9H-fluoren-2-yl)-amide (3id)
Yield: 45.7%. 1H NMR (300 MHz, DMSO-d6) δ 10.33 and 10.13 (s, s, 1H), 10.00 and 9.97 (s, s, 1H), 7.97 and 7.94 (s, br, 1H), 7.84−7.24 (m, 7H), 7.63–7.24 (m, 4H), 5.69 and 5.59 (s, s, 1H), 4.39 and 4.04 (t, J = 5.7 Hz, m, 1H), 3.91 (s, 2H), 3.47−3.29 and 3.13 (m, 2H), 2.05 (s, 3H). MS (ESI) m/z 430.1 [M+H]+. Anal. Calcd for C25H23N3O2S CF3− COOH: C, 56.13; H, 5.89; N, 8.18. Found: C, 56.01; H, 5.87; N, 8.01.
5.3.25. (2RS,4R)-2-(4-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid anthracen-2-yl)-amide (3ie)
Yield: 45.7%. 1H NMR (300 MHz, DMSO-d6) δ 10.49 and 10.27 (s, s, 1H), 10.00 and 9.97 (s, s, 1H), 8.55–8.46 (m, 3H), 8.06−8.03 (m, 3H), 7.68–7.44 (m, 4H), 7.58−7.44 (m, 4H), 5.70 and 5.60 (s, s, 1H), 4.43 and 4.07 (t, br, 1H), 3.49−3.35 and 3.17 (m, t, 2H), 2.04 (s, 3H). MS (ESI) m/z 442.1 [M+H]+. Anal. Calcd for C26H23N3O2SCF3COOH: C, 60.53; H, 4.35; N, 7.56. Found: C, 60.31; H, 4.37; N, 7.71.
5.3.26. (2RS,4R)-2-(4-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid biphenyl-4-ylamide (3if)
Yield: 35.1%. 1H NMR (300 MHz, CDCl3) δ 9.27 and 8.74 (s, s, 1H), 7.83–7.30 (m, 13H), 7.17 and 6.90 (s, br, 1H), 5.68 and 5.40 (s, 0.3H and s, 0.7H), 4.53 and 4.21 (dd, J = 3.9 Hz, t, 1H), 3.84−3.79 and 3.59−3.43 (dd, , J = 3.9 Hz, m, 2H), 2.16 and 2.19 (s, s, 3H). MS (ESI) m/z 440.1 [M+Na]+, 416.0 [M−H]−. Anal. Calcd for C24H23N3O2S: C, 69.04; H, 5.55; N, 10.06. Found: C, 68.87; H, 5.32; N, 10.01.
5.3.27. (2RS,4R)-2-(4-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid benzothiazol-2-ylamide (3ig)
Yield: 37.9%. 1H NMR (300 MHz, CDCl3) δ 10.56 (br, 1H), 7.857.78 (dd, J =14.4 Hz and 7.8 Hz, 2H), 7.55−7.41 (m, 5H), 7.36−7.29 (m, 2H), 5.68 and 5.41 (s, 0.15H and d, J = 9.6 Hz, 0.85H), 4.64 and 4.32 (br, t, 1H), 3.80 and 3.60–3.44 (m, 2H), 2.79 (br, 1H), 2.19 (s, 3H). MS (ESI) m/z 421.1 [M+Na]+. Anal. Calcd for C19H18N4O2S2: C, 57.26; H, 4.55; N, 14.06. Found: C, 57.17; H, 4.32; N, 14.01.
5.3.28. (2RS,4R)-2-(4-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid hexadec-9-cis-enylamide (3ih)
Yield: 18.1%. 1H NMR (300 MHz, CDCl3) δ 7.86−7.41 (m, 4H), 7.30 (br, 1H), 7.09 and 6.42 (br, t, 1H), 5.57 and 5.39 (s, s, 1h), 5.37–5.32 (m, 2H), 4.32 and 3.89 (dd, J = 4.2 Hz, t, J =7.8 Hz, 1h), 3.68 and 3.43−3.36 (dd, J = 4.2 Hz, m, 2H), 3.31–3.20 (m, 2H), 2.51 (br, 1H), 2.18 (s, 3H), 2.01−1.97 (br, 4H), 1.54–1.46 (m, 2H), 1.29 and 1,27 (s, s, 18H), 0.87 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 510.4 [M+Na]+, 486.1 [M−H]−. Anal. Calcd for C25H48N3O2S: C, 68.95; H, 9.30; N, 8.62.
5.3.29. (2RS,4R)-2-(4-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid nonadec-10-ynylamide (3ii)
Yield: 40.5%. 1H NMR (300 MHz, CDCl3) δ 7.83–7.41 (m, 4H), 7.26 (br, 1H), 7.23 and 6.43 (br, br, 1H), 5.57 and 5.31 (s, s, 1h), 4.32 and 3.90 (dd, J = 4.2 Hz, t, J = 7.2 Hz, 1H), 3.68 and 3.43−3.36 (dd, J = 4.2 Hz, m, 2H), 3.32–3.20 (m, 2H), 2.51 (br, 1H), 2.18 (s, 3H), 2.13 (t, 4H), 1.54−1.44 (m, 2H), 1.31 and 1,27 (s, s, 22H), 0.87 (t, 3H, J =6.6 Hz). MS (ESI) m/z 514.3 [M+H]+, 512.1 [M−H]−. Anal. Calcd for C30H47N3O2S: C, 70.13; H, 9.22; N, 8.18. Found: C, 70.21; H, 9.14; N, 8.05.
5.3.30. (2RS,4R)-2-Phenyl-thiazolidine-4-carboxylic acid hexadecylamide (3ja)
Yield: 39.3%. 1H NMR (300 MHz, CDCl3) δ 7.55–7.31 (m, 5H), 7.26 and 6.41 (br, br, 1H), 5.62 and 5.34 (d, J = 9.9 Hz, d, J = 11.4 Hz, 1H), 4.35 and 3.93 (dt, dd, , J =7.5 Hz and 4.2 Hz, 1H), 3.71 and 3.45–3.30 (dd, J = 4.2 Hz m, 2H), 3.30–3.19 (m, 2H), 2.66 and 2.53 (br, 1H), 1.52 (m, 2H), 1.25 (s, 26H), 0.88 (t, 3H, J =6.9 Hz). MS (ESI) m/z 455.4 [M+Na]+, 431.1 [M−H]−. Anal. Calcd for C26H44N2OS: C, 72.17; H, 10.25; N, 6.47. Found: C, 71.98; H, 10.02; N, 6.34.
5.3.31. (2RS,4R)-2-Phenyl-thiazolidine-4-carboxylic acid (Z)-octadec-8-enylamide (3jb)
Yield: 46.1%. 1H NMR (300 MHz, CDCl3) δ 7.53–7.31 (m, 5H), 7.26 and 6.42 (br, br, 1H), 5.61 and 5.34 (s, br, 1H), 5.34 (br, 2H), 4.35 and 3.93 (dd, J = 4.2 Hz, t, J = 7.5 Hz, 0.7H and 0.3H), 3.71 and 3.45–3.39 (dd, J = 4.2 Hz, m, 2H), 3.37−3.19 (m, 2H), 2.54 (br, 1H), 2.01−1.99 (m, 4H), 1.52−1.45 (m, 2H), 1.29 and 1.26 (s, 22H), 0.87 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 459.4 [M+H]+. Anal. Calcd for C28H46N2OS: C, 73.31; H, 10.11; N, 6.11. Found: C, 73.15; H, 10.09; N, 6.14.
5.3.32. (2RS,4R)-2-Phenyl-thiazolidine-4-carboxylic acid (E)-octadec-8-enylamide (3jc)
Yield: 35.7%. 1H NMR (300 MHz, CDCl3) δ 7.47–7.31 (m, 5H), 7.26 and 6.42 (br, br, 1H), 5.61 and 5.37 (br, br, 1H), 5.34−5.33 (br, J = 13.8 Hz, 2H), 4.35 and 3.91 (br, br, 0.7H and 0.3H), 3.71 and 3.45−3.37 (dd, J = 4.2 Hz, m, 2H), 3.34–3.19 (m, 2H), 2.66 and 2.54 (br, 1H), 1.96−1.95 (m, 4H), 1.54−1.42 (m, 2H), 1.26 (s, 22H), 0.87 (t, 3H, J =6.9 Hz). MS (ESI) m/z 459.4 [M+H]+. 457.1 [M−H]−. Anal. Calcd for C28H46N2OS: C, 73.31; H, 10.11; N, 6.11. Found: C, 73.36; H, 10.16; N, 6.12.
5.4. (2RS,4R)-3-Acetyl-2-(4-acetylamino-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide (4)
Under 0 °C, to a mixture of 1b (0.30 g, 0.61 mmol) and pyridine (0.10 g, 1.26 mmol) in 10 mL CH2Cl2, was added acetyl chloride (0.19 g, 2.42 mmol) and stirred at room temperature for 8 h. The reaction mixture was washed with satd NaHCO3 and dried over MgSO4. The solvent was concentrated and purified through column chromatography to yield 4 (0.24 g, 73.6%) as a white solid. 1H NMR (300 MHz, CDCl3) d 7.50 (br, 1H), 7.50−7.34 (d, J = 8.4 Hz, 4H), 7.02 and 6.55 (br, br, 1H), 5.97 (s, 1H), 5.07 and 4.70 (br, br, 1H), 3.67 and 3.14−3.08 (dd,J = 4.2 Hz, br, 2H), 3.31−3.29 (m, 2H), 2.17 (s, 3H), 1.98 (s, 3H), 1.54(m, 2H), 1.25 (s, 26H), 0.87 (t, 3H, J =6.9 Hz). MS (ESI) m/z 554.5 [M+Na]+, 530.2 [M−H]−. Anal. Calcd for C30H49N3O3S: C, 67.76; H, 9.29; N, 7.90. Found: C, 67.49; H, 9.40; N, 7.75.
5.5. (2RS,4R)-2-(4-Acetylamino-phenyl)-thiazolidine-4-carboxylic acid hexadecyl ester (5)
A mixture of Boc-protected carboxylic acid 2i (0.549 g, 1.50 mmol), DCC (0.372 g, 1.8 mmol), DMAP (0.055 g, 0.45 mmol), (1S)-(+)-10-camphorsulfonic acid (0.105 g, 0.45 mmol), and hexadecanol (0.726 g, 3.0 mmol) in CH2Cl2 (15 mL) was stirred at room temperature for 24 h. The reaction mixture was filtered, then washed with satd NaHCO3 and brine (10 mL), and dried over MgSO4. The pure Boc-protected ester was obtained after column chromatography (0.83 g, 93.8%). To a solution of Boc-protected ester (0.83 g, 1.41 mmol) in CH2Cl2 (10 mL) was added TFA (1 mL) and stirred for two days. The reaction mixture was concentrated, washed with satd NaHCO3, and dried over MgSO4. The solvent was removed, and the crude compound was purified by column chromatography using hexane/ethyl acetate gradient elution system to yield 5 as a white solid (0.41 g, 59.5%). 1H NMR (CDCl3) δ 7.52–7.45 (m, 4H), 7.21 and 7.17 (br, br, 1H), 5.79 and 5.52 (s, 1H), 4.19 (t, 2H, J = 6.6 Hz), 4.18−4.14 and 3.96 (m, t, 1H,), 3.493.35 and 3.20−3.07 (m, m, 2H), 2.63 (br, 1H), 2.18 and 2.17 (s, s, 3H), 1.69−1.62 (m, 2H), 1.25 (s, 26H), 0.88 (t, 3H); MS (ESI) m/z 513.3 [M+Na]+, 489.1 [M−H]−. Anal. Calcd for C28H46N2O3S: C, 68.53; H, 9.45; N, 5.71. Found: C, 65.47; H, 9.40; N, 5.75.
5.6. General procedure for preparing (2RS,4R)-alkyl-2-aryl-thiazolidin-4-ylmethyl)-amine 8a-8b
To a mixture of Boc-protected carboxylic acid 2j (5.6 mmol), EDCI (6.7 mmol), HOBt (5.6 mmol), and NMM (13.4 mmol) in CH2Cl2 (20 mL) was added O,N-dimethyl-hydroxylamine hydrochloride (5.9 mmol); the mixture was stirred continuously at room temperature for 10 h. The reaction mixture was diluted with CH2Cl2 (30 mL) and sequentially washed with water, 5% HCl, satd NaHCO3, and brine and dried over MgSO4. The solvent was removed under reduced pressure to yield a crude product meth-oxy-methyl-amide 6, which was recrystallized from ethyl ether/hexane as white crystals and used directly in the next step without purification (71%). Under −40 °C, LAH (1.0 mL, 2 M in THF) was added to a solution of 4-(methoxy-methyl-carbamoyl)-2-phenyl-thiazolidine-3-carboxylic acid tert-butyl ester (0.20 g, 0.57 mmol) in THF (10 mL) and stirred for 30 min. The reaction mixture was quenched with ethyl acetate (10 mL) and satd NH4Cl (10 mL). Then 5% H2SO4 (20 mL) was added under 0 °C. The mixture was extracted with ethyl acetate and dried with MgSO4. The solvent was removed and pure aldehyde 7 (0.14 g, 84.3%) was purified as a colorless oil after column chromatography. To a solution of 7 (0.14 g) in MeOH (3 mL) was charged hexadecylamine (1 equiv) in MeOH (7 mL). After stirring for 10 min, HOAc (1.3 equiv) and NaBH3CN (2.5 equiv) were added and stirred for 2 h. The reaction mixture was quenched with satd NaHCO3 (10 mL), extracted with ethyl acetate, and dried with MgSO4. The solvent was removed under reduced pressure to yield a crude 4-hexadecylaminomethyl-2-phenyl-thiazolidine-3-carboxylic acid tert-butyl ester. Deprotection with TFA yielded product 8a. Then 8a was converted to a corresponding hydrochloride by using 2 M HCl/Et2O. Compound 8b was prepared using the same method.
5.6.1. (2RS,4R)-Hexadecyl-(2-phenyl-thiazolidin-4-ylmethyl)-amine hydrochloride (8a)
Yield: 42.9%. 1H NMR (300 Hz, CDCl3) δ 7.66−7.38 (m, 5H), 5.87 (s, 0.55H), 5.79 (s, 0.45H), 4.34 and 3.96 (br, 1H), 3.34−3.32 (m, 2H), 3.31−3.08 (m, 2H), 2.92 (m, 2H), 1.64 (m, 2H), 1.24 (s, 26H), 0.85 (t, 3H, J =6.6 Hz). MS (ESI) m/z 419.1 [M+H]+). Anal. Calcd for C26H46N2S ■ 2HCl ■ 1/2H2O: C, 62.37; H, 9.86; N, 5.60. Found: C, 62.29; H, 9.91; N, 5.80.
5.6.2. (2RS,4R)-Hexadecyl-(2-(4-acetyl-phenyl-thiazolidin-4-ylmethyl)-amine hydrochloride (8b)
Yield: 48.4%. 1H NMR (300 Hz, CDCl3) δ 7.35–7.57 (m, 4H), 5.52 (s, 0.55H), 5.47 (s, 0.45H), 4.02, 3.52 (br, 1H), 3.00−3.24 (m, 2H), 2.61−2.92 (m, 4H), 2.17 (s, 3H), 1.48−1.61 (m, 2H), 1.25 (s, 26H), 0.87 (t, 3H, J = 6.9 Hz); MS (ESI) m/z 476.4 [M+H]+. Anal. Calcd for C28H49N3OS HCl: C, 65.65; H, 9.84; N, 8.20. Found: C, 65.55; H, 9.73; N, 8.04.
5.7. Synthesis of disulfanyl dimer (9)
Under 0 °C, to a solution of 1b (330 mg, 0.67 mmol) in THF (10 mL) was charged B2H6 (8.0 mL, 1 M in THF) and refluxed for 24 h. Then the reaction mixture was quenched by satd NH4Cl and dried over MgSO4. Disulfanyl dimer 9b was separated from a silica gel column using hexane/ethyl acetate gradient elution system (127 mg, 41%). 1H NMR (500 MHz, CDCl3) δ 7.44 and 7.21 (d, d, 8H), 7.40 (t, 2H, J = 6.0 Hz), 3.69 (q, 4H), 3.37 (q, 2H, J = 4.0 Hz), 3.29−3.18 (m, 4H), 3.13 (dd, 2H, J = 4.0 Hz, J =14.0 Hz), 2.71 (dd, 2H, J =14.0 Hz, J = 8.5 Hz), 2.17 (s, 6H), 1.51−1.48 (m, 4H), 1.25 and 1.29 (s, 52H), 0.87 (t, 6H). MS (ESI) m/z 1003.9 [M+Na]+. Anal. Calcd for C56H96N6O4S2: C, 68.53; H, 9.86; N, 8.56. Found: C, 68.54; H, 9.74; N, 8.31.
5.8. Synthesis of (2RS,4R)-2-(4-acetylamino-phenyl)-thiazolidine-3,4-dicarboxylic acid 3-(9H-fluoren-9-ylmethyl) ester (10)
At 0 °C, to a suspension of (2RS,4R)-2-(4-acetylaminophenyl)-thiazolidine-4-carboxylic acid (1.00 g, 3.76 mmol) in 15 mL CH2Cl2 was added triethylamine (0.38 g, 3.80 mmol) and 9-fluorenylmeth-yl chloroformate (0.98 g, 3.80 mmol). After stirring for 30 min, the mixture was washed with 5% HCl and dried with MgSO4. The solvent was removed, and crude compound was purified by column chromatography using hexane/ethyl acetate gradient elution system to yield Fmoc-protected acid as a white foam 10 (1.82 g, 99%). Yield: 99%. 1H NMR (300 MHz, CDCl3) δ 10.24 (br, 1H), 7.73−7.05 (m, 13H), 6.14−5.85 (m, 1H), 5.05−4.38 (m, 3H), 3.99 (br, 1H), 3.36−3.23 (m, 2H), 2.22 (s, 3H). MS (ESI) m/z 511.1 [M+Na]+, 486.9 [M−H]−.
5.9. Preparation of (2RS,4R)-2-(4-aminophenyl)-thiazolidine-4-carboxylic acid amide 13a-13b
The Fmoc-protected acid 10 (3.97 g, 8.135 mmol), EDCI (1.2 equiv) and HOBT (1.0 equiv) in CH2Cl2 (80 mL) was stirred at room temperature for 10 min. To this solution, dodecylamine or hexadecylamine (1.0 equiv) and Et3N (1.2 equiv) were added and stirred continuously at room temperature for 3.5 h. The reaction mixture was sequentially washed with water, satd NaHCO3, and brine, and then dried over MgSO4. The solvent was removed under reduced pressure to yield a crude oil 11 (4.11 g, 70.98%), which was used in the next step without purification. 11 (2.0 g) was added to a solution of CH3COCl (4.5 mL) in MeOH (10 mL) under 0 °C and stirred at room temperature for four days. The solvent was evaporated on vacuum and diluted with CH2Cl2 (30 mL), then washed with satd NaHCO3, and dried over MgSO4 to obtain the crude compound 12 as a yellow solid (0.9 g, 47.8%). To a solution of 12 (1 equiv) in CH2Cl2 (10 mL) was added 1,8-diazabicycloundec-7-ene (1.5 equiv) and stirred for 30 min to 1 h. The mixture was washed with 2.5% HCl, extracted with CH2Cl2, and dried with MgSO4. The solvent was removed on vacuum and purified by column chromatography using hexane/ethyl acetate gradient elution system to give 13a and 13b as a light yellow solid.
5.9.1. (2RS,4R)-2-(4-Amino-phenyl)-thiazolidine-4-carboxylic acid dodecylamide (13a)
Yield: 65.7%. 1H NMR (300 MHz, CDCl3) δ 7.31−7.24 (m, 4H), 7.29 and 6.39 (br, br, 1H), 5.51 and 5.24 (br, 0.3H, d, J = 9.6 Hz, 0.7H), 4.32 and 3.86 (br, br, 1H), 3.74 (br, 2H), 3.67 and 3.40 (dd, dd, J = 4.2 Hz, J = 8.1 Hz, 2H), 3.29−3.20 (m, 2H), 2.44 (br, 1H), 1.53−1.47 (m, 2H), 1.25 (s, 18H), 0.87 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 392.4 [M+H]+, 390.1 [M−H]−. Anal. Calcd for C22H37N3OS: C, 67.47; H, 9.52; N, 10.73. Found: C, 67.34; H, 9.52; N, 10.65.
5.9.2. (2RS,4R)-2-(4-Amino-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide (13b)
Yield: 53.2%. 1H NMR (300 MHz, CDCl3) δ 7.31−7.25 (m, 4H), 7.26 and 6.39 (br, br, 1H), 5.52 and 5.23 (br, 0.3H, br, 0.7H), 4.32 and 3.86 (br, m, 1H), 3.72 (br, 2H), 3.68 and 3.41 (dd, J = 3.9 Hz, dd, J =8.1 Hz, 2H), 3.29−3.20 (m, 2H), 2.44 (br, 1H), 1.51−1.47 (m, 2H), 1.25 (s, 26H), 0.88 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 448.5 [M+H]+, 446.1 [M−H]−. Anal. Calcd for C26H45N3OS: C, 69.75; H, 10.13; N, 9.39. Found: C, 69.80; H, 10.21; N, 9.16.
5.10. General procedure for preparing (2RS,4R)-2-(4-amido-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide 15a-15c
5.10.1. (2RS,4R)-2-(4-Methanesulfonylaminophenyl)-thiazolidine-4-carboxylic acid hexadecylamide (15a)
At 0 °C, to a solution of 12 (450 mg, 0.672 mmol) in CH2Cl2 (10 mL) were added pyridine (88 mg, 1.11 mmol) and methanesul-fonyl chloride (144 mg, 1.25 mmol) and stirred at room temperature for 10 h. The reaction mixture was washed with satd NaHCO3 and dried over MgSO4. The solvent was removed to yield the crude Fmoc-protected product 14a, which was used in the next step without further purification (79.7%). To the solution of 14a (310 mg, 0.414 mmol) in CH2Cl2 (10 mL) was added 1, 8-diazabicy-cloundec-7-ene (94.5 mg, 0.622 mmol) and stirred for 1 h. The mixture was washed with 2.5% HCl, extracted with CH2Cl2, and dried with MgSO4. The solvent was removed on vacuum and purified by column chromatography using hexane/ethyl acetate gradient elution system to give 15a as a white solid. Yield: 45.9%. 1H NMR (300 MHz, CDCl3) d 7.52−7.46 and 7.24−7.19 (m, 4H), 7.15 and 6.33 (br, 0.3H, br, 0.7H), 6.52 (br, 1H), 5.57 and 5.33 (d, J =10.8 Hz, 0.3H, d, J =11.4 Hz, 0.7H), 4.33 and 3.89 (br, br, 0.7H and 0.3H), 3.69 and 3.42 (dd, J = 4.2 Hz, m, 2H), 3.32–3.22 (m, 2H), 3.02 and 3.01 (s, s, 3H), 2.65 and 2.50 (br, 1H), 1.52−1.48 (m, 2H), 1.25 (s, 26H), 0.87 (t, 3H, J = 6.6 Hz). MS (ESI) m/z 548.4 [M+Na]+, 524.2 [M−H−. Anal. Calcd for C27H47N3O3S2: C, 61.67; H, 9.01; N, 7.99. Found: C, 61.91; H, 9.03; N, 7.91.
5.10.2. (2RS,4R)-2-[4-(2-Chloro-acetylamino)-phenyl]-thiazoli-dine-4-carboxylic acid hexadecylamide (15b)
At 0 °C, to a solution of 12 (390 mg, 0.582 mmol) in CH2Cl2 (10 mL) were added pyridine (72 mg, 9.11 mmol) and chloro-acetyl chloride (122 mg, 1.08 mmol) and stirred at room temperature for 10 h. The reaction mixture was washed with satd NaHCO3 and dried over MgSO4. The solvents were removed to yield the crude Fmoc-protected product 14b, which was used in the next step without further purification (50.7%). To the solution of 14b (220 mg, 0.295 mmol) in CH2Cl2 (10 mL) was added 1, 8-diazabicy-cloundec-7-ene (103 mg, 0.678 mmol) and stirred for 20 min. The mixture was washed with 1 N HCl, extracted with CH2Cl2, and dried with MgSO4. The solvent was removed on vacuum and the resulting crude material was purified by column chromatography using hexane/ethyl acetate gradient elution system to give 15b as a white solid. Yield: 53.2%. 1H NMR (300 MHz, CDCl3) d 8.26 (s, 1H), 7.58−7.46 (m, 4H), 7.19 and 6.38 (br, br, 1H), 5.59 and 5.33 (s, s, 1H), 4.34 and 3.91 (br, br, 0.7H and 0.3H), 4.20 (s, 2H), 3.69 and 3.39 (dd, J = 3.9 Hz, br, 2H), 3.33−3.26 (m, 2H), 2.52 (br, 1H), 1.56 (br, 2H), 1.25 (s, 26H), 0.87 (t, 3H, J = 6.0 Hz). MS (ESI) m/z 541.5 [M+Na]+, 522.3 [M−H]−. Anal. Calcd for C28H46ClN3O3S: C, 64.15; H, 8.84; N, 8.02. Found: C, 64.06; H, 8.88; N, 7.91.
5.10.3. (2RS,4R)-2-(4-Ureido-phenyl)-thiazolidine-4-carboxylic acid hexadecylamide (15c)
At 0 °C, to a solution of 12 (240 mg, 0.32 mmol) in water (5 mL) were added sodium cyanate (64 mg, 0.98 mmol) and acetic acid (0.15 mL). The mixture was stirred at 50 °C for 24 h. It was extracted with ethyl acetate and dried over MgSO4. The solvents were removed to yield the crude Fmoc-protected product 14c, which was used in the next step without further purification (87.7%). To the solution of 14c (200 mg, 0.28 mmol) in CH2Cl2 (10 mL) was added 1,8-diazabicycloundec-7-ene (99 mg, 0.65 mmol) and stirred for 45 min. The mixture was washed with 1 N HCl, extracted with CH2Cl2, and dried with MgSO4. The solvent was removed on vacuum, and the resulting crude material was purified by column chromatography using hexane/ethyl acetate gradient elution system to give 15c. Yield: 51.1%. 1H NMR (300 MHz, CDCl3) δ 7.56−7.33 (m, 4H), 7.28 and 6.46 (br, 1H), 6.82 (br, 1H), 5.57 and 5.33 (s, 0.3H, s, 0.7H), 4.78 (s, 2H), 4.33 and 3.91 (dd, J = 4.2 Hz, t, J = 7.5 Hz, 0.7H and 0.3H), 3.68 and 3.41 (dd, J= 11.1 Hz and 4.2, m, 2H), 3.32−3.25 (m, 2H), 2.60 (br, 1H), 1.56−1.45 (m, 2H), 1.27 (s, 26H), 0.89 (t, 3H, J = 6.9 Hz). MS (ESI) m/z 513.5 [M+Na]+, 491.4 [M+H]+, 489.1 [M−H−. Anal. Calcd for C27H48N4O3S2 H2O: C, 63.74; H, 9.51; N, 11.01. Found: C, 63.89; H, 9.01; N, 10.91.
5.11. (2R)-2-Amino-N-hexadecyl-3-methylsulfanyl-propion-amide (16a)
At 0 °C, (2R)-2-amino-3-methylsulfanyl-propionic acid (2.5 g, 18.5 mmol) was dissolved in 1 N NaOH (20 mL) and 1,4-dioxane (40 mL); then di-tert-butyldicarbonate (8.1 g, 37 mmol) was added slowly and stirred at room temperature overnight. The reaction mixture was concentrated in vacuum and washed with ethyl acetate (20 mL). The aqueous phase was adjusted to a pH 3 by adding 1 N HCl, then extracted with ethyl acetate, dried with magnesium sulfate, filtered, and concentrated on vacuum to gave Boc-protected (2R)-2-amino-3-methylsulfanyl-propionic acid as a white solid (90.96%). A mixture of Boc-protected (2R)-2-amino-3-meth-ylsulfanyl-propionic acid (1.76 g, 7.5 mmol), EDCI (1.2 equiv), and HOBT (1.0 equiv) in CH2Cl2 (35 mL) was stirred at room temperature for 10 min. To this solution, hexadecylamine (1.0 equiv) and Et3N (1.2 equiv) were added and stirring continuously at room temperature for 10 h. The reaction mixture was diluted with CH2Cl2 (50 mL) and sequentially washed with water, satd NaHCO3, and brine, and dried over MgSO4. The solvent was removed under reduced pressure to yield a crude oil, which was stirred with TFA (3 mL) in 20 mL CH2Cl2 at room temperature for 1 h to cleave the Boc group. The reaction mixture was concentrated, diluted with ethyl acetate, washed with satd NaHCO3, and dried over MgSO4. The solvent was removed to yield a crude solid, which was purified by column chromatography using hexane/ethyl acetate gradient elution system as a white solid. Yield: 63.4%. Mp (hexane) 55–56 °C. 1H NMR (300 MHz, CDCl3) d 7.42 (br, 1H), 3.95 and 3.50 (br, dd, 1H, J = J = 3.9 Hz, 9.0 Hz), 3.24 (q, 2H), 3.02 and 2.66 (dd, dd, 2H, J = 3.9 Hz, J = 9.0 Hz, J = 13.8 Hz), 2.10 (s, 3H), 1.52−1.48 (m, 2H), 1.25 (s, 26H), 0.88 (t, 3H). MS (ESI) m/z 359.9 [M+H]+, 357.1 [M−H]−.
5.12. (2R)-2-(4-Acetylamino-benzylamino)-N-hexadecyl-3-methylsulfanyl-propionamide (16b)
To a suspension of 16a (244 mg, 0.728 mmol) in MeOH (10 mL) was added N-(4-formylphenyl)-acetamide (119 mg, 0.728 mmol). After stirring for 5 min, HOAc (75 mg) was charged to the mixture and stirred for 20 min. Then NaBH3CN (115 mg, 1.82 mmol) was added and stirred for 2 h. The reaction mixture was quenched with satd NaHCO3, extracted with ethyl acetate, and dried on MgSO4. The solvent was removed to yield a crude solid, which was purified by column chromatography using hexane/ethyl acetate gradient elution system. Yield: 61.1%. Mp (hexane) 83–84 °C. 1H NMR (300 MHz, CDCls) δ 7.48–7.24 (m, 4H), 7.40 (br, 1H), 7.19 (br, 1H), 3.70 (q, J = 9.0 Hz, 2H), 3.28−3.18 (m, br, 3H), 2.98 (dd, J = 3.6 Hz, 1H), 2.58 (dd, J = 9.6 Hz, J = 13.8 Hz, 1H), 1.49−1.47 (m, 2H), 1.25 (s, 26H), 0.88 (t, 3H, J = 6.3 Hz). MS (eSI) m/z 506.4 [M+H]+, 504.1 [M−H−. Anal. Calcd for C29H51N3O2S: C, 68.86; H, 10.16; N, 8.31 Found: C, 69.98; H, 10.12; N, 8.47.
5.13. (2R)-2-Amino-3-benzylsulfanyl-N-hexadecyl-propionamide (16c)
A mixture of (2R)-3-benzylsulfanyl-2-tert-butoxycarbonylami-no-propionic acid (0.5 g, 1.61 mmol), EDCI (0.371 g 1.93 mmol), and HOBT (0.228 g, 1.69 mmol) in CH2Cl2 (20 mL) was stirred at room temperature for 10 min. To this solution, hexadecylamine (0.409 g, 1.69 mmol) and Et3N (0.195 g, 1.93 mmol) were added and stirred continuously at room temperature for 10 h. The reaction mixture was sequentially washed with water, satd NaHCO3, and brine, and then dried over MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was stirred with TFA (3 mL) in 20 mL CH2Cl2 at room temperature for 2–3 h to cleave the Boc group. The reaction mixture was concentrated, diluted with ethyl acetate, washed with satd NaHCO3, and dried over MgSO4. The solvent was removed to yield a crude solid, which was purified by column chromatography using hexane/ethyl acetate gradient elution system as a white solid. Yield: 53.7%. Mp (hexane) 66–67 °C. 1H NMR (300 MHz, CDCl3 + D2O) δ 7.51 (br, 1H), 7.35−7.20 (m, 5H), 3.75 (s, 2H), 3.70 (br, 1H), 3.28−3.13 (m, 2H), 2.99 (br, 1H), 2.90−2.68 (m, 1H), 1.48–1.46 (m, 2H), 1.25 (s, 26H), 0.88 (t, 3H, J =6.9 Hz). MS (ESI) m/z 435.4 [M+H)+, 433.1 [M−H]−. Anal. Calcd for C26H46N2OS: C, 71.83; H, 10.67; N, 6.44. Found: C, 71.82; H, 10.51; N, 6.51.
5.14. 2-(4-Acetylamino-benzylamino)-N-hexadecyl-acetamide (16d)
At 0 °C, aminoacetic acid (2.08 g, 27.7 mmol) was dissolved in 1 N NaOH (28 mL) and 1,4-dioxane (25 mL); then di-tert-butyldi-carbonate (12.1 g, 55.4 mmol) was added slowly and stirred at room temperature overnight. The reaction mixture was concentrated in vacuum and washed with ethyl acetate (20 mL). The aqueous phase was adjusted to a pH 3 by adding 1 N HCl, then extracted with ethyl acetate, dried with magnesium sulfate, filtered, and concentrated on vacuum to gave Boc-protected aminoacetic acid as a white crystalline solid (92.3%). A mixture of Boc-protected aminoacetic acid (0.87 g, 5 mmol), EDCI (1.152 g, 6 mmol), and HOBT (0.675 g, 5 mmol) in CH2Cl2 (25 mL) was stirred at room temperature for 10 min. To this solution, hexadecylamine (1.21 g, 5 mmol) and Et3N (0.606 g, 6 mmol) were added and stirred continuously at room temperature for 10 h. The reaction mixture was diluted with CH2Cl2 (50 mL) and sequentially washed with water, satd NaHCO3, and brine, and dried over MgSO4. The solvent was removed under reduced pressure to yield a crude oil, which was stirred with TFA (3 mL) in 20 mL CH2Cl2 at room temperature for 1 h to cleave the Boc group. The reaction mixture was concentrated, diluted with EtOAc (50 mL), and washed with satd NaHCO3. The white precipitate 2-amino-N-hexadecylacetamide was obtained by treatment with 1 N HCl (10 mL) (77.7%). To a suspension of 2-amino-N-hexadecylacetamide (200 mg, 0.60 mmol) in MeOH (15 mL) was added N-(4-formylphenyl)-acetamide (100 mg, 0.61 mmol). After stirring for 5 min, HOAc (75 mg) was charged and stirred for 20 min. Then NaBH3CN (93 mg, 1.48 mmol) was added and stirred for 1 h. The reaction mixture was quenched with satd NaHCO3, extracted with ethyl acetate, and dried on MgSO4. The solvent was removed, and 16d was purified by column chromatography using hexane/ethyl acetate gradient elution system as a white solid. Yield: 53.1%. Mp (hexane) 81–83 °C. 1H NMR (300 MHz, CDCl3) d 7.48 (d, 2H, J = 8.4 Hz), 7.24 (d, 2H, J = 8.4 Hz), 7.15 (br, 1H), 7.15 (br, 1H), 3.72 (s, 2H), 3.27 (s, 2H), 3.26−3.22 (m, 2H), 2.17 (s, 3H), 1.51−1.47 (m, 2H), 1.25 (s, 26H), 0.87 (t, 3H, J= 6.6 Hz). MS (ESI) m/z 468.4 [M+Na]+, 444.1 [M−H]−. Anal. Calcd for C29H51N3O2S: C, 72.76; H, 10.63; N, 9.43. Found: C, 73.00; H, 10.77; N, 9.35.
6. Biology
6.1. Cell culture and cytotoxicity assay of melanoma
We examined the antiproliferative activity of ATCAA analogues in two human melanoma cell lines (A375 and WM-164) and one mouse melanoma cell line (B16-F1). We used activity on fibroblast cells as a control to determine the selectivity of these compounds against melanoma. A375 cells and B16-F1 cells were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). WM-164 cells were derived from metastatic melanoma tumors and were a gift from Dr. Meenhard Herlyn (Wistar Institute, Philadelphia, PA, USA). Human dermal fibroblast cells were purchased from Cascade Biologics, Inc., Portland, OR, USA. All cell lines were cultured in DMEM (Cellgro Mediatech, Inc., Herndon, VA, USA), supplemented with 5% FBS (Cellgro Mediatech), 1% antibiotic/antimycotic mixture (Sigma-Aldrich, Inc., St. Louis, MO, USA), and bovine insulin (5 μg/mL; Sigma-Aldrich). Cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Standard sulforhodamine B assay was used. Cells were exposed to a wide range of concentrations for 48 h in round-bottomed 96-well plates. Cells were fixed with 10% trichloroacetic acid and washed five times with water. After cells were air-dried overnight and stained with SRB solution, total proteins were measured at 560 nm with a plate reader. IC50 (i.e., concentration that inhibited cell growth by 50% of no treatment controls) values were obtained by nonlinear regression analysis with GraphPad Prism (GraphPad Software, San Diego, CA).
6.2. Cell culture and cytotoxicity assay of prostate cancer
We examined the antiproliferative activity of ATCAA analogues in four human prostate cancer cell lines (LNCaP, DU 145, PC-3, and PPC-1). We used LPL receptor-negative RH7777 cells as a control to determine the selectivity of these compounds. LNCaP, PC-3, DU 145, and RH7777 cells were purchased from ATCC. Dr. Mitchell Steiner at the University of Tennessee Health Science Center kindly provided PPC-1cells. All prostate cancer cell lines were cultured in RPMI1640 (Cellgro Mediatech), supplemented with 10% FBS (Cellgro Mediatech). RH7777 cells were cultured in DMEM medium with 10% FBS. Cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2.1000 to 5000 cells were plated into each well of 96-well plates depending on growth rate and exposed to different concentrations of a test compound for 96 h in three to five replicates. Cell numbers at the end of the drug treatment were measured by the SRB assay. Briefly, the cells were fixed with 10% of trichloroacetic acid and stained with 0.4% SRB, and the absorbances at 540 nm were measured using a plate reader (DYNEX Technologies, Chantilly, VA). Percentages of cell survival versus drug concentrations were plotted, and the IC50 values were obtained by nonlinear regression analysis using WinNonlin (Pharsight Corporation, Mountain View, CA).
6.3. Cell cycle analysis
Flow cytometry analysis was performed as described else-where25 to study cell cycle phase distribution. Briefly, A375 and LNCaP cells (4 × 106) were seeded in each 10-cm dish; A375 and LNCaP cells were synchronized using 0.5% charcoal-stripped DMEM and RPMI medium for 72 h, respectively. After that, the medium was changed to the media containing 10% FBS with different concentrations (0, 1, 5, 10 and 20 μM) of testing compounds. Cells were incubated for additional 24 h. All cells were collected by trypsinization. Cell pellets were washed by PBS. The supernatant was discarded, and the pellets were fixed in ice-cold 70% ethanol at 4 °C overnight. Ethanol was removed by centrifugation and cell pallets were washed with PBS twice. 1 mL of PBS containing 100 μg/mL RNAase A was then added and cell pallets were incubated for 1 h. Cells were centrifuged and re-suspended in 1 mL of PBS containing propidium iodide (50 μg/mL). Cell cycle analysis was conducted with a FACS Calibur Autometer (Beckton Dickinson, San Diego, CA, USA). Data were analyzed and graphs were prepared using the MODFIT 2.0 program (Verity Software House, Topsham, ME, USA).
6.4. FLIPR intracellular calcium mobilization assay
Chem-1 cells (Chemicon Int., Inc., Billerica, MA) were stable-transfected with LPA1, LPA3, and LPA5, respectively. Cells are maintained in DMEM/10% FBS/100 U/mL penicillin. Cells are passaged by washing with Ca2+ and Mg2+-free HBSS (10 mL/T75) and incubated with 0.05% trypsin/0.2 g/L EDTA (1 mL/T75) for 5–10 min at 37 °C. Cells are loaded with Fluo-4 NW, and the assay was read for 180 s using the FLIPRtetra. The signal is proportional to [Ca2+]i and thus can be used to quantify differences in calcium mobilization in ATCAA-treated and control cells. Percentage activations were determined upon initial addition of 3ad followed by 10-min incubation at 25 °C. To study antagonism of ATCAA on LPAs, cells were incubated with 3ad for 10 min at 25 °C. Following compound incubation, reference agonists (oleoyl-LPA) were added at EC80 to determine percentage inhibition. Compound 3ad was plated in an eight-point, fourfold serial dilution series with a top concentration of 10 μM. The concentration described here reflects the final concentration of the compound during the antagonist assay.
6.5. Colony-formation assay
To measure colony formation and growth of melanoma, A375 cells were plated at a colony-forming density (2000 cells per well on 6-well plates). Cells were grown in DMEM supplemented with FBS (Atlanta Biologicals, Lawrenceville, GA) and an antibiotic-antimycotic solution (Sigma, St. Louis, MO) at 37 °C in an atmosphere of 95% air and 5% CO2.27 Cells were treated with compounds 1b and 3ad at different concentrations (2, 20, and 100 μM). Compounds were added to the medium from 1 mM DMSO stock solutions, and corresponding dilution of DMSO was used as negative (vehicle) control. Cells were grown for 14 days, and colonies were stained with 0.1% crystalline blue for 30 min and rinsed with distilled water to remove excess dye. Plates were photographed, and the number of colonies was measured by Artek 880 Automated Colony Counter (Artek Systems Corporation, Farmingdale, NY).
6.6. Formulation for in vivo studies
Compound 1b was dissolved into a co-solvent system that was composed of 80% Tween 80 (Sigma-Aldrich, St. Louis, MO) and 20% Captex 200 (Abitec Corporation, Columbus, OH). Dacarbazine (DTIC) (Sigma-Aldrich, St. Louis, MO) was dissolved in saline solution.
6.7. Animals
Male athymic nude mice age 4–5 weeks were purchased from Harlan Laboratories (Harlan Laboratories, Indianapolis, IN, USA). The laboratory housing the animals met all Association for Assessment and Accreditation and Laboratory Animal Care specifications. All of the procedures were conducted in accordance with the guidelines of our Institutional Animal Care and Use Committee.
6.8. In vivo evaluation of antitumor efficacy
Logarithmic growth phase A375 cells were prepared in FBS-free DMEM medium (Cellgro Mediatech) at a concentration of 5 × 107 viable cells/mL and placed on ice. The cell suspension was mixed with BD Martigel (BD Biosciences, Waltham, MA, USA) at a 1:1 ratio. This cell suspension (100 μL) was injected subcutaneously in the right dorsal flank of each mouse. When tumor sizes reached about 150 mm3, about seven days after cell inoculation, all mice bearing tumors were divided into control and treatment groups based on tumor size (n = 8 per group). Each group had similar average tumor size. Mice in control groups were injected intraperitoneally with 50 μL vehicle solution only (negative control) or DTIC at 60 mg/kg (positive control) once daily. Tumor volume was measured twice weekly with a Traceable® electronic digital caliper (Fisher Scientific, Inc., Pittsburgh, PA) and calculated using the formula a × b2 × 0.5, where a and b represented the larger and smaller diameters, respectively.32 Tumor volume was expressed as cubic millimeters. Data were expressed as mean ± SE for each group and plotted as a function of time. Percentage tumor reduction at the conclusion of the experiment (22 days after initiating treatment) was calculated from the formula 100 × [(T – T0)/(C – C0)], where T represents mean tumor volume of a treated group on a specific day, T0 represents mean tumor volume of the same group on the first day of treatment, C represents mean tumor volume of a control on a specific day, and C0 represents mean tumor volume of the same group on the first day of treatment.33 Animal activity and average body weight of each group were monitored during the entire experiment period to assess compound toxicity. At the end of treatment, all mice were euthanized by CO2 followed by cervical dislocation, and tumors were harvested for further studies.
NCI-60 cell line cytotoxicity data of 1a and 1b are available via the internet at http://pubs.acs.org.
Supplementary Material
Acknowledgment
This research was supported by funds from Department of Pharmaceutical Sciences, College of Pharmacy, University of Tennessee Health Science Center; the Van Vleet Endowed Professorship; Division of Pharmaceutics, The Ohio State University; USPHS grant 1R15CA125623, and University of Tennessee Technology Maturation Grant. Zhao Wang acknowledges the support from the Alma & Hal Reagan Fellowship. We thank Dr. George Wood’s group in the College of Pharmacy, University of Tennessee Health Science Center, for their initial help with formulations for compound 1b; Dr. Dave Dickason and Dr. Tai Ahn at GTx, Inc., for their help in developing drug formulations in this study; Dr. David Arm-bruster at the University of Tennessee Health Science Center for editorial assistance; and Dr. David Hamilton at the University of Tennessee Health Science Center animal facility for assistance in developing the approved animal protocols.
Abbreviations:
- ATCAA
2-arylthiazolidine-4-carboxylic acid amide
- ATCAs
2-arylthiazolidine-4-carboxylic acids
- CDDP
cisplatin
- DTIC
dacarbazine
- DCC
N,N′-dicyclohexylcarbodiimide
- DMAP
4-dimethylaminopyridine
- DMEM
Dulbecco’s modified Eagle medium
- EDCI
3-ethyl-1(N,N-dimethyl)aminopropylcarbodiimide
- FBS
fetal bovine serum
- FLIPR
fluorescence imaging plate reader
- GPCR
G-protein-coupled receptors
- HBSS
Hank’s buffered salt solution
- HOBT
1-hydroxybenzotri-azole
- LPA
lysophosphatidic acid
- NOE
nuclear Overhauser effect
- Oleoyl-LPA
1-oleoyl lysophosphatidic acid
- SAR
structure-activity relationship
- SRB
sulforho-damine B
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2009.12.020.
References and notes
- 1.Cancer Facts & Figures; American Cancer Society: Atlanta, 2007. [Google Scholar]
- 2.Carlson JA; Ross Jeffery S; Slominski A; Linette G; Mysliborski J; Hill J; Mihm M Jr. J. Am. Acad. Dermatol 2005, 52, 743. [DOI] [PubMed] [Google Scholar]
- 3.Gogas HJ; Kirkwood JM; Sondak VK Cancer (Hoboken, NJ, U.S.A.) 2007,109, 455. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A; Tiwari Ram C; Murray T; Ghafoor A; Samuels A; Ward E; Feuer Eric J CA Cancer J. Clin 2004, 54, 8. [DOI] [PubMed] [Google Scholar]
- 5.Bosland MC; McCormick DL; Melamed J; Walden PD; Zeleniuch-Jacquotte A; Lumey LH Eur. J. Cancer Prev 2002, 11, S18. [PubMed] [Google Scholar]
- 6.Karan D; Holzbeierlein J; Thrasher JB Cancer Res. 2009, 69, 2. [DOI] [PubMed] [Google Scholar]
- 7.Gududuru V; Hurh E; Dalton JT; Miller DD J. Med. Chem 2005, 48, 2584. [DOI] [PubMed] [Google Scholar]
- 8.Li W; Lu Y; Wang Z; Dalton JT; Miller DD Bioorg. Med. Chem. Lett 2007, 17,4113. [DOI] [PubMed] [Google Scholar]
- 9.Chen J; Wang Z; Lu Y; Dalton JT; Miller DD; Li W Bioorg. Med. Chem. Lett 2008, 18, 3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W; Wang Z; Gududuru V; Zbytek B; Slominski AT; Dalton JT; Miller DD Anticancer Res. 2007, 27, 883. [PubMed] [Google Scholar]
- 11.Gududuru V; Zeng K; Tsukahara R; Makarova N; Fujiwara Y; Pigg KR; Baker DL; Tigyi G; Miller DD Bioorg. Med. Chem. Lett 2006, 16, 451. [DOI] [PubMed] [Google Scholar]
- 12.Gududuru V; Hurh E; Sullivan J; Dalton JT; Miller DD Bioorg. Med. Chem. Lett 2005, 15, 4010. [DOI] [PubMed] [Google Scholar]
- 13.Schubert MP J. Biol. Chem 1936, 114, 341. [Google Scholar]
- 14.Zhao L; Huang W; Liu H; Wang L; Zhong W; Xiao J; Hu Y; Li SJ Med. Chem 2006, 49, 4059. [DOI] [PubMed] [Google Scholar]
- 15.Nahm S; Weinreb SM Tetrahedron Lett. 1981, 22, 3815. [Google Scholar]
- 16.Burns CJ; Guitton J-D; Baudoin B; Lelievre Y; Duchesne M; Parker F; Fromage N; Commercon AJ Med. Chem 1997, 40,1763. [DOI] [PubMed] [Google Scholar]
- 17.Kurzer F Org. Synth 1951, 31, 8. [Google Scholar]
- 18.Wilhelm S; Carter C; Lynch M; Lowinger T; Dumas J; Smith RA; Schwartz B; Simantov R; Kelley S Nat. Rev. Drug Disc 2006, 5, 835. [DOI] [PubMed] [Google Scholar]
- 19.McFadyen MCE; Melvin WT; Murray GI Mol. Cancer Ther 2004, 3, 363. [PubMed] [Google Scholar]
- 20.Rooseboom M; Commandeur JNM; Vermeulen NP E. Pharmacol. Rev 2004, 56, 53. [DOI] [PubMed] [Google Scholar]
- 21.Lu Y;Li CM;Wang Z;Ross CR 2nd, Chen J;Dalton JT;Li W;Miller DD J. Med. Chem 2009, 52, 1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brugarolas A; Gosalvez M Lancet 1980, 1, 68. [DOI] [PubMed] [Google Scholar]
- 23.Liu J; Qu S; Li B; Li D Yiyao Gongye 1988, 19, 203. [Google Scholar]
- 24.Ohta H; Sato K; Murata N; Damirin A; Malchinkhuu E; Kon J; Kimura T; Tobo M; Yamazaki Y; Watanabe T; Yagi M; Sato M; Suzuki R; Murooka H;Sakai T;Nishitoba T;Im DS;Nochi H; Tamoto K;Tomura H;Okajima F Mol. Pharmacol 2003, 64, 994. [DOI] [PubMed] [Google Scholar]
- 25.Meyskens FL Jr.; Moon TE; Dana B; Gilmartin E; Casey WJ; Chen HS; Franks DH; Young L; Salmon SE Br. J. Cancer 1981, 44, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Von Hoff DD; Casper J; Bradley E; Sandbach J; Jones D; Makuch R Am. J. Med 1981, 70,1027. [DOI] [PubMed] [Google Scholar]
- 27.Zmijewski MA;Li W; Zjawiony JK; Sweatman TW; Chen J;Miller DD; Slominski AT Steroids 2009, 74, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zmijewski MA;Li W; Zjawiony JK; Sweatman TW; Chen J;Miller DD; Slominski AT Photochem. Photobiol. Sci 2008, 7, 1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zbytek B; Janjetovic Z; Tuckey RC; Zmijewski MA; Sweatman TW; Jones E; Nguyen MN; Slominski AJ Invest. Dermatol 2008, 128, 2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panayiotis P; Constantinides CML J. Controlled Release 1995, 34, 109. [Google Scholar]
- 31.Povlsen CO; Jacobsen GK Cancer Res. 1975, 35, 2790. [PubMed] [Google Scholar]
- 32.Guo W; Wu S; Liu J; Fang B Cancer Res. 2008, 68, 7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen J; Smith M; Kolinsky K; Adames V; Mehta N; Fritzky L; Rashed M; Wheeldon E; Linn M; Higgins B Cancer Chemother. Pharmacol 2007,59, 651. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.