

Abstract
Catechol-bearing polymers form hydrogel networks through cooperative oxidative crosslinking and coordination chemistry. Here we describe the kinetics of cation-dependent electrochemical-mediated gelation of precursor solutions composed of catechol functionalized four-arm poly(ethylene glycol) combined with select metal cations. The gelation kinetics, mechanical properties, crosslink composition, and self-healing capacity is a strong function of the valency and redox potential of metal ions in the precursor solution. Catechol-bearing hydrogels exhibit highly compliant mechanical properties with storage moduli ranging from G’ = 0.1–5 kPa depending on the choice of redox active metal ions in the precursor solution. The gelation kinetics is informed by the net cell potential of redox active components in the precursor solution. Finally, redox potential of the metal ion precursor can differentially alter the effective density of crosslinks in networks and confer properties to hydrogels such as self-healing capacity. Taken together, this parametric study generates new insight to inform the design of catechol-bearing hydrogel networks formed by electrochemical-mediated multimodal crosslinking.
Graphical Abstract
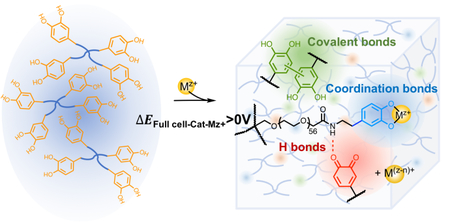
Applying full cell potential estimation in redox pairs of [PEG-Cat]4+Mz+ to elucidate contributions of metal ion precursors on hydrogel properties.
1. Introduction
Catechol-bearing hydrogels are highly tunable polymer networks that have applications ranging from medical materials to soft robotics.1–6 The versatility of catechol-bearing hydrogels is attributed to the following properties of catechols: redox activity, multimodal bonding, metal ion chelation, and thus coordination bond formation between catechols and metal ions.7–10 Furthermore, the properties of coordination bonds can be tuned by controlling metal ion composition. Redox active metal ions such as Fe3+ promote crosslink formation and gelation of catechol-functionalized precursors through multiple mechanisms such as oxidative covalent coupling of o-quinones/semiquinones or coordination bond formation through metal-catechol complexes.7, 11, 12 The redox activity and valency of metal cations can alter the balance between oxidative covalent coupling of catechols relative to other types of bonds such as coordination bonds.11, 13–15 While increasing the concentration of oxidizing agent can promote crosslinking in hydrogel networks,16–18 the composition and stoichiometry of metal ions can also influence network properties through multiple mechanisms.10, 19–21 For example, controlling the stoichiometric ratios of metal ions to catechols along with pH can alter the mechanical properties of the resulting hydrogel by promoting coordination bonds with properties that approximate covalent bonds.8 Complexing catechols with metal ions such as Fe3+ can create mono-, bis-, or tris-catechol-Fe3+ depending on the pH.7, 19 Furthermore, the storage moduli of gels prepared from catechol-bearing polyallylamine crosslinked with multivalent cations such as Fe3+, Al3+, Ga3+, or In3+ is highly pH-dependent. The maximum storage moduli of hydrogels is observed when the pH value approximates the pKa of primary amines.12 Networks composed of catechol-bearing polyallylamine are highly branched and topologically complex since the combination of primary amines and catechols can create covalent bonds through Michael addition and Schiff base formation.12, 22 Catechol-functionalized four-arm poly(ethylene glycol) ([PEG-Cat]4) is potentially a more convenient model precursor polymer to isolate the contributions of metal ion composition on crosslink formation and gelation of hydrogel networks. Catechol-bearing PEG-based networks have been previously used to measure the characteristic relaxation times in hydrogels crosslinked with various metal ions.10 4-arm [PEG-Cat]4 precursors have a tractable molecular weight between crosslinks, which facilitates calculations of swelling and simplifies comparative assessment.23
The multimodal bonding in these networks provides simultaneous tunabilty and complexity as the precise distribution of these crosslinks is difficult to decipher. Understanding the role of metal ion composition on intermolecular bonding, crosslinking, and eventual gelation is important because the distribution of crosslinking chemistry can further influence the physical properties of the hydrogel network including gelation kinetics, mechanical properties, and self-healing capacity. We hypothesize that in low pH environments (pH < pKa,-NH2; pH < pKa,Cat), the full cell potential of metal ion and catechol (Mz+:Cat) redox pairs in polymer precursors can predict network gelation, independent of the cation valency z+. Here, we describe a parametric study to test this hypothesis and elucidate the relative contributions of the valency and redox potential of metal ions on networks prepared from precursor solutions of [PEG-Cat]4 and Mz+.
2. Materials and methods
[PEG-Cat]4 was synthesized as previously described albeit with minor modifications.11, 16, 24 Briefly, under an N2 (g) blanket, four-arm poly(ethylene glycol) succinimidyl carboxymethyl ester (Mw ~10,000 g/mol; Jenkem Technology Ltd, Plano, TX) was combined with dopamine hydrochloride (2:3 mol ratio, neutralized with N-methylmorpholine) in anhydrous N,N-dimethylformamide (DMF) for 18 h. The product was dialyzed against H2O with pH~3.5 (titrated with 1M HCl) for 24 h and then against ddH2O for 2 h. The final product of [PEG-Cat]4 was lyophilized and stored at −15 oC until further use. Purified [PEG-Cat]4 and dehydrated hydrogel samples were characterized using 1H NMR (300 MHz, DMSO-d6, δ, ppm; Bruker Avance 300 MHz), Fourier transform infrared spectroscopy (FTIR; 16 scans at 4 cm−1 resolution; Spectrum 100 ATR-FTIR, PerkinElmer), and UV-vis spectroscopy (model UV-2600, Shimadzu, Tokyo, Japan). UV-vis tests were performed with a sealed cuvette for 3 h with diluted precursor solutions (1:64 with ddH2O) for hydrogel preparation. Prior to measurements, ddH2O was purged with N2 gas for at least 30 min to displace dissolved O2.
[PEG-Cat]4+Mz+ hydrogels were prepared by combining solutions of [PEG-Cat]4 with the appropriate metal ion (Mz+) precursor (see Table 1 and Table S1, concentrations of [PEG-Cat]4 were maintained at 8 mM in mixtures). Gelation kinetics, mechanical properties, and self-healing capacity were characterized using rheometry (Discovery HR-2, TA instrument, New Castle, DE USA). Gelation time τgel was defined as τgel = = 0.9. Storage G’ and loss G” moduli were measured at frequencies of ω = 0.1 – 100 rad/s with maximum strain amplitudes of γ0 = 5%. The self-healing capacity of hydrogel networks was studied with cyclic sweep with strain increased from γ = 1 to 1000% at ω = 5 rad/s over 100 s, following by another 100 s period for healing.
Table 1.
Standard Gibbs free energy calculations and experimentally observed gelation times.
| Mz+ | Standard Reduction Potential (mV) (vs. SHEa | Net Reactionb | Full Cell Potential ΔE0 (mV) | Standard Gibbs Free Energy ΔG 0 (kJ/mol) | Gelation Time (min) |
|---|---|---|---|---|---|
| Fe3+ | 771 | 2Fe3+ + QH2 → 2Fe2+ + Q + 2H+ | 21.0 | −4.05 | 15.0 |
| V5+ | 1000 | 2VO3− + 8H+ + QH2 → 2VO2+ + Q + 4H2O | 250 | −48.3 | 20.7 |
| Ag+ | 799 | 2Ag+ + QH2 → 2Ag + Q + 2H+ | 49.0 | −9.46 | 1080 |
| Au3+ | 930 | 2[AuCl4−] + 3QH2 → 2Au + 3Q + 6HCl +2Cl− | 180 | −104 | 90.0 |
| Cu2+ | 159c | 2 Cu2+ + QH2 → 2Cu+ + Q + 2H+ | −591 | 114 | ∞ |
| Cu2+ | 337d | Cu2+ + QH2 → Cu + Q + 2H+ | −413 | 79.7 | ∞ |
| Al3+ | −1680 | 2Al3+ + 3QH2 → 2Al + 3Q + 6H+ | −2430 | 1407 | ∞ |
| V4+ | 340 | 2VO2+ + 2H+ + QH2 → 2V3+ + 2H2O + Q | −410 | 79.1 | ∞ |
Standard hydrogen electrode.
Note: QH2 - fully reduced catechol; Q - fully oxidized o-quinone. Ered, quinone = 750 mV 33
Reduction of Cu2+ to Cu+
Reduction of Cu2+ to Cu0
The swelling behaviour of hydrogels was characterized in ddH2O (n = 3). The swelling ratio Q of hydrogel was calculated by the mass ratio of samples in swollen and dehydrated state (Q = 𝑤𝑠/𝑤0). Dimensional swelling was defined as q = . The average molecular weight between crosslinks (g/mol) was calculated using the following equation:23, 25
| (1) |
where 𝜈𝑠 is the volume fraction of polymer in the swollen hydrogel, 𝜈𝑟 is the volume fraction of polymer in the initial pre-swollen state. 𝑉1 is molar volume of the solvent (18 mL/mol for H2O), ρp is the mass density of the polymer (ρPEG = 1.123 g/cm3, and χ1 is the polymer-solvent interaction parameter (χPEG-H2O = 0.462).23, 26 The relationship between mechanical properties and effective crosslink density ρx,eff (mol/m3) was determined using a modified rubber elasticity model:27, 28
| (2) |
where Qr is the swelling ratio in reference to the amount of water in sample at initial pre-swollen state, R is the gas constant and T is the temperature in kelvin.
3. Results and Discussion
The redox-mediated gelation of catechol-bearing PEG is largely controlled by the net cell potential of redox active components in the precursor solution. The central redox reaction that precedes gel formation is the reduction of metal ions and oxidation of catechols into 1,2-benzoquinone.11, 14 1,2-benzoquinones subsequently couple oxidatively to create covalent crosslinks.8, 14 The forward reaction proceeds spontaneously if the reduction potential of metal ion is more positive than that of 1,2-benzoquinone such that the full cell potential ΔEfull-Cat-Mz+ > 0. The full cell potentials and changes in Gibbs free energy of each reaction for catechols with select metal ions Mz+ are summarized in Table 1 while the gelation time and mechanical properties are summarized in Fig. 1. Hydrogel networks form when [PEG-Cat]4 precursors are combined with the following metal cations: Fe3+, V5+, Au3+, or Ag+. Notably, monovalent Ag+ (z = 1; ΔEfull-Cat-Ag+ = +49.0 mV) can induce gelation, albeit with slow kinetics (Fig. 1b, Fig. S4c). [PEG-Cat]4+Ag+ hydrogels possess an equilibrium storage modulus of G’ = 603 ± 15 Pa. In comparison, Fe3+ (z = 3; ΔEfull-Cat-Fe3+ = +21.0 mV) combined with [PEG-Cat]4 form hydrogel networks within 30 min. UV-vis spectra have prominent absorption peaks at λ = 390 nm and λ = 280 nm (Fig. 2), which are attributed to o-quinones and catechols/phenolic intermediates, respectively. The concentrations of these species can be estimated from previously reported extinction coefficients (Fig. 2b).29–31 Values for A390nm and A280nm decrease and increase, respectively, as the reaction proceeds. The temporal evolution of these species suggests that the initial bolus of o-quinones relaxes to phenolic intermediates that have similar absorbance peaks as catechols.23, 32 [PEG-Cat]4+Fe3+ hydrogels prepared from [Fe3+]:[Cat] = 2 are mechanically robust with storage moduli of G’ = 4730 ± 210 Pa (Figs. 1 and 3b). Similarly, [PEG-Cat]4 precursors combined with V5+ (VO3-; z = 5; ΔEfull-Cat-V5+ = +250 mV) promote efficient network formation and gelation. Upon initial mixing of the precursors, UV-vis spectra have an absorption shoulder at λ = 270 nm that emerges immediately and persists for 3 h observation (Fig. S4b). [PEG-Cat]4+V5+ hydrogels exhibit a storage modulus of G’ = 1530 ± 46 Pa. Finally, [PEG-Cat]4 precursors combined with Au3+ (AuCl4-; z = 3; ΔEfull-Cat-Au3+ = +180 mV) yield hydrogel networks within ~2 h. Similar trends in the UV-vis spectra of [PEG-Cat]4+Fe3+ and [PEG-Cat]4+Au3+ mixtures were observed. Namely, the concentration of o-quinone decreases while the adsorption shoulder around λ = 258 nm increases during the first 3 h (Fig. 2c). [PEG-Cat]4+Au3+ hydrogels exhibit an equilibrium storage modulus of G’ = 57 ± 8 Pa, an order of magnitude (or more) smaller than hydrogels prepared using metal ion precursors composed of Fe3+ or V5+.
Figure 1.

(a) Gelation kinetics of [PEG-Cat]4+Mz+ for select metal ion compositions. (b) Gelation time for [PEG-Cat]4+Mz+ networks as a function of metal ion composition. (c) Equilibrium storage modulus and physical appearance of [PEG-Cat]4+Mz+ hydrogels.
Figure 2.

(a) UV-vis spectra along with (b) temporal evolution of [H2Q] and [Q] in dilute polymer solutions of [PEG-Cat]4+Fe3+ as calculated from A280nm and A390nm, respectively. (c) Raw UV-vis spectra and (d) temporal evolution of [H2Q] and [Q] in dilute polymer solutions of [PEG-Cat]4+ Au3+ precursors.
Figure 3.

Storage modulus of [PEG-Cat]4+Mz+ hydrogels with (a) amplitude sweep from γ = 1–1000% strain and (b) frequency sweep from ω = 1–100 rad/s.
[PEG-Cat]4 precursors combined with metal ions Mz+ where z ≥ 2, but ΔEfull-Cat-Mz+ < 0 V remained as polymer solutions. For instance, precursor solutions of [PEG-Cat]4+V4+ in the form of VO2+ remained soluble while UV-vis spectra during 3 h observation exhibit only minor changes in redox behavior, thereby confirming slow reaction kinetics. The UV-vis spectra of precursor solutions of [PEG-Cat]4+Cu2+ and [PEG-Cat]4+Al3+ also support the conclusion that ΔEfull-Cat > 0 V is necessary and sufficient for network formation (Fig. S4e, S4f, S4h). Importantly, pH values for each precursor solution studied herein are <5 and thus significantly lower than the pKa of catechol (pKa,Cat = 9.5).34, 35 Previous reports suggest that [PEG-Cat]4/dopamine-grafted polyallylamine combined with Al3+ can form gels under slightly basic conditions (pH ~ pKa,Cat).10, 12 In addition to possible coordination bonds, gelation of dopamine-grafted polyallylamine could be attributed to catechol-primary amine reactions such as Michael addition and Schiff base formation.12, 22 Taken together, this work suggests that redox reactions between [Cat] and Mz+ such that ΔEfull-Cat-Mz+ > 0 V is necessary and sufficient for network formation.
Where applicable, we posit that redox-mediated covalent coupling of pendant catechols is the primary crosslinking mechanism of [PEG-Cat]4 precursors. It follows that [PEG-Cat]4+Mz+ precursors with more positive values for ΔEfull-Cat+Mz+ should assume larger equilibrium concentrations of o-quinones and therefore an increased rate of gelation. The trend in values of ΔEfull-Cat-Mz+ for [PEG-Cat]4+Mz+ precursors as a function of metal ion composition is V5+ > Au3+ > Ag+ > Fe3+. However, neither the observed reaction rate (gelation kinetics) nor the final value of G’ were directly proportional to calculated values for ΔEfull-Cat-Mz+. Rather, the observed trends in storage modulus G’Mz+ of [PEG-Cat]4+Mz+ hydrogels as a function of metal cation assessed by rheometry is as follows: G’Fe3+ > G’V5+ > G’Ag+ > G’Au3+. While all [PEG-Cat]4+Mz+ hydrogels fail at strain amplitudes of γ0 > 1000%, [PEG-Cat]4+Fe3+ hydrogels possess a relatively high torsional toughness compared to other [PEG-Cat]4+Mz+ hydrogels (Fig. 3). Furthermore, the storage modulus of [PEG-Cat]4+Fe3+ hydrogels is largely constant across frequencies of ω = 0.1–100 rad/s suggesting the domination of covalent bonds.36, 37 Other hydrogel network properties such as effective crosslink density ρx,eff and swelling behaviour Q were independent of the expected trends based on values for ΔEfull-Cat-Mz+. Specifically, the estimated crosslink density of [PEG-Cat]4+Fe3+ hydrogels is ρx,eff = 4.38 mol/m3 which is the highest among all [PEG-Cat]4+Mz+ hydrogels. These data coupled with UV-vis spectra support the following observation: the effective crosslinks in [PEG-Cat]4+Fe3+ and [PEG-Cat]4+Au3+ hydrogels prepared using formulations described here are primarily attributed to covalent bonds formed through redox reactions. The UV-vis spectra of [PEG-Cat]4+Au3+ networks exhibit a new absorption peak at λ = 533 nm, which is attributed to the formation of Au0 nanoparticles (Figs. 2c and S5).38–40 Additionally, the absorbance peaks at λ = 264 nm are optical signatures assigned to covalently coupled catechols (Fig. S6).41, 42 [PEG-Cat]4+Ag+ precursor solutions exhibit slow relation kinetics compared to other [PEG-Cat]4+Mz+ formulations that contain transition metal ions. Furthermore, [PEG-Cat]4+Ag+ precursor solutions exhibit a successive change in color from orange, brown to black, then gradually to yellow-green grey throughout the ~18 h gelation period. These observations suggest that slow, but spontaneous redox reactions generate silver nanoparticles.13, 39 Taken together, these observations suggest that Au3+ and Ag+ promote redox-mediated coupling and oligomerization of pendant catechols.
Given the trends in the physical properties of [PEG-Cat]4+Mz+ hydrogels vs. ΔEfull-Cat, other types of bonds likely contribute to crosslinking and network formation. Multivalent cations can create additional types of chemical bonding in network that confers unique mechanical properties.22 For example, coordination bonds confer additional mechanical robustness or self-healing capacity.7, 8, 22 Fe3+ rapidly oxidizes catechols based on both the Gibbs free energy of the full redox reaction and UV-vis data. In addition to covalent coupling of catechols, absorption peaks at λ = 750 nm suggest the formation of mono-catechol-Fe3+ complexes (Fig. 2a), reconfigurable bonds that can confer self-healing properties to crosslinked networks.7, 15, 43, 44 The values of A750nm and A280nm decrease and increase in UV-vis spectra, respectively, suggesting the shift of equilibrium from mono-Cat-Fe3+ complexes to phenolic intermediates.16, 23 Thus, in this stepwise manner, coordination bonds cooperatively enhance the mechanical properties of covalently bound networks. Previous reports of [PEG-Cat]4+Fe3+ hydrogels prepared in basic buffers describe optical signatures that are consistent with bis- and tris-cat-Fe3+ complexes (absorption peak λ = 575 nm and 492 nm respectively).7, 12, 45 The present findings are consistent with previous reports that mono-Cat-Fe3+ complexes form in acidic conditions. Reconfigurable bonds are significant contributors to crosslinks observed in [PEG-Cat]4+V5+ hydrogels as inferred through rheological properties. We posit that crosslinking in [PEG-Cat]4+Fe3+ hydrogels have substantial concentrations of covalent bonds formed through oxidative coupling since networks exhibit incomplete healing after rupture, as measured by the recovered storage modulus G’ (Fig. 4a). In contrast, [PEG-Cat]4+V5+ hydrogels can recover the initial value of G’ restored after stress-free recovery periods of 100 s for each successive cycle (Fig. 4b). This mechanical behaviour is attributed to the reversible self-healing ability of H-bonds and coordination bonds.8, 46, 47 The frequency sweep of rheology test shows frequency-dependent storage modulus first at low angular frequencies and then exhibits largely constant with increasing frequency (Fig. 3b), indicating the presence of reversible bonds in the hydrogel network.10, 36, 48 Furthermore, [PEG-Cat]4+V5+ precursor solutions produce a yellow tint immediately upon combination, suggesting that networks likely form in a redox-independent manner.49 Compared to other species of metal ions, the FTIR spectra of [PEG-Cat]4+V5+ hydrogels exhibit relatively symmetric peaks between peaks at 962 cm−1 (C-H vibration) and 929 cm−1 (V-O stretching) (Fig. 5).50 In addition, spectra recorded from [PEG-Cat]4+V5+ networks exhibit a new peak at 1491 cm−1, which is assigned to benzenoid ring stretching.51–54 Notably, the V5+ (VO3-) induced hydrogel dissolves within 30 min of incubation in water. Taken together, these data suggest that hydrogels prepared from precursor compositions with [VO3-]:[Cat] = 2 are mostly composed of non-covalent coupling between vanadium complexes and catechol groups.21, 55
Figure 4.

Plots of storage G’ and loss G” modulus for [PEG-Cat]4+Mz+ hydrogels with the following program: successive amplitude sweeps of γ = 1–1000% with a 100 s ramp; recovery time of 100 s. This program was repeated a total of four times for each sample including (a) [PEG-Cat]4+Fe3+ and (b) [PEG-Cat]4+V5+ hydrogels. Representative samples of (c) [PEG-Cat]4+Fe3+ hydrogels (d) [PEG-Cat]4+V5+ hydrogels that undergo self-healing after physical damage.
Figure 5.

FTIR spectra of [PEG-Cat]4+Mz+ hydrogels prepared using metal ion precursors of different compositions. The peak indicated by the arrow is observed only in [PEG-Cat]4+V5+ hydrogels and is attributed to benzenoid ring stretching.
In addition to bonding type, coordination chemistry between metal cations and the pendant catechols can also help to explain the mechanical behavior of [PEG-Cat]4+Mz+ hydrogels.12, 22 Based on the Pearson acid-base concept, early and late transition metals are characterized as “hard” and “soft” acids, respectively.56, 57 Accordingly, the order from hard to soft ions of metals in this work is as follows: V, Fe, Ag, and Au. The binding strength of Mz+ to catechol ligands is proportional to the hardness of the ion.56, 57 These trends can partially explain the mechanical robustness in [PEG-Cat]4+Mz+ hydrogels prepared from V5+, Au3+, and Ag+. [PEG-Cat]4+V5+ hydrogels prepared from hard acids exhibit a higher value for G’ and more rapid gelation kinetics compared to [PEG-Cat]4+Mz+ hydrogels prepared from ions that are soft acids such as Ag+ and Au3+. The chemical hardness η of Fe3+ is ηFe3+ = 13.1 eV.57 When placed in context with the UV-vis spectra, Fe3+ functions as a “hard” acid that promotes coordination bond formation. Then the transition from coordination bonds to covalent bonds thereby increasing the G’ of [PEG-Cat]4+Fe3+ hydrogel networks in a stepwise manner. In comparison, weak Lewis acids such as monovalent Ag+ (ηAg+ = 6.9 eV 57) promote network formation in [PEG-Cat]4+Ag+ hydrogel solely through colavent bonds formation.
4. Conclusions
Catechol-functionalized four-arm PEG ([PEG-Cat]4) serves as a convenient model to elucidate the role of metal ion composition on gelation including vanadium (V4+; V5+), iron (Fe3+), gold (Au3+), aluminium (Al3+), copper (Cu2+) and Silver (Ag+). The full cell potential between pendant catechols and metal ions can predict the redox-mediated gelation behavior of [PEG-Cat]4+Mz+ acidic precursor solutions with pH values smaller than pKa,-NH2 and pKa,-Cat. Multivalent redox active metal ion precursors such as Fe3+ produce crosslinks through both covalent bonds and metal-organic coordination bonds while strongly oxidative precursors such as Au3+ and Ag+ create hydrogel networks primarily through covalent bond formation. Therefore, catechol oxidation is necessary and sufficient for gelation of catechol-bearing hydrogel precursors. While coordination bonds are not necessary for hydrogel formation, they can contribute to the increase of crosslinking density and confer properties such as self-healing owing to their reconfigurable nature. This parametric study can provide forward guidance to designing catechol-bearing networks that are formed from metal ion precursors.
Catechol-bearing hydrogels can match the mechanical properties of excitable tissues. As such, this class of materials is a promising substrate material for flexible electronics and biointegrated electronics that can potentially bridge the biotic-abiotic interface for applications ranging from neuromodulation to implantable sensors.
Supplementary Material
Table 2.
Relevant network properties of [PEG-Cat]4+Mz+ hydrogels.
| Mz+ | Q | q | Mc(g/mol) | ρx (mol/m3) |
|---|---|---|---|---|
| Fe3+ | 15.9 ± 0.3 | 2.51 ± 0.02 | 2650 ± 63 | 4.38 ± 0.19 |
| V5+ | —a | — a | — a | 1.41 ± 0.04 |
| Ag+ | 47.5 ± 7.2 | 3.61 ± 0.20 | 4210 ± 290 | 0.56 ± 0.01 |
| Au3+ | 41.8 ± 2.5 | 3.47 ± 0.07 | 4400 ± 60 | 0.05 ± 0.01 |
Hydrogel dissolved within 30 min incubation in H2O.
Acknowledgements
The author would like to thank Wei-Chen Huang and Andy Zhang for helpful discussion. The authors acknowledge the financial support provided by the following organizations: the National Institutes of Health (R21NS095250), the Defense Advanced Research Projects Agency (D14AP00040), the National Science Foundation (DMR1542196), and the Carnegie Mellon University School of Engineering; The authors would like to thank the CMU Thermomechanical Characterization Facility in the Department of Materials Science and Engineering. NMR instrumentation at CMU was partially supported by the National Science Foundation (CHE-0130903 and CHE-1039870).
References
- 1.Lee BP and Konst S, Advanced Materials, 2014, 26, 3415–3419. [DOI] [PubMed] [Google Scholar]
- 2.Li QC, Barret DG, Messersmith PB and Holten-Andersen N, Acs Nano, 2016, 10, 1317–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang WC, Ong XC, Kwon IS, Gopinath C, Fisher LE, Wu HS, Fedder GK, Gaunt RA and Bettinger CJ, Advanced Functional Materials, 2018, 28. [Google Scholar]
- 4.Brubaker CE, Kissler H, Wang LJ, Kaufman DB and Messersmith PB, Biomaterials, 2010, 31, 420–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehdizadeh M, Weng H, Gyawali D, Tang LP and Yang J, Biomaterials, 2012, 33, 7972–7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dang Y, Quan M, Xing CM, Wang YB and Gong YK, Journal of Materials Chemistry B, 2015, 3, 2350–2361. [DOI] [PubMed] [Google Scholar]
- 7.Holten-Andersen N, Harrington MJ, Birkedal H, Lee BP, Messersmith PB, Lee KYC and Waite JH, Proceedings of the National Academy of Sciences of the United States of America, 2011, 108, 2651–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yavvari PS and Srivastava A, Journal of Materials Chemistry B, 2015, 3, 899–910. [DOI] [PubMed] [Google Scholar]
- 9.Guvendiren M, Messersmith PB and Shull KR, Biomacromolecules, 2008, 9, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holten-Andersen N, Jaishankar A, Harrington MJ, Fullenkamp DE, DiMarco G, He LH, McKinley GH, Messersmith PB and Leei KYC, Journal of Materials Chemistry B, 2014, 2, 2467–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang WC, Ali F, Zhao JS, Rhee K, Mou CC and Bettinger CJ, Biomacromolecules, 2017, 18, 1162–1171. [DOI] [PubMed] [Google Scholar]
- 12.Krogsgaard M, Hansen MR and Birkedal H, Journal of Materials Chemistry B, 2014, 2, 8292–8297. [DOI] [PubMed] [Google Scholar]
- 13.Fullenkamp DE, Rivera JG, Gong YK, Lau KHA, He LH, Varshney R and Messersmith PB, Biomaterials, 2012, 33, 3783–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haemers S, Koper GJM and Frens G, Biomacromolecules, 2003, 4, 632–640. [DOI] [PubMed] [Google Scholar]
- 15.Sever MJ, Weisser JT, Monahan J, Srinivasan S and Wilker JJ, Angewandte Chemie-International Edition, 2004, 43, 448–450. [DOI] [PubMed] [Google Scholar]
- 16.Lee BP, Dalsin JL and Messersmith PB, Biomacromolecules, 2002, 3, 1038–1047. [DOI] [PubMed] [Google Scholar]
- 17.Burke SA, Ritter-Jones M, Lee BP and Messersmith PB, Biomedical Materials, 2007, 2, 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Meng H, Konst S, Sarmiento R, Rajachar R and Lee BP, Acs Applied Materials & Interfaces, 2014, 6, 16982–16992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu H, Hu M, Ren KF, Wang JL, Liu XS, Jia F, Zhao YX and Ji J, Thin Solid Films, 2016, 600, 76–82. [Google Scholar]
- 20.Xu ZP, Scientific Reports, 2013, 3. [Google Scholar]
- 21.Park JP, Song IT, Lee J, Ryu JH, Lee Y and Lee H, Chemistry of Materials, 2015, 27, 105–111. [Google Scholar]
- 22.Krogsgaard M, Nue V and Birkedal H, Chemistry-a European Journal, 2016, 22, 844–857. [DOI] [PubMed] [Google Scholar]
- 23.Cencer M, Liu Y, Winter A, Murley M, Meng H and Lee BP, Biomacromolecules, 2014, 15, 2861–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shafiq Z, Cui JX, Pastor-Perez L, San Miguel V, Gropeanu RA, Serrano C and del Campo A, Angewandte Chemie-International Edition, 2012, 51, 4332–4335. [DOI] [PubMed] [Google Scholar]
- 25.Peppas NA and Merrill EW, Journal of Polymer Science Part a-Polymer Chemistry, 1976, 14, 441–457. [Google Scholar]
- 26.Merrill EW, Dennison KA and Sung C, Biomaterials, 1993, 14, 1117–1126. [DOI] [PubMed] [Google Scholar]
- 27.Salinas CN and Anseth KS, Macromolecules, 2008, 41, 6019–6026. [Google Scholar]
- 28.Zhu CC and Bettinger CJ, Macromolecules, 2015, 48, 1563–1572. [Google Scholar]
- 29.Rzepecki LM and Waite JH, Analytical Biochemistry, 1989, 179, 375–381. [DOI] [PubMed] [Google Scholar]
- 30.Rzepecki LM, Nagafuchi T and Waite JH, Archives of Biochemistry and Biophysics, 1991, 285, 17–26. [DOI] [PubMed] [Google Scholar]
- 31.Waite JH, Analytical Chemistry, 1984, 56, 1935–1939. [Google Scholar]
- 32.Andersen SO, Jacobsen JP, Bojesen G and Roepstorff P, Biochimica Et Biophysica Acta, 1992, 1118, 134–138. [DOI] [PubMed] [Google Scholar]
- 33.Leung PK, Martin T, Shah AA, Anderson MA and Palma J, Chemical Communications, 2016, 52, 14270–14273. [DOI] [PubMed] [Google Scholar]
- 34.Fuguet E, Reta M, Gibert C, Roses M, Bosch E and Rafols C, Electrophoresis, 2008, 29, 2841–2851. [DOI] [PubMed] [Google Scholar]
- 35.Slabbert NP, Tetrahedron, 1977, 33, 821–824. [Google Scholar]
- 36.Roberts MC, Hanson MC, Massey AP, Karren EA and Kiser PF, Advanced Materials, 2007, 19, 2503–2507. [Google Scholar]
- 37.Lee F, Chung JE and Kurisawa M, Soft Matter, 2008, 4, 880–887. [DOI] [PubMed] [Google Scholar]
- 38.Daniel MC and Astruc D, Chemical Reviews, 2004, 104, 293–346. [DOI] [PubMed] [Google Scholar]
- 39.Lee PC and Meisel D, Journal of Physical Chemistry, 1982, 86, 3391–3395. [Google Scholar]
- 40.Haiss W, Thanh NTK, Aveyard J and Fernig DG, Analytical Chemistry, 2007, 79, 4215–4221. [DOI] [PubMed] [Google Scholar]
- 41.Mizrahi B, Shankarappa SA, Hickey JM, Dohlman JC, Timko BP, Whitehead KA, Lee JJ, Langer R, Anderson DG and Kohane DS, Advanced Functional Materials, 2013, 23, 1527–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menyo MS, Hawker CJ and Waite JH, Soft Matter, 2013, 9, 10314–10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avdeef A, Sofen SR, Bregante TL and Raymond KN, Journal of the American Chemical Society, 1978, 100, 5362–5370. [Google Scholar]
- 44.Crisponi G, Nurchi VM and Pivetta T, Journal of Inorganic Biochemistry, 2008, 102, 209–215. [DOI] [PubMed] [Google Scholar]
- 45.Krogsgaard M, Behrens MA, Pedersen JS and Birkedal H, Biomacromolecules, 2013, 14, 297–301. [DOI] [PubMed] [Google Scholar]
- 46.Webber MJ, Appel EA, Meijer EW and Langer R, Nature Materials, 2016, 15, 13–26. [DOI] [PubMed] [Google Scholar]
- 47.Lin YL and Li GJ, Journal of Materials Chemistry B, 2014, 2, 6878–6885. [DOI] [PubMed] [Google Scholar]
- 48.Lopez-Perez PM, da Silva RMP, Strehin I, Kouwer PHJ, Leeuwenburgh SCG and Messersmith PB, Macromolecules, 2017, 50, 8698–8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rehder D, Bioinorganic vanadium chemistry, John Wiley & Sons, 2008, Vol. 30, 13. [Google Scholar]
- 50.Reddy CVS, Yeo IH and Mho SI, Journal of Physics and Chemistry of Solids, 2008, 69, 1261–1264. [Google Scholar]
- 51.Choi YK, Kim HJ, Kim SR, Cho YM and Ahn DJ, Macromolecules, 2017, 50, 3164–3170. [Google Scholar]
- 52.Zhao LB, Chen JL, Zhang M, Wu DY and Tian ZQ, Journal of Physical Chemistry C, 2015, 119, 4949–4958. [Google Scholar]
- 53.Furukawa Y, Ueda F, Hyodo Y, Harada I, Nakajima T and Kawagoe T, Macromolecules, 1988, 21, 1297–1305. [Google Scholar]
- 54.Ping Z, Journal of the Chemical Society-Faraday Transactions, 1996, 92, 3063–3067. [Google Scholar]
- 55.Kustin K, Liu ST, Nicolini C and Toppen DL, Journal of the American Chemical Society, 1974, 96, 7410–7415. [Google Scholar]
- 56.Ho TL, Chemical Reviews, 1975, 75, 1–20. [Google Scholar]
- 57.Parr RG and Pearson RG, Journal of the American Chemical Society, 1983, 105, 7512–7516. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.