

Abstract
Adult facial motor neurons continue to express full-length TrkB tyrosine kinase receptor (TrkB.FL), the high-affinity receptor for the neurotrophins BDNF and neurotrophic factor-4/5 (NT-4/5), suggesting that they remain dependent on target-derived and locally produced neurotrophins in adulthood. Studies on the role of TrkB signaling in the adult CNS have been hampered by the early lethality of bdnf, nt-4/5, and trkB knock-out mice. We disrupted TrkB.FL signaling in adult facial motor neurons using adeno-associated viral vector-mediated overexpression of a naturally occurring dominant-negative TrkB receptor, TrkB.T1. Expression of TrkB.T1 resulted in neuronal atrophy and downregulation of NeuN (neuronal-specific nuclear protein) and ChAT expression in facial motor neurons. A subset of transduced neurons displayed signs of motor neuron degeneration that included dendritic beading and rounding of the soma at 2 months of TrkB.T1 expression. Cell counts revealed a significant reduction in motor neuron number in the facial nucleus at 4 months after onset of expression of TrkB.T1, suggesting that a proportion of TrkB.T1-expressing motor neurons became undetectable as a result of severe atrophy or was lost because of cell death. In contrast, overexpression of TrkB.FL did not result in a decrease in facial motor neuron number. Our results indicate that a subset of facial motor neurons remains dependent on TrkB ligands for the maintenance of structural and molecular characteristics in adulthood.
Keywords: degeneration, facial nucleus, AAV, TrkB, plasticity, motor neuron
Introduction
Brain-derived neurotrophic factor (BDNF) regulates neuronal survival and differentiation in the developing nervous system (Ernfors, 2001; Huang and Reichardt, 2001) and synaptic plasticity in adulthood (McAllister et al., 1999; Schinder and Poo, 2000). BDNF exerts its effects via the TrkB tyrosine kinase receptor, which also binds neurotrophic factor-4/5 (NT-4/5) and to a lesser extent NT-3 (Klein et al., 1991; Middlemas et al., 1991; Soppet et al., 1991; Squinto et al., 1991). Several TrkB isoforms are generated through alternative splicing. The full-length TrkB (TrkB.FL) tyrosine kinase receptor is expressed exclusively in neurons (Klein et al., 1990). The truncated isoforms TrkB.T1 and T2 (Klein et al., 1990; Middlemas et al., 1991) lack tyrosine kinase activity and are expressed in both neurons and glia (Frisen et al., 1993; Armanini et al., 1995).
Truncated TrkB receptors restrict TrkB.FL signaling in several ways. First, by sequestering TrkB ligands, truncated TrkB receptors expressed on non-neuronal cells limit the availability of BDNF (Biffo et al., 1995; Fryer et al., 1997). Second, truncated TrkB receptors can act as dominant-negative receptors when coexpressed with TrkB.FL (Eide et al., 1996; Ninkina et al., 1996; Haapasalo et al., 2001). Third, TrkB.T1 can downregulate surface expression of TrkB.FL (Haapasalo et al., 2002).
Developing motor neurons express TrkB.FL (Yan et al., 1993; Escandon et al., 1994; McKay et al., 1996), and expression persists into adulthood (Koliatsos et al., 1993; Yan et al., 1997). The expression of TrkB.T1 in motor neurons increases with age (Escandon et al., 1994; Koliatsos et al., 1994; Armanini et al., 1995). NT-4/5 is the predominant neurotrophin expressed in the targets of mature facial and spinal motor neurons (Koliatsos et al., 1994; Funakoshi et al., 1995; Griesbeck et al., 1995). Adult motor neurons remain responsive to BDNF, because exogenously applied TrkB ligands maintain the cholinergic phenotype of axotomized adult facial, spinal, and hypoglossal motor neurons (Yan et al., 1994; Friedman et al., 1995; Tuszynski et al., 1996; Wang et al., 1997; Fernandes et al., 1998) and rescue avulsed motor neurons from injury-induced cell death (Clatterbuck et al., 1994; Kishino et al., 1997; Novikov et al., 1997).
Little is known about the role of endogenous neurotrophins in motor neurons. Infusion of TrkB–IgG protein into the gastrocnemius muscle of adult rats to scavenge endogenous TrkB ligands induces an axotomy-like effect on motor axonal conduction velocity (Munson et al., 1997). Adenovirus-mediated overexpression of dominant-negative TrkB.T1 in muscle induces disassembly of postsynaptic acetylcholine receptor clusters and retraction of nerve terminals (Gonzalez et al., 1999). Together, these studies suggest a role for TrkB signaling in the maintenance of functional and structural properties of adult motor neurons.
Here, we investigated the effects of adeno-associated viral (AAV) vector-mediated expression of TrkB isoforms on facial motor neurons. Our results indicate that disruption of TrkB signaling by overexpression of truncated TrkB leads to phenotypic and structural alterations in motor neurons and could eventually result in cell loss. This suggests that a subset of facial motor neurons remains dependent on TrkB ligands for the maintenance of molecular and structural characteristics in adulthood.
Materials and Methods
AAV vector production.
The following rat cDNAs were used to construct AAV vector plasmids: N-terminally FLAG (DYKDDDDK)-tagged TrkB.FL and TrkB.T1 and N-terminally enhanced green fluorescent protein (EGFP)-tagged TrkB.T1 cDNAs. These tagged TrkB isoforms have been characterized previously and are functional (Haapasalo et al., 1999, 2001; Kryl et al., 1999). Farnesylated EGFP (EGFP-f), which is targeted to the inner leaflet of the plasma membrane, was used as a control (Clontech, Palo Alto, CA). TrkB and EGFP-f cDNAs were subcloned into an AAV vector plasmid (see Fig. 1A) containing a cytomegalovirus (CMV) promoter, the Woodchuck post-transcriptional regulatory element (WPRE) to enhance transgene expression (Donello et al., 1998; Loeb et al., 1999), and a simian virus 40 polyadenylation signal, flanked by two AAV-2 inverted terminal repeats (ITRs). The helper plasmid pDG, needed for the production of recombinant AAV-2 vector particles, has been described previously (Grimm et al., 1998). A detailed protocol for AAV vector production is provided in Hermens et al. (1999). Briefly, vector plasmids and helper plasmid were cotransfected into human embryonic kidney (HEK) 293T cells using calcium phosphate precipitation. The medium was replaced after 6 h, and cells were grown for another 48 h. Next, the cells were scraped from the culture dishes and freeze-thawed to release AAV particles from the cells. Cell debris was removed using low-speed centrifugation. The supernatant was loaded on a Matrex Cellufine-sulfate bead column (Amicon, Danvers, MA). After several washings with PBS, pH 7.4, the virus was eluted from the column with PBS containing 1 m NaCl. Viral particles were banded on a Iodixanol (Nycomed Pharm, Oslo, Norway) density gradient using ultracentrifugation, and 0.3 ml fractions were collected from the bottom of the tube. AAV-2-containing fractions were diluted 10 times in PBS to reduce viscosity and reconcentrated by centrifugation in a Centricon-100 concentrator (Amicon). The transgene expressing unit titer (tu/ml) was determined by coinfection of HEK 293T cells with serial dilutions of the viral vector and adenovirus with a multiplicity of infection (m.o.i.) of 3. At 24 h after infection, cells were fixed with 4% paraformaldehyde (PFA; Sigma, Zwijndrecht, The Netherlands) in PBS, and transgene-expressing cells were visualized with an antibody directed against the FLAG epitope tag [FLAG M2 monoclonal antibody (mAb); Sigma] or GFP (GFP rabbit polyclonal antibody; Chemicon, Harrow, UK). Titers obtained were in the range of 109 to 1010 tu/ml.
Figure 1.

Characterization of AAV-derived epitope-tagged TrkB isoforms in HEK 293T cells. A, Schematic representation of the AAV vector plasmid used for the generation of AAV vectors. The AAV vector plasmid contains an expression cassette consisting of a human CMV promoter to drive transgene expression, a transgene cDNA encoding TrkB receptor isoforms or marker protein, the cis-acting WPRE to enhance transgene expression, and a downstream polyadenylation (pA) signal. The transgene expression cassette is flanked by two ITRs, essential for packaging of the viral construct. B, Schematic representation of the transgenes used in this study and their orientation in the plasma membrane. Epitope tags are inserted in the N terminus behind the signal sequence. C, Lysate from AAV–FLAGTrkB.FL- and AAV–FLAGTrkB.T1-infected HEK 293T cells probed with FLAG M2 monoclonal antibody. Equal amounts of protein (10 μg) were loaded in each lane. Molecular weight markers are indicated on the left. The arrows indicate the full-length (145 kDa) and truncated (95 kDa) TrkB receptors. Additional bands likely represent underglycosylated TrkB. D, Lysate from AAV–GFP-TrkB.T1- and AAV–GFP-f-infected HEK 293T cells probed with a GFP polyclonal antibody. The GFP–TrkB.T1 fusion protein has an approximate molecular weight of 125 kDa as a result of the GFP tag (27 kDa). GFP-f is not visible on this gel because of its low molecular weight. E, AAV-mediated expression of GFP-f in HEK 293T cells. Noninfected cells were used as a control.
Western blotting.
HEK 293T cells were grown in DMEM (Invitrogen, Breda, The Netherlands) supplemented with 10% fetal bovine serum (FBS) (Invitrogen) and penicillin/streptomycin (Invitrogen) in a humidified incubator at 37°C and 5% CO2. For analysis of transgene expression, 6 cm plates were infected for 24 h with AAV at an m.o.i. of 10. Medium was replaced the next day, and 24 h later, cells were washed twice in ice-cold PBS and lysed in radioimmunoprecipitation assay (RIPA) buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 10 mm Na2HPO4, 1% Triton X-100, 1% Na-desoxychalate, 0.1% SDS, 1 mm EDTA, 1 mm PMSF, 1 mm benzamidine, 50 mm NaF, 250 μm Na3VO4) for 5 min on ice. Lysates were centrifuged for 10 min at 14,000 rpm, and supernatants were transferred to a new Eppendorf tube (Eppendorf Scientific, Westbury, NY). Protein content was determined with a Bradford assay. For each sample, 10 μg of protein was separated on 7.5% SDS-PAGE gels (11% SDS-PAGE gels for AAV–GFP-f), according to standard procedures. Proteins were transferred semidry to nitrocellulose membrane (Schleicher and Schuell, Dassel, Germany). Membranes were washed in H2O, blocked in TBS–MLK [5% milk powder, 0.05% Tween 20 in Tris-buffered saline (TBS), pH 7.4)] for 1 h at room temperature (RT), and incubated overnight at 4°C in primary antibody (FLAG M2, Sigma; GFP rabbit polyclonal, Invitrogen) diluted in TBS–MLK. The next day, membranes were washed four times in TBS and incubated for 1 h at RT with HRP-conjugated secondary antibodies (DakoCytomation, Glostrup, Denmark) diluted in TBS–MLK. After three washes in TBS, blots were visualized using ECL (PerkinElmer, Boston, MA). Blots were quantified using Image Pro Plus software (version 4.5; Media Cybernetics, Silver Spring, MD) to determine the optical density of each band. Samples were normalized against control samples on the same blot.
Live cell labeling and immunocytochemistry.
HEK 293T cells were grown on 0.2% gelatin-coated glass coverslips (Menzel Gläser, Freiburg, Germany). One day after plating, cells were infected with AAV at an m.o.i. of 10. Medium was replaced the next day. Twenty-four hours later, cells were washed three times with warm, serum-free DMEM and incubated subsequently with primary antibodies (FLAG M2, Sigma; GFP, Chemicon) diluted in DMEM for 30 min at 37°C. Subsequently, cells were washed three times in DMEM and fixed in 4% PFA in PBS for 30 min at RT. After three washes in TBS, cells were incubated with cyanine 3 (Cy3)-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) for 1 h at RT. After several washes, coverslips were mounted in mowiol.
For visualization of antigens that required permeabilization, cells were fixed in 4% PFA in PBS, washed in TBS, and blocked in TBS–FBS (TBS containing 5% fetal bovine serum, 0.2% Triton X-100) for 1 h at RT. Primary antibodies TrkB (TK−) sc-119 rabbit polyclonal antibody, which specifically recognizes truncated TrkB, and Trk sc-11 rabbit polyclonal antibody, which recognizes all full-length Trk receptors, were from Santa Cruz Biotechnology (Santa Cruz, CA). Incubation with primary antibodies was done overnight at 4°C. Primary and Cy-3-conjugated secondary antibodies (Jackson ImmunoResearch) were diluted in TBS–FBS. Images were captured using an LSM510 Zeiss (Oberkochen, Germany) confocal laser-scanning microscope.
Receptor phosphorylation assay.
AAV vector plasmids were transfected into HEK 293T cells using calcium phosphate precipitation. Cotransfections of FLAG–TrkB.FL with EGFP-f, GFP–TrkB.T1, or FLAG–TrkB.T1 were done at a molar ratio of 1:3. After 6 h, medium was replaced and cells were cultured for an additional 24 h. Subsequently, cells were washed in serum-free DMEM and incubated in serum-free DMEM for 2 h. Cells were stimulated with 50 ng/ml recombinant human BDNF (Peprotech, London, UK) for 5 min at 37°C, washed in ice-cold PBS, and lysed in RIPA buffer. Lysates were centrifuged for 10 min at 14,000 rpm, and supernatants were transferred to a new Eppendorf tube. A 100 μl sample of supernatant was set aside for analysis of mitogen-activated protein (MAP) kinase phosphorylation (see below). Supernatants were precleared with 20 μl of protein-A-agarose beads (Santa Cruz Biotechnology) for 30 min at 4°C. After centrifugation, supernatants were transferred to a new tube and incubated for 1 h with primary antibody (Trk sc-11 antibody; Santa Cruz Biotechnology). Subsequently, 20 μl of protein-A-agarose beads were added, and samples were rotated overnight at 4°C. The next day, agarose beads were washed three times with PBS and resuspended in 40 μl of 2× sample buffer. Samples were separated on 7.5% SDS-PAGE gels. Immunoblots were probed with Trk sc-11 antibody diluted in TBS–MLK or anti-phosphotyrosine PY20 monoclonal antibody (Becton Dickinson, Franklin Lakes, NJ) in TBS containing 3% bovine serum albumin and 0.05% Tween 20. For the analysis of MAP kinase phosphorylation, the samples containing cell lysates were assayed for protein content, and 10 μg of protein was separated on 11% SDS-PAGE gels and immunoblotted. Blots were probed with an antibody specific for phosphorylated MAP kinase (anti-phospho MAP kinase [Erk1/2 (extracellular signal-regulated kinase 1/2)]; Upstate Biotechnology, Chicago, IL).
Experimental animals and stereotaxic injections.
A total of 78 adult male Wistar rats (200–250 g; Harlan, Horst, The Netherlands) were used in this study. Animals were housed in group cages and had ad libitum access to water and food. Experimental handling and postoperative care were in accordance with the guidelines of the local committee for laboratory animal welfare and experimentation. A detailed description of the experimental protocol for stereotaxic delivery of viral vectors to the brain is provided in Ruitenberg et al. (2002). Direct stereotaxic injections were performed under deep anesthesia with Hypnorm (0.08 ml/100 g of body weight, i.m.; Janssen Pharmaceuticals, Tilburg, The Netherlands) in the facial nucleus [coordinates: anteroposterior (AP), −3.5 mm; lateral (L), +1.5 mm from lambda; dorsoventral (DV), −8.5 mm from dura; according to Paxinos and Watson (1998)]. Stereotaxic coordinates for the hippocampus were: AP, −3.3 mm; L, +1.7 mm, from bregma; DV, −3.5 mm from dura. Injections were performed with a stainless 30-gauge needle attached via polyethylene tubing to a 10 μl Hamilton syringe in a Harvard 22 microinjection pump. Titers of different AAV vectors were matched (3 × 109 tu/ml). One microliter of AAV vector (3 × 106 tu) was gently infused at a rate of 0.2 μl/min. The needle was left in place for 1 min to allow diffusion from the injection site before it was removed from the brain.
Tissue processing and immunohistochemistry.
Rats were anesthetized deeply with Nembutal (sodium pentobarbital; Sanofi Sante, Maassluis, The Netherlands) and perfused transcardially with 0.9% saline, followed by 4% PFA in 0.1 m phosphate buffer, pH 7.4. Brains were removed from the animals and postfixed overnight in 4% PFA solution at 4°C and subsequently processed for vibratome sectioning or cryosectioning. For vibratome sectioning, four series of sections (40 μm) were collected in TBS. For cryosectioning, brains were cryoprotected in 25% sucrose in 0.1 m phosphate buffer for 3 d at 4°C. Brains were embedded in OCT compound 4583 (Tissue-Tek; Sakura, Zoeterwoude, The Netherlands) and frozen rapidly in 2-methyl butane on dry ice. Five series of sections (20 μm) were collected per animal on Superfrost Plus slides (Menzel Gläser) and stored at −80°C until subjected to histochemical procedure. Vibratome sections were subjected to standard immunohistochemical staining procedures to assess transgene expression. Sections were washed three times in TBS, followed by blocking of endogenous peroxidase activity with 10% methanol and 0.3% H2O2 in TBS for 30 min at RT. Subsequently, sections were washed three times and blocked in TBS–FBS for 1 h at RT. Sections were then incubated overnight at 4°C in TBS-FBS containing the primary antibody (GFP pAb, Chemicon; TrkB(TK−) sc-119 pAb and Trk sc-11 pAb, Santa Cruz Biotechnology). The next day, sections were washed three times and incubated with secondary antibody (goat anti-rabbit; Netherlands Institute for Brain Research, Amsterdam, The Netherlands) in TBS–FBS for 1 h at RT. Subsequently, sections were washed three times and incubated with tertiary antibody (peroxidase anti-peroxidase; Netherlands Institute for Brain Research) in blocking solution for 1 h at RT. After several washes, sections were stained with 3,3′-diaminobenzamidine tetrahydrochloride in 50 mm Tris-HCl, pH 7.6, containing 0.01% H2O2 and 0.2 mg/ml (NH4)2·SO4·NISO4, resulting in dark purple precipitate. Sections were washed and dehydrated in a graded series of ethanol, cleared in xylene, and embedded in Entallan (Merck, Darmstadt, Germany). Specificity of immunostainings was confirmed by absence of staining on the contralateral, noninjected side.
Fluorescent double labelings to analyze and quantify neuronal marker expression in transduced cells were performed on cryosections. Cryosections were pretreated with proteinase K (0.01 mg/ml) and formamide at 55°C overnight for antigen retrieval followed by standard immunohistochemistry as described above. Primary antibodies were neuronal-specific nuclear protein (NeuN) mAb and choline acetyl transferase (ChAT) pAb (both from Chemicon). Secondary antibodies were from Jackson ImmunoResearch. Sections were mounted in Vectashield (Vector Laboratories, Burlingame, CA). To minimize variation, all sections were simultaneously processed according to a standard protocol. Specificity of staining was confirmed by omission of the primary antibody. Images were captured using an LSM 510 Zeiss confocal laser scanning microscope. Constant settings for detector gain, offset, and pinhole were used to acquire all NeuN and ChAT images. Fluorescence intensity was quantified using MetaMorph 6.2 software (Universal Imaging Corporation, Downingtown, PA). Separate channels of red–green–blue images were saved as monochrome images. Subsequently, cells expressing GFP- or Trk-labeled transgenes in the green channel were manually outlined, using the transgene staining to obtain a reliable outline. Only cells with a visible nucleus were selected for analysis. The outlined region was copied to the red channel containing the NeuN or ChAT labeling. The NeuN or ChAT image was thresholded, using a constant threshold setting for all images. MetaMorph software was used to determine the average intensity of NeuN and ChAT staining in transduced cells.
Quantification of motor neuron morphology after AAV-mediated transduction.
One series of vibratome sections, immunohistochemically stained for transgene expression, was selected for quantitative analysis of motor neuron morphology. The morphology of transgene-expressing motor neurons was scored as “degenerating” when they displayed a rounded or irregular soma and/or beaded dendrites. Only neurons with a visible nucleus were scored. Neurons were scored in AAV–GFP-f- (n = 4), AAV–GFPTrkB.T1- (n = 4), and AAV–FLAGTrkB.FL- (n = 4) injected animals at 1, 2, and 4 months after injection. The number of degenerating motor neurons was expressed as a percentage of the total number of counted, transduced motor neurons. Statistical significance was determined by comparing the percentage of degenerating neurons in AAV–GFPTrkB.T1- and AAV–FLAGTrkB.FL-injected groups with AAV–GFP-f-injected controls using Student’s t test.
Soma size measurements of transduced motor neurons.
To determine whether overexpression of TrkB isoforms affected motor neuron soma size, high-resolution digital images of immunohistochemically stained vibratome sections were captured with a 10× objective. Only transgene-expressing motor neurons were selected for analysis. Vibratome sections of AAV–GFP-f-, AAV–GFPTrkB.T1-, and AAV–FLAGTrkB.FL- (n = 4 per time point for all groups) injected animals were simultaneously processed for immunohistochemistry, according to a standard protocol to minimize variation in staining. GFP-f and TrkB receptors are targeted to the cell membrane, and truncated and full-length TrkB receptors are not differentially sorted in neurons (Kryl et al., 1999). Immunohistochemical staining of these transgene products can therefore be used to evaluate the soma size of transduced motor neurons. Digital images were imported in Image Pro Plus software, version 4.5. The perimeters of labeled motor neurons with a visible nucleus were traced manually by an observer unaware of the treatment. The surface area of transduced cells was determined and expressed as square micrometers. Soma size histograms were made using 100 μm2 intervals. Statistical analysis of the soma size distributions was done using a Kolmogorov–Smirnov test. Statistical significance of differences in mean soma area was determined using a one-way ANOVA followed by Bonferroni’s post hoc testing (SPSS software, version 11.0; SPSS, Chicago, IL).
Motor neuron counts.
To evaluate the number of motor neurons in the injected facial nucleus, 20 μm cryosections were stained in 0.2% cresyl violet solution (Merck), rinsed in water, dehydrated, and embedded in Entallan. Motor neuron numbers were determined in AAV–GFP-f-, AAV–GFPTrkB.T1-, AAV–FLAGTrkB.T1- and AAV–FLAGTrkB.FL- (n = 4 for all groups) injected animals, at 4 months after injection. Animals were counted in a randomized order by an observer unaware of the treatment. Every fifth section was counted. Digital images were captured with a 2.5× objective and used to manually outline the area of the facial nucleus. A grid was projected over the outlined area, and systematic randomized samples covering 50% of the grid area were counted. Only neurons containing a clearly discernible nucleolus were counted, with a 40× objective. Neurons containing more than one nucleolus were not observed. S-Plus software (Insightful, Seattle, WA) was used to perform one-way ANOVA using an a priori treatment contrast model (Venables and Ripley, 2002).
Results
Characterization of AAV vectors
We used epitope-tagged TrkB.FL and TrkB.T1 constructs to allow reliable detection of viral vector-mediated transgene expression. For TrkB.FL and TrkB.T1, a FLAG epitope tag was used. In addition, we used an enhanced GFP-tagged version of TrkB.T1. Epitope tags were inserted in the N terminus, directly C-terminal of the signal sequence (Fig. 1B). Transgene cDNAs were subcloned into AAV vector plasmids (Fig. 1A), which were used to generate the following AAV vectors: AAV encoding FLAG-tagged TrkB.FL and TrkB.T1 (termed AAV–FLAGTrkB.FL and AAV–FLAGTrkB.T1, respectively) and GFP–TrkB.T1 (AAV–GFPTrkB.T1). An AAV vector encoding a membrane-attached (farnesylated) variant of GFP (termed AAV–GFP-f) was used as a control.
To determine whether epitope-tagged TrkB isoforms were correctly processed, cell lysates from AAV-infected HEK 293T cells were analyzed on Western blot. Immunoblotting with an antibody directed against the FLAG epitope showed a band of ∼140 kDa in AAV–FLAGTrkB.FL-infected HEK 293T cells (Fig. 1C). A band of 95 kDa was observed for FLAG–TrkB.T1 (Fig. 1C). Both are consistent with the expected molecular weights of 145 and 95 kDa for full-length and truncated TrkB, respectively (Klein et al., 1990). In addition to the bands representing full-length and truncated TrkB, an additional isoform was detected (Fig. 1C). Additional bands after expression of wild-type or epitope-tagged TrkB receptors in heterologous cells have been observed previously (Armanini et al., 1995; Kryl et al., 1999) and most likely represent underglycosylated, immature isoforms of TrkB (Watson et al., 1999). Immunoblotting with an antibody directed against GFP revealed a band of ∼125 kDa for GFP–TrkB.T1 (Fig. 1D). Cells infected with the control vector AAV–GFP-f showed a 30 kDa band (Fig. 1E).
To ascertain that AAV-derived full-length and truncated TrkB receptors were expressed at the cell surface, unfixed and unpermeabilized HEK 293T cells infected with AAV-encoding TrkB isoforms were labeled with FLAG and GFP antibodies. Live labeling with a GFP antibody demonstrated cell surface staining for GFP–TrkB.T1-expressing cells (Fig. 2B). Live FLAG staining of AAV–FLAGTrkB.T1-infected cells showed a similar pattern, with strong FLAG immunoreactivity distributed along the cell membrane including small filopodia (Fig. 2C). Cell surface labeling of FLAG-stained AAV–FLAGTrkB.FL-infected cells generally appeared weaker than surface immunoreactivity for FLAG–TrkB.T1 (Fig. 2D). Surface expression of full-length TrkB in neuronal cells is regulated by neuronal activity and stimulation with BDNF (Meyer-Franke et al., 1998; Du et al., 2000; Haapasalo et al., 2002; Du et al., 2003), and in unstimulated cells, most of TrkB.FL is located intracellularly, which might explain the lower amount of TrkB.FL surface staining we observed in HEK 293T cells. Indeed, after permeabilization of cells, staining with an antibody specific for Trk receptors revealed additional intracellular immunoreactivity for AAV–FLAGTrkB.FL-infected cells (Fig. 2F), whereas staining with an antibody specific for TrkB.T1 showed that the distribution of FLAG–TrkB.T1 in permeabilized cells (Fig. 2E) was similar to live-labeled cells (Fig. 2C). Cells expressing the control vector AAV–GFP-f showed typical cell membrane fluorescence (Fig. 2A).
Figure 2.

AAV-derived TrkB isoforms are targeted to the cell surface. A, AAV–GFP-f-infected HEK 293T cells show typical cell membrane-associated fluorescence (arrow). B–D, Live labeling of AAV–TrkB-infected HEK 293T cells. B, AAV–GFPTrkB.T1-infected cells incubated with GFP antibody before fixation show surface expression of GFP–TrkB.T1 (arrow). C, D, FLAG M2 staining of AAV–FLAGTrkB.T1- (C) and AAV–FLAGTrkB.FL- (D) infected cells. FLAG–TrkB.T1 and FLAG–TrkB.FL are targeted to the cell surface in nonpermeabilized cells (arrows). E, F, Immunostaining of permeabilized AAV-infected cells. E, AAV–FLAGTrkB.T1-infected cells stained with TrkB(TK−) polyclonal antibody specific for TrkB.T1 show a similar pattern (arrow) to the nonpermeabilized cells in C. F, AAV–FLAGTrkB.FL-infected cells stained with a Trk polyclonal antibody show additional intracellular immunoreactivity for TrkB.FL (arrowhead). Scale bar: (in F) A–F, 50 μm.
To verify the biological activity of epitope-tagged TrkB isoforms, we analyzed receptor autophosphorylation and activation of downstream signaling pathways in response to stimulation with recombinant BDNF. AAV vector plasmids encoding full-length and truncated TrkB were cotransfected in HEK 293T cells in a 1:3 ratio. Cells were stimulated briefly with BDNF, and TrkB.FL was immunoprecipitated and analyzed on Western blot. Stimulation of FLAG–TrkB.FL coexpressed with GFP-f resulted in phosphorylation of TrkB.FL (Fig. 3A), showing that BDNF can bind to and activate epitope-tagged TrkB.FL. Coexpression of GFP–TrkB.T1 or FLAG–TrkB.T1 strongly reduced BDNF-induced phosphorylation of TrkB.FL (Fig. 3A). Analysis of the same samples with a Trk antibody showed that approximately equal amounts of TrkB.FL were immunoprecipitated for each condition (Fig. 3A). To correct for possible variations in loading of TrkB.FL, the optical density of phosphorylated TrkB.FL (p-TrkB.FL) bands was determined and divided by the optical density of Trk-labeled TrkB.FL bands. The p-TrkB.FL/TrkB.FL ratio was normalized to TrkB.FL:GFP-f coexpressing samples. Quantification showed that BDNF-induced phosphorylation of TrkB.FL was reduced significantly after coexpression of GFP–TrkB.T1 or FLAG–TrkB.T1 (Fig. 3B) (p < 0.01). The observed effects were not caused by endogenously expressed Trk receptors, because no TrkB.FL was detected in immunoprecipitates of cells expressing GFP-f alone (Fig. 3A). Stimulation of TrkB.FL:GFP-f coexpressing cells resulted in the phosphorylation of Erk1/2 (MAP kinase), a downstream target of TrkB (Huang and Reichardt, 2003) (Fig. 3A). BDNF-induced Erk1/2 phosphorylation was strongly reduced after coexpression of GFP–TrkB.T1 or FLAG–TrkB.T1 but not GFP-f (Fig. 3A). Quantification showed that phosphorylation of both Erk1 (Fig. 3C) (p < 0.01) and Erk2 (Fig. 3D) (TrkB.FL:GFP–TrkB.T1, p < 0.01; TrkB.FL:FLAG–TrkB.T1, p < 0.05) was significantly reduced after coexpression of GFP–TrkB.T1 or FLAG–TrkB.T1. We consistently observed that expression of TrkB.FL was slightly more pronounced in the presence of GFP-f than in the presence of truncated TrkB (Fig. 3A). A possible explanation for this may be that the production of a large protein such as TrkB.T1, which requires additional modifications such as glycosylation, is more demanding than the production of a small protein such as GFP-f. This might slow down the production of TrkB.FL when TrkB.FL and TrkB.T1 are coexpressed at a 1:3 ratio. TrkB.FL also displayed some tyrosine phosphorylation in the absence of ligand (Fig. 3A). This might be because of a high density of receptors in the plasma membrane resulting from high levels of expression, causing cross-phosphorylation in the absence of ligand. It should be noted, however, that although the expression and background phosphorylation of TrkB.FL in the presence of GFP-f are somewhat more pronounced, this does not result in increased phosphorylation of downstream Erk1/2 (Fig. 3A). Together, these results show that AAV vector-derived epitope-tagged TrkB isoforms are produced at their expected molecular weights in HEK 293T cells and are correctly targeted to the cell surface. In addition, epitope-tagged TrkB receptors retain their biological activity. Both phosphorylation of TrkB.FL as well as activation of downstream effector Erk1/2 after stimulation with BDNF are significantly reduced after coexpression of truncated TrkB.
Figure 3.
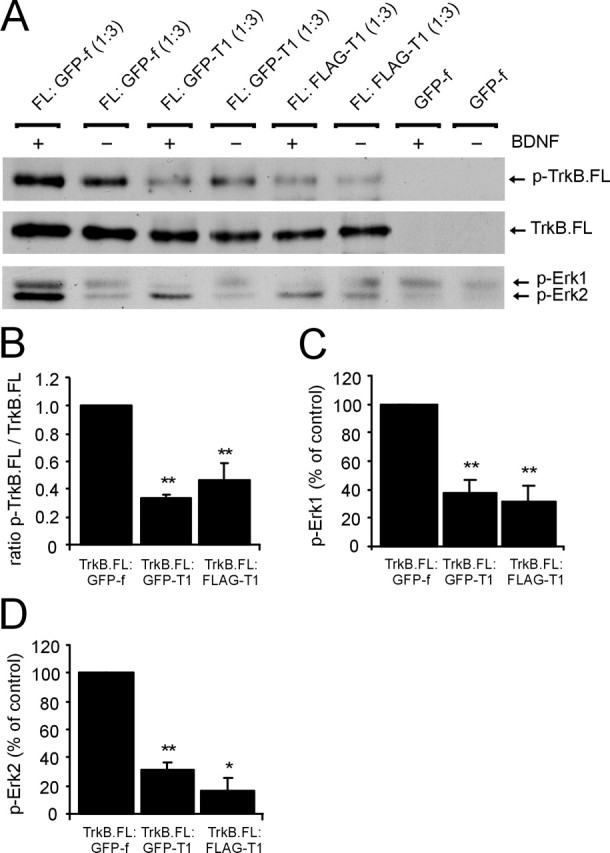
Viral constructs encode functional TrkB isoforms. A, AAV vector plasmid encoding FLAG–TrkB.FL was cotransfected with AAV–GFPTrkB.T1, AAV–FLAGTrkB.T1, or AAV–GFP-f vector plasmids in a 1:3 ratio. Transfected cells were briefly stimulated with 50 ng/ml BDNF, and TrkB.FL was precipitated from HEK 293T cell lysate with an anti-Trk antibody. Blots were probed with an anti-phosphotyrosine antibody. BDNF-induced phosphorylation of TrkB.FL (p-TrkB.FL) is strongly reduced after coexpression with GFP–TrkB.T1 or FLAG–TrkB.T1 but not with GFP-f. Immunoprecipitates from the same samples were probed with Trk antibody to show that equal amounts of TrkB.FL were loaded. Cell lysate from the same samples probed with a phospho-Erk antibody (p-Erk) shows that phosphorylation of Erk1/2 (44 and 42 kDa) in response to BDNF is strongly reduced when GFP–TrkB.T1 or FLAG–TrkB.T1 are coexpressed. Blots representative of three independent experiments are shown. B, Quantification shows significant reduction of BDNF-induced TrkB.FL phosphorylation when GFP–TrkB.T1 or FLAG–TrkB.T1 are coexpressed. To correct for possible variations in loading, the optical density of p-TrkB.FL bands was determined and divided by the optical density of Trk-labeled TrkB.FL bands. Values were normalized to GFP-f-coexpressing controls. C, BDNF-induced phosphorylation of Erk1 is significantly reduced after coexpression of truncated TrkB. D, Phosphorylation of Erk2 in response to BDNF is significantly reduced after coexpression of truncated TrkB. Data are based on three separate experiments. The bar graphs show mean ± SEM. *p < 0.05; **p < 0.01; Student’s t test.
AAV-mediated expression of TrkB isoforms in facial motor neurons
We evaluated the efficiency of AAV-mediated gene delivery to facial motor neurons using immunohistochemical staining to detect transgene-expressing motor neurons 4 weeks after injection. Stereotaxic administration of AAV reproducibly resulted in the transduction of numerous cells expressing high levels of transgene throughout the facial nucleus (Fig. 4). Numerous GFP-f-expressing cells were observed in GFP-stained sections of the facial nucleus of AAV–GFP-f-injected animals (Fig. 4A). In agreement with previous studies, AAV-2-mediated transgene expression in the facial nucleus appeared primarily neuronal (Peel et al., 1997; Bartlett et al., 1998). Transgene-expressing cells showed typical motor neuron morphology, with several dendrites extending from a large soma (Fig. 4A, inset) and immunopositive axons in the facial nerve (Fig. 4C). Injection of AAV–GFPTrkB.T1 resulted in a robust transduction of motor neurons, widely distributed throughout the facial nucleus (Fig. 4B). GFP–TrkB.T1 could be detected on soma and dendrites of motor neurons (Fig. 4B, inset), on axons in the facial nerve (Fig. 4D), and in presynaptic terminals in facial muscle (data not shown). Immunohistochemical detection of the FLAG epitope in AAV–FLAGTrkB.T1- and AAV–FLAGTrkB.FL-injected animals was unsuccessful (data not shown). As a result, we had to resort to the use of antibodies that do not distinguish between transgene-derived and endogenously expressed TrkB receptors. A TrkB.T1-specific antibody (Fig. 2E) detected transduced motor neurons in sections of the facial nucleus of AAV–FLAGTrkB.T1-injected animals (Fig. 4E). At the dilution used, the antibody specifically labeled transgene-expressing motor neurons, because no neuronal staining was detected on the contralateral, noninjected side (Fig. 4G). AAV-mediated expression of FLAG–TrkB.FL was detected with an antibody directed against full-length Trk receptors (Fig. 2F), which specifically detected transduced motor neurons in the facial nucleus of AAV–FLAGTrkB.FL-injected animals (Fig. 4F). Staining was absent on the contralateral side (Fig. 4H).
Figure 4.
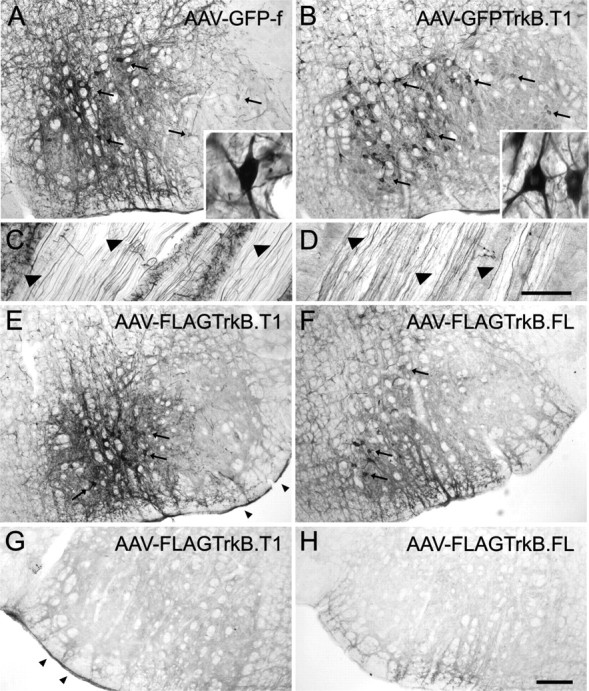
Transgene expression in the facial nucleus 4 weeks after AAV-mediated gene transfer. A, GFP-f-expressing motor neurons (arrows) in AAV–GFP-f-injected facial nucleus. GFP staining labels soma and dendrites of transduced motor neurons (inset). B, AAV–GFPTrkB.T1-transduced facial nucleus. GFP–TrkB.T1 is expressed on cell bodies and dendrites of motor neurons (inset). C, GFP-f-expressing axons in the facial nerve. D, GFP–TrkB.T1-positive axons in the facial nerve. E, Immunohistochemistry with the TrkB(TK−) polyclonal antibody specific for TrkB.T1 shows FLAG–TrkB.T1-expressing motor neurons in an AAV–FLAGTrkB.T1-injected facial nucleus (arrows). G, TrkB(TK−) staining is absent on the contralateral, noninjected side. The arrowheads in E and G indicate endogenous TrkB.T1 in the meninges. F, Trk immunohistochemistry shows FLAG–TrkB.FL-expressing motor neurons in the facial nucleus of an AAV–FLAGTrkB.FL-injected animal (arrows). H, Trk staining was absent on the contralateral, noninjected side. Scale bars: (in H) A, B, E–H, 200 μm; (in D) C, D, 100 μm.
Long-term AAV-mediated expression of GFP–TrkB.T1 in facial motor neurons results in neuronal atrophy and dendritic beading
We next analyzed whether disruption of TrkB.FL signaling in motor neurons by long-term AAV-mediated expression of dominant-negative TrkB.T1 affected the structural characteristics of adult facial motor neurons. To this end, we analyzed the morphology of TrkB.T1-expressing motor neurons at 2 and 4 weeks, and at 2 and 4 months, after injection of AAV. AAV–GFPTrkB.T1 was used because of the sensitivity and specificity of transgene detection.
Two weeks after administration of AAV–GFPTrkB.T1, transduced neurons displayed regular soma contours and several labeled dendrites, sometimes with varicose appearance, characteristic of intact facial motor neurons (Shaw and Baker, 1985; Friauf, 1986) (Fig. 5A). Four weeks after injection, some GFP–TrkB.T1-expressing facial motor neurons exhibited a reduced soma size, suggestive of neuronal atrophy (Fig. 5B). After 2 months of transgene expression, a strikingly abnormal morphology was evident in a subset of transduced facial motor neurons (Fig. 5C–E). The dendrites of these neurons displayed numerous large spherical swellings, connected by thin, atrophied dendritic segments (Fig. 5C–E). Swelling or beading could be observed along the entire length of dendrites of affected motor neurons (Fig. 5C,D). The cell bodies of these motor neurons were intensely labeled, had a shrunken appearance, and showed rounded or irregular contours (Fig. 5C,D). Importantly, only a subset of GFP–TrkB.T1-expressing motor neurons exhibited these structural abnormalities, because unaffected transduced motor neurons could be observed near neurons with beaded dendrites and rounded somata (Fig. 5C). Dendritic beading and abnormal soma contours are characteristics of degenerating motor neurons (Ma and Vacca-Galloway, 1991; Wong et al., 1995). Quantification showed that an average of 16.5 ± 3.7% (mean ± SEM) of GFP–TrkB.T1-expressing motor neurons displayed a degenerative morphology at 2 months after administration of AAV, which was significantly higher than GFP-f controls (3.8 ± 1.4%; n = 4; p = 0.011) (Table 1). AAV–GFP-f-transduced neurons occasionally displayed irregular soma contours, but dendritic beading was never observed in this group (Fig. 5G). At 4 months of GFP–TrkB.T1-expression, the majority of transduced neurons displayed regular soma contours and dendrites (Fig. 5F, Table 1). Qualitative comparison of all time points suggested a gradual decline in the number of GFP–TrkB.T1-expressing facial motor neurons over time and a decrease in the density of stained neurites (Fig. 5A–F), which was not observed in AAV–GFP-f-injected animals (Fig. 5G).
Figure 5.

Long-term overexpression of GFP–TrkB.T1 causes neuronal atrophy and dendritic beading in facial motor neurons. Transgene expression was detected with GFP immunohistochemistry. A, Transduced cells at 2 weeks of expression of GFP–TrkB.T1 show typical motor neuron morphology (arrows). B, At 4 weeks, some GFP–TrkB.T1-expressing neurons exhibit a reduced soma size (arrowheads), whereas other neurons are not affected (arrows). C–E, Transgene expression at 2 months. C, Example of a motor neuron with a rounded soma (open arrowhead) and severe dendritic swelling (indicated with arrowheads). Neighboring transduced neurons display regular soma contours and dendrites (arrows). D, GFP–TrkB.T1-expressing motor neuron with a rounded soma (open arrowhead). Dendrites contain large, spherical swellings (arrowheads) separated by thin, atrophied segments. E, Higher magnification of the boxed area in D. Open arrowheads indicate dendritic varicosities, which appear as elongated enlargements of dendrites connected by segments of normal caliber and also occur in intact motor neurons. F, GFP–TrkB.T1 motor neuron morphology at 4 months (arrows). The open arrowhead indicates transduced motor neuron with rounded soma, eccentric nucleus, and thinned dendrites (arrowheads). G, GFP-f-expressing motor neurons (arrows) show regular soma contours and dendrites (arrowheads) 2 months after injection. Scale bars: (in G) A–C, F, G, 100 μm; D, 25 μm; E, 10 μm.
Table 1.
Quantification of degenerating transduced motor neurons in the facial nucleus
| Time point | Degenerating motor neurons
(%) |
||
|---|---|---|---|
| AAV–GFP-f (n = 4 per time point) | AAV–GFPTrkB.T1 (n = 4 per time point) | AAV–FLAGTrkB.FL (n = 4 per time point) | |
| 4 weeks | 3.8 ± 1.4 | 3.3 ± 1.5 | 5.5 ± 2.0 |
| 2 months | 1.8 ± 1.8 | 16.5 ± 3.7* | 7.6 ± 2.6 |
| 4 months | 2.0 ± 1.2 | 4.1 ± 2.5 | 2.4 ± 2.4 |
Transduced neurons in the facial nucleus were counted at three time points after injection of AAV, four animals per time point (>60 immunoreactive cells). Morphology was scored as degenerating when neurons met one or more of the criteria described in Materials and Methods. Values represent mean ± SEM.
*p < 0.05; significantly different from GFP-f controls; Student’s t test.
To quantify the effects of GFP–TrkB.T1 expression on motor neuron size, we determined the cell size distributions of AAV–GFPTrkB.T1-transduced neurons at four time points after injection and compared these to GFP-f-expressing controls (Fig. 6). At 2 and 4 weeks after injection, the soma size distributions of GFP–TrkB.T1-expressing motor neurons displayed a significant shift toward the smaller size ranges compared with GFP-f control neurons, indicating neuronal atrophy in motor neurons expressing truncated TrkB (p = 0.0121 at 2 weeks; p = 0.000534 at 4 weeks; Kolmogorov–Smirnov test) (Fig. 6A,B). The mean soma area of GFP–TrkB.T1-expressing motor neurons at 4 weeks after injection was significantly smaller than that of GFP-f controls [440 ± 10 vs 530 ± 17 μm2 (mean ± SEM); n = 4; p = 0.003] (Fig. 6E), confirming neuronal atrophy. At 2 and 4 months of transgene expression, soma size distributions and mean soma areas of GFP–TrkB.T1-expressing motor neurons were not significantly different from GFP-f-expressing motor neurons (Fig. 6C–E and data not shown). Note that only neurons with a visible nucleus were measured (see Materials and Methods). Although dendritic beading and rounding of somata were most pronounced at 2 months, the nuclei of these neurons were often no longer visible, which explains the absence of a reduction in mean soma area in GFP–TrkB.T1-expressing motor neurons at this time point. Together, long-term AAV-mediated overexpression of GFP–TrkB.T1 in adult facial motor neurons resulted in neuronal atrophy, followed by rounding of the cell body and dendritic beading in a subset of transduced neurons. The latter two morphological responses were most pronounced at 2 months and had declined at 4 months of transgene expression.
Figure 6.

Effect of TrkB isoform expression on motor neuron soma size distribution. A, 2 weeks; B, 4 weeks; C, 2 months; and D, 4 months of transgene expression. White bars, GFP-f; gray bars, GFP–TrkB.T1; black bars, FLAG–TrkB.FL (n = 4 animals per time point for all experimental groups). E, Average soma areas of GFP–TrkB.T1- (gray bars) and FLAG–TrkB.FL- (black bars) expressing motor neurons, relative to GFP-f-expressing controls (white bars), at four time points after injection. Values are expressed as mean ± SEM. *p < 0.05; **p < 0.01; one-way ANOVA followed by Bonferroni’s post hoc testing.
Long-term expression of FLAG–TrkB.FL in facial motor neurons causes a bimodal distribution of soma sizes
We compared the effects of long-term expression of truncated TrkB on facial motor neuron morphology with the effects of long-term expression of FLAG–TrkB.FL at the same time points. Two and four weeks after injection of AAV–FLAGTrkB.FL, transduced motor neurons showed regular soma contours and dendrites (Fig. 7A,B). At 2 months of transgene expression, FLAG–TrkB.FL-expressing motor neurons occasionally displayed a swollen, rounded soma (Fig. 7C,D). Dendritic beading, however, as observed in GFP–TrkB.T1-expressing neurons at this time point (Fig. 5C–E), was not apparent in FLAG–TrkB.FL-expressing motor neurons at 2 months (Fig. 7E). The percentage of motor neurons with a degenerative morphology in AAV–FLAGTrkB.FL-injected animals after injection was slightly elevated but not significantly different from controls at this time point (Table 1). GFP-f-expressing motor neurons displayed a normal morphology (Fig. 7G). At 4 months, the majority of FLAG–TrkB.FL-expressing motor neurons exhibited regular soma contours and dendrites (Fig. 7F, Table 1).
Figure 7.
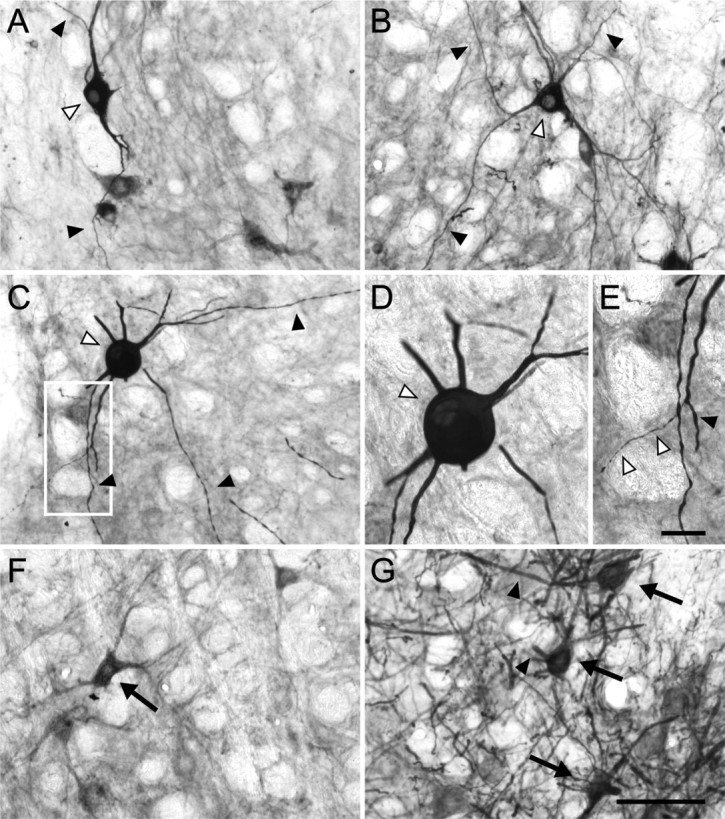
Long-term expression of FLAG–TrkB.FL in facial motor neurons. Trk immunohistochemistry was used to detect transgene expression. A, AAV-mediated expression of FLAG–TrkB.FL, 2 weeks after injection. Trk immunohistochemistry labels cell body (open arrowhead) and dendrites (arrowheads) of the transduced neuron. B, Transduced motor neuron at 4 weeks (open arrowhead). The arrowheads indicate dendrites. C–E, Transgene expression at 2 months. C, Transduced motor neuron with a hypertrophied soma (open arrowhead) and labeled dendrites (arrowheads). D, Higher magnification of the same neuron shown in C. E, Higher magnification of the boxed area in C. Some distal dendritic branches appeared severely thinned (open arrowheads). The arrowhead indicates dendritic varicosity, which also occurs in intact motor neurons. F, FLAG–TrkB.FL transgene expression at 4 months. G, GFP-f-expressing motor neurons (arrows) at 2 months show regular soma contours and dendrites (arrowheads). Scale bar: (in G) A–C, F, G, 100 μm; (in E) D, E, 25 μm.
Quantification of the effect of FLAG-TrkB.FL-expression on motor neuron size showed a bimodal distribution of cell sizes (Fig. 6). Two and four weeks after injection, a decrease in the percentage of cells in the middle ranges was accompanied by an increase in the percentages of cells in the smaller as well as the larger ranges, suggesting that both atrophy and hypertrophy occur in FLAG–TrkB.FL-expressing motor neurons (Fig. 6A,B). Soma size distribution and mean soma areas at these time points, however, were not significantly different from GFP-f-expressing controls (Fig. 6E and data not shown), which may be caused by the simultaneous increase in both the smaller and larger size ranges. At 2 months of transgene expression, the mean soma area of FLAG–TrkB.FL-expressing cells was significantly smaller compared with GFP-f controls (531 ± 31 vs 651 ± 21 μm2; n = 4; p = 0.016) (Fig. 6E). Soma size distributions at this time point were not significantly different from GFP-f controls (p = 0.085; Kolmogorov–Smirnov test). Four months after injection, mean soma area and soma size distribution of FLAG–TrkB.FL-expressing motor neurons were not significantly different from GFP-f-positive cells (Fig. 6E and data not shown). Thus, AAV-mediated overexpression of FLAG–TrkB.FL in facial motor neurons resulted in an irregular distribution of cell sizes, suggesting atrophy as well as hypertrophy of TrkB.FL-expressing neurons.
Downregulation of neuronal marker expression in truncated TrkB-expressing facial motor neurons
Cholinergic marker expression in injured adult motor neurons is regulated by exogenously applied TrkB ligands (Yan et al., 1994; Friedman et al., 1995; Tuszynski et al., 1996; Wang et al., 1997; Fernandes et al., 1998). To determine whether overexpression of TrkB isoforms affected the expression of neuronal markers in mature facial motor neurons, we stained sections of AAV-injected animals with an antibody for NeuN, a nuclear protein with DNA binding properties (Mullen et al., 1992), and an antibody for the neurotransmitter synthesizing enzyme ChAT.
NeuN staining of sections of AAV-GFP-f-injected animals demonstrated that AAV-mediated gene transfer in the facial nucleus was predominantly neuronal (Fig. 8A–C). Unexpectedly, we found that expression of NeuN was reduced or absent in GFP-TrkB.T1-expressing neurons 2 months after injection (Fig. 8D–F). Quantification showed that the average intensity of NeuN staining was significantly reduced in GFP-TrkB.T1-expressing neurons [NeuN intensity, 67.5 ± 5.9% (mean ± SEM) of GFP-f controls; p < 0.001] (Fig. 8J). NeuN expression in FLAG–TrkB.FL-expressing neurons did not differ significantly from GFP-f controls (87.3 ± 3.9% of GFP-f controls; p > 0.05) (Fig. 8G–I,J).
Figure 8.
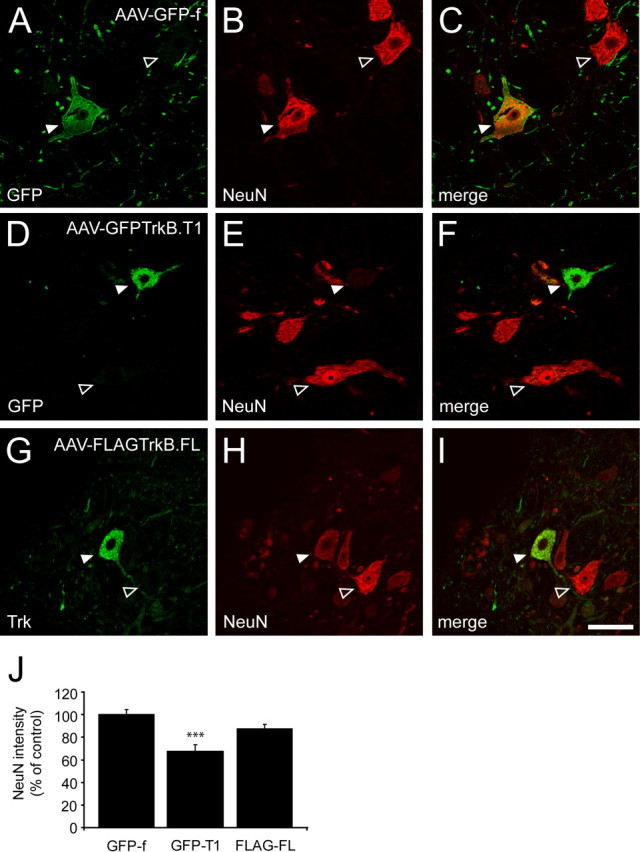
Downregulation of NeuN expression in TrkB.T1-expressing facial motor neurons. A, D, GFP immunohistochemistry; G, Trk immunostaining; B, E, H, NeuN immunohistochemistry. Merged images are shown in C, F, and I. Representative images are shown of AAV–GFP-f- (n = 3), AAV–GFPTrkB.T1- (n = 4), and AAV–FLAGTrkB.FL- (n = 4) injected animals, 2 months after injection. A–C, NeuN expression in GFP-f-expressing motor neuron (arrowhead). The open arrowhead indicates neighboring nontransduced neuron. D–F, Loss of NeuN staining in GFP–TrkB.T1-expressing motor neuron (arrowhead). The open arrowhead indicates normal NeuN expression in nontransduced neighboring neuron. G–I, NeuN expression in FLAG–TrkB.FL-expressing motor neuron (arrowhead). The open arrowhead indicates neighboring nontransduced neuron. Scale bar: (in I) 50 μm. J, Quantification of the average intensity of NeuN staining in transduced motor neurons, normalized to GFP-f-expressing neurons (n = 104 neurons from 3 animals). Expression of GFP–TrkB.T1 results in a significant reduction in the intensity of NeuN labeling (p < 0.001; n = 93 neurons from 4 animals). Expression of NeuN in FLAG–TrkB.FL-expressing neurons does not differ significantly from GFP-f control neurons (p > 0.05; n = 87 neurons from 4 animals). The bar graphs show mean ± SEM. ***p < 0.001; Kruskal–Wallis test.
We next examined the expression of ChAT in TrkB isoform-overexpressing motor neurons at 2 months of transgene expression. ChAT expression was reduced in GFP–TrkB.T1-expressing motor neurons (Fig. 9D–F), compared with GFP-f-expressing motor neurons (Fig. 9A–C). Quantification of the average intensity of ChAT staining showed a significant reduction in ChAT expression in GFP–TrkB.T1-expressing neurons [ChAT intensity, 70.4 ± 3.1% (mean ± SEM) of GFP-f controls; p < 0.001] (Fig. 9J). A decrease in ChAT expression was not observed in FLAG–TrkB.FL-expressing motor neurons (104.8 ± 3.0% of GFP-f controls; p > 0.05) (Fig. 9G–I,J).
Figure 9.

Downregulation of ChAT expression in TrkB.T1-expressing facial motor neurons. A, D, GFP immunohistochemistry; G, Trk immunostaining; B, E, H, ChAT immunohistochemistry. Merged images are shown in C, F, and I. Representative images are shown of AAV–GFP-f- (n = 3), AAV–GFPTrkB.T1- (n = 4), and AAV–FLAGTrkB.FL- (n = 4) injected animals, 2 months after injection. A–C, ChAT expression in GFP-f-transduced motor neurons (arrowheads). The open arrowheads indicate ChAT expression in neighboring nontransduced neurons. D–F, ChAT expression is downregulated in GFP–TrkB.T1-expressing motor neuron (arrowhead). The open arrowhead indicates normal ChAT expression in nontransduced neighboring neuron. G–I, Normal ChAT expression in FLAG–TrkB.FL-expressing motor neuron (arrowhead). The open arrowhead indicates neighboring nontransduced neuron. Scale bar: (in I) 50 μm. J, Quantification of the average intensity of ChAT staining in transduced motor neurons, normalized to GFP-f-expressing neurons (n = 66 neurons from 3 animals). Expression of GFP–TrkB.T1 results in a significant reduction in the intensity of ChAT labeling compared with GFP-f controls (p < 0.001; n = 70 neurons from 4 animals). The average intensity of ChAT labeling is not affected by expression of FLAG–TrkB.FL (p > 0.05; n = 72 neurons from 4 animals). The bar graphs show mean ± SEM. ***p < 0.001; Kruskal–Wallis test.
To determine whether downregulation of NeuN expression after overexpression of GFP–TrkB.T1 is specific for facial motor neurons, we injected AAV–GFP-f and AAV–GFPTrkB.T1 into the hippocampus of adult rats. NeuN immunoreactivity in the majority of GFP–TrkB.T1-expressing granule cells (Fig. 10D–F) and CA1 pyramidal neurons (Fig. 10J–L) was comparable with NeuN expression in GFP-f control neurons 4 weeks after administration of the vector (Fig. 10A–C,G–I). Together, these results show that overexpression of TrkB.T1 resulted in a significant downregulation of expression of the neuronal markers NeuN and ChAT in adult facial motor neurons. Downregulation of NeuN expression was not observed after GFP–TrkB.T1 expression in adult hippocampal granule and CA1 pyramidal cells.
Figure 10.

GFP-TrkB.T1 does not downregulate NeuN expression in hippocampal neurons. A, D, G, J, GFP immunohistochemistry; B, E, H, K, NeuN immunohistochemistry. Merged images are shown in C, F, I, and L. Representative images are shown of AAV–GFP-f- (n = 3) and AAV–GFPTrkB.T1- (n = 4) injected animals, 4 weeks after injection. A–C, NeuN expression in GFP-f-transduced hippocampal granule cells (arrowheads). D–F, NeuN expression in GFP–TrkB.T1-transduced granule cells (arrowheads). G–I, NeuN expression in GFP-f-transduced CA1 pyramidal neuron (arrowhead). J–L, NeuN expression in GFP–TrkB.T1-transduced CA1 pyramidal cells (arrowheads). GCL, Granule cell layer; H, hilus; SR, stratum radiatum; SO, stratum oriens. Scale bar: (in L) A–L, 50 μm.
Long-term expression of truncated TrkB results in a reduction of facial motor neuron number
The swelling of dendrites and rounding of somata observed in GFP–TrkB.T1-expressing motor neurons were most pronounced at 2 months and had declined by 4 months of transgene expression (Fig. 5, Table 1). The diminished expression of NeuN and ChAT in GFP–TrkB.T1-expressing motor neurons persisted until 2 months (Figs. 8, 9 and data not shown) and had returned to normal levels at 4 months after injection (data not shown). Furthermore, there appeared to be a progressive decrease in the number of GFP–TrkB.T1-expressing motor neurons over time (Fig. 5). To determine whether long-term expression of truncated TrkB in adult motor neurons resulted in a reduction in the number of facial motor neurons, we injected four groups of rats with AAV–GFP-f, AAV–GFPTrkB.T1, AAV–FLAGTrkB.T1, and AAV–FLAGTrkB.FL, respectively, and counted the number of cresyl violet-stained neurons in the facial nucleus at 4 months after injection.
Microscopic analysis of cresyl violet-stained sections suggested a modest motor neuron loss in facial nuclei of animals expressing truncated TrkB (Fig. 11B,C) compared with animals expressing full-length TrkB (Fig. 11D) or GFP-f (Fig. 11A). Quantification showed a reduction of ∼20% in mean motor neuron number in both AAV–GFPTrkB.T1-injected animals and AAV–FLAGTrkB.T1-injected animals [81 ± 10% (mean ± SEM) and 78 ± 6% of AAV–GFP-f-injected controls, respectively] (Fig. 11E). The mean number of motor neurons in the facial nucleus of AAV–FLAGTrkB.T1-injected animals was significantly lower than that of AAV–GFP-f-injected animals (2979 ± 174 vs 3839 ± 249 neurons; n = 4; p = 0.035) (Fig. 11E). The reduction in mean number of motor neurons in the facial nucleus of AAV–GFPTrkB.T1-injected animals approached statistical significance [3092 ± 312 vs 3839 ± 249 neurons (mean ± SEM); n = 4; p = 0.061]. When both truncated TrkB-expressing groups were combined, the mean number of motor neurons in animals expressing truncated TrkB was significantly lower than GFP-f-expressing controls (p = 0.019), showing that long-term expression of truncated TrkB resulted in a reduction in facial motor neuron number. AAV–FLAGTrkB.FL-injected animals displayed a slight reduction in mean motor neuron number (87 ± 8% of controls), but this was not significantly different from AAV–GFP-f-injected control animals (3356 ± 268 vs 3839 ± 249 neurons; n = 4; p = 0.206) (Fig. 11E).
Figure 11.

Reduction in facial motor neuron number in animals expressing truncated TrkB. Cresyl violet-stained sections of the facial nucleus from animals injected with AAV–GFP-f (A), AAV–GFPTrkB.T1 (B), AAV–FLAGTrkB.T1 (C), and AAV–FLAGTrkB.FL (D), at 4 months after injection, are shown. The images were taken at the same level of the nucleus. Scale bar (in D) A–D, 400 μm. E, Quantification of neuron numbers in the facial nucleus of AAV-injected animals, 4 months after injection (n = 4 animals per group). Animals expressing truncated TrkB show a significant reduction in facial motor neuron number, which may be attributable either to severe atrophy to a point where motor neurons are no longer detectable or to cell death. Data are represented as mean ± SEM. *p < 0.05. Statistical significance was determined with a one-way ANOVA using an a priori treatment contrast model.
Although the reduction in cresyl violet-stained motor neurons in animals overexpressing truncated TrkB suggests facial motor neuron loss, this reduction could also result from a failure to detect small severely atrophied cells. In an attempt to independently show that motor neurons are lost after overexpression of truncated TrkB, we performed both terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining and immunohistochemistry for cleaved caspase 3 on tissue sections from AAV-injected animals at 1 and 2 months after injection. We were unable to detect TUNEL- or cleaved caspase 3-positive cells in any of the experimental groups (data not shown). Given the relatively small reduction in the number of motor neurons over a time span of 4 months, it is conceivable that the failure to detect apoptotic cells could result from either a gradual loss of TrkB.T1-expressing neurons over a period of several months (Moran et al., 2001) or from a wave of apoptosis at time points not included in our analysis.
Discussion
Adult motor neurons express TrkB.FL (Koliatsos et al., 1993; Yan et al., 1997). Studies on the role of endogenous TrkB ligands in the adult CNS have been hampered by the early lethality of trkB, bdnf, and nt-4/5 null mutants (Klein et al., 1993; Ernfors et al., 1994; Jones et al., 1994; Conover et al., 1995; Liu et al., 1995). Here, we used a viral vector approach to study the role of TrkB signaling in mature facial motor neurons. AAV vectors permit long-term transgene expression in adult neurons in a wild-type environment (Kaplitt et al., 1994; McCown et al., 1996; Peel and Klein, 2000). AAV-mediated expression of dominant-negative TrkB.T1 resulted in a downregulation of neuronal marker expression, neuronal atrophy, rounding of the soma, and dendritic beading in motor neurons. Cell counts revealed a significant reduction in motor neuron number in animals overexpressing truncated TrkB. No significant reduction in motor neuron number was detected in animals expressing TrkB.FL.
Long-term expression of truncated TrkB results in motor neuron degeneration
Long-term expression of TrkB.T1 produced distinct, progressively developing effects on facial motor neuron morphology. Transduced motor neurons displayed significant neuronal atrophy at 2 and 4 weeks of transgene expression, followed 1 month later by dendritic beading and rounding of the soma in ∼17% of GFP–TrkB.T1-expressing neurons. This degenerative morphology was no longer apparent at 4 months of transgene expression, at which time point cell counts showed a 20% reduction in motor neuron number in TrkB.T1-expressing animals. Importantly, expression of TrkB.T1 constructs with different epitope tags (GFP and FLAG) resulted in a comparable reduction in motor neuron number, indicating that these effects did not arise from tag insertion. The gradual development of motor neuron phenotype after overexpression of TrkB.T1 is reminiscent of the phenotype of cortical pyramidal neurons in conditional trkB mutants. Cortical neurons undergo a retraction of dendrites and rounding of the soma, resulting in possible cell death over the course of 6 weeks after postnatal deletion of TrkB (Xu et al., 2000). The structural alterations we observe in TrkB.T1-expressing motor neurons follow a similar course, suggesting that a long-term dominant-negative inhibition of endogenous TrkB.FL by virally expressed TrkB.T1 underlies the degeneration of motor neurons. Several studies have shown a dominant-negative effect of truncated TrkB on TrkB.FL-mediated survival when both isoforms are coexpressed (Ninkina et al., 1996; Haapasalo et al., 2001; Dorsey et al., 2002). Reduced TrkB.FL signaling in facial motor neurons could also result from sequestration of BDNF, NT-4/5, and NT-3 by TrkB.T1 (Biffo et al., 1995). Although overexpression of TrkB.T1 in cortical and hippocampal neurons can produce effects independent of inhibition of TrkB.FL, including distal dendritic growth and filopodia formation (Yacoubian and Lo, 2000; Hartmann et al., 2004), these effects are distinct from the effects we observe after TrkB.T1 overexpression in motor neurons. Thus, our results suggest that a subset of facial motor neurons remains dependent on TrkB.FL signaling in adulthood for the maintenance of structural characteristics.
Analysis of motor neuron development in knock-out mice has suggested that motor neurons develop normally in the absence of neurotrophin signaling, although some discrepancies exist. Single or double mutant mice lacking BDNF and NT-4/5 show little or no motor neuron loss (Ernfors et al., 1994; Jones et al., 1994; Conover et al., 1995; Liu et al., 1995). Mice lacking both TrkB and TrkC develop without deficits in motor neuron number (Silos-Santiago et al., 1997). In striking contrast, neonatal triple mutant mice lacking BDNF, NT-4/5, and NT-3 display a significant 20% reduction in facial motor neuron number (Liu and Jaenisch, 2000). The early lethality of these mutations precluded the analysis of the role of neurotrophins in the maintenance of mature neurons. Interestingly, newborn trkB/trkC double null mutants show normal development of the neocortex (Silos-Santiago et al., 1997), whereas deletion of TrkB in postnatal cortical neurons results in their degeneration (Xu et al., 2000). This suggests that mature neurons can become dependent on neurotrophins for maintenance and survival, despite their normal initial development in the absence of neurotrophin signaling.
Whether degenerating TrkB.T1-expressing motor neurons ultimately die or merely become undetectable as a result of extreme atrophy is uncertain. We were unable to detect apoptotic cell death in TrkB.T1-expressing animals. This may be because of the difficulty in detecting a small number of apoptotic cells over a long time span, because apoptosis is a fast process (Rossiter et al., 1996). A lack of apoptotic cells after adult motor neuron injury has been reported by others (Li et al., 1998; Vanderluit et al., 2000). Adult axotomized rat facial motor neurons may undergo a slow, nonapoptotic form of degeneration (Moran et al., 2001). Alternatively, at least a fraction of axotomized adult mouse facial motor neurons can survive in a state of extreme atrophy in which they are no longer detectable by Nissl stain (McPhail et al., 2004b). A lack of increased apoptotic cell death has also been observed in the cortex of conditional trkB mutants, despite extensive degeneration (Xu et al., 2000). Thus, the degeneration of TrkB.T1-expressing neurons could be followed by either cell death and/or by survival in an extremely atrophied state.
Downregulation of marker expression in TrkB.T1-expressing motor neurons
GFP-TrkB.T1 was expressed on soma, dendrites, and axons. Overexpression of TrkB.T1 likely results in a blockade of target-derived NT-4/5 (Koliatsos et al., 1994), locally produced BDNF (Kobayashi et al., 1996), and BDNF anterogradely transported from other sources (Fawcett et al., 1998). TrkB ligands regulate axonal and dendritic development and synaptic plasticity (for review, see McAllister et al., 1999; Schinder and Poo, 2000). Reduced TrkB.FL signaling in TrkB.T1-expressing motor neurons could induce alterations in dendritic and axonal morphology, which in turn might result in loss of synaptic contacts, leading to decreased marker expression, soma atrophy, and eventually degeneration. Interestingly, not all TrkB.T1-expressing neurons degenerate. The extent of TrkB.T1 overexpression varies between transduced cells and may therefore inhibit TrkB.FL signaling to varying degrees.
Loss of cholinergic phenotype of axotomized mature motor neurons and rescue thereof by exogenous TrkB ligands is well documented (Yan et al., 1994; Friedman et al., 1995; Wang et al., 1997; Fernandes et al., 1998). Loss of NeuN expression occurs in axotomized facial motor neurons but not in axotomized rubrospinal neurons (McPhail et al., 2004a). The TrkB.T1-induced downregulation of NeuN was specific for motor neurons and was not observed in hippocampal neurons. Thus, the TrkB.T1-induced downregulation of ChAT and NeuN expression resembles the downregulation after target deprivation of adult motor neurons. TrkB.T1-induced axotomy-like effects on neuronal marker expression in the cell body could result from reduced retrograde signaling from target-derived TrkB ligands or reflect an actual retraction of motor nerve terminals. We were unable to determine the effects of TrkB overexpression on distal motor axon morphology. The facial nerve innervates many small muscles, making it difficult to locate sufficient numbers of transduced terminals in sections of facial muscle. Intriguingly, postsynaptic TrkB.T1 overexpression in muscle does result in the retraction of nerve terminals (Gonzalez et al., 1999).
Effects of TrkB.FL overexpression in facial motor neurons
Interestingly, overexpression of TrkB.FL caused both atrophy and hypertrophy of motor neurons. An increased sensitivity to TrkB ligands resulting from TrkB.FL overexpression may explain the occurrence of soma hypertrophy in this group. Postnatal overexpression of TrkB.FL in mouse CNS neurons leads to increased phospholipase Cγ phosphorylation, suggesting continuous activation of this pathway (Koponen et al., 2004). Atrophy in TrkB.FL-expressing neurons suggests that overexpression of TrkB.FL can also have inhibitory effects in some motor neurons. However, the effects of TrkB.FL overexpression were distinct from TrkB.T1, because dendritic beading and downregulation of marker expression were not observed. In cultured hippocampal neurons, both overexpression of TrkB.FL and scavenging of endogenous TrkB ligands decreases the number of glutamatergic synapses (Klau et al., 2001). When BDNF is present in limiting amounts, only a fraction of the increased amount of full-length receptors on the surface may bind ligand. Possibly, downstream signaling depends on a balance of liganded and nonliganded receptors (Klau et al., 2001). Alternatively, a continuous activation of TrkB.FL might desensitize downstream signaling pathways, resulting in inhibitory effects of prolonged TrkB.FL expression. In conclusion, it appears that a critical ratio of truncated and full-length TrkB receptors determines the maintenance of molecular and structural characteristics in adult facial motor neurons.
A role for TrkB in motor neuron degenerative diseases?
The morphological alterations after long-term overexpression of TrkB.T1 are reminiscent of mouse models of motor neuron disease (for review, see Wong et al., 2002). Superoxide dismutase 1 mice, a model for amyotrophic lateral sclerosis, and Wobbler mice display dendritic swelling (Ma and Vacca-Galloway, 1991; Wong et al., 1995). Reduced neurotrophin responsiveness, caused by a disturbed balance of full-length and truncated TrkB receptors, might be a general contributing factor to the degeneration of some neuronal populations. An increase in TrkB.T1 expression in mouse trisomy 16 neurons results in a loss of BDNF-dependent survival and in fragmentation of neurites (Dorsey et al., 2002). Increased expression of TrkB.T1 has also been reported in cortical neurons of Alzheimer’s disease patients (Ferrer et al., 1999). Thus, strategies aimed at restoring the balance between full-length and truncated TrkB might constitute an approach to counteract motor neuron degeneration.
Footnotes
J. De Wit’s present address: Center for Neurogenomics and Cognitive Research, Department of Functional Genomics, De Boelelaan 1087, 1081 HV Amsterdam, The Netherlands.
This work was supported by European Community Biotech Grant PL 970259. We thank Ronald Verwer and Floor Stam for expert help with statistical analysis and Joop van Heerikhuize for valuable advice on image analysis.
References
- Armanini MP, McMahon SB, Sutherland J, Shelton DL, Phillips HS (1995). Truncated and catalytic isoforms of trkB are co-expressed in neurons of rat and mouse CNS. Eur J Neurosci 7:1403–1409. [DOI] [PubMed] [Google Scholar]
- Bartlett JS, Samulski RJ, McCown TJ (1998). Selective and rapid uptake of adeno-associated virus type 2 in brain. Hum Gene Ther 9:1181–1186. [DOI] [PubMed] [Google Scholar]
- Biffo S, Offenhauser N, Carter BD, Barde YA (1995). Selective binding and internalisation by truncated receptors restrict the availability of BDNF during development. Development 121:2461–2470. [DOI] [PubMed] [Google Scholar]
- Clatterbuck RE, Price DL, Koliatsos VE (1994). Further characterization of the effects of brain-derived neurotrophic factor and ciliary neurotrophic factor on axotomized neonatal and adult mammalian motor neurons. J Comp Neurol 342:45–56. [DOI] [PubMed] [Google Scholar]
- Conover JC, Erickson JT, Katz DM, Bianchi LM, Poueymirou WT, McClain J, Pan L, Helgren M, Ip NY, Boland P, Friedman B, Wiegand S, Vejsada R, Kato AC, DeChiara TM, Yancopoulos GD (1995). Neuronal deficits, not involving motor neurons, in mice lacking BDNF and/or NT4. Nature 375:235–238. [DOI] [PubMed] [Google Scholar]
- Donello JE, Loeb JE, Hope TJ (1998). Woodchuck hepatitis virus contains a tripartite posttranscriptional regulatory element. J Virol 72:5085–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsey SG, Bambrick LL, Balice-Gordon RJ, Krueger BK (2002). Failure of brain-derived neurotrophic factor-dependent neuron survival in mouse trisomy 16. J Neurosci 22:2571–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Feng L, Yang F, Lu B (2000). Activity- and Ca(2+)-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol 150:1423–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Feng L, Zaitsev E, Je HS, Liu XW, Lu B (2003). Regulation of TrkB receptor tyrosine kinase and its internalization by neuronal activity and Ca2+ influx. J Cell Biol 163:385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide FF, Vining ER, Eide BL, Zang K, Wang XY, Reichardt LF (1996). Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci 16:3123–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P (2001). Local and target-derived actions of neurotrophins during peripheral nervous system development. Cell Mol Life Sci 58:1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R (1994). Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 368:147–150. [DOI] [PubMed] [Google Scholar]
- Escandon E, Soppet D, Rosenthal A, Mendoza-Ramirez JL, Szonyi E, Burton LE, Henderson CE, Parada LF, Nikolics K (1994). Regulation of neurotrophin receptor expression during embryonic and postnatal development. J Neurosci 14:2054–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JP, Bamji SX, Causing CG, Aloyz R, Ase AR, Reader TA, McLean JH, Miller FD (1998). Functional evidence that BDNF is an anterograde neuronal trophic factor in the CNS. J Neurosci 18:2808–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes KJ, Kobayashi NR, Jasmin BJ, Tetzlaff W (1998). Acetylcholinesterase gene expression in axotomized rat facial motoneurons is differentially regulated by neurotrophins: correlation with trkB and trkC mRNA levels and isoforms. J Neurosci 18:9936–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Marin C, Rey MJ, Ribalta T, Goutan E, Blanco R, Tolosa E, Marti E (1999). BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J Neuropathol Exp Neurol 58:729–739. [DOI] [PubMed] [Google Scholar]
- Friauf E (1986). Morphology of motoneurons in different subdivisions of the rat facial nucleus stained intracellularly with horseradish peroxidase. J Comp Neurol 253:231–241. [DOI] [PubMed] [Google Scholar]
- Friedman B, Kleinfeld D, Ip NY, Verge VM, Moulton R, Boland P, Zlotchenko E, Lindsay RM, Liu L (1995). BDNF and NT-4/5 exert neurotrophic influences on injured adult spinal motor neurons. J Neurosci 15:1044–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisen J, Verge VM, Fried K, Risling M, Persson H, Trotter J, Hokfelt T, Lindholm D (1993). Characterization of glial trkB receptors: differential response to injury in the central and peripheral nervous systems. Proc Natl Acad Sci USA 90:4971–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer RH, Kaplan DR, Kromer LF (1997). Truncated trkB receptors on nonneuronal cells inhibit BDNF-induced neurite outgrowth in vitro. Exp Neurol 148:616–627. [DOI] [PubMed] [Google Scholar]
- Funakoshi H, Belluardo N, Arenas E, Yamamoto Y, Casabona A, Persson H, Ibanez CF (1995). Muscle-derived neurotrophin-4 as an activity-dependent trophic signal for adult motor neurons. Science 268:1495–1499. [DOI] [PubMed] [Google Scholar]
- Gonzalez M, Ruggiero FP, Chang Q, Shi YJ, Rich MM, Kraner S, Balice-Gordon RJ (1999). Disruption of Trkb-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron 24:567–583. [DOI] [PubMed] [Google Scholar]
- Griesbeck O, Parsadanian AS, Sendtner M, Thoenen H (1995). Expression of neurotrophins in skeletal muscle: quantitative comparison and significance for motoneuron survival and maintenance of function. J Neurosci Res 42:21–33. [DOI] [PubMed] [Google Scholar]
- Grimm D, Kern A, Rittner K, Kleinschmidt JA (1998). Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther 9:2745–2760. [DOI] [PubMed] [Google Scholar]
- Haapasalo A, Saarelainen T, Moshnyakov M, Arumae U, Kiema TR, Saarma M, Wong G, Castren E (1999). Expression of the naturally occurring truncated trkB neurotrophin receptor induces outgrowth of filopodia and processes in neuroblastoma cells. Oncogene 18:1285–1296. [DOI] [PubMed] [Google Scholar]
- Haapasalo A, Koponen E, Hoppe E, Wong G, Castren E (2001). Truncated trkB.T1 is dominant negative inhibitor of trkB.TK+-mediated cell survival. Biochem Biophys Res Commun 280:1352–1358. [DOI] [PubMed] [Google Scholar]
- Haapasalo A, Sipola I, Larsson K, Akerman KE, Stoilov P, Stamm S, Wong G, Castren E (2002). Regulation of TRKB surface expression by brain-derived neurotrophic factor and truncated TRKB isoforms. J Biol Chem 277:43160–43167. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Brigadski T, Erdmann KS, Holtmann B, Sendtner M, Narz F, Lessmann V (2004). Truncated TrkB receptor-induced outgrowth of dendritic filopodia involves the p75 neurotrophin receptor. J Cell Sci 117:5803–5814. [DOI] [PubMed] [Google Scholar]
- Hermens WT, ter Brake O, Dijkhuizen PA, Sonnemans MA, Grimm D, Kleinschmidt JA, Verhaagen J (1999). Purification of recombinant adeno-associated virus by iodixanol gradient ultracentrifugation allows rapid and reproducible preparation of vector stocks for gene transfer in the nervous system. Hum Gene Ther 10:1885–1891. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2001). Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24:677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2003). Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72:609–642. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF (1994). Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76:989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplitt MG, Leone P, Samulski RJ, Xiao X, Pfaff DW, O’Malley KL, During MJ (1994). Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet 8:148–154. [DOI] [PubMed] [Google Scholar]
- Kishino A, Ishige Y, Tatsuno T, Nakayama C, Noguchi H (1997). BDNF prevents and reverses adult rat motor neuron degeneration and induces axonal outgrowth. Exp Neurol 144:273–286. [DOI] [PubMed] [Google Scholar]
- Klau M, Hartmann M, Erdmann KS, Heumann R, Lessmann V (2001). Reduced number of functional glutamatergic synapses in hippocampal neurons overexpressing full-length TrkB receptors. J Neurosci Res 66:327–336. [DOI] [PubMed] [Google Scholar]
- Klein R, Conway D, Parada LF, Barbacid M (1990). The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 61:647–656. [DOI] [PubMed] [Google Scholar]
- Klein R, Nanduri V, Jing SA, Lamballe F, Tapley P, Bryant S, Cordon-Cardo C, Jones KR, Reichardt LF, Barbacid M (1991). The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell 66:395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M (1993). Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 75:113–122. [PubMed] [Google Scholar]
- Kobayashi NR, Bedard AM, Hincke MT, Tetzlaff W (1996). Increased expression of BDNF and trkB mRNA in rat facial motoneurons after axotomy. Eur J Neurosci 8:1018–1029. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL (1993). Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron 10:359–367. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Cayouette MH, Berkemeier LR, Clatterbuck RE, Price DL, Rosenthal A (1994). Neurotrophin 4/5 is a trophic factor for mammalian facial motor neurons. Proc Natl Acad Sci USA 91:3304–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koponen E, Voikar V, Riekki R, Saarelainen T, Rauramaa T, Rauvala H, Taira T, Castren E (2004). Transgenic mice overexpressing the full-length neurotrophin receptor trkB exhibit increased activation of the trkB-PLCgamma pathway, reduced anxiety, and facilitated learning. Mol Cell Neurosci 26:166–181. [DOI] [PubMed] [Google Scholar]
- Kryl D, Yacoubian T, Haapasalo A, Castren E, Lo D, Barker PA (1999). Subcellular localization of full-length and truncated Trk receptor isoforms in polarized neurons and epithelial cells. J Neurosci 19:5823–5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Houenou LJ, Wu W, Lei M, Prevette DM, Oppenheim RW (1998). Characterization of spinal motoneuron degeneration following different types of peripheral nerve injury in neonatal and adult mice. J Comp Neurol 396:158–168. [PubMed] [Google Scholar]
- Liu X, Jaenisch R (2000). Severe peripheral sensory neuron loss and modest motor neuron reduction in mice with combined deficiency of brain-derived neurotrophic factor, neurotrophin 3 and neurotrophin 4/5. Dev Dyn 218:94–101. [DOI] [PubMed] [Google Scholar]
- Liu X, Ernfors P, Wu H, Jaenisch R (1995). Sensory but not motor neuron deficits in mice lacking NT4 and BDNF. Nature 375:238–241. [DOI] [PubMed] [Google Scholar]
- Loeb JE, Cordier WS, Harris ME, Weitzman MD, Hope TJ (1999). Enhanced expression of transgenes from adeno-associated virus vectors with the woodchuck hepatitis virus posttranscriptional regulatory element: implications for gene therapy. Hum Gene Ther 10:2295–2305. [DOI] [PubMed] [Google Scholar]
- Ma WY, Vacca-Galloway LL (1991). Reduced branching and length of dendrites detected in cervical spinal cord motoneurons of Wobbler mouse, a model for inherited motoneuron disease. J Comp Neurol 311:210–222. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC (1999). Neurotrophins and synaptic plasticity. Annu Rev Neurosci 22:295–318. [DOI] [PubMed] [Google Scholar]
- McCown TJ, Xiao X, Li J, Breese GR, Samulski RJ (1996). Differential and persistent expression patterns of CNS gene transfer by an adeno-associated virus (AAV) vector. Brain Res 713:99–107. [DOI] [PubMed] [Google Scholar]
- McKay SE, Garner A, Caldero J, Tucker RP, Large T, Oppenheim RW (1996). The expression of trkB and p75 and the role of BDNF in the developing neuromuscular system of the chick embryo. Development 122:715–724. [DOI] [PubMed] [Google Scholar]
- McPhail LT, McBride CB, McGraw J, Steeves JD, Tetzlaff W (2004a). Axotomy abolishes NeuN expression in facial but not rubrospinal neurons. Exp Neurol 185:182–190. [DOI] [PubMed] [Google Scholar]
- McPhail LT, Fernandes KJ, Chan CC, Vanderluit JL, Tetzlaff W (2004b). Axonal reinjury reveals the survival and re-expression of regeneration-associated genes in chronically axotomized adult mouse motoneurons. Exp Neurol 188:331–340. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG Jr, Reichardt LF, Barres BA (1998). Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 21:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middlemas DS, Lindberg RA, Hunter T (1991). trkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol 11:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran LB, Kosel S, Spitzer C, Schwaiger FW, Riess O, Kreutzberg GW, Graeber MB (2001). Expression of alpha-synuclein in non-apoptotic, slowly degenerating facial motoneurones. J Neurocytol 30:515–521. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM (1992). NeuN, a neuronal specific nuclear protein in vertebrates. Development 116:201–211. [DOI] [PubMed] [Google Scholar]
- Munson JB, Shelton DL, McMahon SB (1997). Adult mammalian sensory and motor neurons: roles of endogenous neurotrophins and rescue by exogenous neurotrophins after axotomy. J Neurosci 17:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninkina N, Adu J, Fischer A, Pinon LG, Buchman VL, Davies AM (1996). Expression and function of TrkB variants in developing sensory neurons. EMBO J 15:6385–6393. [PMC free article] [PubMed] [Google Scholar]
- Novikov L, Novikova L, Kellerth JO (1997). Brain-derived neurotrophic factor promotes axonal regeneration and long-term survival of adult rat spinal motoneurons in vivo. Neuroscience 79:765–774. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998). In: The rat brain in stereotaxic coordinates New York: Academic.
- Peel AL, Klein RL (2000). Adeno-associated virus vectors: activity and applications in the CNS. J Neurosci Methods 98:95–104. [DOI] [PubMed] [Google Scholar]
- Peel AL, Zolotukhin S, Schrimsher GW, Muzyczka N, Reier PJ (1997). Efficient transduction of green fluorescent protein in spinal cord neurons using adeno-associated virus vectors containing cell type-specific promoters. Gene Ther 4:16–24. [DOI] [PubMed] [Google Scholar]
- Rossiter JP, Riopelle RJ, Bisby MA (1996). Axotomy-induced apoptotic cell death of neonatal rat facial motoneurons: time course analysis and relation to NADPH-diaphorase activity. Exp Neurol 138:33–44. [DOI] [PubMed] [Google Scholar]
- Ruitenberg MJ, Eggers R, Boer GJ, Verhaagen J (2002). Adeno-associated viral vectors as agents for gene delivery: application in disorders and trauma of the central nervous system. Methods 28:182–194. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Poo M (2000). The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci 23:639–645. [DOI] [PubMed] [Google Scholar]
- Shaw MD, Baker R (1985). Morphology of motoneurons in a mixed motor pool of the cat facial nucleus that innervate orbicularis oculis and quadratus labii superioris, stained intracellularly with horseradish peroxidase. Neuroscience 14:627–643. [DOI] [PubMed] [Google Scholar]
- Silos-Santiago I, Fagan AM, Garber M, Fritzsch B, Barbacid M (1997). Severe sensory deficits but normal CNS development in newborn mice lacking TrkB and TrkC tyrosine protein kinase receptors. Eur J Neurosci 9:2045–2056. [DOI] [PubMed] [Google Scholar]
- Soppet D, Escandon E, Maragos J, Middlemas DS, Reid SW, Blair J, Burton LE, Stanton BR, Kaplan DR, Hunter T, Nikolics K, Parada LF (1991). The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell 65:895–903. [DOI] [PubMed] [Google Scholar]
- Squinto SP, Stitt TN, Aldrich TH, Davis S, Bianco SM, Radziejewski C, Glass DJ, Masiakowski P, Furth ME, Valenzuela DM, Distefano PS, Yancopoulos GD (1991). trkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell 65:885–893. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Mafong E, Meyer S (1996). Central infusions of brain-derived neurotrophic factor and neurotrophin-4/5, but not nerve growth factor and neurotrophin-3, prevent loss of the cholinergic phenotype in injured adult motor neurons. Neuroscience 71:761–771. [DOI] [PubMed] [Google Scholar]
- Vanderluit JL, McPhail LT, Fernandes KJ, McBride CB, Huguenot C, Roy S, Robertson GS, Nicholson DW, Tetzlaff W (2000). Caspase-3 is activated following axotomy of neonatal facial motoneurons and caspase-3 gene deletion delays axotomy-induced cell death in rodents. Eur J Neurosci 12:3469–3480. [DOI] [PubMed] [Google Scholar]
- Venables WN, Ripley BD (2002). In: Modern applied statistics with S Ed 4 New York: Springer.
- Wang W, Salvaterra PM, Loera S, Chiu AY (1997). Brain-derived neurotrophic factor spares choline acetyltransferase mRNA following axotomy of motor neurons in vivo. J Neurosci Res 47:134–143. [DOI] [PubMed] [Google Scholar]
- Watson FL, Porcionatto MA, Bhattacharyya A, Stiles CD, Segal RA (1999). TrkA glycosylation regulates receptor localization and activity. J Neurobiol 39:323–336. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL (1995). An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14:1105–1116. [DOI] [PubMed] [Google Scholar]
- Wong PC, Cai H, Borchelt DR, Price DL (2002). Genetically engineered mouse models of neurodegenerative diseases. Nat Neurosci 5:633–639. [DOI] [PubMed] [Google Scholar]
- Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt LF (2000). Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron 26:233–245. [DOI] [PubMed] [Google Scholar]
- Yacoubian TA, Lo DC (2000). Truncated and full-length TrkB receptors regulate distinct modes of dendritic growth. Nat Neurosci 3:342–349. [DOI] [PubMed] [Google Scholar]
- Yan Q, Elliott JL, Matheson C, Sun J, Zhang L, Mu X, Rex KL, Snider WD (1993). Influences of neurotrophins on mammalian motoneurons in vivo. J Neurobiol 24:1555–1577. [DOI] [PubMed] [Google Scholar]
- Yan Q, Matheson C, Lopez OT, Miller JA (1994). The biological responses of axotomized adult motoneurons to brain-derived neurotrophic factor. J Neurosci 14:5281–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC (1997). Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol 378:135–157. [PubMed] [Google Scholar]