

Abstract
We tested the response of interneurons to the absence of Reelin signaling or p35 in the mouse neocortex. We provide three independent strands of evidence to demonstrate that layering of late-born (but not early-born) interneurons is regulated by Reelin signaling. First, early-born and late-born interneurons behaved differently in mice lacking Reelin or disabled 1 (Dab1). Early-born interneurons showed layer inversion, whereas late-born interneurons did not demonstrate layer inversion but were randomly distributed across the cortex. Second, in p35 mutant brains (in which Reelin signaling is intact), late-born interneurons are appropriately positioned in the upper layers despite the malpositioning of all other cortical neurons in these mice. Third, transplanted late-born interneuron precursors (wild type) into Dab1−/− cortices showed appropriate upper layer segregation. Together, these results indicate that, in the absence of Reelin signaling, late-born interneurons fail to laminate properly, and this is restored in an environment in which Reelin signaling is intact. These studies suggest different mechanisms for the stratification of cortical interneurons. Whereas the early-born interneurons appear to be associated with projection neuron layering, late-born interneurons rely on Reelin signaling for their correct lamination.
Keywords: Reelin, disabled-1, p35, interneuron, migration, GABA
Introduction
The correct positioning of both projection neurons and local circuit interneurons is crucial to the proper functioning of the mature cortex. Although GABAergic interneurons comprise a minor component of the total neuronal population, they are distributed in all layers in which they form intricate networks of synaptic contacts with both excitatory projection neurons and other inhibitory neurons (Somogyi et al., 1983; Meskenaite, 1997; Somogyi et al., 1998). Interneuron positioning is an important issue, and aberrant distribution of cortical interneurons is a known association with epilepsy in humans (Hannan et al., 1999; Thom et al., 2004).
Unlike projection neurons, the development of interneuron layering is poorly understood. One reason is the lack of clear-cut relationships between interneuron stratification and interneuron subtypes (anatomical or physiological) (Hendry and Jones, 1981; Lin et al., 1986). This is not to imply that interneurons are randomly inserted into the cortical wall during corticogenesis. A number of investigators point to stratification of nonoverlapping interneuron populations distinguished by neurochemical markers (Kubota et al., 1994; Gonchar and Burkhalter, 1997). In addition, birthdating and transplantation studies have demonstrated robust relationships between the timing of interneuron generation and their layer destinations (Miller, 1986; Peduzzi, 1988; Valcanis and Tan, 2003; Hevner et al., 2004; Yozu et al., 2004). How this layering process is controlled is presently unknown. The question then arises, do interneurons obey the same layering rules that govern projection neuron positioning? This is a reasonable question given that the two different neuronal populations inhabit the same developmental space and their layering sequences are synchronously allied with their sequence of generation (Angevine and Sidman, 1961; Fairen et al., 1986).
The Reelin signaling pathway is the major effector of cortical layering. Mice deficient in genes of the Reelin signaling pathway, which include reelin, disabled-1 (Dab1), very low-density lipoprotein receptor (VLDLR), and apolipoprotein E receptor 2 (ApoER2) exhibit abnormal projection neuron layering (D’Arcangelo et al., 1995; Howell et al., 1997; Sheldon et al., 1997; Trommsdorff et al., 1999). However, it remains unclear whether interneuron layering is also controlled by the Reelin signaling pathway. Components of the Reelin pathway (Dab1, VLDLR, ApoER2) are certainly expressed in the medial ganglionic eminence (MGE) in which interneurons are generated and from where they commence their tangential migration (Rice et al., 1998; Trommsdorff et al., 1999). Blocking Reelin function with antibodies on organotypic slices cultures was able to arrest the tangential migration of interneurons, and postnatal day 0 (P0) reeler mice showed disturbed interneuron packing (Morante-Oria et al., 2003). An opposite conclusion was reached by Hevner et al. (2004) who found that interneuron inversion in P7 reeler mice mirrored that of projection neurons, leading them to conclude that projection neurons, rather than Reelin per se, are the sources of signals for interneuron layering (Hevner et al., 2004).
To address this issue, we examined the layer distribution of interneuron subpopulations in juvenile mice lacking either Reelin or Dab1, two crucial members of the Reelin signaling pathway. For comparison, we also examined interneurons in p35 mutant mice in which the pyramidal layers are known to be inverted despite intact Reelin signaling. Interneuron subpopulations in different layers were marked using two methods: by either bromodeoxyuridine (BrdU) birthdating (reeler, Dab1, p35 mice) or staining for interneuron subpopulations expressing distinctive neurochemical markers (reeler, Dab1). We found that early- and late-born interneurons responded differently to the absence of Dab1, Reelin, or p35. Whereas early-born interneurons behaved like their projection neuron counterparts by showing layer inversion (in all three mutants), late-born interneurons responded to the absence of Reelin or Dab1 (but not p35) by random distribution throughout the cortical thickness. We propose that this indiscriminate distribution is caused by interneuron sensitivity to the absence of either Reelin or Dab1. Restoring Reelin signaling by transplanting late-born Dab1+/+ interneurons into a Dab1−/− environment containing Reelin showed positioning of the transplanted interneurons to the upper layers. Together, these studies point to different mechanisms for the stratification of cortical interneuron populations. Whereas the early-born interneurons appear to receive their layering cues from lower layer projection neurons, the late-born interneurons appear to require a functional Reelin signaling pathway for proper upper layer positioning.
Materials and Methods
BrdU birthdating.
For interneuron birthdating, pregnant females from matings of reeler (orleans allele, BALB/c), Dab1 (C57BL/6J × DBA), and p35 (C57BL/6J × DBA) heterozygous mice were injected with BrdU at a dose of 100 μg/mg body weight (intraperitoneally, dissolved in sterile saline containing 0.007N NaOH). Pregnant females were injected at embryonic day 12.5 (E12.5) and E13.5 to label early-born interneurons and E15.5 and E16.5 to label late-born interneurons. Dab1+/− and p35+/− females were injected at E12.5 and E15.5 and reeler females at E13.5 and E16.5. Labeled neurons take up BrdU during their final cell division.
Genotyping.
To isolate DNA for PCR genotyping, tissue (toe or tail) was digested in 200 μl of PCR lysis buffer (50 mm KCl, 10 mm Tris-HCl, pH 8.3, 2 mm MgCl2, 0.45% Nonidet P-40, and 0.45% Tween 20) with Proteinase K (200 μg/ml) at 56°C overnight. All DNA primers were used at a final reaction concentration of 10 ng/μl. Primers for Dab1 covering the deleted portion of the gene (Howell et al., 1997) and for the neomycin phosphotransferase (neo) gene were used: Dab1, forward, 5′-GCGAAGCCACTTTGATAAAGA-3′; Dab1, reverse 5′-TAACTTGTCTCCCCGAGCTG-3′; neo, forward, 5′-ATCAGGATGATCTGGACG-AAGA-3′; and neo, reverse, 5′-CCACAGTCGATGAATCCAGAA-3′. For genotyping of the p35 mice, primers for p35 covering the deleted portion of the gene (Chae et al., 1997) were used in a multiplex reaction with the neo primers described above: p35, forward, 5′-GGCAATGAAATCTCCTACCCG-3′; and p35, reverse, 5′-ACCCCAAGGAAAAGAACGTG-3′.
For genotyping of the reeler mice, Reelin forward and reverse primers were designed either side of the L1 insertion site. L1 insertion at this site leads to exon skipping and the 220 bp deletion in the Reelin gene that results in the reeler phenotype (Takahara et al., 1996). An additional reverse primer was designed to the 5′ end of the L1 element. Primers for the Reelin gene were as follows: Reelin, forward, 5′-CGACTGCTCTGTCTTCAGTCACG-3′; Reelin, reverse, 5′-GGTGGCAGCTTGCCTTATCTG-3′; and the L1 element reverse, 5′-GCTGCCTCAGTGCCTCTGTG-3′.
Heat-inactivated DNA (2.5 μl) was added to a reaction mix containing primers (10 μg/μl) [100 mm Tris-HCl, pH 8.3, 500 mm KCl (1× PCR Buffer II; PerkinElmer, Wellesley, MA), 1.5 mm MgCl2, 100 μm dNTPs, and 1.25 U of TaqDNA polymerase (PerkinElmer)]. Amplification of the samples was performed after 1 min at 94°C using 40 cycles consisting of annealing at 62°C (for Dab1 and p35), 61°C (reeler) for 30 s, extension at 72°C for 30 s, and denaturing at 94°C for 30 s. PCR products were analyzed on a 2.5% (Dab1 and p35) or 4% (reeler) agarose gel. Product sizes were as follows: 81 bp (wild-type Dab1), 347 bp (wild-type p35), 170 bp (neo gene in mutant Dab1 or p35 alleles), 188 bp (wild-type reeler), and 160 bp (L1 element in mutant Reelin allele).
Histology.
After a lethal injection of Nembutal (Merial Australia, Parramatta, Australia), mice at P16 (Dab1 and p35) or P23 (reeler) were intracardially perfused for 7 min with 4% paraformaldehyde in 0.1 m Sorensen’s phosphate buffer, pH 7.4, and the brains were removed for 7 min after fixation. After cryoprotection with 20% sucrose in phosphate buffer, brains were embedded in OCT (Tissue-Tek, Miles, Torrance, CA) for cryosectioning. Thin coronal sections (14 μm) of the cortex were prepared for immunocytochemistry, and thicker sections (60 μm) were obtained from the transplanted mouse brains for 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) histochemistry to visualize the wild-type cells labeled with β-galactosidase (βgal). Sections for X-gal histochemistry were incubated overnight at 37°C in a solution of 0.1% X-gal containing 2 mm MgCl2, 5 mm EGTA, 0.01% (w/v) sodium deoxycholate, 0.02% (w/v) Nonidet P-40, 5 mm K3Fe(CN)6, and 5 mm K4Fe(CN)6·6H2O in 0.1 m Sorenson’s phosphate buffer. The X-gal was prepared as a 4% stock in dimethylformamide and was added to the mixture just before use.
Immunocytochemistry.
For immunofluorescence studies, 14 μm coronal sections were mounted on 3-aminopropyltreithoxy-silane-coated slides and dried for 2 h before incubation in primary antibodies. To expose the BrdU, tissues were preincubated with 2N HCl at 37°C for 45 min before addition of the primary antibody. All primary antibodies were diluted in 0.1 m PBS with 0.3% Triton X-100. Primary antibodies included a rabbit polyclonal antibody to GABA (1:1000; Sigma, St. Louis, MO), a rabbit polyclonal antibody to calbindin (CB) (1:1000; Swant, Bellizona, Switzerland), a rabbit polyclonal antibody to calretinin (CR) (1:200; Swant), a rabbit polyclonal antibody to neuronal nitric oxide synthase (nNos) (1:200; Zymed, San Francisco, CA), and a mouse monoclonal antibody to BrdU (1:30; Becton Dickinson, San Jose, CA). Secondary antibodies used were a biotinylated goat anti-rabbit (1:200; Vector Laboratories, Burlingame, CA), Alexa Fluor 594-conjugated donkey anti-mouse IgG (1:500; Invitrogen, Carlsbad, CA), and Alexa Fluor 594 conjugated goat anti-rabbit IgG (1:500; Invitrogen). When biotinylated secondaries were used, visualization of a positive signal was achieved with fluorescein-avidin D (1:200; Vector Laboratories). All primary antibody incubations were overnight followed by 1 h incubations in secondary antibodies and 1 h in fluorescein–avidin D when necessary, before coverslipping with 10% (w/v) Mowial (Hoechst, Melbourne, Australia) in 25% glycerol, 0.1 m Tris-HCl, pH 8.5, and 2.5% (w/v) 1,4-diazobicyclo-[2.2.2]-octane (Sigma).
To quantify the layer positions of interneurons, one section from each animal [taken from positions −0.10 and −0.34 mm of the bregma line (Franklin and Paxinos, 1997)] was used for immunocytochemistry (against calbindin, nNos, calretinin, GABA, and double-labeling for calretinin and BrdU). For double labeling of GABA and BrdU, sections were obtained between positions −1.34 and −1.7 mm of the bregma line. Using Microsoft (Seattle, WA) PowerPoint, montages were created from images of fluorescent positive cells within the somatosensory cortex bounded by the primary motor and granular insular cortex. The cortical thickness was divided into 10 bins (bin 10 is closest to the pial surface), and the number of positive cells in each bin was counted. The data (normalized as a percentage of total positive cells per section) were entered into Microsoft SigmaPlot and plotted as a histogram.
In utero cell transplantation.
Isochronic transplantations of E15.5 MGE cells into the lateral ventricles of E15.5 brains (day of vaginal plug detection, E0.5) were performed essentially as described previously (Valcanis and Tan, 2003). Both donor and host embryos were derived from parental strains maintained on a C57BL/6J × DBA hybrid background. Donor cells were obtained from a transgenic mouse line in which all neurons and glia express the lacZ reporter gene (Tan and Breen, 1993), and recipient embryos were derived from matings of mice heterozygous for mutant the Dab1 allele. To isolate the MGE cells, the brains were removed from donor embryos using watchmakers forceps and stored in HBSS (Invitrogen) on ice. The roof of the cerebral hemispheres was then removed exposing the ganglionic eminences (GEs), and the MGE was then dissected, taking care to avoid tissue from the adjacent lateral ganglionic eminence and the interventricular septum. MGE tissue for all littermates was pooled and enzymatically digested in 1 ml of HBSS containing 0.125% trypsin (Thermo Electron Corporation, Waltham, MA) and 0.1% DNase1 (Roche Diagnostics, Mannheim, Germany) at 37°C for 10 min. After digestion, the trypsin was inactivated by the addition of 100 μl of fetal calf serum, and the cells were pelleted. The cells were resuspended in 0.5 ml of HBSS, counted using a hemacytometer, and resuspended at 2 × 105 cells/μl in HBSS containing 0.1% DNase. Pregnant mice containing E15.5 embryos were anesthetized using Nembutal (0.6 mg of sodium pentobarbitone per 10 g of body weight). The uterine horns were exposed and bathed in 0.25% MgSO4 containing penicillin/streptomycin (50 IU/ml and 50 μg/ml, respectively) (Trace Electron). Using a mouth pipette, the cells were collected in a fine glass pipette (tip diameter of 30–60 μm), and, under fiber optic lighting, 1 μl of the cell suspension (2 × 105 cells) was injected through the uterine wall into the lateral ventricles of the embryos. All embryos in both uterine horns were injected except for the two most proximal to the vagina. The recipient female was then sutured and allowed to recover on a 37°C warming tray. The resultant pups were reared until P16.
Data quantification.
For data quantification, 60 μm sections taken ∼200 μm apart were reacted for X-gal histochemistry, mounted on chrome alum (0.05% w/v)/gelatin (0.5% w/v) treated slides, and photographed under bright field using a 4× objective. Montages were produced, the cortical thickness was divided into 10 bins (bin 10 is closest to the pial surface), and the number of βgal-positive cells were counted in each bin. Only labeled cells within the somatosensory cortex bounded by the primary motor and granular insular cortex were recorded, and this process was repeated for all sections between positions +0.74 mm anterior to and −1.34 mm posterior to the bregma line. The data (normalized as a percentage of total positive cells per section) was entered into Microsoft SigmaPlot and plotted as a histogram.
To test the reliability of the dataset, we used the nonparametric bootstrap approach, a method that does not rely on any underlying assumptions about the data (Efron and Tibshirani, 1986). Bootstrapping allows error estimates to be obtained for fitted functions, particularly when the data sample is small (Foster and Bischof, 1987; Bui et al., 1998, 2005). A pseudoreplication of datasets is obtained, and each dataset is fitted to obtain estimates of the data descriptive parameters. Although the distribution of the true dataset is not known, the estimate obtained by the resampling strategy provides an approximation of the empirical distribution (Darvas et al., 2004). However, the accuracy of statistical inferences made about the different data samples is dependent on the relative bias of the samples.
We applied a bootstrap technique on the dataset of the percentage of cells as a function of cortical depth using a straight line fit (y = mx + c) and obtained estimates of the slope parameter and/or used a Gaussian function to the following equation (Treutwein, 1995; Battista and Kalloniatis, 2002):
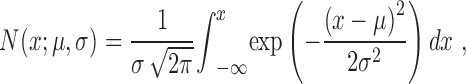 |
The mean parameter value and 95% confidence intervals (CIs) of the mean (μ) and SD (σ) for the Gaussian function or the slope parameter (m) for the straight line fit was obtained after 1000 bootstrap replications. We applied a two-tailed comparison when comparing the parameter estimates for the different experimental conditions, and, when appropriate, we consider significant departure at the (p < 0.05) if the CIs did not overlap. The nature of the fitted equation does not have biological relevance but simply allows the comparison of different experimental manipulations from which statistical inferences can be drawn.
Results
Early-born interneurons in Dab1 mutant and Reeler cortices show layer inversion
The positions of GABA-positive interneurons born at E12.5 or E13.5 were examined in the cortices of both wild-type and Dab1 and reeler mutant brains using double-labeled immunocytochemistry for GABA (green) and BrdU (red) (Figs. 1, 2). In wild-type cortex, these early-born interneurons (double-labeled with GABA and BrdU) are found in the lower layers (layers V/VI), in agreement with previous studies (Miller, 1986; Valcanis and Tan, 2003) (Figs. 1A,B, 2A,B). Summary histograms depict early-born interneurons predominantly in layers V/VI (Figs. 1C, 2C). In contrast, early-born interneurons in Dab1 mutants are distributed mainly in the upper parts of the cortex (Fig. 1D,E, summary histogram in F). This pattern is also seen in the reeler mouse (Fig. 2D,E, summary histogram in F). Previous studies have established that projection neuron layering is inverted in reeler and Dab1 mutants (Howell et al., 1997; Sheldon et al., 1997); hence, the interneuron inversion observed here is consistent with the notion that, without Reelin signaling, both interneurons and deep layer projection neurons are inverted.
Figure 1.
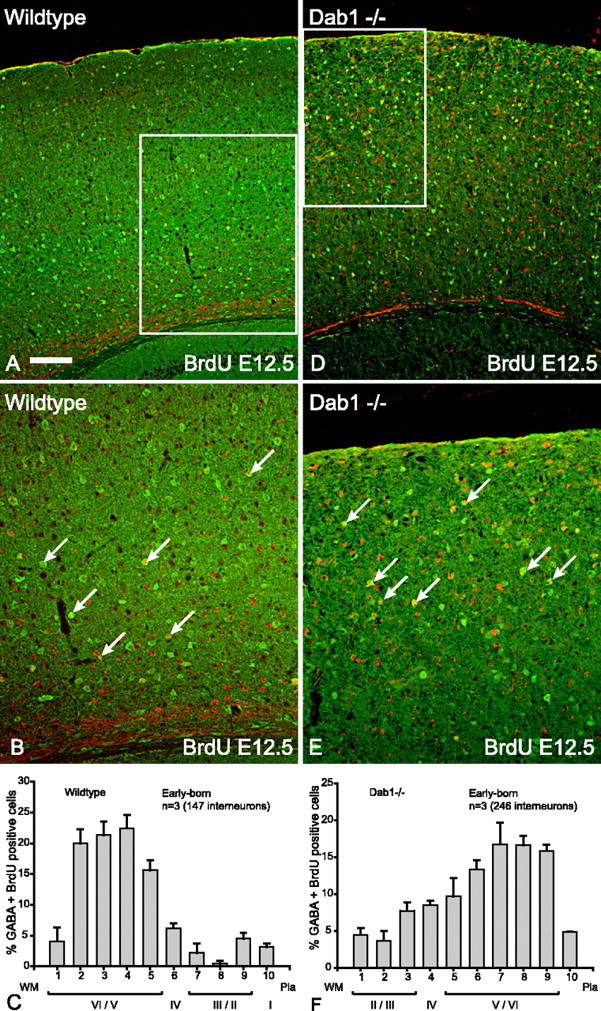
Early-born (E12.5) interneurons in Dab1 mutant brains show layer inversion. A, Double-labeling for GABA (green) and BrdU (red) reveals that the vast majority of double-labeled cells (yellow) in wild-type cortex are in the lower layers at P16. B, Higher magnification of boxed area in A indicating that the majority of double-labeled interneurons (arrows) are in the lower layers of the cortex. C, Histogram depicting the layer positions of GABA–BrdU interneurons (n = 3 animals, 147 cells; mean ± SEM percentage) indicating that majority of early-born interneurons in wild-type cortices are distributed in the lower layers. D, In Dab1 mutants, double-labeled interneurons (yellow) born at E12.5 are found in the upper layers. E, Higher magnification of boxed area in D indicating that the majority of double-labeled interneurons (arrows) are in the upper layers of the cortex. F, Histogram depicting the layer positions of GABA–BrdU interneurons (n = 3 animals, 246 cells; mean ± SEM percentage) indicating that the majority of early-born interneurons in Dab1 mutant cortices are distributed in the upper layers. Scale bar: A, D, 160 μm; B, E, 80 μm. WM, White matter.
Figure 2.
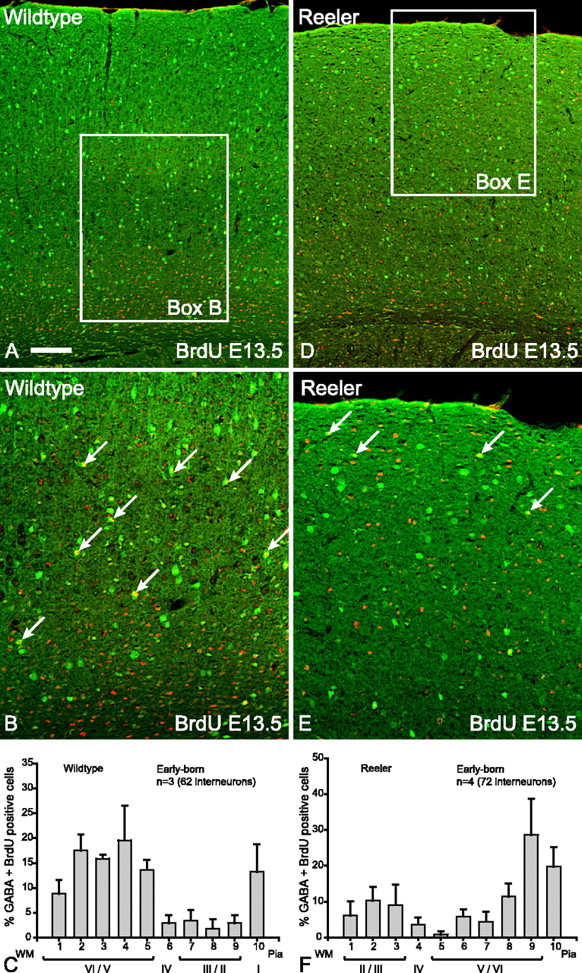
Early-born (E13.5) interneurons in reeler brains show layer inversion. A, Double- labeling for GABA (green) and BrdU (red) reveals that the vast majority of double-labeled cells (yellow) in wild-type cortex are in the lower layers. B, Higher magnification of boxed area in A indicating that the majority of double-labeled interneurons (arrows) are in the lower layers of the cortex. C, Histogram depicting the layer positions of GABA–BrdU interneurons (n = 3 animals, 62 cells; mean ± SEM percentage) indicating that the majority of early-born interneurons in wild-type cortices are distributed in the lower layers. D, In reeler cortex, double-labeled interneurons (yellow) born at E13.5 are found in the upper layers. E, Higher magnification of boxed area in D indicating that the majority of double-labeled (arrows) interneurons are in the upper layers of the cortex. F, Histogram depicting the layer positions of GABA–BrdU interneurons (n = 4 animals, 72 cells; mean ± SEM percentage) indicating that the majority of early-born interneurons in Reelin mutant cortices are distributed in the upper layers. Scale bar: A, D, 160 μm; B, E, 80 μm. WM, White matter.
Quantification of layer distributions was verified using a nonparametric bootstrapping algorithm that permits mean bin (μ) or slope (m) together with CIs to be derived (see Materials and Methods) (Table 1). Figure 1, C and F, was fitted with a Gaussian function, and the nonoverlapping CIs for estimates of μ indicate that early-born neurons in wild type are located at approximately level 33.9% (μ = 3.39; 95% CI, 3.19, 3.61), whereas the Dab1−/− neurons are located at level 68.9% (μ = 6.89; 95% CI, 6. 59, 7.32; p < 0.05). For early-born Reeler interneurons, Figure 2, C and F, was compared using a straight line fit because a Gaussian function did not provide a reasonable fit to the dataset (Table 1). Figure 2C displays a slope of −0.96% (95% CI, −1.80, −0.09), indicating that wild-type interneurons are mainly located in the lower bins (closer to white matter) whereas the early-born Reeler interneurons displayed a positive slope of 2.13 (95% CI, 0.61, 3.62), with higher proportions in the upper bins. The 95% CI of the slope estimates do not overlap, and therefore the two datasets are significantly different (p < 0.05).
Table 1.
Parameter estimates for fitted functions of the distribution of cell density as a function of cortical depth
| n | Gaussian fit [μ (95% CI)] | Straight line fit [m (95% CI)] | CI overlap | |
|---|---|---|---|---|
| Dab1 wild-type GABA + BrdU, E12.5 (1C) | 3 | 3.39(3.19, 3.61) | ||
| Dab1 mutant GABA + BrdU, E12.5 (1F) | 3 | 6.89(6.59, 7.32) | No | |
| Reelin wild-type GABA + BrdU, E13.5 (2C) | 3 | −0.96(−1.80, −0.09) | No | |
| Reelin mutant GABA + BrdU, E13.5 (2F) | 4 | 2.13 (0.61, 3.62) | ||
| Dab1 wild-type GABA + BrdU, E15.5 (3C) | 3 | 2.17 (1.71, 2.71) | No | |
| Dab1 mutant GABA + BrdU, E15.5 (3G) | 3 | 0.29 (0.10, 0.46) | ||
| Reelin wild-type GABA + BrdU, E16.5 (4C) | 3 | 2.52 (1.60, 3.81) | No | |
| Reelin mutant GABA + BrdU, E16.5 (4G) | 4 | 0.45 (−0.37, 1.29) | ||
| Dab1 wild-type nNos (5B) | 5 | −2.28 (−2.86, −1.70) | No | |
| Dab1 mutant nNos (5D) | 5 | 1.44 (0.85, 1.98) | ||
| Dab1 wild-type calbindin (5F) | 4 | 3.14(2.92, 3.36) | No | |
| Dab1 mutant calbindin (5H) | 4 | 8.76(8.49, 9.14) | ||
| Dab1 wild-type calretinin (6B) | 3 | 8.75(7.35, 12.51) | Yes | |
| Dab1 mutant calretinin (6D) | 3 | 7.64 (7.11, 8.19) | ||
| Dab1 wild-type calretinin + BrdU, E15.5 (6E) | 4 | 3.83 (3.20, 4.42) | No | |
| Dab1 mutant calretinin + BrdU, E15.5 (6F) | 4 | 0.07 (−0.99, 0.77) | ||
| p35 wild-type GABA + BrdU, E15.5 (7C) | 2 | 1.32 (1.10, 1.53) | Yes | |
| p35 mutant GABA + BrdU, E15.5 (7F) | 2 | 0.82 (0.57, 1.11) | ||
| p35 mutant total GABA (7H) | 2 | 0.69 (0.51, 0.87) | No | |
| p35 wild-type total GABA (7I) | 2 | −0.46 (−0.57, −0.36) | ||
| Wild-type E15.5 MGE transplanted into wild-type cortex (8C) | 2 | 6.95(6.68, 7.21) | Yes | |
| Wild-type E15.5 MGE transplanted into Dab1 mutant cortex (8F) | 2 | 6.66(6.42, 6.86) |
The mean estimate is followed by the lower and upper limits of the 95% confidence interval (CI). In the first column, figure numbers are indicated in parentheses.
Late-born interneurons in Dab1 mutant and Reeler cortices do not show layer inversion but are uniformly distributed
Late-born interneurons show a different phenotype in the absence of either Dab1 or Reelin. Wild-type interneurons born at either E15.5 or E16.5 are found in the upper layers (layers II/III) (Figs. 3A,B, 4A,B, summary histograms in 3C, 4C). However, late-born interneurons in Dab1 mutants or reeler mice did not show layer inversion; instead, they are evenly distributed throughout the entire cortical depth (Figs. 3D –F, 4D –F, summary histograms in 3G, 4G). Comparison of histogram patterns confirm that the distribution of interneurons is quite different between wild type and mutant (Table 1). For both Dab1 and reeler, the 95% CI of the slope estimates between mutant and wild type do not overlap, and therefore the datasets for each genotype are significantly different (p < 0.05) (Table 1). These results demonstrate that the absence of Reelin signaling causes late-born interneurons to insert randomly into the cortex, although projection neurons born at equivalent stages in the same environment are known to be inverted (Howell et al., 1997; Sheldon et al., 1997). Random distribution is an indication that factors that normally control layering of late-born interneurons may be disturbed.
Figure 3.
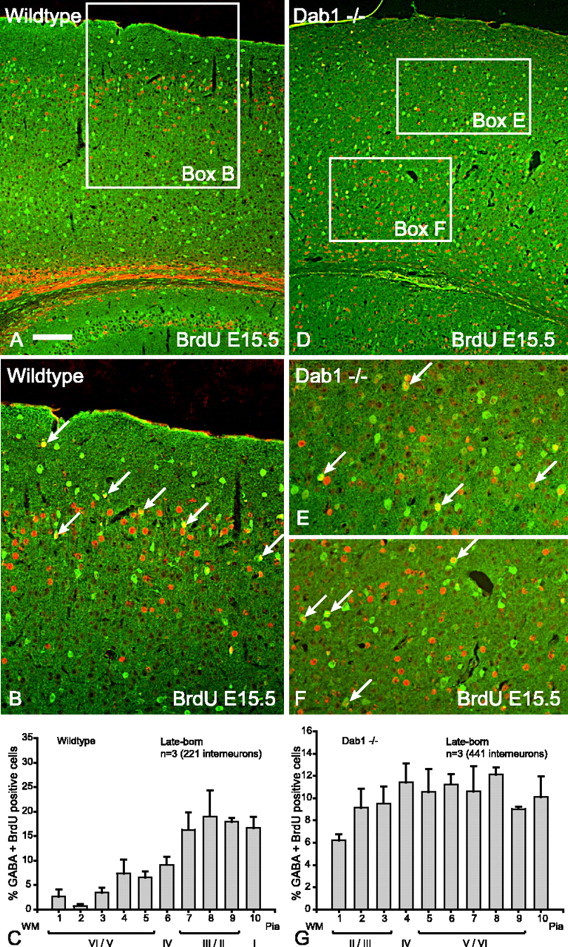
Late-born (E15.5) interneurons in Dab1 mutant brains show random distribution. A, Double-labeling for GABA (green) and BrdU (red) reveals that the vast majority of double-labeled cells (yellow) are in the upper layers of the wild-type cortex. B, Higher magnification of boxed area in A indicating that the majority of double-labeled interneurons (arrows) are in the upper layers of the cortex. C, Histogram depicting the layer positions of GABA–BrdU interneurons (n = 3 animals, 221 cells; mean ¶ SEM percentage) indicating that the majority of late-born interneurons in wild-type cortices are distributed in the upper layers. D, In Dab1 mutants, double-labeled interneurons (yellow) born at E15.5 are found in all layers of the cortex. E, F, Higher magnification of upper and lower (respectively) boxed areas in D indicating that double-labeled interneurons (arrows) are equally distributed to upper and lower layers of the Dab1 mutant cortex. G, Histogram depicting the layer positions of GABA–BrdU interneurons (n = 3 animals, 441 cells; mean ¶ SEM percentage) indicating that the late-born interneurons in Dab1 mutant cortices are randomly distributed throughout all layers of the cortex. Scale bar: A, D, 160 μm; B, E, F, 80 μm. WM, White matter.
Figure 4.
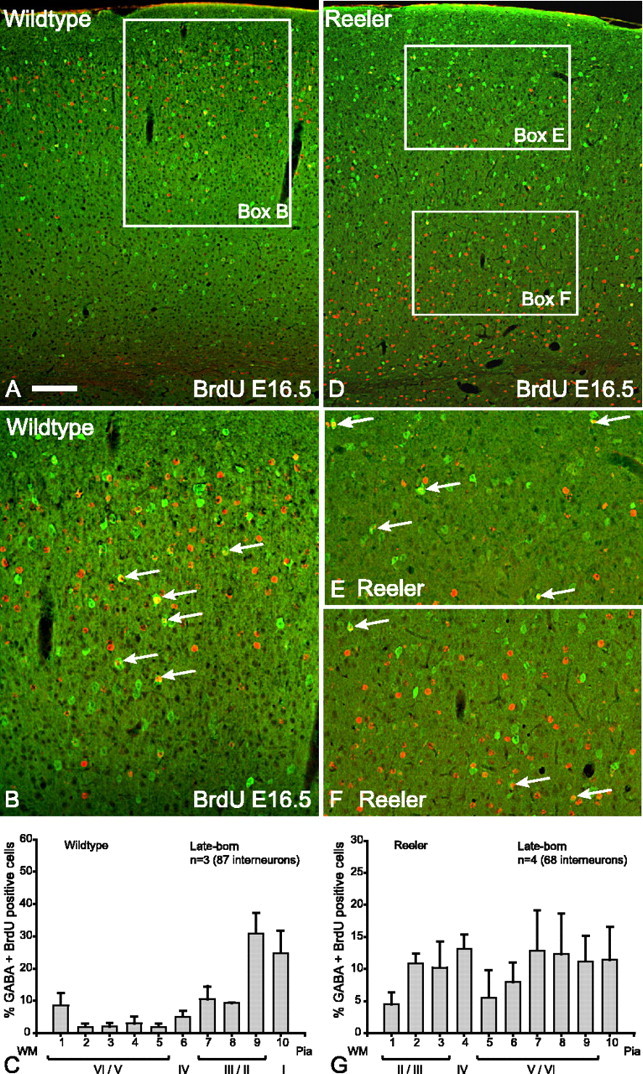
Late-born (E16.5) interneurons in reeler brains are randomly distributed. A, In wild-type context, double-labeling for GABA (green) and BrdU (red) reveals that the vast majority of double-labeled cells (yellow) are in the upper layers. B, Higher magnification of boxed area in A indicating that the majority of double-labeled interneurons (arrows) are in the upper layers of the cortex. C, Histogram depicting the layer positions of GABA–BrdU interneurons (n = 3 animals, 87 cells; mean ¶ SEM percentage) indicating that the majority of late-born interneurons in wild-type cortices are distributed in the upper layers. D, In reeler cortex, double-labeled interneurons (yellow) born at E16.5 are found in all layers. E, F, Higher magnification of upper and lower (respectively) boxed areas in D indicating that double-labeled interneurons (arrows) are in the upper and lower layers of the Reelin mutant cortex. G, Histogram depicting the layer positions of GABA–BrdU interneurons (n = 4 animals, 68 cells; mean ¶ SEM percentage) indicating that the late-born interneurons in reeler cortices are distributed throughout all layers of the cortex. Scale bar: A, D, 160 μm; B, E, F, 80 μm. WM, White matter.
The distribution patterns of interneuron subpopulations are also affected in Reeler and Dab1 mutants
We used BrdU birthdating to show that late-born interneurons respond differently to the lack of Reelin signaling compared with early-born interneurons. An additional way of demonstrating these differences would be to compare interneuron subpopulations (expressing specific markers) that are predominantly found in either upper or lower layers. Interneurons expressing nNos are relatively rare in number, but, in wild-type cortex, they are found in lower layers (V/VI) (Fig. 5 A,B). In mice lacking Dab1, the distribution of nNos interneurons is inverted (Fig. 5C,D), again reflecting the observation that early-born interneurons in these mice are associated with inverted projection neurons. These experiments were repeated in reeler mice in which nNos interneurons were also are inverted (results not shown). CB is marker for deep layer interneurons, and these can be identified by their strong cellular immunoreactivity (as against weak neuropil immunoreactivity surrounding layer II/III projection neurons) (Fig. 5E,F). In the absence of Dab1, CB interneurons are found in upper layer positions (Fig. 5G,H). This was affirmed by the significantly different mean values of the Gaussian fit with nonoverlapping CIs (p < 0.05) (Table 1). A similar result was also seen in reeler mice (data not shown). CR marks a distinct group of interneurons with similar frequency to CB (Gonchar and Burkhalter, 1997), and this group is distributed in all layers with a bias toward the upper layers (Fig. 6A,B). In the absence of Dab1, there appears to be no major change in the overall layer distribution of CR interneurons between wild type and mutant (Fig. 6, compare B, D), indicating that CR interneurons continue to be positioned in the upper layers despite the absence of Dab1 (Fig. 6C,D) (Gaussian fit in Table 1 suggests overlapping CIs and therefore not significantly different). This result appears to contradict data from GABA–BrdU labeling in Dab1 mutants, which demonstrates that upper layer interneurons are evenly distributed across the cortex (Fig. 3G). However, further consideration of Figure 6D suggests that late-born CR interneurons may have been masked by the superimposition of early-born CR interneurons (that are layer inverted), giving rise to the peaks in the upper layer histograms. To test this, we labeled only the late-born CR interneurons with BrdU (injected at E15.5), and the results confirm that late-born CR interneurons (labeled with BrdU) are indeed randomly distributed across the mutant cortex (Fig. 6F). Table 1 confirms a positive slope for wild type, whereas the mutant has effectively a flat slope with nonoverlapping CIs (note that bin 10 was not considered for this particular comparison). Collectively, these results show that interneuron subpopulations that are normally biased toward upper or lower layers reflect the data obtained with GABA–BrdU labeling.
Figure 5.
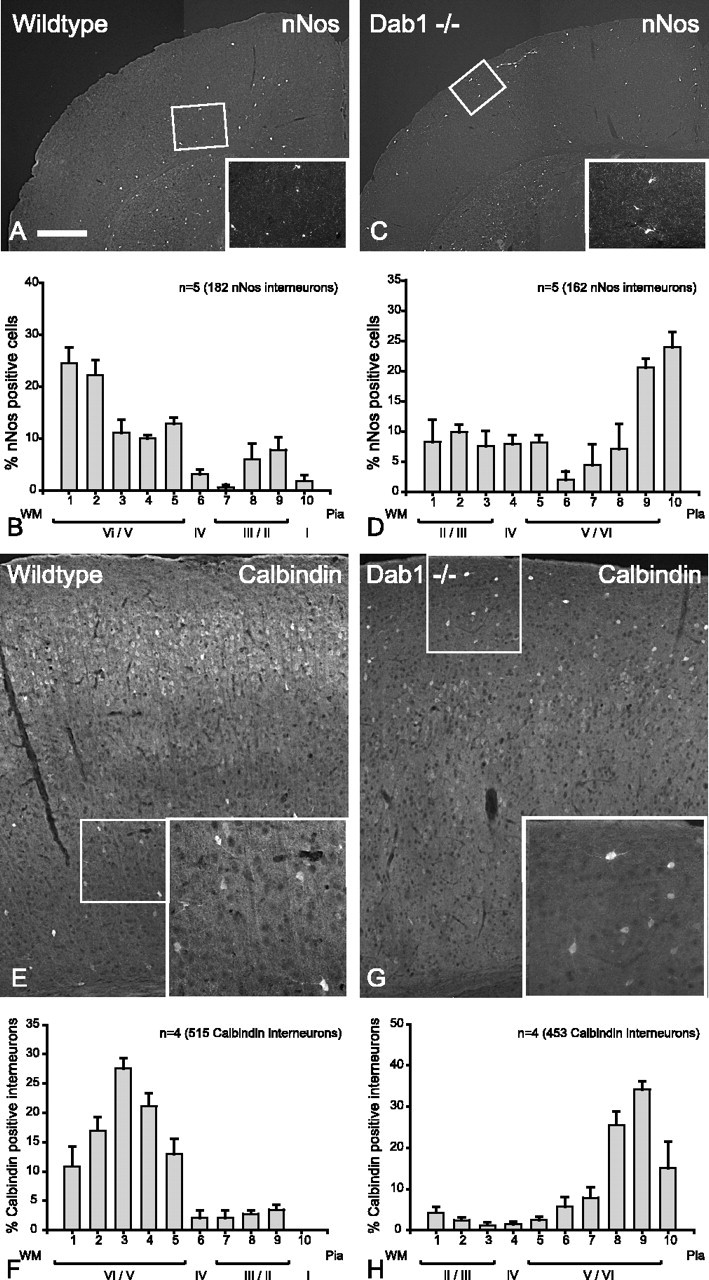
In the Dab1 mutant cortex, nNos and calbindin subpopulations of interneurons are inverted. A, In wild-type cortex, nNos interneurons are found in the lower layers (inset shows interneurons stained with nNos). B, Histogram depicting the layer positions of nNos interneurons (n = 5 animals, 182 cells; mean ± SEM percentage) indicating that majority of nNos interneurons in wild-type cortices are distributed in the lower layers. C, In Dab1 mutant cortex, nNos interneurons are found mostly in the upper layers (inset shows interneurons stained with nNos). D, Histogram depicting the layer positions of nNos interneurons (n = 5 animals, 162 cells; mean ± SEM percentage) indicating that the majority of nNos interneurons in the Dab1 mutant cortices are situated in the upper layers of the cortex. E, Labeling for calbindin in wild-type cortex reveals that the vast majority of strongly immunolabeled cells are in the lower layers (inset shows interneurons reactive for calbindin). F, Histogram depicting the layer positions of calbindin interneurons (n = 4 animals, 515 cells; mean ± SEM percentage) indicating that vast majority of calbindin-positive interneurons in wild-type cortices are distributed in the lower layers. G, In the Dab1 mutant cortex, calbindin interneurons are found mostly in the upper layers (inset shows interneurons staining for calbindin). H, Histogram depicting the layer positions of calbindin interneurons (n = 4 animals, 453 cells; mean ± SEM percentage) indicating that the vast majority of calbindin positive interneurons in the Dab1 mutant cortices are situated in the upper layers of the cortex. Scale bar: A, C, 576 μm; insets, 260 μm; E, G, 160 μm; insets, 80 μm. WM, White matter.
Figure 6.
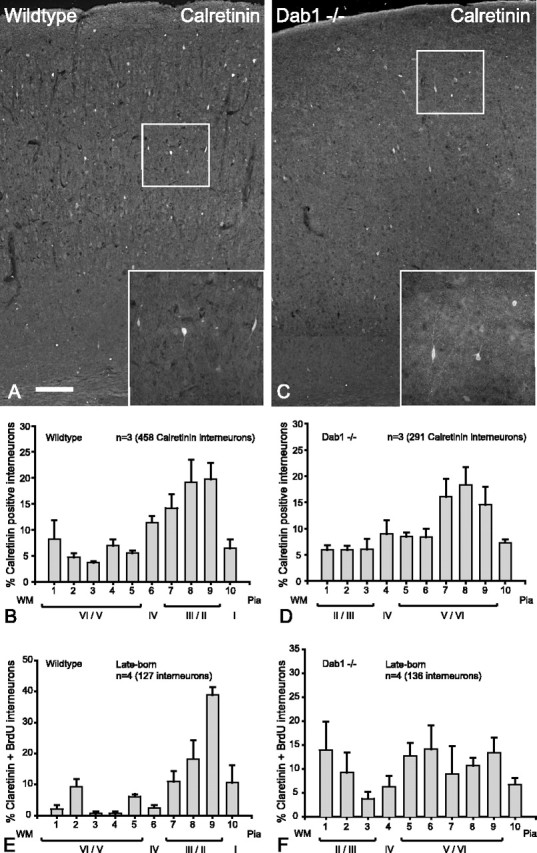
Late-born (but not early-born) calretinin interneurons are randomly distributed. A, In wild-type cortex, calretinin interneurons are found in all layers, with a bias toward the upper layers (inset shows immunoreactivity for calretinin). B, Histogram depicting the layer positions of calretinin interneurons (n = 3 animals, 458 cells; mean ± SEM percentage) indicating that the majority of calretinin interneurons are distributed in the upper layers with a substantial contribution in the lower layers. C, In the Dab1 mutant, calretinin interneurons appear to have a similar distribution to wild-type cortex (inset shows interneuron immunoreactivity for calretinin). D, Histogram depicting the layer positions of all calretinin interneurons (n = 3 animals, 291 cells; mean ± SEM percentage), suggesting that calretinin interneurons in the Dab1 mutant have a similar distribution pattern to that of the wild-type (compare B, D). E, In wild-type cortex, late-born calretinin interneurons labeled with BrdU at E15.5 (n = 4 animals, 127 cells; mean ± SEM percentage) show predominantly upper layer distribution. F, In Dab1 mutant cortex, late-born calretinin interneurons labeled with BrdU at E15.5 (n = 4 animals, 136 cells; mean ± SEM percentage) show a random pattern of distribution. Scale bar: A, C, 160 μm; insets, 80 μm. WM, White matter.
The random distribution of late-born interneurons is not a consequence of an inverted cortex
By the time late-born interneurons arrive in the cortex from the subpallial GE, the cortical wall in reeler and Dab1 mutants is fairly disorganized with projection neurons in wrong-order sequence with some attendant radial glia abnormalities (Hunter-Schaedle, 1997). To determine whether the random distribution of the late-born interneurons is a consequence of projection neuron inversion, we examined the positions of late-born interneurons in p35 mutants in which the cortex is also inverted from a malfunctioning cyclin-dependent kinase 5 (Cdk5)/p35 pathway, but the Reelin signaling system is intact (Chae et al., 1997). The results show that, unlike those in the reeler and Dab1 mutants, late-born interneurons in the p35 mutant cortex are not randomly distributed but are segregated to the upper layers (Fig. 7D–F), just as in the wild-type control (Fig. 7A–C). Table 1 shows that the CIs of the slopes overlap, and therefore they are not significantly different (p > 0.05). Because this observation has not been reported before in p35 mutant mice, we further examined the layering of early-born interneurons in these mice to see whether they are also inverted together with the early-born projection neurons. The results demonstrate that early-born interneurons in p35 mutant mice are inverted to the upper layers (Fig. 7G), similar to mice lacking Reelin or Dab1. These results would predict that most interneurons in p35 mutant mice are distributed to the upper layers from a combination of inverted early-born cells superimposed on normally positioned late-born interneurons. This was validated by a global analysis of all interneurons in the p35 mutant cortex, which showed that most interneurons are distributed to the upper parts of the cortex (Fig. 7, compare mutant in H with wild type in I). Table 1 shows that the CIs of the slopes do not overlap, suggesting significantly different layering patterns for the two genotypes (p < 0.05). Together, these experiments indicate that positioning of late-born interneurons is unaffected by the absence of p35. They also substantiate the argument that random distributions of late-born interneurons in reeler and Dab1 mutants are not consequences of a mislayered cortex. Furthermore, this appears to be a specific effect of Reelin signal malfunction, and, if Reelin signaling is intact as in the case in p35 mutant mice, then the late-born subpopulation can be correctly positioned in the upper layers. Conversely, Reelin signaling appears ineffective for correct layering of the early-born interneurons in p35 mutant animals, suggesting that Reelin signaling has no influence on early-born interneurons in these mice and that their layering cues might reside elsewhere.
Figure 7.
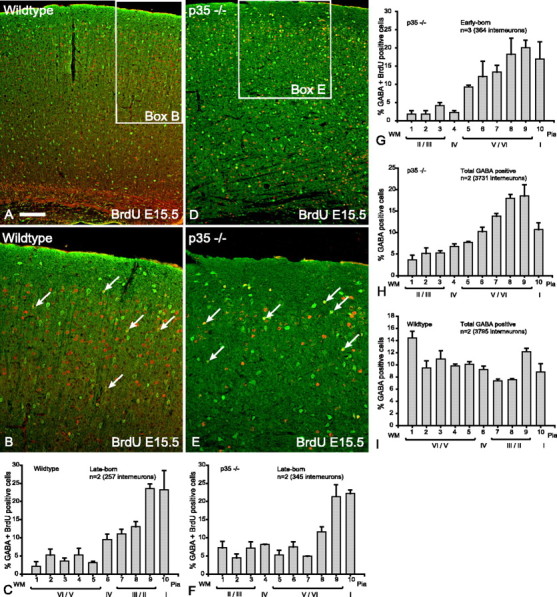
Late-born (E15.5) interneurons in p35 mutant brains are properly layered despite layer inversion of projection neurons. A, In the wild-type cortex, double-labeling for GABA (green) and BrdU (red) reveals that the vast majority of double-labeled interneurons at E15.5 (yellow) are in the upper layers. B, Higher magnification of boxed area in A indicating that the majority of double-labeled interneurons (arrows) are in the upper layers of the cortex. C, Histogram depicting the layer positions of GABA–BrdU interneurons (n = 2 animals, 257 cells; mean ± SEM percentage) indicating that the majority of late-born interneurons in wild-type cortices are distributed in the upper layers. D, In the p35 mutant cortex, double-labeled interneurons (yellow) born at E15.5 are also found in the upper layers. E, Higher magnification of boxed area in D indicating that double-labeled interneurons (arrows) are in the upper layers of the p35 mutant cortex. F, Histogram depicting the layer positions of GABA–BrdU interneurons (n = 2 animals, 345 cells; mean ± SEM percentage) indicating that the majority of late-born interneurons in p35 mutant cortex are situated in the upper layers of the cortex. G, Early-born interneurons (BrdU at E12.5) in the mutant p35 cortex are inverted. Double-labeled interneurons (GABA–BrdU) (n = 3 animals, 364 cells; mean ± SEM percentage) indicate that the majority of early-born interneurons in p35 mutant cortices are situated in the upper layers of the cortex. H, Global analysis of all interneurons in p35 mutant cortex (both early- and late-born interneurons) (n = 2 animals, 3731 cells; mean ± SEM percentage) shows a biased distribution of interneurons to the upper half of the mutant cortex. I, In contrast, a relatively even distribution of interneurons to all cortical layers was seen in wild-type cortex (n = 2 animals, 3795 cells; mean ± SEM percentage). Scale bar: A, D, 160 μm; B, E, 80 μm. WM, White matter.
Transplanted wild-type interneurons are correctly layered in Dab1 mutants
Thus far, the results suggest that late-born (but not early-born) interneurons are sensitive to the absence of Reelin signaling by random distribution throughout the cortical depth. In Dab1 mutants, Reelin protein is expressed (Howell et al., 1997), and hence there is potential for restoration of Reelin signaling if interneurons expressing Dab1 are introduced into this environment. In control transplantations, interneuron precursors (E15.5) from wild-type MGEs were isochronously transplanted into the ventricles of wild-type or heterozygous brains (E15.5). The results show that transplanted cells have integrated into the upper layers of the wild-type cortex (Fig. 8A–C), confirming the results of previous studies (Valcanis and Tan, 2003). When wild-type MGE cells (E15.5) were transplanted into the ventricles of Dab1 mutant embryos (E15.5), the transplanted cells are also found in the upper layers despite a misordered cortical environment in which the projection neurons are inverted (Fig. 8D–F). This impression is confirmed by their Gaussian means showing overlap of the CIs, indicating that they are not significantly different (Table 1). Therefore, it appears that the transplanted cells have responded appropriately to Reelin protein in the mutant hosts by being positioned superficially, although the rest of the mutant cortex is inverted.
Figure 8.

Transplanted wild-type interneurons are correctly layered in Dab1 mutant cortex. A, Control transplantation of wild-type MGE cells (βgal+) into Dab1 wild-type and heterozygous hosts at E15.5 shows predominantly upper layer distribution of transplanted cells. B, Higher power of boxed area in A showing colonization of transplanted cells in upper layers. C, Histograms plotting the layer positions of transplanted cells into wild-type (n = 1) and heterozygous (n = 1) hosts (1150 βgal-positive cells; mean ± SEM percentage) showing upper layer distribution. D, Transplantation of wild-type MGE cells into Dab1 mutant ventricles at E15.5 also shows upper layer colonization of transplanted cells. E, Higher power of boxed area in D showing a similar layer distribution to the upper layers as in the control host (compare B, E). F, Histograms depicting the layer positions of transplanted wild-type cells (n = 2 animals, 490 βgal-positive cells; mean ± SEM percentage) in Dab1 mutant cortex showing that the majority of transplanted cells are situated in the upper layers. Scale bar: A, D, 290 μm; B, E, 100 μm. WM, White matter.
Discussion
We considered the issue of how cortical interneurons, despite their generation in a subcortical site, are able to find their proper cortical locations after long-range tangential migration. Our working hypothesis sets out to question whether or not Reelin signaling is a major determinant of interneuron positioning and whether early-born and late-born interneurons have equivalent responses to this signal. For comparison, we also examined interneuron positions in p35 mutants because disruptions to this signaling pathway also produces an inverted cortex. Surprisingly, the results demonstrate a nonequivalent pattern of interneuron response in the absence of Reelin signaling. Whereas early-born interneurons behaved similarly to projection neurons and showed layer inversion, late-born interneurons were not inverted but indiscriminately distributed throughout the cortical depth. This disturbed pattern of late-born interneuron positioning was restored when interneurons were introduced into an environment in which the potential for Reelin signaling was restored. These studies suggest that separate mechanisms may be at work for interneuron stratification. Whereas late-born interneurons are responsive to Reelin signaling, the mechanisms controlling early-born interneurons appear to be tightly bound with their projection neuron counterparts. Based on the current results, we propose that early-born interneurons receive their laminar cues from their isochronic projection neurons, whereas late-born interneuron position is dependent on Reelin signaling.
Compared with projection neurons, the notion of Reelin signaling as an active determinant of interneuron positioning has been ambiguous. There are a number of reasons for this. First, a hallmark of Reelin signal malfunction is the failure of preplate splitting to allow projection neurons to form a cortical plate (CP), and this process is crucial to subsequent layer ordering of radially migrating neurons (Caviness, 1982). Because interneurons do not participate in preplate splitting, it is generally assumed that Reelin has little or no impact on interneuron layering. Second, because interneurons originate from an extracortical location, it has been difficult to ascertain whether aberrant interneuron positioning is intrinsic to a failure of Reelin signaling or extrinsic to interneurons arriving at a vastly disorganized cortex. In the current study, we show that a disorganized cortical environment is not a barrier to proper interneuron layering provided that Reelin signaling is present (p35 mutant) or restored (transplantation of wild-type interneurons into Dab1 mutant). These results clearly point to Reelin signaling as the primary driver of interneuron layering but only in the late-born population. The components of Reelin signaling are known to be present in interneuron precursors, including Dab1 and the Reelin receptors (VLDLR and ApoER2) (Rice et al., 1998; Trommsdorff et al., 1999), and future biochemical studies will be required to demonstrate that they are appropriately active.
It should be noted that, although the present study is focused on juvenile stages (P16–P23), other studies dealing with earlier time points (P0–P9) have observed substantial remodeling of interneuron positions at postnatal stages, and this remodeling appears to be influenced by the absence of Reelin (Hevner et al., 2004). Although postnatal remodeling could be one mechanism for the action of Reelin on interneurons, this study did not demonstrate a distinction between early- versus late-born interneurons. On that basis, it was suggested that all interneurons take their laminar cues from pyramidal neurons (Hevner et al., 2004). In this regard, it is important to note that we examined only interneurons in the somatosensory cortex in which the absence of Reelin is strongly correlated with layer inversion of pyramidal neurons. It is now apparent that the positioning of layer V pyramidal neurons in response to Reelin is not straightforward, with certain cortical areas showing complex layering patterns (Polleux et al., 1998; Yamamoto et al., 2003). Additional studies would need to inquire whether or not early-born interneurons in these different areas are associated with their pyramidal counterparts.
So why does Reelin signaling only affect the late-born and not the early-born interneurons in the somatosensory cortex? The finding that the early-born interneurons are unresponsive to the absence of Reelin signaling requires the consideration of other possible mechanisms that might influence interneuron positioning. Projection neurons have been proposed to be one possible source of laminar cues (Nadarajah et al., 2002; Ang et al., 2003; Hevner et al., 2004), although the present study would suggest that they may affect only the early-born population. The nature of these cues remain unknown. The migratory behavior of early-born interneurons would also discount Reelin as a major influence. Most early-born interneurons penetrate the marginal zone (MZ) and then descend into the CP (Ang et al., 2003; Tanaka et al., 2003; Hevner et al., 2004). The MZ is rich in Reelin, and, if this protein is considered instrumental for driving early-born interneurons to their deep positions in the CP, it would suggest a chemorepellent role for Reelin. However, a chemorepellent role is not compatible with the fact that early-born interneurons readily enter the Reelin-rich MZ from the subpallial origin. Additional evidence, that Reelin signaling is not a key effector of early-interneuron ordering, is provided by p35 mutant mice in which the early-born interneurons are displaced and inverted despite the presence of intact Reelin signaling. However, this conclusion needs to be tempered by the fact that there are subtle differences in layer inversions produced by reeler and p35 mice, and a strict comparison between the two cortices requires additional qualifications, for example, the p35 mutant cortex has a properly positioned subplate. This notwithstanding, the present study provides new perspectives on how interneuron positioning in these mutants may be interpreted. Clearly, late-born interneuron positioning in p35 mutants is unaffected despite the inversion of all pyramidal neuron layers. This confirms previous observations in p35 chimeras wherein all interneurons appear to be randomly distributed despite genotype-specific segregation of pyramidal neurons (Hammond et al., 2004). Similar conclusions, that interneuron positioning is unaffected by the Cdk5/p35 pathway, has been suggested previously in chimeric studies using Cdk5 mutants (Gilmore and Herrup, 2001).
Conversely, the finding that late-born interneurons are responsive to the presence of Reelin signaling is supported by their behavior in time-lapse studies. For example, late-born interneurons appear more likely to exhibit ventricle-directed migration in which they dive down toward the cortical ventricular zone before reversing direction and migrating superficially (Nadarajah et al., 2002). This is consistent with a range of studies showing that late-born interneurons tend to track deeper pathways in the intermediate zone and subventricular zone (Anderson et al., 2001; Wichterle et al., 2001; Polleux et al., 2002; Tanaka et al., 2003).
Time-lapse imaging of these deeply situated interneurons report 90° turning of migrating interneurons to ascend radially into the CP (Polleux et al., 2002; Tanaka et al., 2003). It is likely that these late-born interneurons migrate radially toward the CP in response to Reelin. Additional support may be found in p35 mutant cortex in which Reelin signaling is intact and late-born interneurons are appropriately positioned in the upper layers despite the fact that all other cortical neurons are inverted.
What might be the evolutionary implications of these findings? Our results point to different mechanisms for the stratification of cortical interneuron populations and would suggest that these developmental adaptations are necessitated by the outsourcing of interneuron production to a subcortical location. The developmental constraints of tangential migration from a remote location, coupled with a need to integrate precisely with projection neuron layering, requires ingenious modes of migration. Broadly speaking, early-born interneurons tend to take the superficial route into the MZ in which they find themselves in close proximity for integration with layer V/VI projection neurons. Conversely, late-born interneurons are generated at later stages of the expanding cortex when the MZ is situated farther and farther from the MGE, and, as a consequence, interneurons are forced to migrate into deeper aspects of the expanding cortex. From these deeper reaches, the late-born interneurons have to scale the cortical wall along with the projection neurons generated at the same time. It is therefore plausible that interneurons in these positions have co-opted Reelin signaling as a mechanism for migration toward the upper layers of the CP. In conclusion, the demarcation of interneurons into early- and late-born populations reflects not only the age of their genesis but also different migratory strategies (Anderson et al., 2001; Wichterle et al., 2001; Polleux et al., 2002; Tanaka et al., 2003) and differential molecular properties (Anderson et al., 1997; Casarosa et al., 1999; Marin et al., 2000). Being of subpallial origin and faced with the need to adapt to the developmental agenda of the pallium, it is not surprising that these two subpopulations have crafted separate strategies to ensure their proper integration.
Footnotes
This work was supported by a program grant from the National Health and Medical Research Council, a Kenneth Myer Foundation grant, and a professorship funded by the Robert G. Leitl estate (M.K.). We thank Andre Goffinet for the gift of Reeler mice, Brian Howell for the gift of Dab1 mutant mice, Li-Huei Tsai for the p35 mutant mice, Frank Weissenborn for expert technical assistance, and Fiona Christensen for assistance with the transplantations. We also thank Dr. Bang Bui for providing the bootstrap algorithm.
References
- Anderson SA, Qiu M, Bulfone A, Eisenstat DD, Meneses J, Pedersen R, Rubenstein JL (1997). Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late born striatal neurons. Neuron 19:27–37. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Marin O, Horn C, Jennings K, Rubenstein JL (2001). Distinct cortical migrations from the medial and lateral ganglionic eminences. Development 128:353–363. [DOI] [PubMed] [Google Scholar]
- Ang ES Jr, Haydar TF, Gluncic V, Rakic P (2003). Four-dimensional migratory coordinates of GABAergic interneurons in the developing mouse cortex. J Neurosci 23:5805–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angevine JB, Sidman RL (1961). Autoradiographic study of cell migration during histogenesis of the cerebral cortex in the mouse. Nature 192:766–768. [DOI] [PubMed] [Google Scholar]
- Battista J, Kalloniatis M (2002). Left-right word recognition asymmetries in central and peripheral vision. Vision Res 42:1583–1592. [DOI] [PubMed] [Google Scholar]
- Bui B, Weisinger H, Sinclair A, Vingrys A (1998). Comparison of guinea pig electroretinograms measured with bipolar corneal and unipolar intravitreal electrodes. Doc Ophthalmol 95:15–34. [DOI] [PubMed] [Google Scholar]
- Bui BV, Edmunds B, Cioffi GA, Fortune B (2005). The gradient of retinal functional changes during acute intraocular pressure elevation. Invest Ophthalmol Vis Sci 46:202–213. [DOI] [PubMed] [Google Scholar]
- Casarosa S, Fode C, Guillemot F (1999). Mash1 regulates neurogenesis in the ventral telencephalon. Development 126:525–534. [DOI] [PubMed] [Google Scholar]
- Caviness VS (1982). Neocortical histogenesis in normal and reeler mice. A developmental study based upon 3H-thymidine autoradiography. Dev Brain Res 4:293–302. [DOI] [PubMed] [Google Scholar]
- Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH (1997). Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 18:29–42. [DOI] [PubMed] [Google Scholar]
- Darvas F, Pantazis D, Kucukaltun-Yildirim E, Leahy RM (2004). Mapping human brain function with MEG and EEG: methods and validation. NeuroImage 23:S289–S299. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T (1995). A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374:719–723. [DOI] [PubMed] [Google Scholar]
- Efron B, Tibshirani RJ (1986). Bootstrap methods for standard errors, confidence intervals and other measures of statistical accuracy. Stat Sci 1:54–77. [Google Scholar]
- Fairen A, Cobas A, Fonseca M (1986). Times of generation of glutamic acid decarboxylase immunoreactive neurons in mouse somatosensory cortex. J Comp Neurol 251:67–83. [DOI] [PubMed] [Google Scholar]
- Foster D, Bischof W (1987). Bootstrap variance estimators for the parameters of small-sample sensory-performance functions. Biol Cybern 57:341–347. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G (1997). In: The mouse brain in stereotaxic coordinates San Diego: Academic.
- Gilmore EC, Herrup K (2001). Neocortical cell migration: GABAergic neurons and cells in layers I and VI move in a cyclin-dependent kinase 5-independent manner. J Neurosci 21:9690–9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonchar Y, Burkhalter A (1997). Three distinct families of GABAergic neurons in rat visual cortex. Cereb Cortex 7:347–358. [DOI] [PubMed] [Google Scholar]
- Hammond V, Tsai LH, Tan SS (2004). Control of cortical neuron migration and layering: cell and non-cell-autonomous effects of p35. J Neurosci 24:576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan AJ, Servotte S, Katsnelson A, Sisodiya S, Blakemore C, Squier M, Molnar Z (1999). Characterization of nodular neuronal heterotopia in children. Brain 122:219–238. [DOI] [PubMed] [Google Scholar]
- Hendry SH, Jones EG (1981). Sizes and distributions of intrinsic neurons incorporating tritiated GABA in monkey sensory-motor cortex. J Neurosci 1:390–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevner RF, Daza RA, Englund C, Kohtz J, Fink A (2004). Postnatal shifts of interneuron position in the neocortex of normal and reeler mice: evidence for inward radial migration. Neuroscience 124:605–618. [DOI] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA (1997). Neuronal position in the developing brain is regulated by mouse disabled-1. Nature 389:733–737. [DOI] [PubMed] [Google Scholar]
- Hunter-Schaedle KE (1997). Radial glial cell development and transformation are disturbed in reeler forebrain. J Neurobiol 33:459–472. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Hattori R, Yui Y (1994). Three distinct subpopulations of GABAergic neurons in rat frontal agranular cortex. Brain Res 649:159–173. [DOI] [PubMed] [Google Scholar]
- Lin CS, Lu SM, Schmechel DE (1986). Glutamic acid decarboxylase and somatostatin immunoreactivities in rat visual cortex. J Comp Neurol 244:369–383. [DOI] [PubMed] [Google Scholar]
- Marin O, Anderson SA, Rubenstein JL (2000). Origin and molecular specification of striatal interneurons. J Neurosci 20:6063–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meskenaite V (1997). Calretinin-immunoreactive local circuit neurons in area 17 of the cynomolgus monkey, Macaca fascicularis. J Comp Neurol 379:113–132. [PubMed] [Google Scholar]
- Miller MW (1986). The migration and neurochemical differentiation of γ-aminobutyric acid (GABA)-immunoreactive neurons in rat visual cortex as demonstrated by a combined immunocytochemical-autoradiographic technique. Dev Brain Res 28:41–46. [DOI] [PubMed] [Google Scholar]
- Morante-Oria J, Carleton A, Ortino B, Kremer EJ, Fairen A, Lledo PM (2003). Subpallial origin of a population of projecting pioneer neurons during corticogenesis. Proc Natl Acad Sci USA 100:12468–12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadarajah B, Alifragis P, Wong RO, Parnavelas JG (2002). Ventricle-directed migration in the developing cerebral cortex. Nat Neurosci 5:218–224. [DOI] [PubMed] [Google Scholar]
- Peduzzi JD (1988). Genesis of GABA-immunoreactive neurons in the ferret visual cortex. J Neurosci 8:920–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, Dehay C, Kennedy H (1998). Neurogenesis and commitment of corticospinal neurons in reeler. J Neurosci 18:9910–9923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, Whitford KL, Dijkhuizen PA, Vitalis T, Ghosh A (2002). Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development 129:3147–3160. [DOI] [PubMed] [Google Scholar]
- Rice DS, Sheldon M, D’Arcangelo G, Nakajima K, Goldowitz D, Curran T (1998). Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development 125:3719–3729. [DOI] [PubMed] [Google Scholar]
- Sheldon M, Rice DS, D’Arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T (1997). Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 389:730–733. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Kisvarday ZF, Martin KAC, Whitteridge D (1983). Synaptic connections of morphologically identified and physiologically characterized large basket cells in the striate cortex of cat. Neuroscience 10:261–294. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Tamas G, Lujan R, Buhl EH (1998). Salient features of synaptic organisation in the cerebral cortex. Brain Res Brain Res Rev 26:113–135. [DOI] [PubMed] [Google Scholar]
- Takahara T, Ohsumi T, Kuromitsu J, Shibata K, Sasaki N, Okazaki Y, Shibata H, Sato S, Yoshiki A, Kusakabe M, Muramatsu M, Ueki M, Okuda K, Hayashizaki Y (1996). Dysfunction of the Orleans reeler gene arising from exon skipping due to transposition of a full-length copy of an active L1 sequence into the skipped exon. Hum Mol Genet 5:989–993. [DOI] [PubMed] [Google Scholar]
- Tan S-S, Breen SJ (1993). Radial mosaicism and tangential dispersion both contribute to mouse neocortex development. Nature 362:638–640. [DOI] [PubMed] [Google Scholar]
- Tanaka D, Nakaya Y, Yanagawa Y, Obata K, Murakami F (2003). Multimodal tangential migration of neocortical GABAergic neurons independent of GPI-anchored proteins. Development 130:5803–5813. [DOI] [PubMed] [Google Scholar]
- Thom M, Martinian L, Parnavelas JG, Sisodiya SM (2004). Distribution of cortical interneurons in grey matter heterotopia in patients with epilepsy. Epilepsia 45:916–923. [DOI] [PubMed] [Google Scholar]
- Treutwein B (1995). Adaptive psychophysical procedures. Vision Res 35:2503–2522. [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J (1999). Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97:689–701. [DOI] [PubMed] [Google Scholar]
- Valcanis H, Tan S-S (2003). Layer specification of transplanted interneurons in developing mouse neocortex. J Neurosci 23:5113–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A (2001). In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development 128:3759–3771. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Sakakibara S, Mikoshiba K, Terashima T (2003). Ectopic corticospinal tract and corticothalamic tract neurons in the cerebral cortex of yotari and reeler mice. J Comp Neurol 461:61–75. [DOI] [PubMed] [Google Scholar]
- Yozu M, Tabata H, Nakajima K (2004). Birth-date dependent alignment of GABAergic neurons occurs in a different pattern from that of non-GABAergic neurons in the developing mouse visual cortex. Neurosci Res 49:395–403. [DOI] [PubMed] [Google Scholar]