

Abstract
Huntington’s disease (HD) is caused by an abnormal expanded polyglutamine (polyQ) repeat in the huntingtin protein. Insulin-like growth factor-1 acting through the prosurvival kinase Akt mediates the phosphorylation of huntingtin at S421 and inhibits the toxicity of polyQ-expanded huntingtin in cell culture, suggesting that compounds enhancing phosphorylation are of therapeutic interest. However, it is not clear whether phosphorylation of S421 is crucial in vivo. Using a rat model of HD based on lentiviral-mediated expression of a polyQ-huntingtin fragment in the striatum, we demonstrate here that phosphorylation of S421 is neuroprotective in vivo. We next demonstrate that calcineurin (CaN), a calcium/calmodulin-regulated Ser/Thr protein phosphatase, dephosphorylates S421 in vitro and in cells. Inhibition of calcineurin activity, either by overexpression of the dominant-interfering form of CaN or by treatment with the specific inhibitor FK506, favors the phosphorylation of S421, restores the alteration in huntingtin S421 phosphorylation in HD neuronal cells, and prevents polyQ-mediated cell death of striatal neurons. Finally, we show that administration of FK506 to mice increases huntingtin S421 phosphorylation in brain. Collectively, these data highlight the importance of CaN in the modulation of S421 phosphorylation and suggest the potential use of CaN inhibition as a therapeutic approach to treat HD.
Keywords: Huntington disease, polyglutamine, protein phosphatase 2B, lentiviruses, rat model, calcium, neuroprotection
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder characterized by motor and cognitive impairments (Young, 2003). The neuropathology of HD is a marked neuronal death in the striatum, whereas other brain structures are selectively spared. HD is caused by an abnormally expanded CAG tract in the coding region of the IT15 gene, which encodes a polyglutamine tract in the protein huntingtin (htt) (Gusella and MacDonald, 1995). There is currently no effective treatment to prevent or delay disease progression.
Huntingtin becomes toxic when it contains an abnormal polyglutamine (polyQ) expansion. PolyQ-huntingtin is cleaved in the cytoplasm and thereafter translocates to the nucleus, where it forms ubiquitin-immunopositive nuclear aggregates (Davies et al., 1998; DiFiglia, 2002; Gafni and Ellerby, 2002; Wellington et al., 2002). When in the nucleus, polyQ-huntingtin induces transcriptional dysregulation and neuronal death through a gain-of-function mechanism (Saudou et al., 1998; Ross, 2002; Sugars and Rubinsztein, 2003; Landles and Bates, 2004). A loss of the protective functions of huntingtin could act concomitantly and/or synergistically with the gain of new toxic functions (Cattaneo et al., 2001). In agreement, dysregulation of BDNF transcription is linked to the loss of huntingtin normal function (Zuccato et al., 2001). In addition, when huntingtin contains the polyQ expansion, its ability to transport BDNF-containing vesicles and to promote neuronal survival is lost (Gauthier et al., 2004). Finally, aggregates are also found in neurites and could also participate in neuronal dysfunction by altering the microtubule network dynamics and/or axonal transport (Li et al., 2000, 2003; Gunawardena et al., 2003; Szebenyi et al., 2003; Guzik and Goldstein, 2004; Lee et al., 2004; Trushina et al., 2004; Charrin et al., 2005).
Several posttranslational modifications such as proteolysis, ubiquitination, and sumoylation modify the toxicity of huntingtin (Kalchman et al., 1996; Saudou et al., 1998; Kim et al., 2001; Mende-Mueller et al., 2001; Gafni and Ellerby, 2002; Lunkes et al., 2002; Wellington et al., 2002; Huang et al., 2004; Steffan et al., 2004). Another posttranslational modification that plays an important role in disease is phosphorylation. In particular, the Ser/Thr kinases, Akt and the serum and glucocorticoid-induced kinase (SGK), phosphorylate huntingtin at serine 421 (S421) (Humbert et al., 2002; Rangone et al., 2004; Warby et al., 2005). Phosphorylation at S421 subsequently abolishes polyQ-huntingtin-induced toxicity in a cellular model of HD (Humbert et al., 2002; Rangone et al., 2004). In addition, huntingtin is phosphorylated at serine 434 by the cyclin-dependent kinase Cdk5, and this reduces its cleavage by caspases (Luo et al., 2005). These findings not only reveal that elements outside the polyQ expansion are important during the course of the disease but also that identifying enzymes that modulate the phosphorylation status of huntingtin is crucial to understanding the cellular mechanisms that control neuronal death in HD.
Because phosphatase activities certainly allow a dynamic regulation of S421 phosphorylation, we aimed to identify phosphatases that act on S421. Calcineurin (CaN), also known as protein phosphatase 2B (PP2B), is a phosphoprotein Ser/Thr phosphatase activated physiologically by Ca2+/calmodulin (for review, see Mansuy, 2003). Therefore, it couples intracellular calcium to the dephosphorylation of selected substrates, which include transcription factors [nuclear factor of activated T-cells (NFAT)], ion channels (inositol-1,4,5 triphosphate receptor), proteins involved in vesicular trafficking (amphyphysin, dynamin), scaffold proteins (AKAP79), and phosphatase inhibitors (DARPP-32, inhibitor-1) (Aramburu et al., 2000; Rusnak and Mertz, 2000). CaN is present in all tissues in mammals, with notably high levels in brain, some studies indicating that it may account for 1% of the total protein content of the brain (Shenolikar, 1994). The catalytic subunit is mainly expressed in the cortex, striatum, and the hippocampus (Sola et al., 1999). Within the CNS, CaN activity has been involved in synaptic plasticity, as it is considered a negative constraint for long-term memory (Mansuy et al., 1998; Malleret et al., 2001). In nerve terminals, CaN triggers synaptic vesicle endocytosis by dephosphorylating vesicular and plasma membrane proteins in response to Ca2+ (Cousin and Robinson, 2001). Finally, CaN is inhibited by drugs such as cyclosporin A (CsA) and FK506 through the binding of these drugs to their appropriate receptors (immunophilins) (Snyder et al., 1998; Hamawy, 2003). Although initially used after organ transplantation because of their immunosuppressive properties, immunophilin ligands have been recently a center of attention as a putative therapeutic strategy for neuroregeneration and neuroprotection (Klettner and Herdegen, 2003; Pong and Zaleska, 2003). Furthermore, FK506 and its derived immunophilin ligands, unlike cyclosporin, can readily cross the blood–brain barrier (Pong and Zaleska, 2003).
In this study, we report that phosphorylation of polyQ-huntingtin at S421 is neuroprotective in vivo. We demonstrate that CaN dephosphorylates phosphorylated S421 in vitro and in cells. Importantly, inhibition of CaN activity either by overexpression of a dominant-interfering form, by RNA interference, or by the specific inhibitor FK506, leads to an increased phosphorylation of huntingtin at S421 and prevents polyQ-mediated death of striatal neurons. Of particular interest, we found that administration of FK506 to mice increases phosphorylation of endogenous cerebral huntingtin at S421. Together, we demonstrate that CaN regulates phosphorylation at S421 and suggest that inhibition of this phosphatase may have therapeutic implications as it decreases polyQ-induced toxicity.
Materials and Methods
Constructs.
The pSIN-480-17Q, pSIN-480-68Q, pSIN-480-68Q-S421A, and pSIN-480-68Q-S421D were generated from the 480-17Q, 480-68Q, 480-68Q-S421A, and 480-68Q-S421D plasmids, respectively (Humbert et al., 2002). These plasmids encode the first 480 aa fragment of huntingtin with 17 or 68 glutamines and a serine-to-alanine mutation (S421A) or a serine to aspartic acid mutation (S421D) at position 421. First, a PCR strategy was used to modify the C-terminal part of the 480 constructs (QuickChange site-directed mutagenesis; Stratagene, La Jolla, CA) using the forward primer, 5′-GCAGCTCACTCTGGTTCAAGAAGAG-3′, and the reverse primer, 5′-CTCGAGTTAAGCGTAATCTGGAACATCGTATGGGTAGGATCTAGGCTGCTCAGTG-3′, containing a hemagglutinin (HA) tag. The various fragments were cloned into the parental 480-17Q/68Q plasmids resulting in the generation of the vectors 480-17Q/68Q with S421, S421A, and S421D mutation with a C-terminal HA tag and then subcloned in the SIN-W-PGK-GDNF (BamHI-XhoI) (Deglon et al., 2000).
The lentiviral particles were produced in 293T cells and resuspended in PBS/1% bovine serum albumin (BSA) as reported previously (Hottinger et al., 2000). The particle content of viral batches was determined by p24 antigen ELISA (PerkinElmer Life and Analytical Sciences, Boston, MA).
The vectors encoding wild-type Akt, constitutively active Akt (Akt c.a.), wild-type CaNA (CaNA), a constitutively active (Ca2+-insensitive) form of CaNA (CaNA-ΔCaM), a catalytic-dead dominant-interfering form of CaNA (CaNA-D130N) and CaNB have been described previously (Bellacosa et al., 1998; Lilienbaum and Israel, 2003).
Animals.
Adult female Wistar rats (Iffa Credo/Charles River, Les Oncins, France) weighing ∼180–200 g were used. The animals were housed in a controlled temperature room that was maintained on a 12 h light/dark cycle. Food and water were available ad libitum.
FK506 (Alexis, Lausen, Switzerland) was administered to C57BL/6 male mice aged 5–6 weeks (Iffa Credo/Charles River) orally or by intraperitoneal injections (5 mg/kg) (Dunn et al., 1999; Singh et al., 2003). For oral administration, FK506 was dissolved in 100 μl of 0.5% carboxymethylcellulose (Sigma, St. Louis, MO). For intraperitoneal injections, FK506 was dissolved in 200 μl of Cremophor (Sigma). Mice were killed at indicated times after administration. The whole brain was homogenized in 1% Triton X-100 lysis buffer (see below, Western blot analysis) supplemented with 1 μm okadaic acid (Sigma), 1 μm FK506 (Alexis), 1 μm cyclosporine A (Sigma), and 40 nm tautomycin (Calbiochem, Darmstadt, Germany) and centrifuged at 20,000 × g (5 min; 4°C). An aliquot of the supernatant was resolved by 6% SDS-PAGE.
Studies using animals were performed in accordance with the Declaration of Helsinki and with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Injection of the lentiviruses.
Concentrated viral stocks were thawed and resuspended by repeated pipetting. Lentiviral vectors were stereotaxically injected (David Kopf Instruments, Tujunga, CA) into the striatum of ketamine (75 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) anesthetized animals using a Hamilton syringe (Reno, NV) with a 34-gauge blunt-tip needle. For each vector, particle content was matched to 200,000 ng of p24 per milliliter. The viral suspensions (4 μl) were injected at 0.2 μl/min by means of an automatic injector (Stoelting, Wood Dale, IL), and the needle was left in place for an additional 5 min. The stereotaxic coordinates were as follows: 0.5 mm rostral to bregma 3 mm lateral to midline, and 5 mm from the skull surface. The skin was closed using a 6-0 Prolene suture (Ethicon; Johnson and Johnson, Brussels, Belgium).
Histological processing.
After lentiviral injection (1, 12, and 24 weeks), the animals were given a sodium pentobarbital overdose and were transcardially perfused with saline and 4% paraformaldehyde/10% picric acid. The brains were removed and postfixed in 4% paraformaldehyde/10% picric acid for ∼24 h and finally cryoprotected in 25% sucrose/0.1 m phosphate buffer for 48 h. The brains were frozen in dry ice and 25 μm coronal sections were cut on a sliding microtome cryostat (Cryocut 1800; Leica Microsystems, Glattbrugg, Switzerland) at −20°C. Slices throughout the entire striatum were collected and stored in 48-well trays (Costar, Cambridge, MA) as free-floating sections in PBS supplemented with 0.12 μm sodium azide. The trays were stored at 4°C until immunohistochemical processing.
Striatal sections from injected rats were processed by immunochemistry for dopamine and cAMP-regulated phosphoprotein of a molecular mass of 32 kDa (DARPP-32; 1:7500; Chemicon, Temecula, CA), and for HA tag (monoclonal anti-HA antibody; 1:1000; Covance Research Products, Berkeley, CA) as described previously (Bensadoun et al., 2001). Sections were subsequently incubated with the biotinylated secondary goat anti-mouse or goat anti-rabbit antibodies (Vector Laboratories, Burlingame, CA), and the visualization was done as described previously (de Almeida et al., 2002).
Quantification of the DARPP-32 depleted regions and statistical analyses.
The downregulation of DARPP-32 was measured with an image analysis program (NIH Image 1.63) in each section containing the lesion. For each section, the optical density is first determined in the targeted region, the striatum, to obtain the T value and, in a noninfected region of the striatum (NI) that did not received the virus. A background value is then obtained from a DARPP-32 nonexpressing region (B, cortex). The percentage of lesion is then calculated using the following formula: % of lesion = 100 − ((NI − B)/(T − B) × 100) and is expressed as a percentage of lesion; 100% lesion corresponding to the lesion induced by the 480-68Q construct. Comparisons between two constructs injected in the same animal were made using paired t tests. At 24 weeks, the striatal volume is not modified by the various lentiviruses (data not shown).
In vitro phosphorylation/dephosphorylation assays.
Kinase assays were performed as described previously (Rangone et al., 2004) using recombinant Akt (Upstate, Charlottesville, VA) and a purified truncated form of huntingtin protein as a substrate [amino acids 384–467 of human huntingtin fused with glutathione S-transferase (GST); 1 h incubation, 30°C]. Purified calcineurin (native protein isolated from bovine brain; Upstate) was added as stated. The reaction products were resolved by 12% SDS-PAGE.
Cell culture, transfection, and drug treatments.
Primary cultures of striatal neurons were prepared from embryonic day 17 Sprague Dawley rats and transfected at 4 d in vitro by a modified calcium phosphate technique (Saudou et al., 1998). Mouse neuronal cells derived from wild-type huntingtin mice (neuronal cells, +/+) and from HdhQ109 knock-in mice (109Q/109Q) were cultured as described previously (Trettel et al., 2000) and transfected with Lipofectamine 2000 (Invitrogen, Breda, The Netherlands). When cotransfected (see Fig. 3A), the ratio 480-17Q/Akt/CaNA-ΔCaM/CaNB was 1:0.5:1:1. Human embryonic kidney 293 (HEK293) cells were cultured in DMEM supplemented with 10% bovine calf serum (BCS) and were transfected by the calcium phosphate technique. SHSY-5Y cells and Jurkat T-cells were grown in Roswell Park Memorial Institute media supplemented with 10% BCS. Jurkat T-cells were transferred to Tyrode’s buffer (137 mm NaCl, 3 mm KCl, 20 mm HEPES, 2 mm MgCl2, 1 mm CaCl2, 5.6 mm glucose, and 0.3 mg/ml BSA; 4 × 106 cells/ml) with previous addition of FK506 (1 μm) and/or ionomycin (2.5 μm).
Figure 3.

Calcineurin dephosphorylates S421 of huntingtin in cells. A, Mouse striatal cells (+/+ cells) were transfected with an N-terminal fragment of huntingtin (480-17Q), Akt, a constitutive active form of calcineurin (CaNA-ΔCaM/CaNB), or the corresponding empty vector, and were processed for Western blot analysis with anti-phospho-huntingtin-S421-714 or anti-huntingtin (1259) antibodies. Data from three independent experiments revealed that constitutive active form of calcineurin significantly reduced the phosphorylation of S421 triggered by Akt (Student’s t test; t(3) = 3.63; *p < 0.05). B, SHSY-5Y cells were transfected with a catalytic-dead form of calcineurin (CaNA-D130N), Akt, or the corresponding empty vector and were analyzed with anti-phospho-huntingtin-S421-763. Data are from three independent experiments (ANOVA; F(2,6) = 17.31; p = 0.032). The phosphorylation of endogenous huntingtin on S421 was significantly increased by the catalytic-dead construct of calcineurin (Student’s t test; t(4) = 4.34; *p < 0.05) and by Akt (t(4) = 5. 26; **p < 0.01). Error bars represent SEM.
Western blot analysis.
After transfection/incubation with drugs, cells were washed with ice-cold PBS before scraping and lysis. Lysis buffer consisted of 50 mm Tris-HCl, pH 7.5, containing 0.1% Triton X-100, 2 mm EDTA, 2 mm EGTA, 50 mm NaF, 10 mm β-glycerophosphate, 5 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 0.1% (v/v) β-mercaptoethanol, 250 μm PMSF, and 10 mg/ml aprotinin and leupeptin. Cell lysates were centrifuged at 20,000 × g for 5 min at 4°C. Equal amounts of protein (40 μg) were subjected to SDS-PAGE and electrophoretically transferred to PVDF membranes (Immobilon-P; Millipore, Bedford, MA). Blots were blocked in 5% BSA/TBST buffer (20 mm Tris-HCl, 0.15 m NaCl, 0.1% Tween 20) and immunoblotted with anti-α-tubulin (1:1000; DM1A; Sigma), anti-calcineurin Pan A (1:1000; Chemicon), anti-phospho-huntingtin-S421–763 (Humbert et al., 2002), anti-phospho-huntingtin-S421-714 (see below), anti-huntingtin 1259 (1:1000) (Lunkes et al., 2002), and MAB2166 (1:5000; Chemicon) antibodies for 1 h. Blots were then labeled with anti-rabbit IgG/HRP (Jackson ImmunoResearch, West Grove, PA) and washed and incubated for 5 min with SuperSignal West Pico Chemiluminescent Substrate (Pierce, Erembodegem, Belgium) according to the instructions of the supplier. Membranes were exposed to Kodak (Rochester, NY) BioMax films and then developed. Quantification of the signal was performed by densitometric scanning of the film using NIH ImageJ software.
Immunofluorescence experiments.
SHSY-5Y cells were grown on uncoated glass coverslips, transfected with wild-type CaNA/CaNB for 24 h, and treated for 15 min with 1 μm ionomycin (Sigma) or left untreated. Cells were fixed with 4% paraformaldehyde-PHEM buffer for 20 min (in mm: 120 PIPES, 50 HEPES, 20 EGTA, and 4 Mg-acetate) and incubated with anti-phospho-S421-huntingtin-763 (1:100) and anti-CaNA (1:200; catalog #C1956; Sigma) antibodies.
Pictures were captured with a three-dimensional deconvolution imaging system as described previously (Gauthier et al., 2004). Mean fluorescence intensity of the phospho-S421 signal was next quantified using MetaMorph software (Universal Imaging, Princeton, NJ). For each pair of cells (transfected/nontransfected), signal from the transfected cell was standardized by giving a value of 100 to the nontransfected cell. Only nonmitotic cells of the same approximate size were considered. More than 30 cells in total were analyzed in two independent experiments.
Neurons were grown on laminin and poly-d-lysine-coated glass coverslips, fixed as described before, and incubated with the following primary antibodies: anti-phospho-S421-huntingtin-714 (1:100), N-ter 1259 (1:500), and CaNA (1:200). Secondary antibodies were anti-mouse Alexa Fluor 488 (1:200) and anti-rabbit Alexa Fluor 555 (1:200; Invitrogen, Eugene, OR). Pictures were captured with a three-dimensional deconvolution imaging system.
Measurement of neuronal survival.
Four days after plating, primary cultures of striatal neurons were transfected with wild-type or polyQ-huntingtin and green fluorescent protein (GFP) to identify the transfected cells. To be certain that each neuron synthesizing GFP also expressed the huntingtin construct, transfections were performed using a derived phosphate calcium method with a high ratio of huntingtin DNA to GFP DNA (10:1 ratio) (Humbert et al., 2002). Under these conditions, >95% of the GFP-positive neurons also express the huntingtin construct (data not shown). GFP-positive neurons were scored using fluorescence microscopy in a blinded manner 16 and 36 h after transfection. Cell death occurring within the GFP-positive cells was determined as the difference in the number of surviving neurons between the two time points and expressed as a fold increase in neuronal cell death relative to the death induced by the 480-17Q construct. Each graph represents two to four independent experiments performed in triplicate. Each bar in a given graph corresponds to the scoring of ∼2000 neurons. For RNA interference experiment, neuronal cell death is expressed as the number of 480-68Q transfected cells that survived in the presence of the various small interfering RNA (siRNA) relative to the 480-68Q/scrambled condition. Data were submitted to complete statistical analysis.
siRNA against calcineurin.
The siRNA sequences targeting rat CaNAα and CaNAβ correspond to the coding regions 677–695 (GenBank accession number NM_017041) and 448–466 (GenBank accession number NM_017042), respectively. siRNA (3 μg) or scrambled RNA were mixed with 4 × 106 freshly isolated striatal neurons, nucleofected following instructions of the manufacturer (Amaxa Biosystems, Cologne, Germany), plated onto 12-well plates and incubated for 40 h. The scrambled RNA has the same nucleotide composition than the CaNAα siRNA but lacks a significant sequence homology to any other gene.
Antibodies against huntingtin phosphorylated on S421.
Human specific anti-phospho-huntingtin-S421-763 antibody was described previously (Humbert et al., 2002). Generation of mouse specific anti-phospho-huntingtin-S421-714 antibody: phosphopeptide corresponding to mouse huntingtin sequence (CARGRSGS[PO3H2]IVELL) was synthesized, coupled to keyhole limpet hemocyanin (NeoMPS, Strasbourg, France), and injected into rabbits. Polyclonal antibody was obtained from serum and affinity-purified with a phospho-peptide column. Briefly, the serum was filtered (0.22 μm filter), and, after addition of 1 m Tris, pH 8.0, up to a final concentration of 100 mm, it was applied to a Sulfolink column (Pierce) coupled to the phosphorylated peptide. Retained antibodies were eluted with 100 mm glycine buffer, pH 2.7, and pH was rapidly neutralized with 1 m Tris, pH 9. Antibodies were concentrated (Vivaspin concentrator 10,000 molecular weight; VivaScience, Hannover, Germany) and stored in 50% glycerol.
Results
Phosphorylation of polyQ-huntingtin at serine 421 is neuroprotective in vivo
We demonstrated previously that the IGF1/Akt pathway is neuroprotective in a cellular model of HD (Humbert et al., 2002). Indeed, after IGF-1 activation, Akt phosphorylates polyQ-huntingtin at S421 and blocks its toxicity in primary cultures of striatal neurons. To examine whether phosphorylation at S421 plays a role in vivo and could therefore be a therapeutic target, we used a rat model of HD based on lentiviral-mediated expression of polyQ-huntingtin in the striatum (de Almeida et al., 2002). This model recapitulates several features observed in HD patients, such as the presence of neuritic and intranuclear inclusions, neuronal dysfunction, and death. We generated HA-tagged lentiviral constructs encoding the first 480 aa of huntingtin containing 17 (wild-type, 480-17Q) or 68 glutamine residues (mutant, 480-68Q) with either an intact S421, an S421 to alanine (S421A) or an S421 to aspartic acid (S421D) mutation. Lentiviruses were then injected into rat striatum to allow direct comparison for a given rat between the 480-17Q/480-68Q, 480-68Q/480-68Q-S421A, and 480-68Q/480-68Q-S421D constructs (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). One week after injection, proper expression of the various huntingtin constructs was controlled by anti-huntingtin immunostaining and showed no differences in their expression levels (data not shown). According to an intermediate evaluation of the lesions at 12 weeks (data not shown), we analyzed the polyQ-huntingtin-induced lesions in rat striatum 24 weeks after injection. Immunohistochemical analysis revealed that all constructs were expressed at similar levels and that expression of the transgenes were sustained at 24 weeks (Fig. 1). At this stage, no significant cell death was detected as indicated by the presence in the injected region of HA- and Akt-immunopositive neurons (Fig. 1 and supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Lesions were then evaluated using DARPP-32 as a marker. DARPP-32 is a regulator of the dopamine signaling that is present in ∼96% of the striatal medium-sized spiny neurons. DARPP-32 is a marker of the dysfunction of these projecting neurons in HD as downregulation of DARPP-32 is observed in various models of the disease (Bibb et al., 2000; de Almeida et al., 2002; Canals et al., 2004). Immunohistochemical analysis with DARPP-32 antibody was performed, and the size of the lesions was determined for all constructs and expressed as a percentage of the lesions induced by the 480-68Q constructs. As expected, the 480-68Q construct induced a significant lesion in contrast to wild-type huntingtin. We found that absence of phosphorylation at S421 resulted in a strong increase (twofold) in the size of the DARPP-32-depleted region induced by the 480-68Q construct. In contrast, constitutive phosphorylation at S421 led to a significant reduction (∼30%) in the polyQ-induced DARPP-32-depleted region. Together, we conclude that phosphorylation of S421 in huntingtin is crucial to regulate disease progression in vivo.
Figure 1.

Phosphorylation of polyQ-huntingtin at S421 is neuroprotective in vivo. Rat striata were injected with 480-17Q (wild-type), 480-68Q (polyQ), 480-68Q-S421A, or 480-68Q-S421D lentiviruses. The polyQ-huntingtin-induced lesions in rat striatum were analyzed 24 weeks after injection. Immunohistochemical analysis with DARPP-32 antibody were performed, and the size of the lesions were determined for all constructs and expressed as a percentage of the lesion induced by the 480-68Q construct. As expected, a drastic loss of DARPP-32-immunoreactive neurons was observed in the 480-68Q-infected striatum, whereas expression of the wild-type protein had no effect (paired Student’s t test; t(7) = 6.95; ***p < 0.001). Absence of phosphorylation at S421 (480-68Q-S421A) resulted in an increased size of the DARPP-32-immunodepleted region (paired Student’s t test; t(6) = 3.17; *p < 0.05). In contrast, constitutive phosphorylation at S421 (480-68Q-S421D) significantly reduced the size of the DARPP-32-immunodepleted region (paired Student’s t test; t(7) = 3.46; *p < 0.05). Anti-HA immunohistochemical analysis revealed that all constructs were expressed at similar levels (bottom panels). Scale bars: DARPP-32, 500 μm; HA, 1000 μm. Error bars represent SEM.
Calcineurin dephosphorylates phospho-S421 of huntingtin in vitro
Because phosphatase activities could allow a dynamic regulation of S421 phosphorylation, we aimed to identify phosphatases that would dephosphorylate S421. Calcineurin, also known as PP2B, is a calcium- and calmodulin-dependent phosphatase (for review, see Rusnak and Mertz, 2000; Mansuy, 2003). To assess whether calcineurin acts on S421, we first performed dephosphorylation experiments in vitro. For this purpose, we used a polyclonal antibody that only binds to the phospho-serine at S421 (anti-phospho-huntingtin-S421-763) on the huntingtin protein (Humbert et al., 2002). In addition, we previously generated a GST-fused form of huntingtin (GST-huntingtin, amino acids 384–467 of human huntingtin fused to GST) that is phosphorylated by Akt and SGK on S421 (Humbert et al., 2002). We incubated this huntingtin fragment with a constitutive active form of Akt (Akt c.a.) resulting in the phosphorylation of GST-huntingtin at S421 as detected with the anti-phospho-huntingtin-S421-763 antibody (Fig. 2A,B). This phosphorylated fragment of huntingtin was next incubated with purified calcineurin for different times (Fig. 2A) and for 60 min with different concentrations of calcineurin (Fig. 2B). As observed, calcineurin dephosphorylates S421 of huntingtin in vitro in a time- and dose-dependent manner.
Figure 2.

Calcineurin dephosphorylates S421 in vitro. A, Time course. B, Dose-dependent dephosphorylation of S421 by purified calcineurin. A GST-coupled fragment of human huntingtin (amino acids 384–467) was phosphorylated with recombinant Akt (0.2 μg/sample) and then incubated with 2 μg (A) or different quantities (B) of purified calcineurin for the indicated times (A) or 60 min (B). Immunoblotting experiments were performed using anti-phospho-huntingtin-S421-763 or anti-huntingtin (1259) antibodies. Inc.time, Incubation time.
Calcineurin dephosphorylates phospho-S421 of huntingtin in cells
Calcineurin is a heterodimer composed of a 60 kDa catalytic subunit (CaNA) and a 19 kDa regulatory subunit (CaNB) (Rusnak and Mertz, 2000). Heterodimeric holoenzyme is necessary for its phosphatase activity. To determine whether calcineurin dephosphorylation of huntingtin at S421 occurs in cells, we cotransfected immortalized mouse striatal cells (+/+ cells) (Trettel et al., 2000) with 480-17Q, a constitutive active form of calcineurin A (CaNA-ΔCaM), CaNB, and/or Akt (Fig. 3A). As expected, Akt induced the phosphorylation of huntingtin on S421 as detected by Western blotting with our newly generated anti-phospho-huntingtin-S421-714 antibody that recognizes preferentially the mouse sequence. The phosphorylation of S421 triggered by Akt was reduced by the cotransfection of active calcineurin (CaNA-ΔCaM/CaNB). Both calcineurin constructs were validated previously using a luciferase reporter of NFAT activity and induced a 15-fold NFAT activation when CaNA-ΔCaM/CaNB were cotransfected in HEK293 cells (data not shown). Thus, we demonstrate that calcineurin dephosphorylates the 480-17Q form of huntingtin at Akt-phosphorylated S421.
To test whether endogenous huntingtin is the target of calcineurin, we used a dominant-interfering form of CaNA in which aspartic acid 130 is mutated into an asparagine (CaNA-D130N). Cotransfection of this construct with an NFAT reporter in HEK293 cells reduced endogenous NFAT activity by 60% (data not shown). When CaNA-D130N encoding construct was transfected in the human neuroblastoma SHSY-5Y cell line, phosphorylation of endogenous huntingtin at S421 was increased (Fig. 3B). Therefore, phosphorylated S421 of endogenous huntingtin is a physiological substrate of calcineurin.
To further demonstrate that calcineurin indeed dephosphorylates huntingtin at S421 in cells, we transfected neuronal cells with wild-type CaNA/CaNB and analyzed phosphorylation of endogenous huntingtin by immunofluorescence using the anti-phospho-huntingtin-S421-763 antibody in control conditions or after activation of calcineurin by the calcium ionophore ionomycin (Fig. 4A, top). Although huntingtin remained phosphorylated in the presence of calcineurin, we observed a reduction of huntingtin phosphorylation at S421 when calcineurin-transfected cells were treated with ionomycin (Fig. 4A, bottom). Quantification of fluorescence intensity in >30 cells from two independent experiments revealed a statistically significant decrease in endogenous huntingtin phosphorylation at S421 (Fig. 4, bar graph). This demonstrates that activation of calcineurin by Ca2+ in neuronal cells leads to the dephosphorylation of S421 of huntingtin.
Figure 4.

Huntingtin colocalizes with calcineurin and is dephosphorylated during Ca2+-induced calcineurin activation. A, Ionomycin (Io; bottom panels) decreased huntingtin S421 phosphorylation in SHSY-5Y cells transfected with wild-type CaNA/CaNB compared with nontreated cells (N.T.; top panels). The graph corresponds to the quantification of the mean fluorescence intensity of the phospho-S421-htt (763 antibody) signal issued from 32 cells (ANOVA; F(3,28) = 7,05; p = 0.0011) and shows that ionomycin-induced dephosphorylation of huntingtin was significant (Fisher’s post hoc test; ***p < 0.001). B, Huntingtin colocalized with the catalytic subunit of calcineurin (CaNA) to vesicular structures. Immunostaining is shown for huntingtin (1259 antibody), phospho-S421-huntingtin (714 antibody), and CaNA in striatal neurons. A substantial colocalization was observed in vesicular structures along neurites (see enlargements). Scale bars, 5 μm. Error bars represent SEM.
Finally, we confirmed the relevance of our findings by verifying that, in our experimental system, calcineurin and huntingtin are present in the same striatal neurons. We prepared primary cultures of striatal neurons that were subsequently immunostained for CaNA, huntingtin, and phospho-S421-huntingtin. We found that most, if not all, calcineurin-immunopositive striatal cells were also immunoreactive for total and phosphorylated huntingtin at S421 (Fig. 4B). Subcellularly, phospho-huntingtin and huntingtin were found in vesicular structures spread all along the cell body and the neurites. In addition to this prominent vesicular localization, huntingtin was also found in the cis-Golgi as demonstrated by the colocalization with GM130 (data not shown). Interestingly, we observed a partial colocalization of endogenous huntingtin and calcineurin that was particularly evident in vesicles along the neurites (Fig. 4B, enlargements).
A dominant-interfering form of calcineurin decreases polyQ-huntingtin induced toxicity
We showed previously that phosphorylation of huntingtin at S421 is neuroprotective. We therefore investigated whether a dominant-interfering form of calcineurin possesses neuroprotective properties by studying a neuronal model of HD that recapitulates the main features of the disease (Fig. 5A). We transfected primary cultures of striatal neurons with constructs 480-17Q and 480-68Q, either alone or in the presence of CaNA-D130N, and analyzed neuronal death 24 h after transfection. As expected, the 480-68Q construct induced a statistically significant increase in neuronal death compared with the 480-17Q construct. Interestingly, transfection of CaNA-D130N decreased neuronal death induced by the 480-68Q fragment of huntingtin (Fig. 5A). These findings indicate that a dominant-interfering form of calcineurin that increases phosphorylation of huntingtin on S421 exerts a neuroprotective effect on neuronal death induced by polyQ-huntingtin.
Figure 5.

A dominant-interfering form of calcineurin inhibits polyQ-huntingtin-induced toxicity in striatal neurons in a S421 phosphorylation-dependent manner. A, Wild-type huntingtin (480-17Q) or polyQ-huntingtin (480-68Q) were cotransfected with expression vectors encoding a catalytic-dead form of calcineurin (CaNA-D130N) or the corresponding empty vector in striatal neurons. Data from four independent experiments (ANOVA; F(3,56) = 6.99; p = 0.0004) revealed that CaNA-D130N significantly reduced polyQ-huntingtin-induced cell death (Fisher’s post hoc test, *p < 0.05; ***p < 0.0001). B, Striatal neurons were nucleofected with siRNA directed against CaNAα, CaNAβ, or both isoforms and 30 h later transfected with 480-68Q, as in A. Combination of siRNA directed against both isoforms of CaN efficiently reduced CaNA expression as detected by Western blot analysis. Data from three independent experiments (ANOVA, F(3,6) = 5.33, p = 0.039) demonstrated a protective effect toward polyQ-dependent toxicity (Fisher’s post hoc test, *p < 0.05). C, Striatal neurons were cotransfected with 480-68Q or a nonphosphorylable form (480-68Q-S421A) and CaNA-D130N. Data from four independent experiments (ANOVA, F(2,41) = 4.30, p = 0.0004) demonstrated that the catalytic-dead form of calcineurin did not prevent from polyQ-dependent cell death triggered by the 480-68Q-S421A construct (Fisher’s post hoc test, *p < 0.05, **p < 0.01), indicating that neuroprotection was dependent on the phosphorylation of S421. Error bars represent SEM.
To further confirm the role of calcineurin inhibition on polyQ-huntingtin-induced cell death, we decreased the levels of calcineurin by RNA interference. Two isoforms of calcineurin A subunits can be found in the brain, CaNAα and CaNAβ (Rusnak and Mertz, 2000). We therefore targeted both α and β subunits by RNA interference and found that the presence of both siRNA were necessary to ensure a significant decrease in the levels of CaNA (Fig. 5B). In these conditions, we observed a significant reduction in polyQ-huntingtin-induced cell death, further supporting the role of calcineurin in this process.
Does the neuroprotection mediated by CaNA-D130N depend on S421? To answer this question, we cotransfected CaNA-D130N in striatal neurons either with the 480-68Q construct or with a 480-68Q construct, where S421 has been mutated into an alanine, 480-68Q-S421A (Fig. 5C). As mentioned previously, the CaNA-D130N construct protects neurons against polyQ-huntingtin-induced toxicity, but it was unable to do so when position 421 could not be phosphorylated.
Therefore, calcineurin exerts its effect, at least in part, through the dephosphorylation of S421 on polyQ-huntingtin.
Inhibition of calcineurin increases phosphorylation of huntingtin at S421 in cells.
FK506 is an immunosuppressant drug used routinely in human treatment after transplantation. This compound mediates immunosuppression by inhibiting CaN-mediated dephosphorylation of NFAT, a transcription factor that regulates the expression of IL-2, which in turn regulates T-lymphocyte proliferation (Aramburu et al., 2004). Jurkat T-cell line is a T-lymphocyte cell line extensively used to study calcium signaling pathways in which calcineurin participates (Lilienbaum and Israel, 2003). To determine the effect of inhibiting calcineurin on the phosphorylation of huntingtin at S421, we treated Jurkat T-cells with FK506. As seen in Figure 6A, FK506 increased the phosphorylation of S421 on endogenous huntingtin. We next activated calcineurin by treating Jurkat T-cells with the calcium ionophore ionomycin (Fig. 6A, bottom panels). This resulted in a rapid dephosphorylation of huntingtin on S421, which was blocked by FK506.
Figure 6.
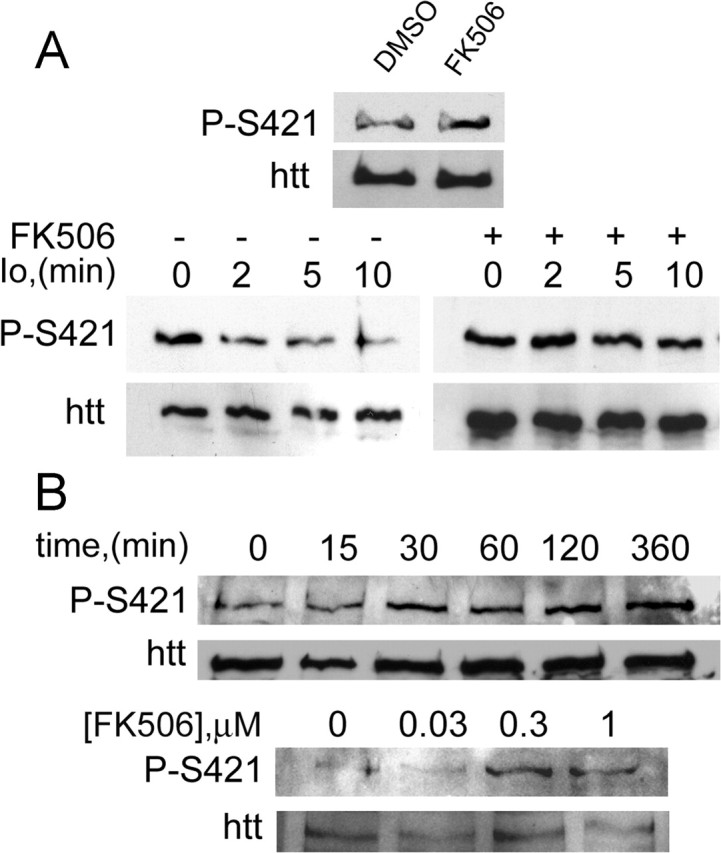
Inhibition of calcineurin by FK506 prevents the dephosphorylation of S421. A, Treatment with FK506 (20 min, 1 μm) in Jurkat cells increased the phosphorylation of endogenous huntingtin at S421 (left). The calcium ionophore ionomycin (Io; 2.5 μm) induced a rapid dephosphorylation of S421 in Jurkat cells. Io-elicited S421 dephosphorylation was prevented by FK506 (1 μm) (right). Whole-cell extracts of treated Jurkat T-cells were analyzed with the anti-phospho-huntingtin-S421-763 and the anti-huntingtin (1259) antibodies. B, Time-course- and dose-dependent phosphorylation of S421 after FK506 treatment of rat striatal neurons. Neurons were treated with 1 μm FK506 for different times (top) or with the indicated concentrations for 2 h (bottom). Whole-cell extracts were analyzed with the mouse phospho-specific (anti-phospho-huntingtin-S421-714) and the anti-huntingtin (1259) antibodies.
We next tested whether FK506 could also induce S421 phosphorylation in a cell type more relevant to the disease. We therefore treated primary cultured striatal neurons with FK506 for different times and concentrations (Fig. 6B). As for Jurkat cells, phosphorylation of endogenous huntingtin on S421 was increased over time and remained sustained for at least 6 h. We also found that increasing concentrations of FK506 resulted in an increase in S421 phosphorylation. Together, these data show that inhibition of CaN activity with FK506 results in an increased phosphorylation of endogenous huntingtin on S421.
PolyQ expansion leads to reduced huntingtin phosphorylation at S421 that can be increased by FK506: consequences on polyQ-induced neuronal toxicity
We next assessed the level of phosphorylation of huntingtin in the pathological situation. We used mouse neuronal cells derived from knock-in mice in which a CAG expansion, encoding 109 glutamine residues, was inserted into the endogenous mouse huntingtin gene (109Q/109Q) (Trettel et al., 2000). This cell line closely resembles the situation in HD patients because, in these cells, polyQ-huntingtin is expressed at endogenous levels. We performed immunoblotting experiments on +/+ or 109Q/109Q cells using our anti-phospho-huntingtin-S421-714 antibody and observed that phosphorylation of polyQ-huntingtin was drastically reduced compared with wild-type huntingtin (Fig. 7A). This agrees with previous work on transiently transfected HEK293 cells showing that S421 phosphorylation of polyQ-huntingtin was lower than that of wild-type huntingtin (Warby et al., 2005). We then asked whether inhibiting calcineurin would increase phosphorylation of polyQ-huntingtin on S421. Interestingly, treatment of 109Q/109Q cells with FK506 resulted in an increased S421 phosphorylation (Fig. 7B). We demonstrated previously that phosphorylation of huntingtin at S421 is neuroprotective. Therefore, because FK506 increases polyQ-huntingtin phosphorylation, we tested the effect of this immunosuppressive agent on polyQ-huntingtin-induced cell death (Fig. 7C). Primary cultures of striatal neurons were transfected with 480-68Q construct and treated with the vehicle or various doses of FK506. Treatment of cells with 0.3 and 1 μm of FK506 resulted in a decreased polyQ-induced neuronal death. Together, our data demonstrate that FK506, by preventing dephosphorylation of polyQ-huntingtin on S421, is neuroprotective in a cellular model of HD.
Figure 7.
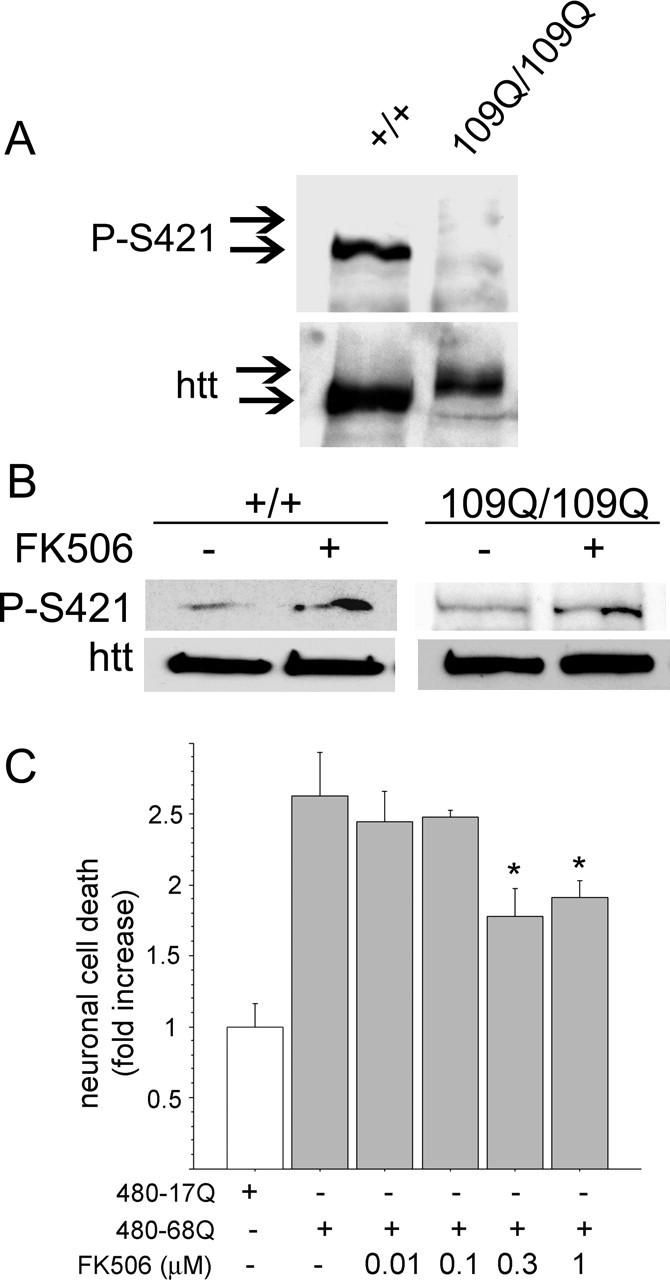
109Q/109Q cells show reduced phosphorylation of huntingtin on S421 that can be increased by FK506. A, Whole-cell extracts of immortalized knock-in cells issued from wild-type (+/+) or mutant mice (109Q/109Q) were analyzed by Western blot with the mouse phospho-specific and anti-huntingtin (1259) antibodies. B, FK506 (0.2 μm, 48 h) increased the phosphorylation of S421 on wild-type and polyQ-huntingtin. C, FK506 protects neurons from polyQ-mediated cell death. Striatal neurons were transfected with 480-68Q in the presence of different doses of FK506 or vehicle. Data from three independent experiments (ANOVA; F(5,68) = 8.02; p < 0.0001) revealed that FK506 significantly reduced neuronal death induced by 480-68Q (Fisher’s post hoc test, *p < 0.05). Error bars represent SEM.
A single administration of FK506 increases phosphorylation of huntingtin S421 in brain.
Because phosphorylation of huntingtin is crucial to regulating polyQ-huntingtin-induced toxicity in vitro and in vivo and considering that phosphorylation is reduced in disease models, we aimed to determine whether FK506 administration could induce the phosphorylation of huntingtin in vivo. Mice were treated intraperitoneally or orally with FK506 and killed at different times after administration. Brains were processed for immunoblotting and analyzed for huntingtin phosphorylation at S421 (Fig. 8). Strikingly, both intraperitoneal and oral administration of FK506 induced a sustained phosphorylation of endogenous htt on S421 in the brain (twofold/threefold increase). In agreement with a specific effect of calcineurin on huntingtin phosphorylation, we found, as reported previously (Yoshimoto et al., 2001), that the level of Akt phosphorylation at S473 is not regulated by FK506. These results demonstrate that inhibition of calcineurin by administration of FK506 leads to the phosphorylation of endogenous huntingtin in vivo.
Figure 8.
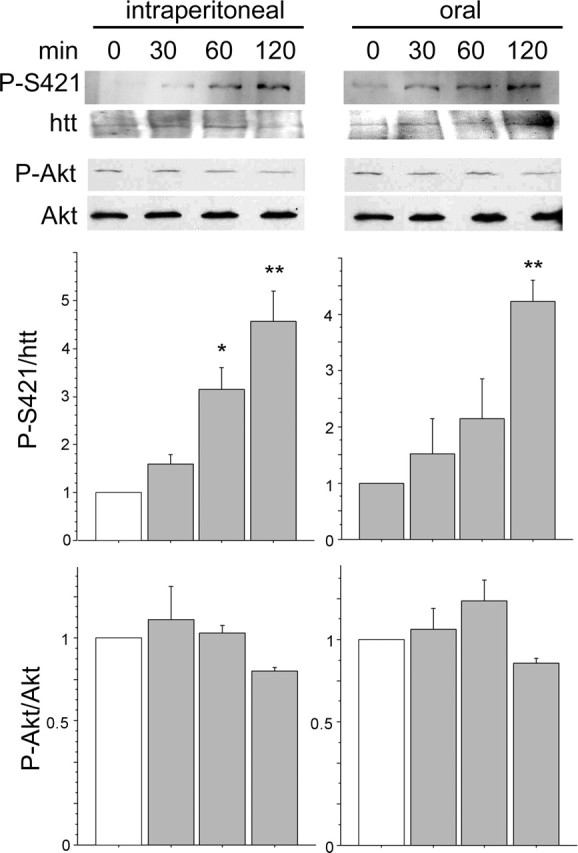
Administration of FK506 to mice results in increased phosphorylation of S421 in the brain. FK506 was administered intraperitoneally or orally to mice (5 mg/kg). Animals were killed at the indicated times after administration, and then brains were dissected, homogenized, and processed for Western blot analysis. Whole brain extracts were probed with anti-phospho-S421, anti-phospho-S473-Akt, anti-total huntingtin and anti-total Akt antibodies. Bottom panels correspond to the densitometric analysis of two independent experiments performed in duplicate. A total of 18 mice were killed. From top to bottom and from left to right, the following are shown: ANOVA, F(3,4) = 11.52, p = 0.019; ANOVA, F(3,4) = 8.694, p = 0.031; ANOVA, F(3,4) = 1.668, p = 0.31; ANOVA, F(3,4) = 0.97, p = 0.4. *p < 0.05; **p < 0.01. Error bars represent SEM.
Discussion
We show here that phosphorylation of huntingtin at S421 is crucial to regulating disease progression in vivo. Using a lentiviral approach to deliver N-terminal fragments of huntingtin directly to the striatum, we demonstrate that a construct encoding polyQ-huntingtin that is constitutively phosphorylated (480-68Q-S421D) induces a smaller DARPP-32-depleted region compared with a polyQ-huntingtin with an intact serine 421. Conversely, a polyQ-huntingtin that cannot be phosphorylated (480-68Q-S421A) is more toxic. We identified previously Akt and SGK as being able to act positively on S421 (Humbert et al., 2002; Rangone et al., 2004). We also reported that Akt is cleaved in postmortem brain samples from HD patients (Humbert et al., 2002) and found that, during disease progression, Akt activity is downregulated (Colin et al., 2005), further indicating that a reduced phosphorylation of huntingtin by Akt could occur in HD. In agreement, a decreased phosphorylation of polyQ-huntingtin at S421 is observed in YAC transgenic mice containing the polyQ expansion and in a cellular model of HD (Warby et al., 2005) (Fig. 7A). This implies that, during disease progression, the phosphorylation of huntingtin at S421 diminishes, thereby increasing the toxicity of polyQ-huntingtin. Therefore, identifying the cellular mechanisms that control huntingtin phosphorylation at S421 could help to prevent excessive dephosphorylation of huntingtin during disease. In the present investigation, we demonstrate that calcineurin (CaN) is a phosphatase that targets huntingtin at S421. This indicates that calcineurin is a new cellular element that controls huntingtin phosphorylation at S421 and that could participate in HD pathogenesis.
The phosphorylation of a particular residue usually depends on a balance in the activities of tightly regulated kinases and phosphatases. The observed reduction in huntingtin phosphorylation could result from a reduction in Akt activity (Colin et al., 2005) but also from an excess of calcineurin phosphatase activity. Indeed, calcineurin phosphatase is highly expressed in the striatum and in particular in the medium-size spiny neurons (MSNs), the first cells to degenerate in HD (Goto et al., 1989), and could therefore predispose huntingtin to dephosphorylation in these cells. Calcineurin is activated by Ca2+ (Mansuy, 2003), and several studies indicate that excessive Ca2+ entry in striatal neurons could play a role in HD (Bezprozvanny and Hayden, 2004). polyQ-huntingtin facilitates the activity of type I inositol 1,4,5-triphosphate receptor (InsP3R1) and NR2B subtype NMDA receptor (Zeron et al., 2002; Tang et al., 2003), thereby leading to increased cytosolic Ca2+ levels. In MSNs, the effect of mutant huntingtin on InsP3R requires HAP1 (Huntingtin-associated protein-1) (Tang et al., 2003; Tang et al., 2005). Because the connection between disturbed Ca2+ signaling and apoptosis is well established (Orrenius et al., 2003), it was proposed that glutamate released from corticostriatal projecting neurons elicits a supranormal Ca2+ response in MSNs from HD patients, leading to the opening of the mitochondria permeability transition pore and the activation of the caspase-dependent apoptotic cascade (Tang et al., 2003, 2005). Here, we propose that, in addition to this mechanism, Ca2+, by activating calcineurin, leads to dephosphorylation of huntingtin at S421 and participates in the pathogenic mechanism. In agreement, we showed that an increase in cytosolic Ca2+ concentration leads to dephosphorylation of huntingtin that can be blocked by FK506, demonstrating that the Ca2+-dependent dephosphorylation of S421 is dependent on CaN activity. Interestingly, another pathway by which calcineurin is activated in HD could involve calpain. Calpain is activated in HD (Kim et al., 2001; Gafni and Ellerby, 2002; Goffredo et al., 2002; Bizat et al., 2003). Calpain can regulate calcineurin activity either directly by cleaving calcineurin into an active form (Wu et al., 2004) or by cleaving and inactivating Cabin1/cain, a potent endogenous inhibitor of calcineurin (Kim et al., 2002). Together, for the first time in HD, our study links excitotoxicity and Ca2+ to the phosphorylation of huntingtin.
Our findings on the beneficial role of huntingtin phosphorylation at S421 on disease progression in vivo extend our previous findings (Humbert et al., 2002; Rangone et al., 2004) and further indicate that promoting huntingtin phosphorylation might have an impact on disease progression in HD patients because huntingtin phosphorylation is decreased when huntingtin contains the pathological polyQ expansion (Warby et al., 2005) (Fig. 7A). One strategy for increasing huntingtin phosphorylation could involve drugs that activate the Akt or SGK pathway. However, hyperactivation of kinases such as Akt is associated with cancers (Franke et al., 2003), and drugs activating these pathways could have deleterious effects by inducing unwanted proliferative effects. Therefore, a more reasonable pharmacological approach to increase the phosphorylation of S421 is the inhibition of the phosphatase, once its identity is known. Many calcineurin inhibitors have been described, of which CsA and FK506 are the most potent, specific, and well known.
In this study, we focused on FK506 and found that FK506 is effective in inducing phosphorylation of huntingtin at S421 both in vitro and in vivo and in blocking polyQ-huntingtin-induced neuronal death. This suggests that FK506 is of therapeutic interest for HD patients. In addition, as discussed above, calcineurin inhibitors will have protective effects on mechanisms relevant to HD such as Ca2+-induced excitotoxicity and calpain activation. Furthermore, increasing experimental data indicate mitochondrial failures in patients and cellular/animal models of HD (Panov et al., 2002; Milakovic and Johnson, 2005). Interestingly, FK506 protects mitochondrial respiration and blocks ATP depletion during ischemia (Nakai et al., 1997) and prevents mitochondrial-dependent neuronal death in the 3-nitropropionic acid rat model of HD (Almeida et al., 2004). Finally, calcineurin inhibitors have a general protective role by acting on other substrates involved in cell death process. For example, calcineurin leads to dephosphorylation of Bad at S112 and S136, leading to its translocation to mitochondria and apoptosis (Wang et al., 1999).
As indicated previously, FK506 crosses the blood–brain barrier and, unlike CsA, does not require direct brain delivery of the compound. Moreover, several studies have highlighted the potential therapeutic role of immunophilin ligands in the treatment of neurodegenerative disorders. These include not only acute diseases such as cerebral ischemia, spinal cord, and traumatic brain injuries but also chronic diseases such as Parkinson’s disease and amyotrophic lateral sclerosis (for review, see Pong and Zaleska, 2003). Clinical trials for such diseases and the fact that FK506 is routinely used in grafting procedures have established the safety and tolerability of such compounds and therefore could serve as a basis for similar therapeutic trials in HD.
Footnotes
This work was supported by grants from Association pour la Recherche sur le Cancer (ARC, 4807, F.S., and 3665, S.H.), Fondation pour la Recherche Médicale (FRM) and Fondation BNP Paribas (F.S.), Association Française contre les Myopathies (AFM, F.S.), Provital–P. Chevalier (F.S. and S.H.), and Swiss National Science Foundation (N.D.). R.P. was supported by a fellowship from FRM and is currently supported by a Ministerio de Ciencia y Tecnologia fellowship. E.C. is supported by a MRT doctoral fellowship. S.H. is an INSERM investigator. F.S. is a recipient from an EMBO Young Investigator award and an INSERM/AP-HP investigator. We greatly acknowledge E. Bryson for generating lentiviral constructs; A. Israel, A. Lilienbaum, M. Kobr and N. Mermod for kindly providing us with CaN constructs; Y. Trottier and D. Devys for huntingtin antibodies; the Institut Curie Imaging Facility; members of the Saudou/Humbert’s laboratory for help and comments and I. Mansuy for discussions.
References
- Almeida S, Domingues A, Rodrigues L, Oliveira CR, Rego AC (2004). FK506 prevents mitochondrial-dependent apoptotic cell death induced by 3-nitropropionic acid in rat primary cortical cultures. Neurobiol Dis 17:435–444. [DOI] [PubMed] [Google Scholar]
- Aramburu J, Rao A, Klee CB (2000). Calcineurin: from structure to function. Curr Top Cell Regul 36:237–295. [DOI] [PubMed] [Google Scholar]
- Aramburu J, Heitman J, Crabtree GR (2004). Calcineurin: a central controller of signalling in eukaryotes. EMBO Rep 5:343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P (1998). Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene 17:313–325. [DOI] [PubMed] [Google Scholar]
- Bensadoun JC, de Almeida LP, Dreano M, Aebischer P, Deglon N (2001). Neuroprotective effect of interleukin-6 and IL6/IL6R chimera in the quinolinic acid rat model of Huntington’s syndrome. Eur J Neurosci 14:1753–1761. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Hayden MR (2004). Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun 322:1310–1317. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC, Messer A, Greengard P (2000). Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc Natl Acad Sci USA 97:6809–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizat N, Hermel JM, Boyer F, Jacquard C, Creminon C, Ouary S, Escartin C, Hantraye P, Kajewski S, Brouillet E (2003). Calpain is a major cell death effector in selective striatal degeneration induced in vivo by 3-nitropropionate: implications for Huntington’s disease. J Neurosci 23:5020–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martin-Ibanez R, Munoz MT, Mengod G, Ernfors P, Alberch J (2004). Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J Neurosci 24:7727–7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S (2001). Loss of normal huntingtin function: new developments in Huntington’s disease research. Trends Neurosci 24:182–188. [DOI] [PubMed] [Google Scholar]
- Charrin BC, Saudou F, Humbert S (2005). Axonal transport failure in neurodegenerative disorders: the case of Huntington’s disease. Pathol Biol (Paris) 53:189–192. [DOI] [PubMed] [Google Scholar]
- Colin E, Regulier E, Perrin V, Durr A, Brice A, Aebischer P, Deglon N, Humbert S, Saudou F (2005). Akt is altered in an animal model of Huntington’s disease and in patients. Eur J Neurosci 21:1478–1488. [DOI] [PubMed] [Google Scholar]
- Cousin MA, Robinson PJ (2001). The dephosphins: dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci 24:659–665. [DOI] [PubMed] [Google Scholar]
- Davies SW, Beardsall K, Turmaine M, DiFiglia M, Aronin N, Bates GP (1998). Are neuronal intranuclear inclusions the common neuropathology of triplet-repeat disorders with polyglutamine-repeat expansions? Lancet 351:131–133. [DOI] [PubMed] [Google Scholar]
- de Almeida LP, Ross CA, Zala D, Aebischer P, Deglon N (2002). Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J Neurosci 22:3473–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deglon N, Tseng JL, Bensadoun JC, Zurn AD, Arsenijevic Y, Pereira de Almeida L, Zufferey R, Trono D, Aebischer P (2000). Self-inactivating lentiviral vectors with enhanced transgene expression as potential gene transfer system in Parkinson’s disease. Hum Gene Ther 11:179–190. [DOI] [PubMed] [Google Scholar]
- DiFiglia M (2002). Huntingtin fragments that aggregate go their separate ways. Mol Cell 10:224–225. [DOI] [PubMed] [Google Scholar]
- Dunn SE, Burns JL, Michel RN (1999). Calcineurin is required for skeletal muscle hypertrophy. J Biol Chem 274:21908–21912. [DOI] [PubMed] [Google Scholar]
- Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C (2003). PI3K/Akt and apoptosis: size matters. Oncogene 22:8983–8998. [DOI] [PubMed] [Google Scholar]
- Gafni J, Ellerby LM (2002). Calpain activation in Huntington’s disease. J Neurosci 22:4842–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118:127–138. [DOI] [PubMed] [Google Scholar]
- Goffredo D, Rigamonti D, Tartari M, De Micheli A, Verderio C, Matteoli M, Zuccato C, Cattaneo E (2002). Calcium-dependent cleavage of endogenous wild-type huntingtin in primary cortical neurons. J Biol Chem 277:39594–39598. [DOI] [PubMed] [Google Scholar]
- Goto S, Hirano A, Rojas-Corona RR (1989). An immunohistochemical investigation of the human neostriatum in Huntington’s disease. Ann Neurol 25:298–304. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS (2003). Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40:25–40. [DOI] [PubMed] [Google Scholar]
- Gusella JF, MacDonald ME (1995). Huntington’s disease: CAG genetics expands neurobiology. Curr Opin Neurobiol 5:656–662. [DOI] [PubMed] [Google Scholar]
- Guzik BW, Goldstein LS (2004). Microtubule-dependent transport in neurons: steps towards an understanding of regulation, function and dysfunction. Curr Opin Cell Biol 16:443–450. [DOI] [PubMed] [Google Scholar]
- Hamawy MM (2003). Molecular actions of calcineurin inhibitors. Drug News Perspect 16:277–282. [DOI] [PubMed] [Google Scholar]
- Hottinger AF, Azzouz M, Deglon N, Aebischer P, Zurn AD (2000). Complete and long-term rescue of lesioned adult motoneurons by lentiviral-mediated expression of glial cell line-derived neurotrophic factor in the facial nucleus. J Neurosci 20:5587–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Yanai A, Kang R, Arstikaitis P, Singaraja RR, Metzler M, Mullard A, Haigh B, Gauthier-Campbell C, Gutekunst CA, Hayden MR, El-Husseini A (2004). Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron 44:977–986. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F (2002). The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev Cell 2:831–837. [DOI] [PubMed] [Google Scholar]
- Kalchman MA, Graham RK, Xia G, Koide HB, Hodgson JG, Graham KC, Goldberg YP, Gietz RD, Pickart CM, Hayden MR (1996). Huntingtin is ubiquitinated and interacts with a specific ubiquitin-conjugating enzyme. J Biol Chem 271:19385–19394. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Jo DG, Hong GS, Kim BJ, Lai M, Cho DH, Kim KW, Bandyopadhyay A, Hong YM, Kim do H, Cho C, Liu JO, Snyder SH, Jung YK (2002). Calpain-dependent cleavage of cain/cabin1 activates calcineurin to mediate calcium-triggered cell death. Proc Natl Acad Sci USA 99:9870–9875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Yi Y, Sapp E, Wang Y, Cuiffo B, Kegel KB, Qin ZH, Aronin N, DiFiglia M (2001). Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci USA 98:12784–12789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klettner A, Herdegen T (2003). FK506 and its analogs—therapeutic potential for neurological disorders. Curr Drug Targets CNS Neurol Disord 2:153–162. [DOI] [PubMed] [Google Scholar]
- Landles C, Bates GP (2004). Huntingtin and the molecular pathogenesis of Huntington’s disease. Fourth in molecular medicine review series. EMBO Rep 5:958–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Yoshihara M, Littleton JT (2004). Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc Natl Acad Sci USA 101:3224–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li SH, Johnston H, Shelbourne PF, Li XJ (2000). Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet 25:385–389. [DOI] [PubMed] [Google Scholar]
- Li H, Wyman T, Yu ZX, Li SH, Li XJ (2003). Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum Mol Genet 12:2021–2030. [DOI] [PubMed] [Google Scholar]
- Lilienbaum A, Israel A (2003). From calcium to NF-kappa B signaling pathways in neurons. Mol Cell Biol 23:2680–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y (2002). Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell 10:259–269. [DOI] [PubMed] [Google Scholar]
- Luo S, Vacher C, Davies JE, Rubinsztein DC (2005). Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J Cell Biol 169:647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM (2001). Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell 104:675–686. [DOI] [PubMed] [Google Scholar]
- Mansuy IM (2003). Calcineurin in memory and bidirectional plasticity. Biochem Biophys Res Commun 311:1195–1208. [DOI] [PubMed] [Google Scholar]
- Mansuy IM, Mayford M, Jacob B, Kandel ER, Bach ME (1998). Restricted and regulated overexpression reveals calcineurin as a key component in the transition from short-term to long-term memory. Cell 92:39–49. [DOI] [PubMed] [Google Scholar]
- Mende-Mueller LM, Toneff T, Hwang SR, Chesselet MF, Hook VY (2001). Tissue-specific proteolysis of huntingtin (htt) in human brain: evidence of enhanced levels of N- and C-terminal htt fragments in Huntington’s disease striatum. J Neurosci 21:1830–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milakovic T, Johnson GV (2005). Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem 280:30773–30782. [DOI] [PubMed] [Google Scholar]
- Nakai A, Kuroda S, Kristian T, Siesjo BK (1997). The immunosuppressant drug FK506 ameliorates secondary mitochondrial dysfunction following transient focal cerebral ischemia in the rat. Neurobiol Dis 4:288–300. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P (2003). Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4:552–565. [DOI] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT (2002). Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci 5:731–736. [DOI] [PubMed] [Google Scholar]
- Pong K, Zaleska MM (2003). Therapeutic implications for immunophilin ligands in the treatment of neurodegenerative diseases. Curr Drug Targets CNS Neurol Disord 2:349–356. [DOI] [PubMed] [Google Scholar]
- Rangone H, Poizat G, Troncoso J, Ross CA, MacDonald ME, Saudou F, Humbert S (2004). The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur J Neurosci 19:273–279. [DOI] [PubMed] [Google Scholar]
- Ross CA (2002). Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron 35:819–822. [DOI] [PubMed] [Google Scholar]
- Rusnak F, Mertz P (2000). Calcineurin: form and function. Physiol Rev 80:1483–1521. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95:55–66. [DOI] [PubMed] [Google Scholar]
- Shenolikar S (1994). Protein serine/threonine phosphatases—new avenues for cell regulation. Annu Rev Cell Biol 10:55–86. [DOI] [PubMed] [Google Scholar]
- Singh A, Kumar G, Naidu PS, Kulkarni SK (2003). Protective effect of FK506 (tacrolimus) in pentylenetetrazol-induced kindling in mice. Pharmacol Biochem Behav 75:853–860. [DOI] [PubMed] [Google Scholar]
- Snyder SH, Sabatini DM, Lai MM, Steiner JP, Hamilton GS, Suzdak PD (1998). Neural actions of immunophilin ligands. Trends Pharmacol Sci 19:21–26. [DOI] [PubMed] [Google Scholar]
- Sola C, Tusell JM, Serratosa J (1999). Comparative study of the distribution of calmodulin kinase II and calcineurin in the mouse brain. J Neurosci Res 57:651–662. [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL (2004). SUMO modification of huntingtin and Huntington’s disease pathology. Science 304:100–104. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC (2003). Transcriptional abnormalities in Huntington’s disease. Trends Genet 19:233–238. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST (2003). Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 40:41–52. [DOI] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I (2003). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinas R, Kristal BS, Hayden MR, Bezprozvanny I (2005). Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc Natl Acad Sci USA 102:2602–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME (2000). Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 9:2799–2809. [DOI] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD II, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, Chacko C, McMurray CT (2004). Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol 24:8195–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC (1999). Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 284:339–343. [DOI] [PubMed] [Google Scholar]
- Warby SC, Chan EY, Metzler M, Gan L, Singaraja RR, Crocker SF, Robertson HA, Hayden MR (2005). Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum Mol Genet 14:1569–1577. [DOI] [PubMed] [Google Scholar]
- Wellington CL, Ellerby LM, Gutekunst CA, Rogers D, Warby S, Graham RK, Loubser O, van Raamsdonk J, Singaraja R, Yang YZ, Gafni J, Bredesen D, Hersch SM, Leavitt BR, Roy S, Nicholson DW, Hayden MR (2002). Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington’s disease. J Neurosci 22:7862–7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, Li ST, Moriwaki A, Matsui H (2004). Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem 279:4929–4940. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Uchino H, He QP, Li PA, Siesjo BK (2001). Cyclosporin A, but not FK506, prevents the downregulation of phosphorylated Akt after transient focal ischemia in the rat. Brain Res 899:148–158. [DOI] [PubMed] [Google Scholar]
- Young AB (2003). Huntingtin in health and disease. J Clin Invest 111:299–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA (2002). Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron 33:849–860. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E (2001). Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 293:493–498. [DOI] [PubMed] [Google Scholar]