

Abstract
The timing and strength of synaptic transmission is profoundly dependent on temperature. However, the temperature dependence of the multiple mechanisms that contribute to short-term synaptic plasticity is poorly understood. Here, we use voltage-clamp recordings to quantify the temperature dependence of exocytosis at the calyx of Held synapse. EPSC and miniature EPSC amplitudes were larger at physiological temperature, but quantal content during low-frequency (0.05 Hz) stimulation was constant after temperature jumps from 22–24°C to 35–37°C. The initial degree of EPSC depression during 100 Hz stimuli trains was unchanged with temperature, as were estimates of release probability and vesicle pool size. In contrast, physiological temperatures dramatically relieved depression measured after 40 stimuli at 100 Hz by increasing twofold the rate of recovery from depression. Presynaptic calyx recordings revealed that physiological temperature increased capacitance jumps resulting from 0.5 and 1 ms depolarizations by increasing Ca2+ influx. When Ca2+ entry was equalized at the two temperatures, exocytosis exhibited little temperature dependence for brief depolarizations. However, in response to longer depolarizations, raising temperature increased a slow phase of exocytosis, without affecting overall Ca2+ entry or the size of the readily releasable pool of vesicles. Higher temperatures also increased the rate of presynaptic Ca2+ current inactivation; nevertheless, the degree of steady-state EPSC depression was greatly reduced. Our results thus suggest that changes in steady-state EPSCs during stimulus trains at physiological temperature reflect larger quantal amplitudes and faster refilling of synaptic vesicle pools, leading to reduced short-term depression during prolonged high-frequency firing.
Keywords: auditory brainstem, development, calyx of Held, MNTB, exocytosis, glutamate release, short-term synaptic plasticity, AMPA receptor desensitization, presynaptic Ca2+ current
Introduction
The calyx of Held nerve terminal operates within a sound localization circuit (Grothe, 2003), and its giant size permits a direct study of presynaptic aspects of mammalian neurotransmission (Forsythe, 1994). Morphological and electrophysiological data obtained from this synapse have been used to successfully constrain models that account for its behavior over a broad range of experimental conditions (Weis et al., 1999; Meinrenken et al., 2002; Wong et al., 2003). However, these models generally rely on data obtained at ambient room temperature (RT), usually 12–15°C below physiological temperature (PT). Because many steps in excitation–secretion coupling may be temperature sensitive (Cao and Oertel, 2005), a detailed study of basic synaptic parameters closer to mammalian body temperature is needed to understand how synapses behave under physiological conditions.
When the calyx of Held is stimulated at high frequencies (e.g., 100 Hz), EPSCs exhibit marked short-term depression (Borst et al., 1995; von Gersdorff et al., 1997). Depletion of synaptic vesicle pools has emerged as a leading candidate mechanism for this form of short-term plasticity (Schneggenburger et al., 2002). It was thus surprising to find that during a brief stimulus train at 100 Hz, raising temperature increased the amplitude of the first EPSC while at the same time decreasing depression (Taschenberger and von Gersdorff, 2000). A uniform increase in quantal size does not explain this observation. Nor does increased release probability (pr), because vesicle pool depletion models predict greater depression with increased pr. Extensive effort has been made to understand short-term depression, and heterogeneous release probability among distinct vesicle pools has been invoked as a mechanism to explain deviations from simple depletion models (Sakaba and Neher, 2001a; Trommershauser et al., 2003). Likewise, we hypothesize that the differential effects of temperature at the beginning of a train and at a depressed steady state may reflect differential effects of temperature on different pools of vesicles or on specific phases of vesicular recycling.
Release probability and quantal size control EPSC amplitudes. At the calyx of Held, shifting temperature to near-physiological values slightly reduces the total Ca2+ charge entry during a single action potential (AP) (Borst and Sakmann, 1998), but corresponding variations of pr with temperature have not been measured. Previous electrophysiological studies examining the effect of temperature on pr at other CNS synapses have led to different results (Allen and Stevens, 1994; Hardingham and Larkman, 1998; Pyott and Rosenmund, 2002; Volgushev et al., 2004) and have not distinguished the effects of temperature on presynaptic Ca2+ entry from downstream effects on vesicle fusion. Recent imaging studies have shown that the kinetics of synaptic vesicle exocytosis and endocytosis is strongly temperature dependent (Yang et al., 2005), underscoring its fundamental importance for synaptic function. However, one study indicated higher temperatures slow exocytosis (Fernandez-Alfonso and Ryan, 2004), whereas another indicated a 2.6-fold faster rate of exocytosis (Micheva and Smith, 2005). Here, we used recordings of EPSCs, presynaptic Ca2+ current, and capacitance measurements to explore the multiple effects of temperature at the calyx of Held synapse.
Materials and Methods
Slice preparation.
Sprague Dawley rat pups (Charles River, Wilmington, MA) aged postnatal day 8 (P8) to P11 and P16–P18 were used in this study. After rapid decapitation, the brainstem was removed quickly from the skull and immersed in ice-cold (for P8–P11) or warm (for P16–P18) artificial CSF (aCSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 3 MgCl2, 0.2 CaCl2, 25 glucose, 25 NaHCO3, 1.25 NaHPO4, 0.4 ascorbic acid, 3 myo-inositol, and 2 Na-pyruvate, pH 7.3–7.5 when bubbled with carbogen (95% O2, 5% CO2) and osmolarity of 310–320 mOsm. Cyanoacrylic glue (World Precision Instruments, Sarasota, FL) was used to glue the brainstem to the stage of a vibratome slicer (VT1000; Leica, Bannockburn, IL), and 180- to 200-μm-thick transverse slices were made of the region containing the medial nucleus of the trapezoid body (MNTB). Slices were then transferred to an incubation chamber containing normal aCSF (P8–P11) or a low-sodium high-magnesium incubation aCSF (for P16–P18), bubbled with carbogen, maintained for 30–45 min at 35°C, and thereafter at RT (22–24°C). Normal aCSF was the same as slicing aCSF, but with 1 mm MgCl2 and 2 mm CaCl2. Incubation aCSF for P16–P18 contained (in mm) 85 NaCl, 7 MgCl2, and 0.5 CaCl2, to inhibit endogenous synaptic activity, and sucrose (75 mm) was added to maintain osmolarity (310–320 mOsm).
Electrophysiology.
Slices were placed in a 1 ml chamber perfused with normal aCSF at 1–3 ml/min and visualized using infrared-differential interference contrast microscopy (Leica) and a 40× water-immersion objective. EPSCs were stimulated with a bipolar platinum/iridium electrode (FHC, Bowdoinham, ME) placed near the midline spanning the afferent fiber tract of the MNTB. An Iso-Flex stimulator driven by a Master 8 pulse generator (A.M.P.I., Jerusalem, Israel) was used to deliver step pulses (100 μs, <15 V DC). EPSCs were recorded in normal aCSF containing d-APV (50 μm) and MK-801 ((+)-5-methyl-10, 11-dihydro-5H-dibenzo {a, d} cyclohepten-5, 10-imine maleate; 5 μm) to isolate AMPA receptor currents and strychnine (0.5 μm) to eliminate inhibitory transmission present, especially in older MNTB (Awatramani et al., 2004). Recordings of miniature EPSCs (mEPSCs) also contained tetrodotoxin (TTX; 1 mm) in some cases, although results were identical with or without TTX. The pipette internal solution for postsynaptic recordings contained the following (in mm): 130 Cs-gluconate, 10 CsCl, 5 Na2-phosphocreatine, 10 HEPES, 5 EGTA, 10 tetraethylammonium chloride (TEA-Cl), 4 Mg-ATP, and 0.3 GTP, pH adjusted to 7.3 with CsOH. Presynaptic recordings of capacitance and Ca2+ currents used a pipette solution of the following (in mm): 130 Cs-gluconate, 15 CsCl, 5 Na2-phosphocreatine, 10 HEPES, 0.2 EGTA, 20 TEA-Cl, 4 Mg-ATP, and 0.3 GTP, pH adjusted to 7.3 with CsOH. For presynaptic recordings, TEA (10 mm) and TTX (1 μm) were added to the bath aCSF to block voltage-activated potassium and sodium channel currents, respectively.
Pipettes were pulled from borosilicate glass (World Precision Instruments) with a Sutter P-97 electrode puller (Sutter Instruments, Novato, CA) to open tip resistances of 1.6–2.5 MΩ for postsynaptic recordings and 3.5–4.5 MΩ for presynaptic recordings. Access resistance (Rs) was ≤6 MΩ for postsynaptic recordings and ≤20 MΩ for presynaptic recordings. Rs was compensated >85% for postsynaptic recordings and ∼50% for presynaptic recordings. Principal cells were voltage clamped at a holding potential of −70 mV, and presynaptic terminals were held at −80 mV. Occasionally, the EPSC amplitude in older (P16–P18) principal cells exceeded −20 nA, saturating the recording amplifier. In these instances, the holding potential was changed to −40 mV to reduce the driving force, or the cells were rejected from analysis.
Capacitance recordings were calculated from a 1 kHz, 40 mV amplitude sine wave on the holding potential of −80 mV using the software lock-in capability of the EPC-9 amplifier (sine + DC method) (Gillis, 1995), except for 0.5 ms depolarizing pulses in which a 2 kHz sine wave was used. The reversal potential was assumed to be 0 mV. Membrane capacitance (Cm) was not measured during depolarizations, and 30–60 s were allowed between depolarizations to allow complete recovery of exocytosis. Baseline was measured for 100–1000 ms before depolarization and corrected for baseline drift in Cm. ΔCm was calculated after membrane conductance changes had settled (100–200 ms after the depolarization) (Yamashita et al., 2005). Presynaptic calcium currents (ICa) were leak subtracted using the P/n method.
Data was acquired at 10–25 μs sampling rate using an EPC-9 amplifier (HEKA Elektronik, Lambrecht, Germany) controlled by Pulse 8.4 software (Instrutech, Port Washington, NY) and filtered on-line at 2.9 kHz. Software was controlled by a Power Macintosh G3 or G4 computer (Apple Computers, Cupertino, CA). Data were analyzed off-line and presented using Igor Pro (WaveMetrics, Lake Oswego, OR). All traces for kinetic analysis and display were corrected off-line for holding potential and series resistance errors {see the methods of Schneggenburger et al. (1999)}. Data presented were the average of several (usually three to five) sweeps under the conditions described.
Depletion model and statistical analysis.
We assumed a pool of releasable vesicles from which each AP releases a fraction determined by the release probability pr and which refills between APs as a pseudo-first-order process (i.e., assuming an infinite “reserve” pool of vesicles) with rate constant r. Rather than fix the recovery rate constant at its value measured between trains, we allow for use-dependent acceleration of recovery between APs during a train, as proposed by Weis et al. (1999). However, different from the model by Weis et al. (1999), here we do not explicitly model Ca2+ in the terminal because, although there is solid evidence that a rise in Ca2+ concentration can trigger an acceleration of recovery rate (Wang and Kaczmarek, 1998), we note that higher temperature leads to slightly less Ca2+ influx per AP (Borst and Sakmann, 1998) and to a twofold increase in the rate of Ca2+ extrusion (Helmchen et al., 1997). Thus, the changes we report at physiological temperature are unlikely to be caused by an increase in residual Ca2+. Quantal size may become depressed during 100 Hz trains in younger animals as a result of postsynaptic desensitization or saturation (Scheuss et al., 2002; Wong et al., 2003). In the older animals used here, AMPA receptor desensitization and saturation during 100 Hz trains are greatly reduced (Taschenberger et al., 2002; Renden et al., 2005), and thus we assumed constant quantal size. The normalized EPSC amplitudes, E(i), are then given by the following: E(i) = pr × N(i); i = 1, 2, …, where N(i) is the fraction remaining of the releasable vesicle pool just before the ith AP, and pr is the vesicle release probability.
The N(i) is then given by the following recursive relationship: N(i + 1) = N(i) × (1 − pr) + {N(1) − N(i) × (1− pr)} × {1 − exp(−r Δt)}, where N(1) = 1 by definition, and Δt = 10 ms, the interval between APs at 100 Hz. Note that this equation for N(i + 1) has two parts: a depletion term, N(i) × (1 − pr), and a first-order recovery term, {N(1) − N(i) − (1 − pr)} − {1 − exp(−r Δt)}. This model was programmed in Igor software (WaveMetrics). To determine the two free parameters, we adjusted pr to fit the first three data points and adjusted r to fit the observed steady-state degree of depression.
Curve fitting shown in Figure 8 was achieved using Igor. Statistical comparison of parameters was achieved based on the SD of these parameters calculated by the nonlinear curve fitting routines in Igor. Statistical comparison was made by calculating the overlap integral of normal distributions, the mean and SD of which were determined by the best-fit parameter values and its SD, respectively.
Figure 8.
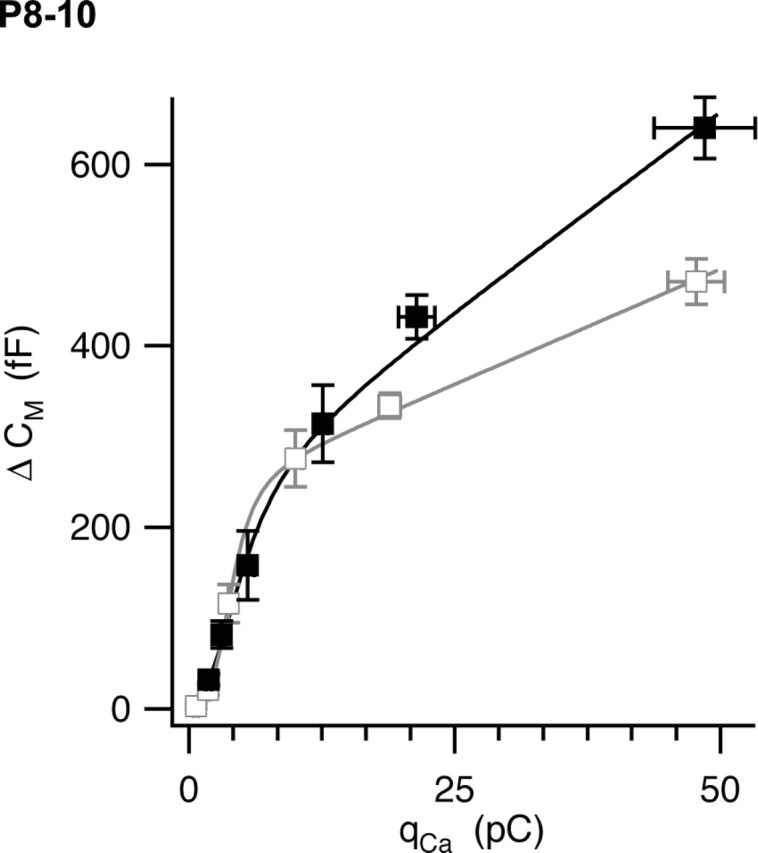
Ca2+–exocytosis coupling in the calyx of Held at room temperature (open symbols; gray) and physiological temperature (filled symbols; black). The calyx was depolarized under voltage clamp for variable times ranging from 0.5 to 30 ms. Resulting capacitance jumps (ΔCm) were plotted against the integral of the Ca2+ current (qCa). Error bars represent SEM (n = 9–25 measurements per point). The solid curves represent fits of a model equation described in Results.
Drugs and reagents.
All salts were purchased from Sigma (St. Louis, MO). All pharmacological reagents were purchased from Tocris Cookson (Ellisville, MO). Tetrodotoxin was purchased from Alomone Labs (Jerusalem, Israel).
Results
Effects of temperature on EPSC amplitude, kinetics, and short-term depression
We studied presynaptic and postsynaptic changes that occur at the synapse formed by the calyx of Held as temperature was changed from RT (22–24°C) to near-PT (35–37°C). All reported effects of temperature on the presynaptic AP, EPSC size, kinetics, depression, and recovery from depression reversed fully after return to RT.
Raising temperature to PT resulted in a reduction of the presynaptic calyceal AP amplitude and half-width, as illustrated in Figure 1A. This confirms the findings of Borst and Sakmann (1998), who report an average AP amplitude change from 117 to 110 mV and a half-width reduction from 0.54 to 0.26 ms. These changes in AP waveform with temperature are similar to those observed in the squid giant axon (Hodgkin and Katz, 1949) and in neurons of the cochlear nucleus (Cao and Oertel, 2005). At fixed temperature, modulation of AP half-width alters EPSC amplitude by changing presynaptic Ca2+ entry (Augustine, 1990) (for review, see Sabatini and Regehr, 1999). The situation is more complicated when the waveform of the AP changes in response to temperature jumps, because compensatory changes in Ca2+ channel kinetics may occur and it is thus difficult to predict the effect of reduced AP half-width on neurotransmission under these conditions.
Figure 1.

The presynaptic action potential (A), EPSC (B), and average mEPSC (C) recorded in the MTNB from a P10 animal at RT (gray) or PT (black). The data shown in B and C were recorded from the same cell. The mEPSCs shown in C are the average of 74 and 231 miniature events recorded at room temperature or physiological temperature, respectively.
To explore the effect of temperature changes on glutamate release, we measured EPSC amplitude and charge integrals at the two temperatures (Fig. 1B). The amplitude of EPSCs was increased at PT by 75 ± 15% (Table 1). Raising the temperature also accelerated EPSC kinetics. The EPSC 10–90% rise time decreased by 39 ± 2%, and the half-width of the EPSC was shortened by 49 ± 3%. The onset of the EPSC after the stimulus artifact also occurred earlier at PT, which reflects an earlier triggering of calyx AP (as shown in Fig. 1A) and thus results in a decrease in synaptic delay with higher temperature, as documented previously at this synapse (Borst et al., 1995; Taschenberger and von Gersdorff, 2000).
Table 1.
Effects of temperature at the calyx of Held synapse
| Parameter | Average across cells
|
Paired |
||
|---|---|---|---|---|
| RT | PT | Percentage change | p (paired t test) | |
| P8–P10 | ||||
| EPSC (N = 9) | ||||
| Amplitude (nA) | −8.0 ± 1.26 | −13.8 ± 1.9 | 75 ± 15 | <0.001 |
| 10–90% rise time (ms) | 0.33 ± 0.025 | 0.22 ± 0.016 | −39 ± 2 | <0.001 |
| Half-width (ms) | 1.2 ± 0.12 | 0.71 ± 0.073 | −49 ± 3 | <0.001 |
| Charge (pC) | −16 ± 2.8 | −16 ± 2.5 | 0 ± 10 | >0.05 |
| Quantal content | 279 ± 50 | 284 ± 44 | 4.0 ± 9.2 | >0.05 |
| mEPSC (N = 9) | ||||
| Frequency (s−1) | 4.7 ± 1.3 | 9.3 ± 1.8 | 167 ± 38 | <0.05 |
| Quantal size (pA) | −53.1 ± 7.5 | 83.3 ± 12.3 | 62 ± 10 | <0.001 |
| 10–90% rise time (ms) | 0.30 ± 0.04 | 0.21 ± 0.02 | −26 ± 4 | <0.001 |
| Half-width (ms) | 0.69 ± 0.09 | 0.46 ± 0.06 | −32 ± 3 | <0.001 |
| Charge (fC) | −57 ± 9.3 | −57 ± 8.7 | 5 ± 7 | >0.05 |
| Depression (100 Hz; N = 9) | ||||
| Paired-pulse ratio | 0.53 ± 0.10 | 0.52 ± 0.07 | 7 ± 11 | >0.05 |
| Steady-state EPSC (nA) | −0.31 ± 0.08 | −1.8 ± 0.49 | 486 ± 82 | <0.001 |
| Steady-state depression (%) | 95 ± 3.3 | 84 ± 3.8 | −12 ± 4 | <0.001 |
| qSteady-state (pC) | 1.1 ± 0.28 | 3.0 ± 0.78 | 198 ± 25 | <0.001 |
| Recovery from depression (N = 10) | ||||
| Percentage fast | 16.7 ± 2.0 | 34.1 ± 4.5 | 127 ± 30 | <0.05 |
| Rate (s−1) | 0.61 ± 0.09 | 1.2 ± 0.14 | 104 ± 21 | <0.01 |
| P16–P18 | ||||
| EPSC (N = 5) | ||||
| Amplitude (nA) | −8.6 ± 1.9 | −14.5 ± 3.0 | 82 ± 35 | <0.05 |
| Charge (pC) | −7.1 ± 2.0 | −6.2 ± 1.5 | −6 ± 12 | >0.05 |
| mEPSC (N = 3) | ||||
| Quantal size (pA) | −62 ± 16 | −88 ± 15 | 51 ± 18 | <0.05 |
| Charge (fC) | −34 ± 8.6 | −29 ± 4.7 | −10 ± 9 | >0.05 |
| Depression (100 Hz; N = 5) | ||||
| Paired-pulse ratio | 0.83 ± 0.08 | 0.77 ± 0.03 | −5 ± 7 | >0.05 |
| Steady-state EPSC (nA) | −1.4 ± 0.43 | −4.3 ± 1.1 | 219 ± 45 | <0.01 |
| Steady-state depression (%) | 83 ± 2.8 | 70 ± 2.5 | −15 ± 2 | <0.01 |
| qSteady-state (pC) | 2.4 ± 0.61 | 3.7 ± 0.94 | 58 ± 21 | <0.05 |
| Refilling rate (model fit; s−1) | 4.3 ± 0.29 | 10.9 ± 0.8 | 157 ± 20 | <0.01 |
For each parameter, the mean ± SEM value taken across cells is given for each temperature condition (RT and PT). On the right, we calculated the average percentage change of each parameter after raising temperature from RT to PT based on a paired comparison within each cell.
We next examined spontaneous mEPSCs to determine to what extent the observed changes in evoked EPSCs can be explained by changes in the underlying quantal currents. As illustrated in Figure 1C, the effect of temperature on mEPSC amplitude and kinetics was similar to the effects on evoked EPSCs (Table 1). The amplitude of mEPSCs increased by 62 ± 10%, and the rise time and half-width were shortened by 26 ± 4 and 32 ± 3.5%, respectively (mean data from nine cells; 27–348 mEPSCs per cell at each temperature, RT and PT). Thus, a large part of the effects of temperature on the size and shape of EPSCs is attributable to corresponding changes in postsynaptic quantal currents, suggesting that only modest changes in the probability and time course of vesicle release occur when temperature is varied from RT to PT. Raising temperature also caused a large (170 ± 40%) increase of mEPSC frequency.
To compare quantal content at RT and PT, we calculated the ratio of the amplitudes and charge integrals of EPSCs to that of mEPSCs at these two temperatures. Despite the significant increases in EPSC and mEPSC amplitudes resulting from increased temperature, the total charge entry during EPSCs or mEPSCs was constant (Table 1). Dividing the mean EPSC charge by the mean quantal charge gives an estimate of the content of the EPSC, which averaged 280 quanta at both temperatures tested. This value is higher than reported previously (Schneggenburger et al., 1999). However, our estimate was calculated using the total charge of the EPSC, integrated until the current returned to baseline. We note that in calyxes of this age group (P8–P10), the decay of the EPSC contained a prominent slow component lasting several milliseconds. We observed little difference in this component of the EPSC at RT and PT, suggesting it could be caused by slow diffusion of glutamate from the synaptic cleft (Sakaba and Neher, 2001b), because aqueous diffusion has low temperature dependence (Finkelstein, 1987). Alternatively, this current could reflect asynchronous release, because it is absent from mEPSCs (Fig. 1C).
We included the slow component of the EPSC in our estimate of quantal content because, although such current does not contribute to the peak amplitude of the EPSC, it would be relevant to the physiological situation in which the MNTB neuron is not voltage clamped (Trommershauser et al., 2003), and if it represents asynchronous release, it is relevant to considerations of depression resulting from vesicle pool depletion (Otsu et al., 2004). We also calculated quantal content as the ratio of EPSC to mEPSC amplitudes. Temperature increased the amplitude of EPSCs and mEPSCs to the same extent (difference not statistically significant) (Table 1, Fig. 1B, C). Thus, quantal content during low-frequency stimulation, measured as the ratio of either amplitudes or charges of EPSCs to mEPSCs, was unchanged across temperature jumps from RT to PT.
EPSCs display strong short-term depression in response to high-frequency stimulation, yet the synapse formed by the calyx of Held is able to maintain transmission at remarkably high frequencies even at RT (von Gersdorff and Borst, 2002). As temperature is raised, transmission improves further still, and at near-body temperature, the rat calyx of Held synapse can follow brief trains of stimuli at up to 800 Hz (Taschenberger and von Gersdorff, 2000). To explore temperature-dependent changes in high-frequency transmission, we next examined the response to long trains of stimuli at 100 Hz (Fig. 2). Surprisingly, the effect of temperature at steady-state conditions during such trains was very different from that of the first few EPSCs of the train.
Figure 2.

Synaptic depression recorded in P8–P10 MNTB neurons during stimulation of the calyx of Held with 100 Hz trains. A, B, The first five EPSCs (A) or last five EPSCs (B) recorded from a P10 MNTB neuron during a train of 50 stimuli at RT (gray) or PT (black). In B, stimulus artifacts were removed for clarity. C, Summary (n = 9 cells) of EPSC amplitudes (C1) or scaled amplitudes (C2) during 100 Hz stimulus train at room temperature (open symbols; gray) or physiological temperature (filled symbols; black). The error bars represent the SEM. In C2, EPSC amplitudes for each cell were scaled to the first EPSC in the respective train, and then mean and SEM of the normalized data set was calculated.
As shown in Figure 2, in response to 100 Hz stimuli, EPSCs recorded in the P8–P10 MNTB depressed rapidly during the first several stimuli and then reached an apparent steady-state by the 10th stimuli that continued through at least the 50th stimuli. Figure 2A illustrates a typical response to 100 Hz stimuli during the first five EPSCs of a train. When the temperature was raised, the amplitude of the first EPSC increased, as described above, but initial depression of the EPSC during the train was similar at RT or PT. In contrast, the steady-state response recorded at the end of the train (e.g., stimuli 46–50) was greatly increased at PT compared with RT (Fig. 2B). Thus, although initial depression was similar at RT and PT, the steady-state output of the synapse was increased greatly by raising temperature. A similar effect of temperature on steady-state EPSCs also occurs at other synapses (Brenowitz et al., 1998; Pyott and Rosenmund, 2002).
The differential effect of temperature on EPSC amplitudes during a train can be seen clearly in the summary data (mean ± SEM) presented in Figure 2C. Raising temperature increased the amplitude of the first EPSC but also raised the steady-state output. To separate these phenomena, we normalized data from individual cells to the first EPSC in the train (Fig. 2C2). The paired-pulse ratio (ratio of the second to the first EPSC) was unchanged by raising temperature (Table 1), suggesting little effect on release probability during the initial phase of depression. The steady-state EPSC during a 100 Hz train at RT decayed to <4% of initial EPSC amplitude (Table 1). Raising temperature markedly reduced this depression, resulting in a steady-state EPSC amplitude of >12% of the initial EPSC. Comparing the effect of temperature on the first EPSC and at the end of a train, we observed that raising temperature to PT increased the amplitude of the steady-state EPSC by 490 ± 83%, or 6.5-fold larger than the effect of PT on the first EPSC (Fig. 2A). As mentioned above, raising temperature also lead to EPSCs with faster kinetics. To measure the effect of temperature on steady-state charge per EPSC, we averaged 30 steady-state responses from each cell (stimuli 20–50) and calculated the charge integral. Increasing temperature from RT to PT increased steady-state charge per EPSC by 198%.
Effect of temperature on recovery from short-term depression
The data above suggest that although the initial pr of vesicles was unchanged with temperature, going from RT to PT greatly increases steady-state release recorded late during a train. In vesicle depletion models that include pool refilling during the train (Weis et al., 1999), the amount of steady-state release depends on the rate of vesicle pool refilling. The increase in steady-state output we observed at PT could thus reflect accelerated refilling kinetics compared with RT. To test this, we measured recovery from short-term depression by applying two trains (20 stimuli at 100 Hz) separated by a variable recovery time (ranging from 0.1 to 13 s). After each pair of trains, the calyx was allowed to recover for 20 s, sufficient for complete recovery from short-term depression (von Gersdorff et al., 1997; Billups et al., 2005) (Fig. 3). No over-filling of the vesicle pool was observed in these experiments (Dinkelacker et al., 2000).
Figure 3.
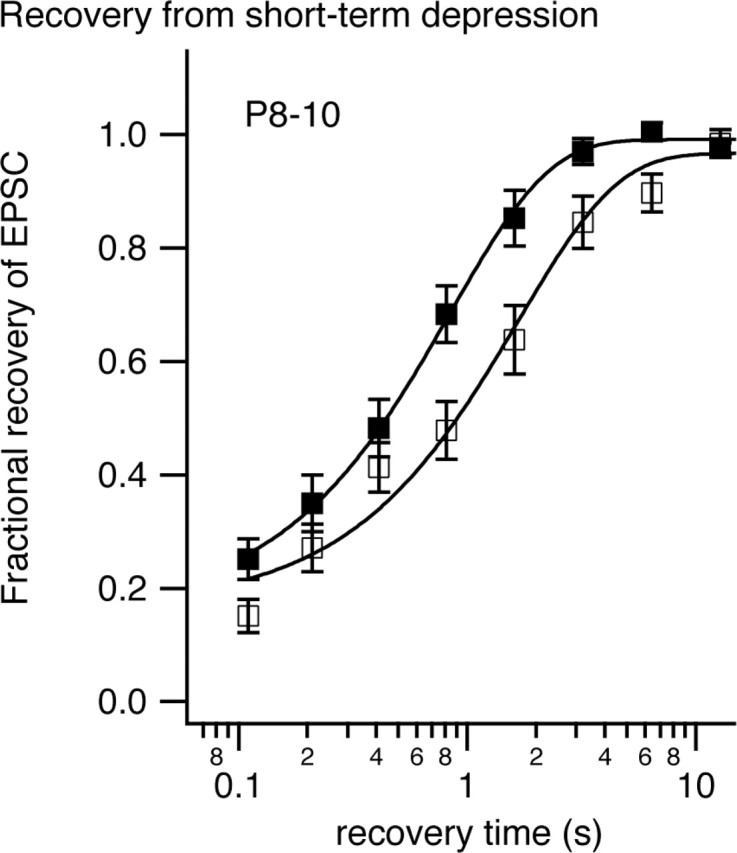
Recovery from synaptic depression recorded in P8–P10 MNTB neurons at room temperature (open symbols) or physiological temperature (filled symbols). A conditioning train of 20 stimuli at 100 Hz was delivered followed by a variable recovery time before a test stimuli was delivered. The interval between successive conditioning trains was 20 s. Fractional recovery was calculated as follows: (A − Ass)/(Ao − Ass), where A is the amplitude of the test EPSC after a given recovery time, Ao is the amplitude of the first EPSC in the conditioning train, and Ass is the steady-state EPSC amplitude at the end of the conditioning train. The solid curves represent exponential fits to the data. Error bars represent SEM.
Recovery from depression occurred in two phases. A fast phase, which occurred within 100 ms, may represent resensitization of AMPA receptors (Otis et al., 1996) or a Ca-dependent fast phase of recovery (Wang and Kaczmarek, 1998) or preferential recovery of a subset of vesicles (Trommershauser et al., 2003). The fraction of recovery that occurred during this fast phase increased from 15 to 25% when temperature was changed to PT (Table 1). The remainder of the recovery followed an exponential time course that was accelerated 2.1-fold when temperature was raised to PT (rate constants of 0.64 s−1 at RT and 1.34 s−1 at PT). Recovery was 97–98% complete at 3.2 s in PT but took 12.8 s in RT. Higher temperatures thus accelerate the recovery from short-term depression. This result is similar to that reported by Pyott and Rosenmund (2002) for recovery of the pool of vesicles releasable by hypertonic solution.
AMPA receptor desensitization does not explain the effects of temperature
Raising temperature could decrease postsynaptic depression, because AMPA receptor desensitization contributes to short-term depression at this synapse during high-frequency stimulation in MNTB from P8–P13 rats (Wong et al., 2003). Furthermore, glutamate transporters operate more efficiently at physiological temperatures, and they could influence the degree of AMPA receptor desensitization. To test this possibility, we compared the effect of temperature on depression in control and in the presence of 4 mm γ-d-glutamylglycine (γ-DGG). This rapidly dissociating AMPA receptor antagonist is thought to protect a large fraction of the total number of receptors from entering a desensitized state (Wong et al., 2003). γ-DGG effectively makes the synapse resistant to saturation and desensitization (Wadiche and Jahr, 2001).
Figure 4 summarizes results from six cells from P8–P10 animals challenged with increased temperature in control aCSF and in the presence of γ-DGG. In the controls for this set of experiments, 20 stimuli at 100 Hz stimulation reduced EPSC amplitude to 6.1 ± 1.1% of initial amplitude at RT, and steady-state depression in control aCSF was reduced at PT (steady-state EPSC amplitude at PT, 23 ± 3.9% of initial amplitude; an increase of 17% of initial amplitude over RT). In the presence of γ-DGG, the first EPSC in a train was reduced by 94 ± 1.1% at RT and 81 ± 2.0% at PT. As reported previously, in this condition, EPSCs exhibited marked initial facilitation followed by depression (Wong et al., 2003). The extent of depression at RT with γ-DGG was less than in control (steady-state EPSC amplitude at RT with γ-DGG, 14 ± 1.8% of initial amplitude), consistent with previous studies and reflecting a decrease in postsynaptic depression (Wong et al., 2003; Renden et al., 2005). Despite reduced postsynaptic depression in the presence of γ-DGG, raising temperature decreased depression further (steady-state EPSC amplitude at PT with γ-DGG, 31 ± 4% of initial amplitude; an increase of 17% over γ-DGG steady-state at RT). The increases in steady-state EPSC amplitude obtained either by raising temperature or relieving postsynaptic depression with γ-DGG were thus additive, and therefore reduction in AMPA receptor desensitization cannot explain the reduced depression seen at PT.
Figure 4.

Protecting AMPA receptors from desensitization with 4 mm γ-DGG does not antagonize effects of temperature on steady-state depression in MNTB from P8–P10 animals. The mean normalized EPSC amplitudes (n = 6 cells) recorded in either normal aCSF (open symbols; ctrl) or in the presence of 4 mm γ-DGG (filled symbols; DGG) and at RT (squares) or PT (circles) are shown. Error bars represent SEM. Stim, Stimulus.
Temperature effects at more mature P16–P18 synapses
Several studies have shown that the contribution of postsynaptic mechanisms to short-term depression in the MNTB changes during the first 3 weeks of postnatal development (Joshi and Wang, 2002; Taschenberger et al., 2005). Low-affinity AMPA receptor antagonists (e.g., kynurenate and γ-d-glutamylglycine) relieve AMPA receptor desensitization (Wong et al., 2003), and the sensitivity of short-term depression (100 Hz trains of stimuli) to γ-d-glutamylglycine is lost by P16–P18 (Renden et al., 2005), suggesting that in these older animals, the contribution of AMPA receptor desensitization to depression is greatly reduced. We thus reasoned that recordings from older animals (P16–P18) would allow us to examine better the effects of temperature on presynaptic components of depression and may shed light on the physiology of the more mature synapse. In addition, unlike P8–P10 synapses, P16–P18 calyxes show very little synaptic inhibition via metabotropic glutamate receptors (Renden et al., 2005), thus simplifying an analysis of the mechanisms of short-term depression.
As shown in Figure 5A–C, responses to trains and corresponding changes with temperature in recordings from P16–P18 animals were qualitatively similar to those from P8–P10 animals (Fig. 2). Initial EPSC amplitudes were the same in the two age groups (Taschenberger and von Gersdorff, 2000), as were increases in initial EPSC amplitude when temperature was raised from RT to PT (Table 1). The extent of steady-state depression was decreased at RT in these older animals, consistent with a reduction in postsynaptic components of depression. The paired-pulse ratio was also higher, again indicating less depression and suggesting that release probability may be lower in the older animals. As in the P8–P10 group (Fig. 2), recording at physiological temperature increased steady-state EPSC amplitude but did not change the paired-pulse ratio of the first two EPSCs (Table 1).
Figure 5.
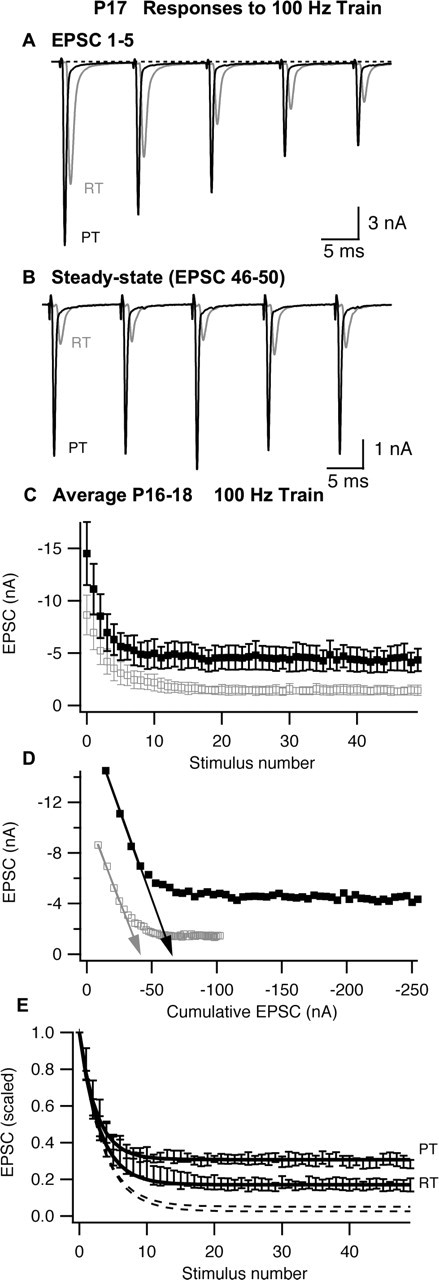
Synaptic depression recorded in P16–P18 MNTB neurons during stimulation of the calyx of Held with 100 Hz trains. A, B, The first five EPSCs (A) or last five EPSCs (B) recorded from a P17 MNTB neuron during a train of 50 stimuli at RT or PT. In B, stimulus artifacts were removed for clarity. C, Summary (n = 5 cells) of EPSC amplitudes. D, Determination of vesicle pool size and release probability by linear extrapolation (arrows) of EPSC amplitude versus cumulative EPSC amplitude. E, Normalized EPSC amplitudes. The dashed lines represent model predictions (see Materials and Methods) based on vesicle refilling rates obtained from measurements of recovery from synaptic depression (see Fig. 3) at RT or PT, and the solid lines represent model predictions when the recovery rate was adjusted to fit observed steady-state depression. EPSC amplitudes for each cell were scaled to the first EPSC in the respective train, and the mean and SEM of the normalized data set were calculated. In C and D, the open gray symbols represent data recorded at room temperature, and filled black symbols represent data recorded at physiological temperature; in E, symbols were omitted for clarity. Error bars represent SEM.
We calculated the charge integral of EPSCs obtained from P16–P18 animals as described above for P8–P10. In these older animals, the EPSC decayed rapidly, and the slow current that was prevalent at P8–P10 was mostly absent (compare Figs. 2A, 5A). This may be attributable to improved clearance of glutamate because, as the calyx terminal matures, its morphology changes from a cup-like structure to a series of stalks and stems (Kandler and Friauf, 1993; Rowland et al., 2000). These finger-like extensions may favor rapid diffusion of glutamate (Taschenberger et al., 2005). Moreover, the kinetics of EPSCs were faster at P16–P18 compared with P8–P10. Faster EPSC kinetics and the absence of slow current after the EPSC resulted in EPSC charge that was significantly smaller in P16–P18 animals compared with P8–P10 but independent of temperature (Table 1). In these older animals, mEPSC frequency was very low at RT, and we could not always obtain a reliable measure of quantal size in every cell. In cells from which we could obtain sufficient numbers of mEPSCs at both RT and PT, quantal charge was not affected by temperature (three cells; 21–67 mEPSCs per cell). Quantal content was also quantified based on EPSC and mEPSC amplitudes. The effect of temperature on EPSC and mEPSC amplitudes was not significantly different. Thus, similar to results obtained in P8–P10 animals, lack of effect of temperature on EPSC charge and quantal charge suggests no change in quantal content and, together with the lack of change in paired-pulse ratio, suggest little change in release probability.
We next examined changes in the number of vesicles available for release at the beginning of a train. We calculated the cumulative EPSCs (EPSCcum) for subsequent stimuli in the train, using the method of Elmqvist and Quastel (1965). We restricted analysis to the first four EPSCs, in which the relationship between EPSC amplitude versus cumulative EPSC was linear (Fig. 5D). Extrapolation of this portion of the data permitted calculation of an effective total EPSCcum available at the beginning of the train. As shown in Figure 5D, the extrapolated EPSCcum increased by 61% when temperature was raised from RT to PT. To test whether facilitation of release probability (from the second EPSC on) distorted this result, we also analyzed changes in pool size estimates obtained in the same manner but omitting the first EPSC. The percentage of effects of temperature on EPSCcum calculated by these two methods was not different when applied to the averaged data (Fig. 5D) nor when each cell was analyzed separately (data not shown). The increase in EPSCcum size with temperature was similar to that observed for quantal size in P8–P10 animals (62% increase) or P16–P18 animals (51% increase), suggesting little or no change in the number of vesicles available for release at the beginning of a train that could explain the large decrease in depression that occurs later in the train when temperature is raised to physiological levels.
Normalized data from P16–P18 animals (Fig. 5E) show that although raising temperature did not affect initial depression, it greatly decreased depression at the end of the train, raising normalized steady-state ESPC amplitude by 219%. The increased steady-state synaptic output was much larger than the effect of temperature on the initial EPSC, suggesting that raising temperature specifically increases the steady-state rate at which vesicles are released from the calyx. The curves in Figure 5E represent the prediction of a simple model whereby each AP depletes a constant fraction, pr, of available vesicles and between each AP the pool of available vesicles refills as a first-order process with rate r (see Materials and Methods). The parameter pr was set to 0.25 to fit the first three EPSCs. The dashed lines in Figure 5E show the model prediction when the parameter r was set to 0.64 and 1.34 s−1, which are the recovery rates between trains after 100 Hz stimulation at RT and PT respectively (Fig. 3, Table 1). As has been shown previously for stimulation at 10 Hz (von Gersdorff et al., 1997; Weis et al., 1999), this model predicts much stronger depression than is actually observed. We then adjusted r to fit the observed steady-state depression (solid lines). To obtain an adequate fit to initial depression and steady-state amplitude, it was necessary to raise r to 4.3 s−1at RT and to 10.9 s−1 at PT (Fig. 5, solid lines). Thus, recovery of the EPSC between each stimulus during a train was much faster than between trains (Fig. 3) and was more than twofold faster at PT than RT.
The simple model used to fit the data in Figure 5E assumes constant pr and refilling rates, both of which determine steady-state depression. However, both of these parameters may change during the course of the train. For example, facilitation (Wong et al., 2003) and Ca-dependent acceleration of recovery rates (Wang and Kaczmarek, 1998) have been reported at the calyx of Held. Nevertheless, we do not expect that neglecting facilitation affects our conclusion, because facilitation was not temperature dependent either when observed directly after unmasking with γ-DGG (Fig. 4) or inferred by the lack of change of paired-pulse ratio in MNTB from either P8–P10 or P16–P18 animals (Table 1). Likewise, acceleration of the refilling rates during the train do not affect the main conclusion that refilling was increased twofold at PT, because the refilling rate reported was obtained by adjusting to the steady-state portion of the curve where the refilling rate is presumably constant. It is unknown whether the high rate of recovery of EPSCs during a train represents an acceleration of the basal recovery mechanism active between trains or additional recovery mechanisms that operate in parallel. In the later case, the parameter r determined here would represent the sum of rate constants for all processes contributing to recovery from depression of the EPSC.
Effect of temperature on Cm jumps: short depolarizations
We examined the effects of temperature on presynaptic capacitance jumps resulting from short depolarizations of 0.5–1 ms, with the intent of simulating the physiologically relevant calcium influx equivalent to one or two action potentials at RT or PT in P8–P10 rats. For these short depolarizations, effects of temperature on Ca2+ channel kinetics dominated. After a 0.5 ms depolarization, essentially no capacitance jump was recorded at RT (2.9 ± 2.8 fF; n = 10) despite measurable ICa, whereas after raising temperature, the much larger ICa gave rise to a robust capacitance jump of 32 ± 6.2 fF (n = 10), as illustrated in Figure 6 and Table 2.
Figure 6.
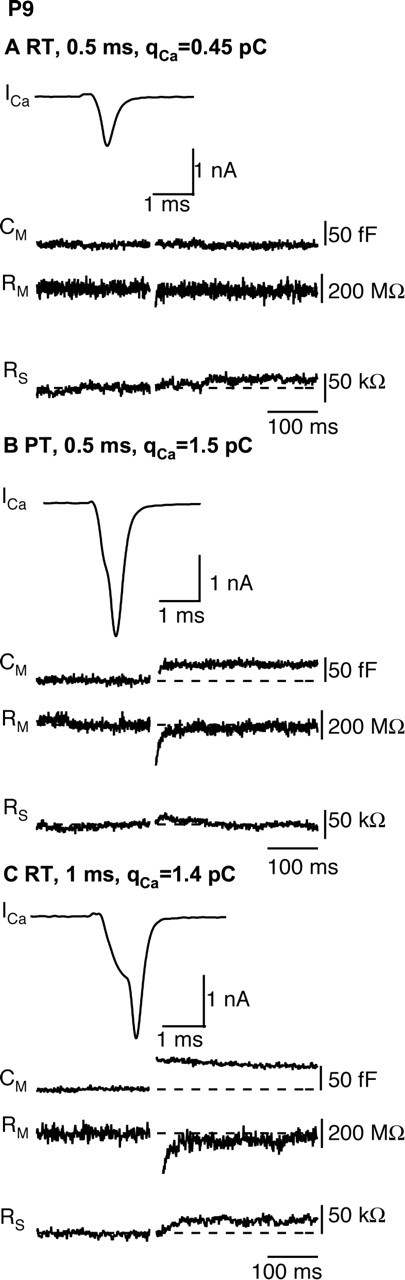
Temperature dependence of exocytosis during brief depolarizations. A, B, A 0.5 ms depolarization to 0 mV evokes a capacitance jump at physiological temperature but not at room temperature. Voltage-clamp recordings of ICa, Cm, membrane resistance (Rm), and series resistance (RS) in response to 0.5 ms depolarization to 0 mV at room temperature (A) or physiological temperature (B) are shown. C, A 1.0 ms depolarization at room temperature generates robust ICa and Cm jump. The data in A–C were obtained from the same calyx of Held.
Table 2.
Capacitance jumps for P8–P10 calyxes in response to depolarizations that give similar Ca entry at RT and PT
| Parameter | RT | PT | p (t test) |
|---|---|---|---|
| Brief depolarizations (1 ms RT, 0.5 ms PT) | |||
| qCa (pC) | 1.7 ± 0.13 (n = 13) | 1.62 ± 0.13 (n = 9) | >0.05 |
| ΔCm (fF) | 21 ± 5.4 (n = 15) | 32 ± 6.2 (n = 9) | >0.05 |
| ΔCm/ qCa (fF/pC) | 16 ± 3.2 (n = 12) | 20 ± 3.4 (n = 9) | >0.05 |
| Long depolarizations (10 and 30 ms) | |||
| 10 ms depolarization | |||
| qCa (pC) | 19 ± 1.0 (n = 25) | 21 ± 1.7 (n = 17) | >0.05 |
| ΔCm (fF) | 335 ± 14 (n = 25) | 432 ± 24 (n = 17) | <0.01 |
| 30 ms depolarization | |||
| qCa (pC) | 48 ± 3 (n = 25) | 49 ± 5 (n = 16) | >0.05 |
| ΔCm (fF) | 471 ± 25 (n = 25) | 640 ± 34 (n = 16) | <0.01 |
| Steady-state exocytosis | |||
| ΔCm 30 ms − ΔCm 10 ms (fF) | 148 ± 22 (n = 25) | 233 ± 21 (n = 12) | <0.05 |
This apparent threshold effect could reflect cooperativity between calcium entry (qCa) and exocytosis or a temperature dependence on exocytosis downstream of calcium entry. To determine whether factors other than increased calcium entry contributed to the increase in capacitance jump at higher temperature, we devised voltage protocols that resulted in similar calcium entry at RT and PT (Fig. 6B, C). Specifically, we depolarized the calyx to 0 mV for 1 ms at RT and 0.5 ms at PT. The resultant calcium influxes were 1.73 ± 0.13 pC (n = 13) and 1.62 ± 0.13 pC (n = 9) at RT and PT, which agree with previous reports for 1 ms depolarizations of the calyx at RT (Taschenberger et al., 2002) and are approximately two times the calcium entry that occurs during a single AP (Borst and Sakmann, 1998). With these protocols, capacitance jumps were not statistically different (21 ± 5.4 fF at RT and 32 ± 6.2 fF at PT). We further quantified this result by normalizing the capacitance jump to the corresponding calcium charge entry (integral of ICa) in that cell. At RT, normalized capacitance jumps were 16 ± 3.2 fF/pC (n = 12), whereas at PT, the value was 20 ± 3.4 fF/pC (n = 9). Thus, at PT, there was a tendency for larger capacitance jumps per picocoulomb of Ca entry. However, this difference is not statistically significant (p > 0.4) and is quite small (25% change) given the rather large temperature step from RT to PT. Thus, for brief depolarizations, exocytosis is not very sensitive to temperature once effects of temperature on Ca2+ influx are taken into account. Raising temperature thus increases release probability during these short depolarizing pulses via an increase in Ca2+ influx.
We considered the possibility that raising temperature changed the specific capacitance of neuronal cell membranes, usually taken to be ∼1 μF/cm2. We thus measured the calyx resting capacitance just before and just after a temperature jump from RT to PT. For 10 calyces studied with this protocol, membrane capacitance was 20 ± 1.5 pF at RT and 21 ± 1.6 pF at PT (p > 0.05).
Effect of temperature on Cm jumps: long depolarizations
As described above, raising temperature greatly increased the steady-state EPSC amplitude during 100 Hz trains. To test whether this reflects increased exocytosis at higher temperature, we examined capacitance jumps resulting from 10 and 30 ms step depolarizations to 0 mV. Previous reports indicate that a 10 ms depolarization of the calyx depletes the releasable pool of vesicles (Wu and Borst, 1999; Sun and Wu, 2001). We thus reasoned that the difference in capacitance jumps in response to 10 and 30 ms depolarizations would reflect exocytosis of vesicles recruited during this period in response to vesicle pool depletion and would give a measure of the rate of vesicle pool refilling. Raising temperature increased peak ICa by 23 ± 6% but also increased the extent of inactivation during a 30 ms depolarization from 39 ± 2.7% at RT to 50 ± 3.1% at PT, as illustrated in Figure 7A. Because of these antagonistic effects of temperature, and in contrast to the effect of temperature on calcium entry during short depolarizations, total calcium charge entry during prolonged depolarizations was not temperature dependent. Mean values of calcium entry during 10 and 30 ms depolarizations at RT were 19 ± 1 and 48 ± 3 pC, respectively. The corresponding values at PT were 21 ± 1.7 and 49 ± 5 pC. Thus, any effects of temperature on exocytosis during these long depolarizations should most likely be downstream of calcium entry.
Figure 7.
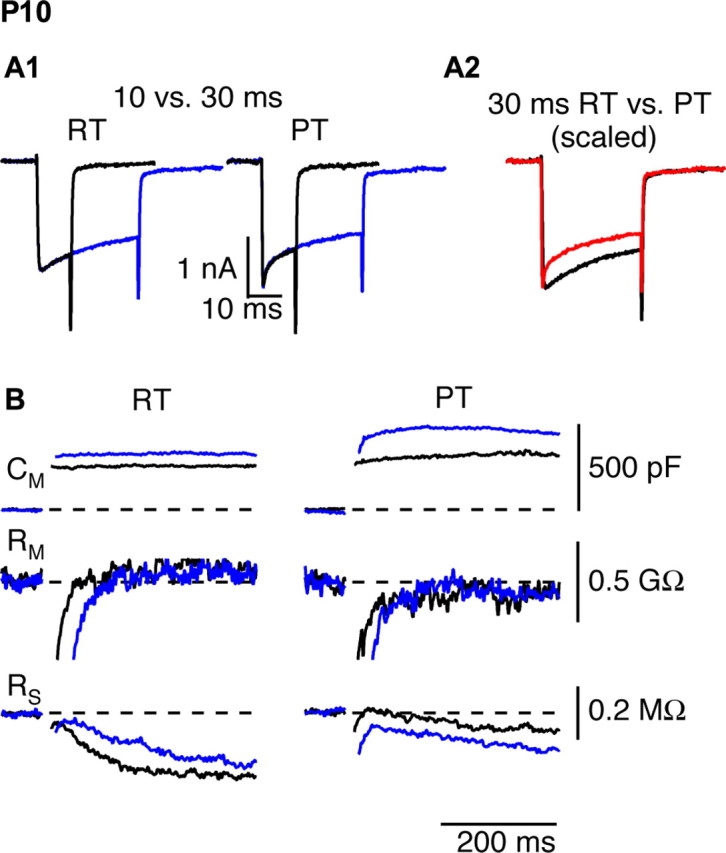
The late phase of exocytosis during prolonged depolarizations is temperature sensitive. The calyx was depolarized for 10 ms (black) or 30 ms (blue) from −80 to 0 mV at room temperature (left) or physiological temperature (right). A, Presynaptic Ca2+ currents. A2, Ca2+ current inactivation was more pronounced at PT (red) than at RT (black), as seen after scaling the currents by their peak amplitudes. The activation kinetics of the Ca2+ current was faster at PT. B, Changes in Cm, membrane resistance (Rm), and series resistance (RS) in response to 10 and 30 ms depolarizations at RT and PT {depolarizing step pulses for 10 ms (black) or 30 ms (blue) as shown in A1}.
In all calyces examined, we observed a greater capacitance jump after a 30 ms depolarization compared with a 10 ms depolarization in the same cell (Fig. 7B, Table 2); this result did not depend on the order in which the depolarizations were delivered. During a sustained depolarization at RT, exocytosis thus continued beyond 10 ms, which we propose represents a correlate of steady-state EPSC output during a prolonged train of stimuli. At RT, 10 and 30 ms depolarizations resulted in capacitance jumps of 335 ± 14 and 471 ± 25 fF, respectively. At PT, the corresponding jumps were 432 ± 24 and 640 ± 36 fF. The mean difference in exocytosis generated by 10 and 30 ms depolarizations in the same cell was 148 ± 22 fF at RT and was significantly higher at PT (233 ± 21 fF; p < 0.05). Therefore, similar to the effect of temperature on steady-state EPSC output during an afferent fiber train, raising temperature increased a late phase of exocytosis during sustained stimulation.
Efficiency of exocytosis at RT versus PT
We varied the duration of depolarization from 0.5 to 30 ms and measured the resulting calcium-dependent capacitance jumps at RT and PT (Fig. 8). The relationship between qCa and resulting capacitance jump was well fit by a Hill equation for short-duration stimuli. For longer stimuli (5, 10, and 30 ms), capacitance jumps were linear with calcium entry. We therefore modeled the data as a fixed-size readily releasable pool (RRP) of vesicles that were released according to the Hill equation, plus a linear phase as new vesicles become available for release, as follows:
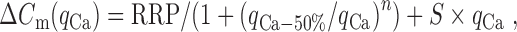 |
where qCa-50% is the calcium entry required to release 50% of the pool, n is the Hill coefficient for cooperativity between calcium entry and exocytosis for the RRP, and S is the slope of the late phase of release. The slope of the late linear phase of release was 5.1 ± 0.3 fF/pC at RT and increased to 8.7 ± 1.12 fF/pC at PT. This correlates to an increase in the linear phase of release from 7.5 ± 0.78 fF/ms at RT to 12.3 ± 2.2 fF/ms at PT. In contrast, the estimate of the readily releasable pool was unchanged at the two temperatures (RT, RRP = 231 ± 10 fF; PT, RRP = 221 ± 35 fF). However, there was a small increase in the value of the parameter qCa-50% from 4.0 ± 0.1 pC at RT to 5.1 ± 0.4 pC at PT and also a statistically significant (p < 0.02) decrease in the Hill coefficient, from 3.9 ± 0.45 at RT to 2.6 ± 0.56 at PT (Fedchyshyn and Wang, 2005). These results suggest that synaptic vesicles can be mobilized more quickly to release sites at PT. A similar proposal has been made for dense core granules from capacitance measurements in pancreatic β-cells (Renstrom et al., 1996) and for synaptic vesicles in inner hair cells (Johnson et al., 2005). More direct evidence for temperature-dependent synaptic vesicle movement has been reported recently in cultured hippocampal neurons (Shtrahman et al., 2005).
Discussion
We examined quantal size, quantal content, vesicle pool size and refilling kinetics, and release probability at the calyx of Held to understand why raising temperature decreases steady-state depression while simultaneously increasing the amplitude of the first EPSC in a train. To separate presynaptic and postsynaptic mechanisms of depression, we adopted three different experimental approaches: (1) protection of AMPA receptors from desensitization with γ-DGG, (2) comparison of results obtained from P8–P10 rats with those from P16–P18 rats (an age at which AMPA receptor desensitization is absent for 100 Hz trains), and (3) direct assay of presynaptic exocytosis via Cm measurements. All three approaches pointed to a presynaptic locus for the decrease in steady-state depression when temperature is raised, because of an increase in the rate of vesicle pool refilling. Surprisingly, temperature had very little effect on release probability after a rest period.
Single EPSCs and the temperature sensitivity of pr
Changes to EPSCs evoked at 0.05 Hz, a stimulus frequency that does not generate short-term depression (von Gersdorff et al., 1997), were closely mirrored by corresponding changes in underlying quantal currents, with little effect on total charge entry during either event, and hence little effect on quantal content. According to quantal theory, the lack of effect of temperature on quantal content implies the product of release probability and releasable pool size remained constant. We examined the effect of temperature on pr and N separately using EPSCs recorded during 100 Hz trains of stimuli. We observed that the initial descent into short-term depression was unchanged, suggesting little effect of temperature on pr. These results imply that the supply of vesicles available during this initial phase of a train is insensitive to temperature.
We used changes in membrane capacitance, a measurement of exocytosis that does not depend on a postsynaptic response, to determine the effect of temperature on vesicle release as a function of presynaptic Ca2+ influx. Although not all Cm jumps necessarily reflect secretion (Xu et al., 1998), at the calyx of Held, there is good correlation between EPSC size and Cm jump if AMPA receptor saturation is avoided (Xu and Wu, 2005). A step depolarization to 0 mV lasting 1 ms at RT or 0.5 ms at PT generated similar qCa values at the two temperatures (Table 2), equivalent to approximately twice the Ca2+ entry that occurs during a single AP (Borst and Sakmann, 1998). These stimulus protocols generated robust Cm jumps at either RT or PT. Exocytosis per unit calcium entry at PT was also statistically indistinguishable from that at RT. Thus, we conclude that once temperature effects on Ca2+ entry are accounted for, exocytosis evoked by very brief depolarizations has very little intrinsic dependence on temperature.
The lack of effect of temperature on exocytosis (elicited by AP stimulation or short voltage-clamp depolarization of the calyx) was in marked contrast to the effect of temperature on the frequency of spontaneous mEPSCs. Similar to the neuromuscular junction (Fatt and Katz, 1952), we observed that the frequency of spontaneous vesicle release at the calyx of Held was highly temperature dependent. The low rate, and high temperature dependence, of synaptic vesicle exocytosis at rest suggests a large energy barrier for this spontaneous process at resting levels of intraterminal Ca2+. After a presynaptic AP at the calyx of Held, exocytosis is accelerated ∼105-fold, because hundreds of synaptic vesicles are released within 1 ms (Bollmann et al., 2000; Schneggenburger and Neher, 2000). A lack of temperature effect on exocytosis after an AP suggests that calcium entry removes essentially all energy barriers for vesicle fusion.
Steady-state EPSCs and vesicle pool size
Raising temperature reduced EPSC depression during trains of 50 stimuli. In P16–P18 animals, in which postsynaptic mechanisms of depression are greatly reduced (Renden et al., 2005), steady-state EPSC size during a train increased more than twofold when temperature was raised from RT to PT. Steady-state EPSC charge was also elevated at PT, and given the temperature independence of quantal charge (Table 1), this indicates an increase in steady-state quantal content. In P8–P10 animals, the effect of temperature on steady-state depression was more pronounced than at P16–P18. Our experiments with γ-DGG suggest that this difference is not caused by relief of AMPA receptor desensitization at physiological temperature. We speculate it may be caused in part by a more synchronized release at PT in P8–P10 animals, as a result of a faster presynaptic AP (Fig. 1A), whereas release may already be highly synchronized at P16–P18, so the margin for improvement by physiological temperature is somewhat reduced.
We used 10 and 30 ms depolarizations of the calyx to measure steady-state exocytosis as Cm jumps, a presynaptic correlate to steady-state output during a prolonged high-frequency train. The values of qCa elicited by these stimuli were equivalent to 20 and 50 times the qCa caused by a single presynaptic AP, respectively. These qCa values also did not change from RT to PT (in part because of faster ICa inactivation at PT) (Forsythe et al., 1998). Because membrane potential does not directly affect release from the calyx of Held (Felmy et al., 2003), steady-state exocytosis may be compared with EPSC charge between the 20th and 50th stimuli of a high-frequency train. Steady-state exocytosis was substantially increased at PT, but to a lesser extent than steady-state EPSC charge in P8–P10 animals. In contrast, EPSC recordings from P16–P18 animals revealed that the increase in steady-state charge during a train was very similar to the increase in steady-state exocytosis seen with capacitance measurements. We used measurements of EPSC charge instead of amplitude for comparison to Cm jump data, because charge measurements do not depend on the synchrony of release. However, the slow component of the EPSC observed in P8–P10 animals may result in an overestimate of release if it is caused by slow diffusion of glutamate from the cleft. The absence of this slow current at P16–P18 may explain why EPSC charge correlates better with capacitance measurements in these more mature animals.
One caveat to our γ-DGG experiments is the difference in blocking efficiency of this drug at RT and PT. We consistently observed slightly less inhibition of the EPSC by 4 mm γ-DGG when measured in the same cell at PT versus RT. However, we do not expect this fact to affect our conclusions, because in all cases inhibition was >80%, which is sufficient to protect AMPA receptors at this synapse from desensitization (Wong et al., 2003).
Although we observed an increase in the capacitance jump resulting from a 10 ms depolarization at PT, we do not interpret this as an increase in the pool of releasable vesicles. Rather, our results suggest that steady-state exocytosis contributes to the jump after a 10 ms depolarization and that a more accurate estimate of initial pool size must take steady-state exocytosis into account, especially at PT. Using this method, we estimate a releasable vesicle pool size of ∼230 fF at both RT and PT, significantly smaller than reported previously (Sun and Wu, 2001). For 50-nm-diameter synaptic vesicles (Sätzler et al., 2002; Taschenberger et al., 2002), this capacitance increase corresponds to the fusion of 2500–3000 vesicles, similar to the estimates of Sakaba and Neher (2001b).
Temperature-dependent exocytosis at other synapses
It was shown previously that calcium charge influx elicited by AP waveforms under voltage clamp is reduced at higher temperature (0.92 pC at RT vs 0.81 pC at PT; P8–P10) (Borst and Sakmann, 1998). Although EPSCs recorded at the calyx of Held vary with the third or higher power of calcium influx (Schneggenburger et al., 1999), measurements herein of quantal content, paired-pulse ratio, or analysis of cumulative EPSCs failed to provide evidence for reduced pr at PT. ICa in the immature calyx of Held enters through different calcium channel types, with differential coupling to glutamate release (Iwasaki and Takahashi, 1998; Wu et al., 1999). One possibility is that calcium entry through channels tightly coupled to release may be spared more when total qCa is slightly reduced at PT. Additionally, raising temperature decreased the cooperativity in coupling between calcium influx and EPSC size (Fig. 8) (Fedchyshyn and Wang, 2005), and the reduced qCa–EPSC relationship may explain why we observed little to no reduction of pr when temperature was increased.
At cultured hippocampal neurons, vesicle pool size remained unchanged and the refilling rate measured with hypertonic solution increased threefold from 25 to 35°C, whereas quantal content decreased slightly (Pyott and Rosenmund, 2002). In contrast, studies in cortical synapses have concluded that pr increases at >32°C (Hardingham and Larkman, 1998; Volgushev et al., 2004). Synapses in different locations in the brain may thus behave differently to increased temperature. We found that pr and the initial vesicle pool size do not change from RT to PT, but the rate of refilling increases twofold, leading to reduced depression. The calyx of Held synapse is specialized for high-speed relay of auditory information (Carr et al., 2001). Mechanisms underlying rapid vesicle recruitment at physiological temperature contribute to this goal by maintaining the supply of readily releasable vesicles during high-frequency firing.
Footnotes
*C.K. and R.R. contributed equally to this work.
This work was supported by grants from the National Institutes of Health–National Institute on Deafness and Other Communication Disorders and the Human Frontier Science Program (H.v.G.) and by a National Research Service Award postdoctoral fellowship (R.R.). C. K. was supported by the Conselho de Aperfeiçoamento de Pessoal de Nível Superior (Brazil), Conselho Nacional de Desenvolvimento Científico e Tecnológico (Brazil), and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (Brazil).
References
- Allen C, Stevens CF (1994). An evaluation of causes for unreliability of synaptic transmission. Proc Natl Acad Sci USA 91:10380–10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine G (1990). Regulation of transmitter release at the squid giant synapse by presynaptic delayed rectifier potassium current. J Physiol (Lond) 431:343–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awatramani GB, Turecek R, Trussell LO (2004). Inhibitory control at a synaptic relay. J Neurosci 24:2643–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billups B, Graham BP, Wong AY, Forsythe ID (2005). Unmasking group III metabotropic glutamate autoreceptor function at excitatory synapses in the rat CNS. J Physiol (Lond) 565:885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollmann JH, Sakmann B, Borst JG (2000). Calcium sensitivity of glutamate release in a calyx-type terminal. Science 289:953–957. [DOI] [PubMed] [Google Scholar]
- Borst JG, Sakmann B (1998). Calcium current during a single action potential in a large presynaptic terminal of the rat brainstem. J Physiol (Lond) 506:143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Helmchen F, Sakmann B (1995). Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J Physiol (Lond) 489:825–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz S, David J, Trussell L (1998). Enhancement of synaptic efficacy by presynaptic GABAB receptors. Neuron 20:135–141. [DOI] [PubMed] [Google Scholar]
- Cao X-J, Oertel D (2005). Temperature affects voltage-sensitive conductances differentially in octopus cells of the mammalian cochlear nucleus. J Neurophysiol 94:821–832. [DOI] [PubMed] [Google Scholar]
- Carr CE, Soares D, Parameshwaran S, Perney T (2001). Evolution and development of time coding systems. Curr Opin Neurobiol 11:727–733. [DOI] [PubMed] [Google Scholar]
- Dinkelacker V, Voets T, Neher E, Moser T (2000). The readily releasable pool of vesicles in chromaffin cells is replenished in a temperature-dependent manner and transiently overfills at 37°C. J Neurosci 20:8377–8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmqvist D, Quastel DMJ (1965). A quantitative study of the end plate potentials in isolated human muscle. J Physiol (Lond) 178:505–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatt P, Katz B (1952). Spontaneous subthreshold activity at motor nerve endings. J Physiol (Lond) 117:109–128. [PMC free article] [PubMed] [Google Scholar]
- Fedchyshyn MJ, Wang LY (2005). Developmental transformation of the release modality at the calyx of Held synapse. J Neurosci 25:4131–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmy F, Neher E, Schneggenburger R (2003). The timing of phasic transmitter release is Ca2+-dependent and lacks a direct influence of presynaptic membrane potential. Proc Natl Acad Sci USA 100:15200–15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Alfonso T, Ryan TA (2004). The kinetics of synaptic vesicle pool depletion at CNS synaptic terminals. Neuron 41:943–953. [DOI] [PubMed] [Google Scholar]
- Finkelstein A (1987). Water movement through lipid bilayers, pores, and plasma membranes: theory and reality. In: Distinguished lecture series of the society of general physiologists Vol 4:New York: Wiley. [Google Scholar]
- Forsythe ID (1994). Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS, in vitro. J Physiol (Lond) 479:381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Banes-Davies M, Cuttle MF, Takahashi T (1998). Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron 20:797–807. [DOI] [PubMed] [Google Scholar]
- Gillis KD (1995). Techniques for membrane capacitance measurements. In: Single channel recording (Sakmann B, Neher E, eds) , pp. 155–198. New York: Plenum.
- Grothe B (2003). New roles for synaptic inhibition in sound localization. Nat Rev Neurosci 4:540–550. [DOI] [PubMed] [Google Scholar]
- Hardingham NR, Larkman AU (1998). The reliability of excitatory synaptic transmission in slices of rat visual cortex in vitro is temperature dependent. J Physiol (Lond) 507:249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Borst JGG, Sakmann B (1997). Calcium dynamics associated with a single action potential in a CNS presynaptic terminal. Biophys J 72:1458–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Katz B (1949). The effect of temperature on the electrical activity of the giant axon of the squid. J Physiol (Lond) 109:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T (1998). Developmental changes in calcium channel types mediating synaptic transmission in rat auditory brainstem. J Physiol (Lond) 509:419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Marcotti W, Kros CJ (2005). Increase in efficiency and reduction in Ca2+ dependence of exocytosis during development of mouse inner hair cells. J Physiol (Lond) 563:177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi I, Wang LY (2002). Developmental profiles of glutamate receptors and synaptic transmission at a single synapse in the mouse auditory brainstem. J Physiol (Lond) 540:861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandler K, Friauf E (1993). Pre- and postnatal development of efferent connections of the cochlear nucleus in the rat. J Comp Neurol 328:161–184. [DOI] [PubMed] [Google Scholar]
- Meinrenken CJ, Borst JG, Sakmann B (2002). Calcium secretion coupling at calyx of Held governed by nonuniform channel-vesicle topography. J Neurosci 22:1648–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Smith SJ (2005). Strong effects of subphysiological temperature on the function and plasticity of mammalian presynaptic terminals. J Neurosci 25:7481–7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis T, Zhang S, Trussell LO (1996). Direct measurement of AMPA receptor desensitization induced by glutamatergic synaptic transmission. J Neurosci 16:7496–7504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu Y, Shahrezaei V, Li B, Raymond LA, Delaney KR, Murphy TH (2004). Competition between phasic and asynchronous release for recovered synaptic vesicles at developing hippocampal autaptic synapses. J Neurosci 24:420–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyott SJ, Rosenmund C (2002). The effects of temperature on vesicular supply and release in autaptic cultures of rat and mouse hippocampal neurons. J Physiol (Lond) 539:523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renden R, Taschenberger H, Puente N, Rusakov DA, Duvoisin R, Wang L-Y, Lehre KP, von Gersdorff H (2005). Glutamate transporter studies reveal the pruning of metabotropic glutamate receptors and absence of AMPA receptor desensitization at mature calyx of Held synapses. J Neurosci 25:8482–8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renstrom E, Eliasson L, Bokvist K, Rorsman P (1996). Cooling inhibits exocytosis in single mouse pancreatic B-cells by suppression of granule mobilization. J Physiol (Lond) 494:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland KC, Irby NK, Spirou GA (2000). Specialized synapse-associated structures within the calyx of Held. J Neurosci 20:9135–9144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG (1999). Timing of synaptic transmission. Annu Rev Physiol 61:521–542. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E (2001a). Preferential potentiation of fast-releasing synaptic vesicles by cAMP at the calyx of Held. Proc Natl Acad Sci USA 98:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E (2001b). Quantitative relationship between transmitter release and calcium current at the calyx of Held synapse. J Neurosci 21:462–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sätzler K, Sohl LF, Bollmann JH, Borst JG, Frotscher M, Sakmann B, Lubke JH (2002). Three-dimensional reconstruction of a calyx of Held and its postsynaptic principal neuron in the medial nucleus of the trapezoid body. J Neurosci 22:10567–10579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuss V, Schneggenburger R, Neher E (2002). Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPSCs at the calyx of held synapse. J Neurosci 22:728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E (2000). Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406:889–893. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E (1999). Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23:399–409. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E (2002). Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci 25:206–212. [DOI] [PubMed] [Google Scholar]
- Shtrahman M, Yeung C, Nauen DW, Bi G-q, Wu X-l (2005). Probing vesicle dynamics in single hippocampal synapses. Biophys J 89:3615–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JY, Wu LG (2001). Fast kinetics of exocytosis revealed by simultaneous measurements of presynaptic capacitance and postsynaptic currents at a central synapse. Neuron 30:171–182. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H (2000). Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci 20:9162–9173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, Leão RM, Rowland KC, Spirou GA, von Gersdorff H (2002). Optimizing synaptic architecture and efficiency for high-frequency firing. Neuron 36:1127–1143. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Scheuss V, Neher E (2005). Release kinetics, quantal parameters and their modulation during short-term depression at a developing synapse in the rat CNS. J Physiol (Lond) 568:513–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trommershauser J, Schneggenburger R, Zippelius A, Neher E (2003). Heterogeneous presynaptic release probabilities: functional relevance for short-term plasticity. Biophys J 84:1563–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volgushev M, Kudryashov I, Chistiakova M, Mukovski M, Niesmann J, Eysel UT (2004). Probability of transmitter release at neocortical synapses at different temperatures. J Neurophysiol 92:212–220. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Borst JGG (2002). Short-term plasticity at the calyx of Held. Nat Rev Neurosci 3:53–64. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Schneggenburger R, Weis S, Neher E (1997). Presynaptic depression at a calyx synapse: the small contribution of metabotropic glutamate receptors. J Neurosci 17:8137–8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE (2001). Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron 32:301–313. [DOI] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK (1998). High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394:384–388. [DOI] [PubMed] [Google Scholar]
- Weis S, Schneggenburger R, Neher E (1999). Properties of a model of Ca2+-dependent vesicle pool dynamics and short-term synaptic depression. Biophys J 77:2418–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AY, Graham BP, Billups B, Forsythe ID (2003). Distinguishing between presynaptic and postsynaptic mechanisms of short-term depression during action potential trains. J Neurosci 23:4868–4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Borst JG (1999). The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron 23:821–832. [DOI] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JG, Catterall WA, Sakmann B (1999). Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci 19:726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wu LG (2005). The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron 46:633–645. [DOI] [PubMed] [Google Scholar]
- Xu T, Binz T, Niemann H, Neher E (1998). Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat Neurosci 1:192–200. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Hige T, Takahashi T (2005). Vesicle endocytosis requires dynamin-dependent GTP hydrolysis at a fast CNS synapse. Science 307:124–127. [DOI] [PubMed] [Google Scholar]
- Yang XF, Ouyang Y, Kennedy BR, Rothman SM (2005). Cooling blocks rat hippocampal neurotransmission by a presynaptic mechanism: observations using 2-photon microscopy. J Physiol (Lond) 567:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]