

Abstract
Estrogen receptor β (ERβ) and androgen receptor (AR) are found in high levels within populations of neurons in the hypothalamus. To determine whether AR or ERβ plays a role in regulating hypothalamo–pituitary–adrenal (HPA) axis function by direct action on these neurons, we examined the effects of central implants of 17β-estradiol (E2), 5α-dihydrotestosterone (DHT), the DHT metabolite 5α-androstan-3β, 17β-diol (3β-diol), and several ER subtype-selective agonists on the corticosterone and adrenocorticotropin (ACTH) response to immobilization stress. In addition, activation of neurons in the paraventricular nucleus (PVN) was monitored by examining c-fos mRNA expression. Pellets containing these compounds were stereotaxically implanted near the PVN of gonadectomized male rats. Seven days later, animals were killed directly from their home cage (nonstressed) or were restrained for 30 min (stressed) before they were killed. Compared with controls, E2 and the ERα-selective agonists moxestrol and propyl-pyrazole-triol significantly increased the stress induced release of corticosterone and ACTH. In contrast, central administration of DHT, 3β-diol, and the ERβ-selective compound diarylpropionitrile significantly decreased the corticosterone and ACTH response to immobilization. Cotreatment with the ER antagonist tamoxifen completely blocked the effects of 3β-diol and partially blocked the effect of DHT, whereas the AR antagonist flutamide had no effect. Moreover, DHT, 3β-diol, and diarylpropionitrile treatment significantly decreased restraint-induced c-fos mRNA expression in the PVN. Together, these studies indicate that the inhibitory effects of DHT on HPA axis activity may be in part mediated via its conversion to 3β-diol and subsequent binding to ERβ.
Keywords: hypothalamo–pituitary–adrenal axis, estrogen receptor, hypothalamus, dihydrotestosterone, stress, rat
Introduction
The hypothalamo–pituitary–adrenal (HPA) axis is the major neuroendocrine axis responding to stress. Stressful events, actual or perceived, activate neurons within the paraventricular nucleus (PVN) of the hypothalamus, which results in the enhanced synthesis and secretion of hypothalamic neuropeptides (Spiess et al., 1981). The major secretogogues regulating the HPA axis are corticotropin-releasing hormone (CRH) and arginine vasopressin. These neuropeptides can subsequently act alone or in concert to stimulate the synthesis and release of adrenocorticotropin hormone (ACTH) from anterior pituitary corticotrophs (Spiess et al., 1981; Muglia et al., 1994, 2000; Whitnall et al., 1985). ACTH drives adrenal corticosterone hormone secretion. In turn, HPA axis activity is terminated by a negative feedback loop in which the major inhibitory tone comes from circulating corticosterone (De Kloet et al., 1983; Reul and de Kloet, 1985; Reul et al., 1987).
There exists a sex difference in HPA function attributable, in part, to circulating sex steroid hormones (Kornstein, 1997; Wilhelm et al., 1998a; Viau and Meaney, 1991; Burgess and Handa, 1992; Handa et al., 1994b; Suzuki et al., 2001; Lund et al., 2004a). When stressed, adult female rodents display a more robust HPA response than adult males (Gaskin and Kitay, 1970; Viau and Meaney, 1991; Burgess and Handa, 1992; Handa et al., 1994b; Lund et al., 2004a). It appears that in males, androgens inhibit, whereas in females, estrogens function to enhance (Handa et al., 1994a, b; Viau and Meaney, 1996; Lund et al., 2004a), the activity of the HPA axis.
Within the PVN of both male and female rats, androgen receptor (AR) (Huang and Harlan, 1994; Zhou et al., 1994) and estrogen receptor β (ERβ) are found in relatively high levels (Hrabovszky et al., 1998; Somponpun and Sladek, 2003). However, the role that these receptors play in regulating PVN function are not well established. Interestingly, the PVN is essentially devoid of ERα (Shughrue et al., 1997a; Suzuki and Handa, 2005).
ERβ possesses a relative binding affinity (RBA) for several compounds that different from that of ERα. The RBAs of propyl-pyrazole-triol (PPT) and moxestrol are several-fold greater for ERα than for ERβ, whereas the binding to ERβ relative to ERα is greater for diarylpropionitrile (DPN) (Kuiper et al., 1998; Stauffer et al., 2000; Meyers et al., 2001; Harris et al., 2002; Sun et al., 2002). In addition, ERβ also binds the metabolite of 5α-dihydrotestosterone (DHT), 5α-androstane-3β, 17β-diol (3β-diol) (Kuiper et al., 1998). The binding of 3β-diol to ERβ can activate transcription with a potency equivalent to that of 17β-estradiol (E2) (Pak et al., 2005), and in the prostate gland, this binding is sufficient to regulate growth (Weihua et al., 2002).
To examine the regulatory influence that AR and ERβ might exert on PVN neurons of the male rat in controlling the gain of the HPA axis, we implanted pellets containing steroid hormones or selective ER agonists directly above the PVN. Subsequently, the corticosterone and ACTH response to immobilization stress was measured and correlated with changes in the activity of PVN neurons as monitored by examining c-fos mRNA expression.
Materials and Methods
Animals.
Male Sprague Dawley rats (250–300 g) were obtained from Charles River Laboratories (Wilmington, MA). Animals were caged in pairs and housed in the Colorado State University laboratory animal research facility and maintained on a 12 h light/dark schedule (lights on at 7:00 A.M.) with ad libitum access to food and water. The Animal Care and Use Committee at Colorado State University approved all animal protocols.
Bilateral cannulation of the PVN.
One week after arrival, animals were gonadectomized (GDX) after anesthetization with ketamine (100 mg/ml), xylazine (100 mg/ml), and acepromazine (10 mg/ml). After gonadectomy, rats were fitted bilaterally with two 22 gauge stainless-steel cannulas (Small Parts, Miami Lakes, FL) with the aid of a small animal stereotaxic instrument. The tips of the cannulas were packed previously with one of the following compounds: DHT (Steraloids, Newport, RI), E2, 5α-androstane-3β, 17β-diol (3β-diol), moxestrol (Sigma, St. Louis, MO), PPT, or DPN (Tocris Cookson, Ellisville, MO), dissolved into warmed beeswax (VWR International, Bristol, CT) to a final concentration of 0.5 μm, and packed to a height of 2 mm within the end of the cannulas. This concentration and packed height were determined empirically by implanting a wax pellet containing radiolabeled [125I]E2 to establish acceptable/optimal spread of compound (see below, Verification of steroid diffusion and cannula placement). Controls received cannulas packed with wax alone.
Compounds used in these studies were specifically chosen based on their unique binding characteristics. E2 binds with similar affinity to both ERα and ERβ. The RBA of PPT and moxestrol is several-fold greater for ERα than for ERβ, whereas the binding to ERβ relative to ERα is greater for DPN (Table 1) (Kuiper et al., 1998; Meyers et al., 2001; Sun et al., 2002). Additionally, 3β-diol also binds ERβ (Kuiper et al., 1998), is a potent modulator of ERβ mediated transcription (Pak et al., 2005), and can have physiological actions in prostate (Weihua et al., 2001, 2002). Table 1 lists these compounds and their binding to ERs relative to E2.
Table 1.
Binding affinities of selected compounds for ERα and ERb
| Compound |
Ki (nm) |
|
|---|---|---|
| ERα | ERβ | |
| E2 | 0.12 | 0.15 |
| DHT | 221 | 73 |
| 3β-Diol | 6 | 1.7 |
| DPN | 195 | 2.5 |
| PPT | 0.50 | 700 |
| Moxestrol | 0.50 | 2.6 |
| 4-OH-tamoxifen | 0.09 | 0.04 |
Binding affinities (Ki values in nm) of estrogen receptor subtype-selective ligands compared with 17β-estradiol. The values were determined by competitive binding assay using 100 μl aliquots of rabbit reticulocyte lysate supernatant incubated at optimal time and temperature: 90 min at room temperature (ERβ) and 18 h at 4°C (ERα), with increasing (0.01–50 nm) concentrations of [3H]estrogen.
Stereotaxic coordinates to allow placement of the cannula tip to the region just above the PVN were based on previous work (Glass et al., 2000) and modified to accommodate a lateral 10° insertion angle to 1.9 mm posterior and 2.0 mm lateral to bregma and 7.0 mm below the skull surface; trephining was accomplished using a Dremel (Mount Prospect, IL) Moto-Flex tool. A 28 gauge stainless-steel wire (Small Parts), cut to extend 1 mm past the length of the cannulas, was inserted into the cannulas and the pellet expelled. The cannulas were then retracted and the surgery incision closed. For all cannulations, the incisor bar was set at 0.5 mm above the ear bars. Animals were killed 7 d after stereotaxic surgery. To establish the capacity by which androgens exert their effects locally and under more normal conditions, the approach of delivering androgen receptor antagonist directly into the PVN in gonadal-intact animals was initially considered; however, this is not the most informative approach to determine whether androgen could act at the level of the PVN. The data in the literature (Viau and Meaney, 1996) show that androgen implants into the medial preoptic area and bed nucleus of the stria terminalis could inhibit HPA responses to stress. Thus, there may be multiple sites for the actions of androgen that would not necessarily be discerned by implants of androgen receptor antagonists (such as flutamide) into the PVN region. Therefore, the approach used in the studies presented herein is more easily interpretable. Removing all androgen stimulation (GDX), followed by implants of hormone into the PVN, would establish a role for androgen that is independent of androgen actions at other brain sites.
Verification of steroid diffusion and cannula placement.
The distance of diffusion of hormones from the pellets was established using [125I]E2 or [3H]E2. A subset of male rats was cannulated, as described above, with a wax pellet containing either the [125I]E2 or [3H]E2. After 7 d, the brains were removed, sectioned, mounted, placed on film, or emulsion coated and allowed to develop for 7 and 60 d, respectively, and then films were analyzed to determine location and the diffusion distance from the implantation site.
In the studies presented here, confirmation of pellet placement was always made in cresyl-violet-stained sections, by identifying pellet location relative to the desired region (Kim et al., 2000; Dhillo et al., 2003). Figure 1A shows representative pellet placement. Placement was assessed by an observer blind to the intended pellet placement in slices in which the desired nucleus (PVN) and pellet were identifiable. Animals with pellet placement >0.5 mm away from the top of the PVN were excluded from all data analysis based on the average diffusion of radiolabeled E2 from the pellet in brain tissue. Diffusion of the steroid was mostly contained within a 0.5 mm area surrounding the pellet (Fig. 1B–D shows representative pellet locations). This amounted to a total of five animals that were removed from the analysis.
Figure 1.
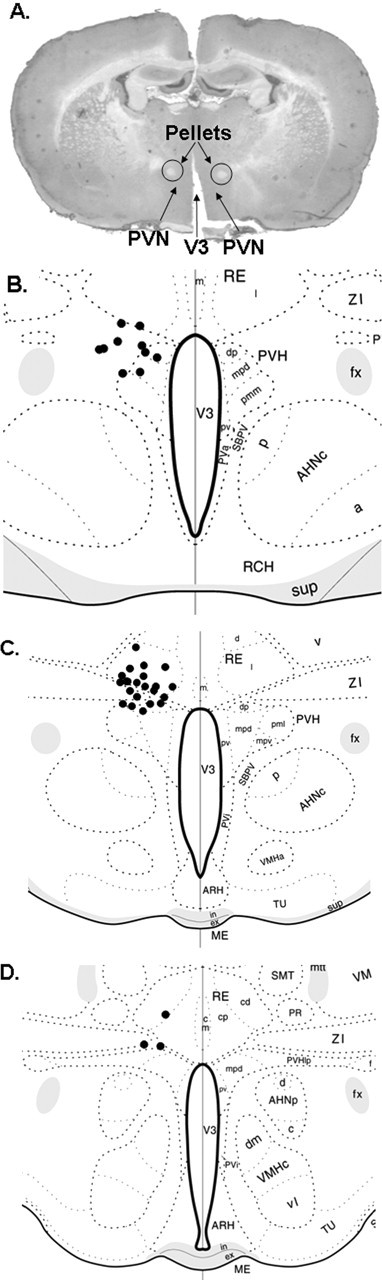
Verification of ligand pellet placement. The locations of pellets were localized in cresyl-violet-stained sections by identifying pellet location relative to the desired region. A, Typical histology with hormone-containing pellets indicated. B–D, Placement of the pellets from a representative experiment with locations plotted onto atlas diagrams of the brain in the region of the PVN [diagrams adapted from Swanson (1998/1999)]. Each black dot represents the center of a hormone pellet. V3, Third ventricle; PVH, paraventricular hypothalamic nucleus; PVHdp, paraventricular hypothalamic nucleus, dorsal parvicellular part; PVHmpd, paraventricular hypothalamic nucleus, medial parvicellular part; PVHpml, paraventricular hypothalamic nucleus, posterior magnocellular part, lateral zone; PVHpmm, paraventricular hypothalamic nucleus, posterior magnocellular part, medial zone; PVHpv, paraventricular hypothalamic nucleus, periventricular part; PVHlp, paraventricular hypothalamic nucleus, lateral pariventricular part; RE, nucleus reunions; REd, nucleus reunions, rostral division, dorsal part; REl, nucleus reunions, rostral division, lateral part; REm, nucleus reunions, rostral division, median part; PVa, periventricular hypothalamic nucleus anterior part; PVi, periventricular hypothalamic nucleus, intermediate part; SBPV, subparaventricular zone hypothalamus; ZI, zona incerta; fx, columns of the fornix; AHNa, anterior hypothalymic nucleus, anterior part; AHNc, anterior hypothalymic nucleus, central part; AHNd, anterior hypothalymic nucleus, dorsal part; AHNp, anterior hypothalymic nucleus, posterior part; RCH, retrochiasmatic area; sup, supraoptic commissures; ARH, arcuate hypothalamic nucleus; VMHa, ventromedial hypothalamic nucleus, anterior part; VMHc, ventromedial hypothalamic nucleus, central part; VMHdm, ventromedial hypothalamic nucleus, dorsomedial part; VMHvl, ventromedial hypothalamic nucleus, ventrolateral part; MEin, median eminence, internal lamina; MEex, median eminence, external lamina; TU, tuberal nucleus; VM, ventromedial nucleus; SMT, submedial nucleus thalamus; PR, perireuniens nucleus; mtt, mammillothalamic tract.
Flutamide and tamoxifen injections.
A subset of animals, implanted with treatments of DHT, 3β-diol, DPN, or empty wax pellet (control) were given concurrent subcutaneous injections of flutamide [50 mg/kg body weight (BW)], an AR antagonist, for 7 d after surgery. Another group, consisting of the same treatments (DHT, 3β-diol, DPN, control), were injected subcutaneously for the 7 d after surgery with the ER antagonist tamoxifen (1 mg/kg BW).
One week after surgery, animals were assigned randomly to stress or nonstress groups. Animals in the nonstress group were killed by decapitation after brief halothane anesthesia directly after removal from their home cages. Animals within the stress groups were restrained for 30 min in a flat bottom Plexiglas restrainer (Plas-Labs, Lansing, MI) and then quickly anesthetized (halothane) and killed by decapitation (all animals were killed between 8:00 and 10:00 A.M.). Trunk blood and brains were collected for later analysis.
Plasma hormones.
When the rats were killed, trunk blood was collected into ice-chilled tubes containing 0.5 m EDTA (200 μl) and 4 μg/ml Aprotinin (100 μl). Blood was centrifuged, and plasma was removed and stored at −20°C until assayed for corticosterone and ACTH by RIA.
Corticosterone was measured as described previously (Handa et al., 1994a). Briefly, plasma was diluted 1:25 in PBS, and binding proteins were heat denatured at 60°C for 1 h. Rabbit anti-corticosterone serum (ICN Biomedicals, Costa Mesa, CA) was used at a final dilution of 1: 2000, according to manufacturer’s protocol. Standard curves were constructed from dilutions of corticosterone (4-pregnen-11β, 21-diol-3, 20-dione; 5–500 ng/ml; Steraloids). The interassay and intraassay coefficients of variation were 4.9 and 8.8%, respectively
ACTH was measured using a RIA kit (DiaSorin, Stillwater, MN) according to manufacturer’s instructions. The assay sensitivity was 15 pg/ml. The intraassay and interassay coefficients of variation were 4 and 7.1%, respectively.
In situ hybridization. To compare changes in hormone secretory patterns with changes in cellular activity within the PVN, we measured c-fos mRNA using a 259 bp cRNA probe corresponding to the C-terminal end of the rat c-fos gene (cDNA kindly provided by T. Curran, University of Tennessee, Memphis, TN). The cRNA probe was reverse transcribed in vitro in the presence of 35S-UTP (1000 Ci mmol−1), as described previously (Nagahara and Handa, 1997)
Brains were sectioned at 16 μm on a Leitz 1720 digital cryostat, thaw mounted onto Superfrost plus slides (VWR Scientific, West Chester, PA), and stored at −80°C until assayed. For assay, tissue sections were thawed at room temperature, fixed with 10% formaldehyde, acetylated with 0.25% acetic anhydride, dehydrated in graded alcohols, and air-dried. Sections were incubated in a hybridization solution (50% formamide, 0.60 m NaCl, 0.02 m Tris, 0.01 m EDTA, 10% dextran sulfate, 2× Denhart’s solution, 50 mm dithiothreitol, 0.2% SDS, 100 mg/ml salmon sperm DNA, 500 mg/ml total yeast RNA, and 50 mg/ml yeast transfer RNA) containing the radiolabeled cRNA at a concentration of 2 × 107 cpm/ml at 60°C overnight. After hybridization, the slides were rinsed in 2× SSC. Nonhybridized RNA was digested with 30 mg/ml RNase A for 30 min at 37°C. The final wash stringency was 0.1× SSC at 60°C. For autoradiographic detection of hybridization, slides were exposed to Kodak BioMax MR autoradiographic film (Amersham Biosciences, Piscataway, NJ) for 10 d.
Image analysis.
Hybridization density per unit area was quantified using Scion (Frederick, MD) Image software. Images were captured using a Sony (Tokyo, Japan) XC-77 CCD camera with a Nikon (Melville, NY) 55 mm lens and a Dell Precision 3340 computer (Dell Computer Company, Round Rock, TX). Background hybridization was first determined in an adjacent region devoid of labeling. The integrated optical density was calculated for the PVN, and background was subtracted. For PVN measurements, a circular template of fixed area, encompassing most of the PVN but not extending beyond its boundaries, was used. Bilateral measurements were taken from the PVN of five different sections for each animal. These measurements were then averaged together to give the mean value for that animal.
Total RNA isolation.
Total RNA was isolated from microdissected PVN according to the method outlined by Chomczynski and Sacchi (1987). The PVN was micropunched from frozen hypothalamic slices as described previously (Price et al., 2000). Quantification of RNA was done with a Beckman Instruments (Fullerton, CA) DU 530 spectrophotometer. Only those samples with a 260:280 ratio of >1.6 were used in these studies.
Reverse transcriptase-PCR.
Two micrograms of total RNA were brought up to 11 μl with RNase-free water. One microliter of oligo d(T) at 0.5 μg/μl (Invitrogen, San Diego, CA) was added, then the reaction was heated to 65°C to allow the RNA to unfold. The reaction was cooled on ice, then RNA mix was combined with M-MLV buffer (50 mm Tris-HCl, pH 8.3, 75 mm KCl, 3 mm MgCl2), 10 mm DTT, and 0.5 mm each dNTP and M-MLV reverse transcriptase (RT) (Invitrogen). Reverse transcriptase reaction was for 10 min at room temperature and 50 min at 42°C and then heated to 95°C for 5 min to denature the reverse transcriptase.
PCR was performed using a DNA Master SYBR Green I kit and a Roche (Indianapolis, IN) Lightcyler. MgCl2 concentration was determined empirically to be optimal at 5 mm final concentration. DNA-free water, MgCl2, and SYBR Green mix were prepared according to kit directions, then 0.8 U of Platinum Taq antibody (Invitrogen) was added and allowed to incubate at 4°C for ∼10 min. Five picomoles of forward and reverse primer per reaction were aliquotted into capillary tubes (Roche), and 1/20th of the reverse transcription mix was added to each sample, except for the control tubes, which received DNA-free water of the same volume. Capillary tubes were heated to 95°C for 2 min, then a repeated cycle of 95°C for 2 s, annealing temperature (Table 2) for 10 s, and 72°C for extension time (Table 2) with fluorescence detection at the end of each 72°C step. Hybrids were then melted with continuous florescence detection to 95°C. PCR for 3α-hydroxysteroid dehydrogenase (3α-HSD) was performed with an Eppendorf Scientific (Westbury, NY) Mastercycler Personal thermocycler. Tubes were heated for 2 min at 95°C, then a repeated cycle of 95°C for 45 s, 60°C for 30 s, and 72°C for 1 min and 15 s. Samples were subjected to electrophoresis in a 2% agarose matrix in Tris borate–EDTA alongside a 100 bp ladder (Promega, Madison, WI).
Table 2.
Primer sequences and annealing temperatures used for amplification of mRNAs for various steroid metabolizing enzymes
| Gene | Primers | Annealing temperature | Extension time |
|---|---|---|---|
| 17β-HSD | Forward primer, 5′-GTGCGAGAGTCTGGCGATCCTG-3′ | 68°C | 12 s |
| Reverse primer, 5′-GGGTAGGAAGCGGTTCGTGGAG-3′ | |||
| Aromatase | Forward primer, 5′-CCTGGCAAGCACTCCTTATCAA-3′ | 64°C | 22 s |
| Reverse primer, 5′-CCTGTGCATTCTTCCGATGTTC-3′ | |||
| 5α-Reductase 1 | Forward primer, 5′-CGCGTCCTGCTGGCTATGTT-3′ | 66°C | 19 s |
| Reverse primer, 5′-CTGATGGTGCTGCTTCGCTCTG-3′ | |||
| CYP7B | Forward primer, 5′-AGCTATGGAAGTCCTGCGTGA-3′ | 62°C | 22 s |
| Reverse primer, 5′-GCCCAGAAACATGCGACTGT-3′ | |||
| 3α-HSD | Forward primer, 5′-GCAAGATTGAAGACGGCACTG-3′ | 60°C | 1:15 |
| Reverse primer, 5′-AGCTGGTAGCGAAGGGCAACTA-3′ | |||
| 3β-HSD | Forward primer, 5′-GGCCAGAGGATCATCCGGATGTT-3′ | 67°C | 10 s |
| Reverse primer, 5′-TGTCCCGATCCACTCCGAGGTTT-3′ |
Synthesis of hormone receptor proteins.
Full-length human ERα expression vector (pcDNA-ERα; R.H. Price, University of California, San Francisco, San Francisco, CA) and rat ERβ expression vector (pcDNA-ERβ; T.A. Brown, Pfizer, Groton, CT) were synthesized in vitro using the TnT-coupled reticulocyte lysate system (Promega) with T7-RNA polymerase, during a 90 min reaction at 30°C. Translation reaction mixtures were stored at −80°C until further use.
Saturation isotherms.
To calculate and confirm the binding affinity of DPN and PPT for ER, 100 μl aliquots of reticulocyte lysate supernatant were incubated at optimal time and temperature, 90 min at room temperature (ERβ) and 18 h at 4°C (ERα), with increasing (0.01–50 nm) concentrations of [3H]E2. These times were determined empirically and represent optimal binding of receptor with estrogen. Nonspecific binding was assessed using a 200-fold excess of the ER agonist diethylstilbestrol in parallel tubes. After incubation, bound and unbound [3H]E2 were separated by passing the incubation reaction through a 1 ml lipophilic Sephadex LH-20 (Sigma) column. Columns were constructed by packing a disposable pipette tip (1 ml; Labcraft; Curtin Matheson Scientific, Houston, TX) with TEGMD (10 mm Tris-Cl, 1.5 mm EDTA, 10% glycerol, 25 mm molybdate, and 1 mm dithiothreitol, pH 7.4)-saturated Sephadex according to previously published protocols (Handa et al., 1986; O’Keefe and Handa, 1990). For chromatography, the columns were re-equilibrated with TEGMD (100 μl), and the incubation reactions were added individually to each column and allowed to incubate on the column for an additional 30 min. After this incubation, 600 μl of TEGMD were added to each column, flow-through was collected, 4 ml of scintillation fluid was added, and samples were counted (5 min each) in an 2900 TR Packard scintillation counter (Packard Bioscience, Meriden, CT).
Statistics and analysis.
Where appropriate, data were analyzed by ANOVA followed by Newman–Keuls post hoc tests. Significance was set at p < 0.05. Curve fitting, scientific graphing, and analysis were completed using GraphPad Prism 3.0 software (GraphPad Software, San Diego, CA).
Results
DHT- and ERβ-selective ligands suppress the hormonal response to restraint stress
Wax pellets containing E2, DHT, 3β-diol, DPN, moxestrol, or PPT were implanted directly above the PVN to determine the direct influence of these compounds on the hormonal response to restraint stress. Two-way ANOVA of plasma corticosterone levels in restrained and nonrestrained animals revealed a significant treatment (F(6, 66) = 6.83; p < 0.01), stress (F(1, 66) = 882.48; p < 0.01), and treatment-by-stress (F(6, 66) = 6.99; p < 0.01). Using post hoc analysis, it was determined that corticosterone levels in stressed animals were significantly reduced by central treatment with DHT or the ERβ-selective compounds 3β-diol and DPN compared with vehicle controls (p < 0.05). On the other hand, plasma corticosterone levels were significantly increased after central treatment with E2 or the ERα-selective ligands moxestrol and PPT, when compared with controls (p < 0.05). These data are shown in Figure 2A. Similarly, as shown in Figure 2B, significant treatment (F(6, 66) = 8.36; p < 0.01), stress (F(1, 66) = 645.66; p < 0.01), and treatment-by-stress (F(6, 66) = 7.36; p < 0.01) effects were seen in the ACTH response to restraint. Post hoc analysis of ACTH levels showed a pattern that was similar to that exhibited by plasma corticosterone. ACTH levels after restraint stress were significantly reduced in DHT, 3β-diol, and DPN treated males (p < 0.05), whereas ACTH levels in E2-, moxestrol-, and PPT-treated animals were significantly increased relative to controls (p < 0.05).
Figure 2.

DHT- and ERβ-selective ligands suppress the hormonal response to restraint stress. Wax pellets containing E2, DHT, 3β-diol, DPN, moxestrol (Mox), or PPT were implanted directly above the PVN, and the hormonal response to a 30 min restraint stress was measured. A, Corticosterone levels. B, ACTH levels in stressed animals after central treatment with DHT, the ERβ-selective compounds 3β-diol and DPN with E2, or the ERα-selective ligands moxestrol and PPT. The asterisk indicates groups that were significantly different from vehicle controls (p < 0.05). Each bar represents the mean ± SEM of six or seven animals.
Androgen receptor antagonism does not block the effects of centrally administered DHT or 3β-diol
Previous research has shown that androgens act to inhibit the activity of the HPA axis (Gaskin and Kitay, 1970; Handa et al., 1994a; Viau and Meaney, 1996; Lund et al., 2004a), and androgen receptors are found in neurons of the PVN (Bingaman et al., 1994; Zhou et al., 1994). Therefore, the inhibitory influence of DHT, 3β diol, and DPN on corticosterone and ACTH secretion could be androgen receptor mediated. Consequently, we administered the androgen receptor antagonist flutamide to animals centrally implanted with DHT, 3β-diol, or DPN. Examination of corticosterone levels before and after restraint stress showed a significant treatment (F(3, 77) = 6.47; p < 0.01), stress (F(1, 77) = 533.31; p < 0.01), and treatment-by-stress (F(3, 77) = 6.68; p < 0.01) effect. However, no flutamide (F(1, 77) = 0.2; p = 0.21), treatment-by-flutamide (F(3, 77) = 0.9; p = 0.45), stress-by-flutamide (F(1, 77) = 0.45; p = 0.5), or treatment-by-stress-by-flutamide (F(3, 77) = 0.62; p = 0.6) effects were seen (Fig. 3A). Similar differences were identified in ACTH levels. Significant ACTH treatment (F(3, 77) = 5.97; p < 0.01), stress (F(1, 77) = 517.11; p < 0.01), and treatment-by-stress (F(3, 77) = 7.04; p < 0.01), but no flutamide (F(1, 77) = 0.2; p = 0.21), treatment-by-flutamide (F(3, 77) = 0.9; p = 0.45), stress-by-flutamide (F(1, 77) = 0.45; p = 0.5), or treatment-by-stress-by-flutamide (F(3, 77) = 0.62; p = 0.6), effects were identified (Fig. 3B). In concert with our previously described observations, DHT, 3β-diol, and DPN significantly decreased corticosterone plasma levels compared with control animals (p < 0.05), regardless of whether the animal was treated with flutamide. Interestingly, although both stressed males treated with DHT and DHT plus flutamide showed a significant decrease in corticosterone and ACTH levels compared with control-treated, stressed males, those animals treated with flutamide had significantly higher corticosterone and ACTH levels than did DHT males not treated with flutamide (p < 0.05). Such data suggest that DHT could be working partially through an AR-mediated mechanism.
Figure 3.

Androgen receptor antagonism does not block the effects of centrally administered DHT 3β-diol or DPN on plasma corticosterone response to restraint stress. Corticosterone (A) and ACTH (B) levels were determined 30 min after restraint stress in animals treated centrally with DHT, 3β-diol, or DPN and treated concomitantly with flutamide. *p < 0.05 significant difference compared with control treatment. **p < 0.05 significantly higher corticosterone levels than DHT males not treated with flutamide. Each bar represents the mean ± SEM of 8–10 animals.
Although it is possible that the dose of flutamide used was insufficient to block AR in the PVN, similar doses of flutamide have been used to block AR-mediated behaviors (Mathias et al., 1999; Lund et al., 2000), and this dose inhibited prostate growth in the DHT-treated males (data not shown).
An estrogen receptor antagonist prevents the effects of centrally administered DHT and 3β-diol
Because the inhibitory effects of 3β-diol or DPN on the hormonal stress response do not seem to be mediated by androgen receptor, we next sought to determine whether the effects are ER mediated. To accomplish this, the ER antagonist tamoxifen was administered to GDX males implanted with carrier alone or DHT, 3β-diol, or DPN. Examination of corticosterone levels before and after restraint stress revealed a significant treatment (F(3, 75) = 6.8; p < 0.01), stress (F(1, 75) = 936.57; p < 0.01), and treatment-by-stress (F(3, 75) = 6.69; p < 0.01) effect. Furthermore, a significant tamoxifen (F(1, 75) = 17.65; p < 0.01), treatment-by-tamoxifen (F(3, 75) = 2.75; p < 0.05), and stress-by-tamoxifen (F(1, 75) = 16.38; p < 0.01) effect was also found. Similarly, ACTH levels before and after restraint stress revealed a significant treatment (F(3, 75) = 7.27; p < 0.01), stress (F(1, 75) = 898.73; p < 0.01), and treatment-by-stress (F(3, 75) = 7.11; p < 0.01) effect as well as a significant tamoxifen (F(1, 75) = 15.98; p < 0.01), treatment-by-tamoxifen (F(3, 75) = 4.83; p < 0.01), and stress-by-tamoxifen (F(1, 75) = 18.45; p < 0.01) effect. In confirmation of the studies presented above, DHT, 3β-diol, and DPN again significantly decreased poststress plasma corticosterone and ACTH levels compared with control males (p < 0.05). However, when males implanted with DHT, 3β-diol, or DPN were injected with tamoxifen, the inhibitory effects of these compounds on poststress corticosterone and ACTH were eliminated. Plasma corticosterone and ACTH levels in stressed males implanted with DHT, 3β-diol, or DPN and then treated with tamoxifen did not differ from controls (p > 0.05). These data are presented graphically in Figure 4, A and B, respectively.
Figure 4.

The estrogen receptor antagonist tamoxifen blocks the effects of centrally administered DHT, 3β-diol, and DPN. Plasma corticosterone (A) and ACTH (B) levels were determined in restrained (30 min) or nonrestrained males with PVN implants containing DHT, 3β-diol, or DPN and then treated with tamoxifen did not differ from controls. *p < 0.05, significant difference compared with control treatment. Double asterisks indicate groups treated with tamoxifen that had significantly higher corticosterone levels than did comparably treated group but without tamoxifen (p < 0.05). Each bar represents the mean ± SEM of 8–10 animals.
E2 and DHT differently influence restraint induced c-fos expression in the PVN
In situ hybridization was used to detect c-fos mRNA levels and to examine the influence of various compounds on the activation of PVN neurons after stress. Significant treatment (F(2, 60) = 3.2; p < 0.05) and stress (F(3, 60) = 13.9; p < 0.05) effects were identified. c-fos mRNA expression was low in all unstressed animals and was significantly elevated in control animals 30 min after restraint. Post hoc analysis showed that restraint-induced c-fos mRNA expression was significantly greater in E2- and PPT-treated animals relative to the stressed control group. In contrast, after restraint, c-fos mRNA in the PVN of DHT-, 3β-diol-, and DPN-treated animals was significantly lower relative to stressed controls (p < 0.05). Graphic representation of these data is presented in Figure 5.
Figure 5.

E2 and DHT influence restraint induced c-fos mRNA expression in the PVN. A, In situ hybridization was used to semiquantitate c-fos mRNA levels in the PVN and to examine the influence of various compounds on the activation of PVN neurons after stress. * indicates groups that were significantly greater relative to the stressed control group (p < 0.05). ** indicates groups that, after restraint, showed c-fos mRNA levels in the PVN that were significantly lower relative to stressed controls (p < 0.05). B, Representative photomicrographs from each of these groups. Each bar represents the mean ± SEM of 8–10 animals. 3V, Third ventricle.
The PVN contains relevant steroid hormone metabolizing enzymes
In order for DHT to be converted to ERβ-binding ligands such as 3β-diol, it is necessary for the appropriate steroid metabolizing enzymes to be present in or around the PVN. Given the relatively short half-life of 3β-diol in peripheral circulation (Grover and Odell, 1975), coupled with the fact that we administered 3β-diol directly above the PVN, these data suggest that local synthesis of the hormone is an absolute requirement for its regulatory action. Previous studies have demonstrated that DHT is produced in the brain by the 5α-reduction of testosterone (Krieger et al., 1983). Subsequently, DHT can be metabolized to either 3α-diol or 3β-diol by the actions of 3α-HSD or 3β-HSD, respectively. Although previous studies indicated two distinct fates of DHT, recent studies indicate that the metabolism to 3α-diol is a reversible reaction (Steckelbroeck et al., 2004). 3β-Diol is also produced from DHT through the actions of 3α-HSD and 17β-HSD (Gangloff et al., 2003; Torn et al., 2003; Steckelbroeck et al., 2004).
Using RT-PCR, we determined that the mRNAs of enzymes critical for the metabolism of testicular androgens to endogenous ER ligands are present within cells of the PVN (see supplemental material, available at www.jneurosci.org). Specifically, mRNAs for aromatase (Fig. 6A), 5α-reductase (Fig. 6B), 17β-HSD (Fig. 6C), 3α-HSD (Fig. 6D), and cytochrome p450 7b (CYP7B) (Fig. 6F) enzymes were found to be expressed in microdissected PVN. 3β-HSD mRNA, however, was not detected in the PVN (Fig. 6E). These data provide for the possibility that the regulation of these enzymes can tightly coordinate the absolute level of ligand delivered to ERβ.
Figure 6.
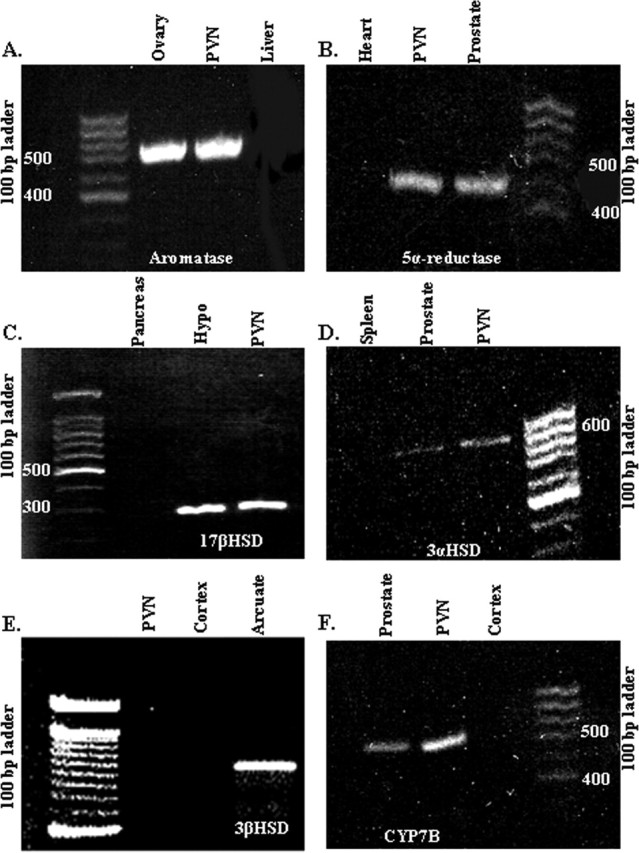
The PVN contains relevant steroid hormone metabolizing enzyme mRNAs. Photomicrographs of agarose gels after electrophoresis of RT-PCR products from microdissected PVN of gonadectomized male rats are shown. Bands represent mRNAs for aromatase (A), 5α-reductase (B), 17β-HSD (C), 3α-HSD (D), 3β-HSD (E), and CYP7B (F). Hypo, Hypothalamus.
Discussion
Sex steroid hormones are primarily responsible for sex difference in adult HPA function; androgens inhibit whereas estrogens enhance HPA axis activation after a stressor (Viau and Meaney, 1991, 1996; Burgess and Handa, 1992; Handa et al., 1994a, b; Lund et al., 2004a). Whether these hormones act directly on hypothalamic neurons to alter HPA axis reactivity to stress is unknown.
It is of interest that the PVN contains relatively high levels of AR (Bingaman et al., 1994; Zhou et al., 1994) and ERβ (Alves et al., 1998; Hrabovszky et al., 1998; Somponpun and Sladek, 2003) but is essentially devoid of ERα (Shughrue et al., 1997a), thus implicating the PVN as a potential site for the integration of sex steroid effects on HPA reactivity. Here, we sought to determine the actions of estrogen or androgen on AR- and ERβ-containing neurons in the PVN and whether these actions might regulate HPA axis function. This was accomplished by directly introducing compounds, via stereotaxic surgery, to a region near the PVN. This approach was used previously by Kovacs and Mezey (1987) and Sawchenko (1987) to evaluate the direct actions of glucocorticoids on the regulation of PVN function. The hormone was not introduced directly into the PVN to prevent mechanical lesions, which could disrupt HPA axis function.
With these studies, we demonstrated that the nonaromatizable androgen DHT and the nonselective ER ligand E2 influence HPA reactivity by acting on neurons within or surrounding the PVN. This inhibitory action of DHT is detectable at both the level of hormone secretion as well as PVN c-fos mRNA expression. Furthermore, the inhibition can be mimicked by the DHT metabolite 3β-diol and by the subtype selective ERβ agonist DPN. In contrast, E2 acts to enhance HPA reactivity to restraint, a response that is imitated by the selective ERα agonists moxestrol and PPT. Finally, the ability of the ER antagonist tamoxifen, but not the AR antagonist flutamide, to block the inhibitory actions of DHT, speaks to the intracellular mechanism by which this inhibitory signal might be transduced.
We found that the DHT metabolite 3β-diol and the ERβ-subtype-selective agonist DPN suppressed ACTH, corticosterone, and c-fos mRNA responses to restraint stress in a manner similar to DHT. This finding seemingly indicates that metabolism of DHT to 3β-diol and subsequent binding to ERβ can be inhibitory to HPA reactivity, and this is one possible mechanism for the action of DHT. This finding is similar to previously published findings in mice in which peripheral 3β-diol injections inhibited the corticosterone response to restraint stress, similar to the effect of DHT in adult male mice (Lund et al., 2004b).
To verify that the actions of DHT, 3β-diol, and DPN were ERβ not AR mediated, the AR antagonist flutamide was injected into animals centrally implanted with these compounds. Plasma corticosterone levels were decreased compared with control animals, regardless of whether the animal was treated with flutamide. Interestingly, although both stressed DHT and DHT plus flutamide males showed a significant decrease in corticosterone levels compared with stressed males, those males treated with flutamide had significantly higher corticosterone levels than did DHT males not treated with flutamide, suggesting that the inhibitory effects of 3β-diol or DPN on the hormonal stress response are not mediated by AR and that the effect of DHT is only partially AR mediated.
Because the inhibitory effects of DHT, 3β-diol, or DPN on the hormonal stress response do not appear to be solely mediated by AR, the effect of the ER antagonist tamoxifen was examined. Again, DHT, 3β-diol, and DPN treatment significantly decreased stress induced circulating corticosterone levels; however, this effect was blocked by tamoxifen. Together, these studies suggest that the inhibitory effects of DHT on HPA axis activity are mediated via its conversion to 3β-diol and subsequent binding to ERβ. Evidence of the effectiveness of DPN in binding hypothalamic ER has been identified previously (Lund et al., 2005).
Our data also suggest that E2 enhances the reactivity of the HPA axis to stress by acting on or near neurons of the PVN. Furthermore, the actions of E2 appear to be through an ERα-dependent mechanism, because PPT mimicked the actions of E2. Although the PVN has been reported to be devoid of ERα-containing neurons (Shughrue et al., 1997a), the population of neurons surrounding the PVN is ERα immunoreactive (Suzuki and Handa, 2005). The phenotypic identity of these neurons is not known; however, given the large number of GABA-containing interneurons surrounding the PVN (Boudaba et al., 1996), one could hypothesize that E2 acts via these ERα-containing neurons to inhibit GABAergic inhibition of the PVN and consequently enhance HPA reactivity. This mechanism has been proposed for other estrogen-sensitive systems, including the actions of estrogen on dendritic spine growth (Murphy et al., 1998) and the positive-feedback response of gonadotropin-releasing hormone neurons (Grattan et al., 1996).
Using c-fos mRNA as a marker for neuronal activation, we demonstrated that local administration of estrogens and androgens can alter the stress-induced activity of PVN neurons in a manner corresponding to the amplitude of the hormonal response to restraint. Such data indicate not only that the activity of PVN neurons is closely related to the amplitude of the stress response, but that the actions of estrogens and androgens in regulating HPA axis activity are by modulating synaptic activity in relevant neuronal populations of the PVN. Regarding the enhancement of c-fos mRNA expression by ERα, this observation is consistent with the reported inhibitory effects of estrogen on GABA function, as described above.
The neuroanatomical basis for ERβ inhibition of PVN neuronal activity is perhaps more complicated. Within the PVN, ERβ is expressed predominantly in magnocellular neurons containing oxytocin and vasopressin and in preautonomic projecting neurons (Somponpun and Sladek, 2003; Stern and Zhang, 2003; Suzuki and Handa, 2005). Although the possibility exists that 3β-diol and DPN can directly activate ERβ in neuroendocrine neurons of the PVN to inhibit their activity, only a small population of neuroendocrine CRH neurons in the parvocellular PVN appear to contain ERβ (Laflamme et al., 1998; Suzuki and Handa, 2005). An alternative hypothesis regarding the mechanism for ERβ inhibition of PVN neuron activity could be that ERβ acts via those ERβ-containing PVN neurons that project to brainstem nuclei involved in autonomic regulation. Subsequently, via reciprocal connections, such nuclei could act to inhibit the activity of the PVN. A third possibility could be that ERβ directly activates oxytocin- and vasopressin-containing magnocellular neurons in the PVN, and through a paracrine action, these neurons inhibit activity of adjacent parvocellular neuroendocrine neurons. In support of this, vasopressin has been shown to be released within the PVN in response to a stressor (Wotjak et al., 1996), and intra-PVN vasopressin and oxytocin are thought to be inhibitory to PVN tone (Kalsbeek et al., 1996; Wotjak et al., 1996; Windle et al., 2004). Whether one or all of these potential regulatory mechanisms are involved remains to be determined.
Although the verification criteria for cannula placement was stringent, the likelihood still exists that steroid, from the pellets, diffused to areas outside of the PVN, which may result in alternative explanations for the findings presented herein. Of particular interest, because of its proximity to the PVN and ERβ expression, is the dorsomedial nucleus (Shughrue et al., 1997b). Because the dorsomedial nucleus also projects to the PVN, it is possible that DPN and 3β-diol act on ERβ cells in the dorsomedial nucleus. Lesions of the DMN have been shown to cause increases in corticosterone and CRH mRNA response to osmotic stress but not basal CRH mRNA levels (Kiss and Jezova, 2001). This suggests that the dorsomedial nucleus is inhibitory and activation of the dorsomedial nucleus could be responsible for inhibition of PVN activity. However, the dorsomedial nucleus is >0.5 mm away from the PVN and the location of the pellets. In addition, projections from the dorsomedial nucleus to the PVN are predominantly to preautonomic cells (Thompson et al., 1996).
Finally, our data regarding ERβ and HPA reactivity are in potential contrast to those of Isgor et al. (2003), who demonstrated that administration of the nonselective ER antagonist ICI182, 760 (7α[9-(4, 4, 5, 5, 5-pentafluoro-pentylsulphinyl)nonyl]oestra 1, 3, 5(10)-triene-3, 17β-diol) to the PVN of intact female rats causes a reduction in HPA reactivity to restraint. It was hypothesized that this response was caused by blockade of ERβ in the PVN, seemingly indicating that ERβ activation by estrogen was HPA enhancing. However, such treatment would also block ERα-containing neurons surrounding the PVN, which, according to our data, would be consistent with the reduction in HPA reactivity observed. Furthermore, it should be emphasized that the studies described in this report used male rats, whereas those of Isgor et al. (2003) examined intact female rats.
In summary, these studies suggest that ERβ, within the male hypothalamus, acts to inhibit the HPA axis and that the inhibitory effects of DHT may be, at least in part, via its intracellular conversion to 3β-diol and subsequent binding to ERβ. Consequently, the possibility that ERβ is an important receptor in the male brain for sensing levels of androgen metabolites such as 3β-diol is an attractive hypothesis that must be further explored.
Footnotes
This work was supported by National Institutes of Health Grant R01 NSO39951.
References
- Alves SE, Lopez V, McEwen BS, Weiland NG (1998). Differential colocalization of estrogen receptor beta (ERbeta) with oxytocin and vasopressin in the paraventricular and supraoptic nuclei of the female rat brain: an immunocytochemical study. Proc Natl Acad Sci USA 95:3281–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingaman EW, Baeckman LM, Yracheta JM, Handa RJ, Gray TS (1994). Localization of androgen receptor within peptidergic neurons of the rat forebrain. Brain Res Bull 35:379–382. [DOI] [PubMed] [Google Scholar]
- Boudaba C, Szabo K, Tasker JG (1996). Physiological mapping of local inhibitory inputs to the hypothalamic paraventricular nucleus. J Neurosci 16:7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess LH, Handa RJ (1992). Chronic estrogen-induced alterations in adrenocorticotropin and corticosterone secretion, and glucocorticoid receptor-mediated functions in female rats. Endocrinology 131:1261–1269. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Versteeg DH, Kovacs GL (1983). Aldosterone blocks the response to corticosterone in the raphe-hippocampal serotonin system. Brain Res 264:323–327. [DOI] [PubMed] [Google Scholar]
- Dhillo WS, Small CJ, Jethwa PH, Russell SH, Gardiner JV, Bewick GA, Seth A, Murphy KG, Ghatei MA, Bloom SR (2003). Paraventricular nucleus administration of calcitonin gene-related peptide inhibits food intake and stimulates the hypothalamo-pituitary-adrenal axis. Endocrinology 144:1420–1425. [DOI] [PubMed] [Google Scholar]
- Fischette CT, Komisaruk BR, Edinger HM, Feder HH, Siegel A (1980). Differential fornix ablations and the circadian rhythmicity of adrenal corticosteroid secretion. Brain Res 195:373–387. [DOI] [PubMed] [Google Scholar]
- Gangloff A, Shi R, Nahoum V, Lin SX (2003). Pseudo-symmetry of C19 steroids, alternative binding orientations, and multispecificity in human estrogenic 17beta-hydroxysteroid dehydrogenase. FASEB J 17:274–276. [DOI] [PubMed] [Google Scholar]
- Gaskin JH, Kitay JI (1970). Adrenocortical function in the hamster. Sex differences and effects of gonadal hormones. Endocrinology 87:779–786. [DOI] [PubMed] [Google Scholar]
- Glass MJ, Billington CJ, Levine AS (2000). Naltrexone administered to central nucleus of amygdala or PVN: neural dissociation of diet and energy. Am J Physiol Regul Integr Comp Physiol 279:R86–R92. [DOI] [PubMed] [Google Scholar]
- Grattan DR, Rocca MS, Strauss KI, Sagrillo CA, Selmanoff M, McCarthy MM (1996). GABAergic neuronal activity and mRNA levels for both forms of glutamic acid decarboxylase (GAD65 and GAD67) are reduced in the diagonal band of Broca during the afternoon of proestrus. Brain Res 733:46–55. [DOI] [PubMed] [Google Scholar]
- Grover PK, Odell WD (1975). Correlation of in vivo and in vitro activities of some naturally occurring androgens using a radioreceptor assay for 5alpha-dihydrotestosterone with rat prostate cytosol receptor proteins. J Steroid Biochem 6:1373–1379. [DOI] [PubMed] [Google Scholar]
- Handa RJ, Reid DL, Resko JA (1986). Androgen receptor in brain and pituitary of female rats: cyclic changes and comparisons with the male. Biol Reprod 34:293–303. [DOI] [PubMed] [Google Scholar]
- Handa RJ, Nunley KM, Lorens SA, Louie JP, McGivern RF, Bollnow MR (1994a). Androgen regulation of adrenocorticotropin and corticosterone secretion in the male rat following novelty and foot shock stressors. Physiol Behav 55:117–124. [DOI] [PubMed] [Google Scholar]
- Handa RJ, Burgess LH, Kerr JE, O’Keefe JA (1994b). Gonadal steroid hormone receptors and sex differences in the hypothalamo-pituitary-adrenal axis. Horm Behav 28:464–476. [DOI] [PubMed] [Google Scholar]
- Harris HA, Katzenellenbogen JA, Katzenellenbogen BS (2002). Characterization of the biological roles of the estrogen receptors, ERalpha and ERbeta, in estrogen target tissues in vivo through the use of an ERalpha-selective ligand. Endocrinology 143:4172–4177. [DOI] [PubMed] [Google Scholar]
- Hrabovszky E, Kallo I, Hajszan T, Shughrue PJ, Merchenthaler I, Liposits Z (1998). Expression of estrogen receptor-beta messenger ribonucleic acid in oxytocin and vasopressin neurons of the rat supraoptic and paraventricular nuclei. Endocrinology 139:2600–2604. [DOI] [PubMed] [Google Scholar]
- Huang X, Harlan RE (1994). Androgen receptor immunoreactivity in somatostatin neurons of the periventricular nucleus but not in the bed nucleus of the stria terminalis in male rats. Brain Res 652:291–296. [DOI] [PubMed] [Google Scholar]
- Isgor C, Cecchi M, Kabbaj M, Akil H, Watson SJ (2003). Estrogen receptor beta in the paraventricular nucleus of hypothalamus regulates the neuroendocrine response to stress and is regulated by corticosterone. Neuroscience 121:837–845. [DOI] [PubMed] [Google Scholar]
- Kalsbeek A, van der Vliet J, Buijs RM (1996). Decrease of endogenous vasopressin release necessary for expression of the circadian rise in plasma corticosterone: a reverse microdialysis study. J Neuroendocrinol 8:299–307. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Kim Y, Choe BK, Kim SA, Lee HJ, Kim JW, Huh Y, Kim C, Chung JH (2000). Differential expression of nicotinamide adenine dinucleotide phosphate-diaphorase in hypothalamic areas of obese Zucker rats. Neurosci Lett 292:60–62. [DOI] [PubMed] [Google Scholar]
- Kiss A, Jezova D (2001). Lesions of central part of the dorsomedial nucleus alters vasopressin but not corticotropin releasing hormone mRNA levels in the rat hypothalamic paraventricular nucleus. Gen Physiol Biopys 20:393–400. [PubMed] [Google Scholar]
- Kornstein SG (1997). Gender differences in depression: implications for treatment. J Clin Psychiatry 58:12–18. [PubMed] [Google Scholar]
- Kovacs KJ, Mezey E (1987). Dexamethasone inhibits corticotropin-releasing factor gene expression in the rat paraventricular nucleus. Neuroendocrinology 46:365–368. [DOI] [PubMed] [Google Scholar]
- Krieger NR, Scott RG, Jurman ME (1983). Testosterone 5 alpha-reductase in rat brain. J Neurochem 40:1460–1464. [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B, Gustafsson JA (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 139:4252–4263. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Nappi RE, Drolet G, Labrie C, Rivest S (1998). Expression and neuropeptidergic characterization of estrogen receptors (ERalpha and ERbeta) throughout the rat brain: anatomical evidence of distinct roles of each subtype. J Neurobiol 36:357–378. [DOI] [PubMed] [Google Scholar]
- Levine S (2001). Primary social relationships influence the development of the hypothalamic-pituitary-adrenal axis in the rat. Physiol Behav 73:255–260. [DOI] [PubMed] [Google Scholar]
- Lund TD, Salyer DL, Fleming DE, Lephart ED (2000). Pre- or postnatal testosterone and flutamide effects on sexually dimorphic nuclei of the rat hypothalamus. Brain Res Dev Brain Res 120:261–266. [DOI] [PubMed] [Google Scholar]
- Lund TD, Munson DJ, Haldy ME, Handa RJ (2004a). Androgen inhibits, while oestrogen enhances, restraint-induced activation of neuropeptide neurones in the paraventricular nucleus of the hypothalamus. J Neuroendocrinol 16:272–278. [DOI] [PubMed] [Google Scholar]
- Lund TD, Munson DJ, Haldy ME, Handa RJ (2004b). Dihydrotestosterone may inhibit hypothalamo-pituitary-adrenal activity by acting through estrogen receptor in the male mouse. Neurosci Lett 365:43–47. [DOI] [PubMed] [Google Scholar]
- Lund TD, Rovis T, Chung WC, Handa RJ (2005). Novel actions of estrogen receptor-beta on anxiety-related behaviors. Endocrinology 146:797–807. [DOI] [PubMed] [Google Scholar]
- Mathias LJ, Jacobson NA, Rhees RW, Lephart ED (1999). Brain aromatase in control versus castrated Norway Brown, Sprague-Dawley and Wistar adult rats. Proc Soc Exp Biol Med 2:126–130. [DOI] [PubMed] [Google Scholar]
- Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA (2001). Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem 44:4230–4251. [DOI] [PubMed] [Google Scholar]
- Muglia LJ, Jenkins NA, Gilbert DJ, Copeland NG, Majzoub JA (1994). Expression of the mouse corticotropin-releasing hormone gene in vivo and targeted inactivation in embryonic stem cells. J Clin Invest 93:2066–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muglia LJ, Jacobson L, Luedke C, Vogt SK, Schaefer ML, Dikkes P, Fukuda S, Sakai Y, Suda T, Majzoub JA (2000). Corticotropin-releasing hormone links pituitary adrenocorticotropin gene expression and release during adrenal insufficiency. J Clin Invest 105:1269–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Cole NB, Greenberger V, Segal M (1998). Estradiol increases dendritic spine density by reducing GABA neurotransmission in hippocampal neurons. J Neurosci 18:2550–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Handa RJ (1997). Age-related changes in c-fos mRNA induction after open-field exposure in the rat brain. Neurobiol Aging 18:45–55. [DOI] [PubMed] [Google Scholar]
- O’Keefe JA, Handa RJ (1990). Transient elevation of estrogen receptors in the neonatal rat hippocampus. Brain Res Dev Brain Res 57:119–127. [DOI] [PubMed] [Google Scholar]
- Pak TR, Chung WC, Lund TD, Hinds LR, Clay CM, Handa RJ (2005). The androgen metabolite, 5alpha-androstane-3beta, 17beta-diol (3betaAdiol), is a potent modulator of estrogen receptor-beta1-mediated gene transcription in neuronal cells. Endocrinology 146:147–155. [DOI] [PubMed] [Google Scholar]
- Price RH Jr, Lorenzon N, Handa RJ (2000). Differential expression of estrogen receptor beta splice variants in rat brain: identification and characterization of a novel variant missing exon 4. Brain Res Mol Brain Res 80:260–268. [DOI] [PubMed] [Google Scholar]
- Reul JM, de Kloet ER (1985). Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 117:2505–2511. [DOI] [PubMed] [Google Scholar]
- Reul JM, van den Bosch FR, de Kloet ER (1987). Relative occupation of type-I and type-II corticosteroid receptors in rat brain following stress and dexamethasone treatment: functional implications. J Endocrinol 115:459–467. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE (1987). Evidence for a local site of action for glucocorticoids in inhibiting CRF and vasopressin expression in the paraventricular nucleus. Brain Res 403:213–223. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Lane MV, Merchenthaler I (1997a). Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol 388:507–525. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Scrimo P, Lane M, Askew R, Merchenthaler I (1997b). The distribution of estrogen receptor-β mRNA in forbrain regions of the estrogen receptor-α knockout mouse. Endocrinology 138:5649–5652. [DOI] [PubMed] [Google Scholar]
- Somponpun SJ, Sladek CD (2003). Osmotic regulation of estrogen receptor-β in rat vasopressin and oxytocin neurons. J Neurosci 23:4261–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess J, Rivier J, Rivier C, Vale W (1981). Primary structure of corticotropin-releasing factor from ovine hypothalamus. Proc Natl Acad Sci USA 78:6517–6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA (2000). Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-alpha-selective agonists. J Med Chem 43:4934–4947. [DOI] [PubMed] [Google Scholar]
- Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM (2004). Human cytosolic 3alpha-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3beta-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem 279:10784–10795. [DOI] [PubMed] [Google Scholar]
- Stern JE, Zhang W (2003). Preautonomic neurons in the paraventricular nucleus of the hypothalamus contain estrogen receptor beta. Brain Res 975:99–109. [DOI] [PubMed] [Google Scholar]
- Sun J, Huang YR, Harrington WR, Sheng S, Katzenellenbogen JA, Katzenellenbogen BS (2002). Antagonists selective for estrogen receptor alpha. Endocrinology 143:941–947. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Handa RJ (2005). Estrogen receptor (ER)-β, but not ER-α is expressed in prolactin neurons of the female rat paraventricular and supraoptic nuclei: a comparison with other neuropeptides. J Comp Neurol 484:28–42. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Lund TD, Price RH, Handa RJ (2001). Sex differences in the hypothalamo-pituitary-adrenal axis: novel roles for androgen and estrogen receptors. In: Recent research developments in endocrinology pp. 69–86. Kerala, India: Transworld Research Network.
- Swanson LW (1998/1999). In: Brain maps: structure of the rat brain Ed 2 Amsterdam: Elsevier.
- Thompson RH, Canieras NS, Swanson LW (1996). Organization of projections from the dorsomedial nucleus of the hypothalamus: a PHAL study in the rat. J Comp Neurol 376:143–173. [DOI] [PubMed] [Google Scholar]
- Torn S, Nokelainen P, Kurkela R, Pulkka A, Menjivar M, Ghosh S, Coca-Prados M, Peltoketo H, Isomaa V, Vihko P (2003). Production, purification, and functional analysis of recombinant human and mouse 17beta-hydroxysteroid dehydrogenase type 7. Biochem Biophys Res Commun 305:37–45. [DOI] [PubMed] [Google Scholar]
- Turner BB (1992). Sex differences in the binding of type I and type II corticosteroid receptors in rat hippocampus. Brain Res 581:229–236. [DOI] [PubMed] [Google Scholar]
- Viau V (2002). Functional cross-talk between the hypothalamic-pituitary-gonadal and -adrenal axes. J Neuroendocrinol 14:506–513. [DOI] [PubMed] [Google Scholar]
- Viau V, Meaney MJ (1991). Variations in the hypothalamic-pituitary-adrenal response to stress during the estrous cycle in the rat. Endocrinology 129:2503–2511. [DOI] [PubMed] [Google Scholar]
- Viau V, Meaney MJ (1996). The inhibitory effect of testosterone on hypothalamic-pituitary-adrenal responses to stress is mediated by the medial preoptic area. J Neurosci 16:1866–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viau V, Soriano L, Dallman MF (2001). Androgens alter corticotropin releasing hormone and arginine vasopressin mRNA within forebrain sites known to regulate activity in the hypothalamic-pituitary-adrenal axis. J Neuroendocrinol 13:442–452. [DOI] [PubMed] [Google Scholar]
- Weihua Z, Makela S, Andersson LC, Salmi S, Saji S, Webster JI, Jensen EV, Nilsson S, Warner M, Gustafsson JA (2001). A role for estrogen receptor beta in the regulation of growth of the ventral prostate. Proc Natl Acad Sci USA 98:6330–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihua Z, Lathe R, Warner M, Gustafsson JA (2002). An endocrine pathway in the prostate, ERbeta, AR, 5alpha-androstane-3beta, 17beta-diol, and CYP7B1, regulates prostate growth. Proc Natl Acad Sci USA 99:13589–13594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitnall MH, Mezey E, Gainer H (1985). Co-localization of corticotropin-releasing factor and vasopressin in median eminence neurosecretory vesicles. Nature 317:248–250. [DOI] [PubMed] [Google Scholar]
- Wilhelm K, Parker G, Asghari A (1998a). Sex differences in the experience of depressed mood state over fifteen years. Soc Psychiatry Psychiatr Epidemiol 33:16–20. [DOI] [PubMed] [Google Scholar]
- Wilhelm K, Parker G, Dewhurst J (1998b). Examining sex differences in the impact of anticipated and actual life events. J Affect Disord 48:37–45. [DOI] [PubMed] [Google Scholar]
- Windle RJ, Kershaw YM, Shanks N, Wood SA, Lightman SL, Ingram CD (2004). Oxytocin attenuates stress-induced c-fos mRNA expression in specific forebrain regions associated with modulation of hypothalamo-pituitary-adrenal activity. J Neurosci 24:2974–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotjak CT, Kubota M, Liebsch G, Montkowski A, Holsboer F, Neumann I, Landgraf R (1996). Release of vasopressin within the rat paraventricular nucleus in response to emotional stress: a novel mechanism of regulating adrenocorticotropic hormone secretion? J Neurosci 16:7725–7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Blaustein JD, De Vries GJ (1994). Distribution of androgen receptor immunoreactivity in vasopressin- and oxytocin-immunoreactive neurons in the male rat brain. Endocrinology 134:2622–2627. [DOI] [PubMed] [Google Scholar]