

Abstract
Endocannabinoid signaling couples activity-dependent rises in postsynaptic Ca2+ levels to decreased presynaptic GABA release. Here, we present evidence from paired recording experiments that cannabinoid-mediated inhibition of GABA release depends on the firing rates of the presynaptic interneurons. Low-frequency action potentials in post hoc identified cholecystokinin-positive CA1 basket cells elicited IPSCs in the postsynaptic pyramidal cells that, as expected, were fully abolished by the exogenous application of the cannabinoid receptor agonist WIN55,212-2 [R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrol[1,2,3-de]-1,4-benzoxazin-6-yl)(1-naphthalenyl) methanone monomethanesulfonate] at 5 μm. However, the presynaptic basket cells recovered from the cannabinoid agonist-induced inhibition of GABA release when the presynaptic firing rate was increased to ≥20 Hz. Pharmacological experiments showed that the recovered transmission was exclusively dependent on presynaptic N-type Ca2+ channels. Furthermore, the increased presynaptic firing could also overcome even complete depolarization-induced suppression of inhibition, indicating that the magnitude of DSI markedly depends on the activity levels of basket cells. These results reveal a new locus of activity-dependent modulation for endocannabinoid signaling and suggest that endocannabinoid-mediated inhibition of GABA release may differ in distinct behavioral states.
Keywords: cannabinoid, IPSC, paired recording, plasticity, hippocampus, GABA
Introduction
Cannabinoid type 1 (CB1) receptors participate in a variety of synaptic plasticity processes in several brain areas (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). In the hippocampus, CB1 receptors are highly expressed on the axonal membranes of cholecystokinin-positive (CCK+) interneurons (Katona et al., 1999), where their activation by exogenous or endogenous cannabinoid ligands decreases GABA release (Wilson et al., 2001). Postsynaptic depolarization is an efficient trigger for engaging the endocannabinoid system, with increases in postsynaptic Ca2+ levels eliciting a cascade of events that ultimately result in a transient depression of GABA release from CB1-expressing interneurons. This retrograde short-term plasticity mechanism, named depolarization-induced suppression of inhibition (DSI) (Llano et al., 1991; Pitler and Alger, 1992), is abolished in the presence of CB1 receptor antagonists. DSI depends on the activity level in the postsynaptic cells (e.g., on the duration of the depolarizing pulse used to elicit DSI) and is subject to significant modulation at the postsynaptic site, for example, by muscarinic and metabotropic glutamate receptors (Varma et al., 2001; Ohno-Shosaku et al., 2003). Here, we tested the specific hypothesis that cannabinoid-mediated inhibition of GABA release depends not only on postsynaptic firing and depolarization but also on the level of activity in the presynaptic interneurons. The paired recording data from cholecystokinin-positive presynaptic basket cells and postsynaptic CA1 pyramidal cells show that the presynaptic, cannabinoid-mediated inhibition of GABA release strongly depends on interneuronal activity levels, both after the exogenous application of synthetic CB1 ligands and after the endogenous mobilization of endocannabinoids.
Materials and Methods
All protocols were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
Electrophysiology.
Transverse hippocampal slices (350 μm) were prepared from 18- to 21-d-old Sprague Dawley rats. Slices were incubated in sucrose-containing artificial CSF (ACSF) for 1 h (in mm: 85 NaCl, 75 sucrose, 2.5 KCl, 25 glucose, 1.25 NaH2PO4, 4 MgCl2, 0.5 CaCl2, and 24 NaHCO3). After the initial incubation period, slices were transferred in the same ACSF solution that was used for recordings (in mm: 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, and 10 glucose). All electrophysiological recordings were made at 33°C.
Slices were visualized with an upright microscope (BX-50; Olympus, Tokyo, Japan) with infrared differential interference contrast optics. Whole-cell recordings were obtained from the interneurons with patch pipettes (3–5 MΩ) filled with internal solution containing 126 mm K-gluconate, 4 mm KCl, 10 mm HEPES, 4 mm ATP-Mg, 0.3 mm GTP-Na, 10 mm phosphocreatine, and 0.2% biocytin, pH 7.2 (270–290 mOsm). The interneurons (all CCK-positive basket cells) were located in stratum radiatum, close to the pyramidal cell layer–stratum radiatum border. Pyramidal cells (whole-cell current-clamp configuration; the holding potential was −70 mV) were recorded with internal solution containing the following (in mm): 40 CsCl, 90 K-gluconate, 1.8 NaCl, 1.7 MgCl2, 3.5 KCl, 0.05 EGTA, 10 HEPES, 2 Mg-ATP, 0.4 Na2-GTP, and 10 phosphocreatine (pH 7.2; 270–290 mOsm). Recordings were made using MultiClamp 700A and MultiClamp 700B amplifiers (Molecular Devices, Union City, CA). Signals were filtered at 4 kHz using a Bessel filter and were digitized at 10 kHz with a Digidata 1320A analog–digital interface (Molecular Devices). Series resistances were carefully monitored, and the recordings were discarded if the series resistance changed >30% (average change in series resistance, 2.7 ± 0.2 MΩ) or reached 25 MΩ. Trains of presynaptic firing were always kept short (<200 pulses) to avoid depletion of the releasable pool of vesicles. For the DSI experiments, the presynaptic interneurons continued to discharge during the postsynaptically applied depolarizing pulse at the same rate as that before and after the pulse, and at least 2 min was allowed between two successive DSI-inducing depolarizing pulses. The recorded traces were analyzed using the Clampfit 9.0 software (Molecular Devices). IPSCs were individually inspected and included in analysis based on their onset latency after the presynaptic action potential. The analysis focused on the synchronous IPSCs (Hefft and Jonas, 2005), i.e., on those that were time locked to the action potentials (we considered an event to be time locked if it occurred within a time window defined by the mean latency to IPSC onset after a presynaptic spike, plus or minus 0.5 ms; see Results for latency values). IPSC decay time constants are the amplitude-weighted means of time constants in double-exponential fits. Data are presented as mean ± SEM. R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrol[1,2,3-de]-1,4-benzoxazin-6-yl)(1-naphthalenyl) methanone monomethanesulfonate [WIN55,212-2 (WIN)], nifedipine, ω-conotoxin GVIA and N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251) were purchased from Tocris Bioscience (Ellisville, MO).
Immunochemistry and neuroanatomy.
After recording, all slices were transferred into a fixative solution containing 4% paraformaldehyde and 0.2% picric acid in 0.1 m phosphate buffer. Slices were resectioned into 50-μm-thick sections and immunoreactivity for CCK was revealed with a mouse monoclonal antibody [mAb 9303, generously provided by the Antibody/Radioimmunoassay Core of the CURE/Digestive Diseases Research Center (University of California, Los Angeles), National Institutes of Health Grant DK41301; diluted 1:1000]; immunoreactivity for parvalbumin (PV) was tested with a rabbit polyclonal antibody [PV-28 (Swant, Bellinzona, Switzerland); diluted 1:1000 in Tris-buffered saline containing 2% normal goat serum]. The reactions were visualized with a goat anti-rabbit IgG conjugated to Alexa 488 (diluted 1:500 in Tris-buffered saline containing 2% normal goat serum; Invitrogen, Eugene, OR) and a goat anti-mouse IgG conjugated to Alexa 594 (diluted 1:500), streptavidin conjugated to Alexa 350 for biocytin (diluted 1:500). The sections were then mounted in Vectashield (Vector Laboratories, Burlingame, CA) and analyzed with a fluorescent microscope. To reveal the axonal and dendritic arbors of the presynaptic basket cell in detail, the biocytin-filled cells were subsequently visualized with 3,3′-diaminobenzidine tetrahydrochloride (0.015%) using a standard ABC kit (Vector Laboratories).
Results
Paired recordings of CCK+ basket cells and pyramidal cells in CA1
Action potentials in the post-hoc-identified presynaptic CCK+ basket cells (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) evoked IPSCs (eIPSCs) (peak amplitudes, 58.98 ± 7.09 pA) with a latency to onset of 1.8 ± 0.25 ms, fast kinetics (rise time constant, 0.58 ± 0.06 ms; decay time constant, 6.89 ± 1.0 ms), and frequent failures (proportion of failures, 59.8 ± 9.2%) (Fig. 1A, Control) in the postsynaptic pyramidal cells. All presynaptic cells were tested for immunoreactivity for both CCK and PV, and the coexpression of the two markers was never observed.
Figure 1.
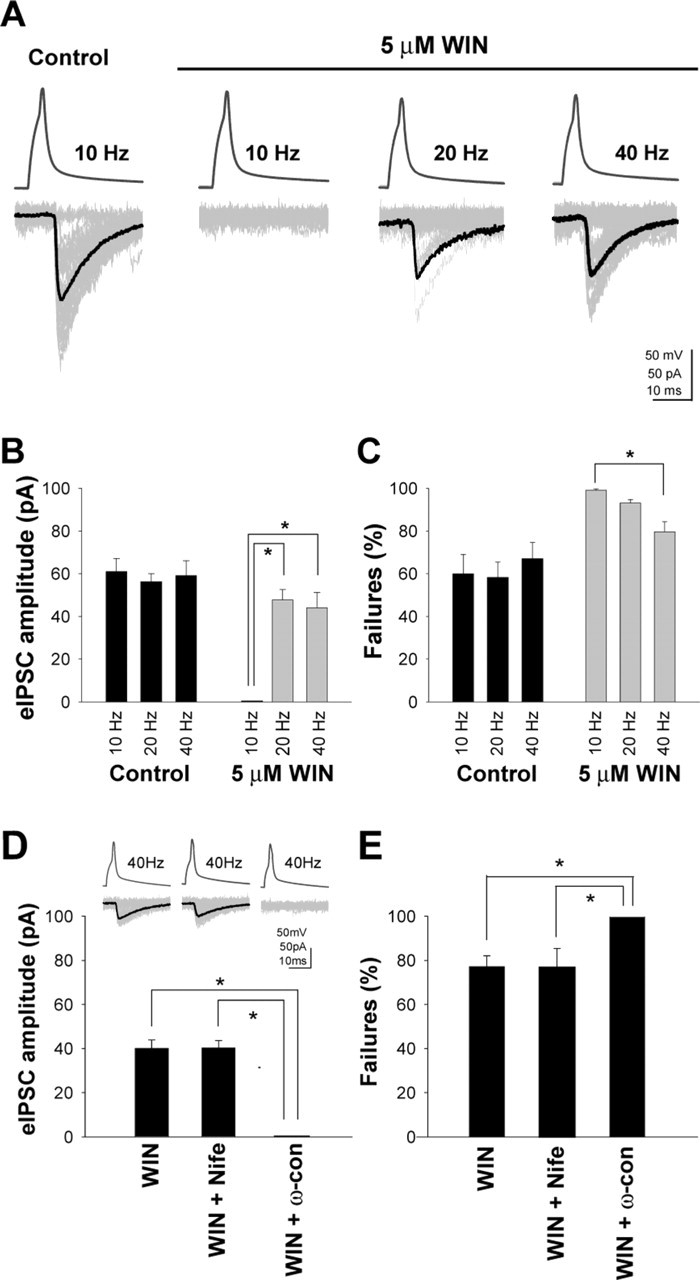
Presynaptic activity modulates CB1-receptor-mediated inhibition of GABAergic transmission in the presence of exogenously applied cannabinoid agonists. A, In control ACSF (control), action potentials at 10 Hz in the presynaptic CCK+ basket cell (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material) evoked IPSCs (eIPSCs) in the postsynaptic pyramidal cell (upper traces, presynaptic spikes; gray traces, eIPSCs, including failures; black traces, averaged eIPSCs without failures). As expected, these eIPSCs were abolished by the application of the CB1 receptor agonist 5 μm WIN (10 Hz). However, when the presynaptic firing was increased to 20 or 40 Hz in the same cell, the transmission reappeared, despite the continued presence of the agonist (WIN, 20 Hz; WIN, 40 Hz). B, C, Summary data showing the frequency-dependent changes in eIPSC amplitude (B) and failure rates (C) in control ACSF and in WIN. The data were obtained from responses elicited by ∼100 presynaptic action potentials in each cell in control ACSF and in WIN (note that this analysis actually underestimates the recovery in WIN, because it considers all responses, including those at beginning of the 100 action-potential-long trials when the eIPSCs appeared from the WIN-induced blockade of GABA release; see supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). D, E, Summary data (with example traces as insets) illustrating that GABAergic transmission observed in WIN at 40 Hz firing is dependent on N-type Ca2+ channels. Nife, Nifedipine; ω-con, ω-conotoxin GVIA. The asterisks indicate significant differences (paired t test, p < 0.05). Error bars represent SEM.
Presynaptic activity relieves inhibition of GABA release induced by exogenous cannabinoid agonist
As expected from previous studies in the hippocampus (Katona et al., 1999; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001; Wilson et al., 2001), the eIPSCs elicited by action potentials in the CCK-positive basket cells were abolished by the application of the CB1 receptor agonist WIN [5 μm (Fig. 1A); n = 9; the CB1 receptor antagonist 10 μm AM251 reversed the WIN blockade of eIPSCs; n = 8 (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material)]. However, the degree of CB1-mediated inhibition was strongly dependent on the presynaptic firing frequency (Fig. 1A). When the presynaptic firing rate was 10 Hz, the transmission showed 100% failures in the presence of WIN. In contrast, when the presynaptic interneuronal firing rate was increased to 20 or 40 Hz, the eIPSCs reappeared, despite the continuous presence of the agonist, in all cell pairs tested (n = 9). Therefore, these data demonstrate that CCK+ basket cells can substantially recover from CB1-receptor-mediated inhibition of GABA release in an activity-dependent manner. The quantification of these observations (Fig. 1B,C) (the course of recovery from WIN-induced inhibition is shown in supplemental Fig. 2B, available at www.jneurosci.org as supplemental material) also illustrates that the amplitude (Fig. 1B) and probability of failures (Fig. 1C) of the eIPSCs in the absence of the agonist (control) were similar at 10, 20, and 40 Hz, but the eIPSC amplitudes and the probability of failure were strongly dependent on the presynaptic firing rate in WIN.
Figure 2.
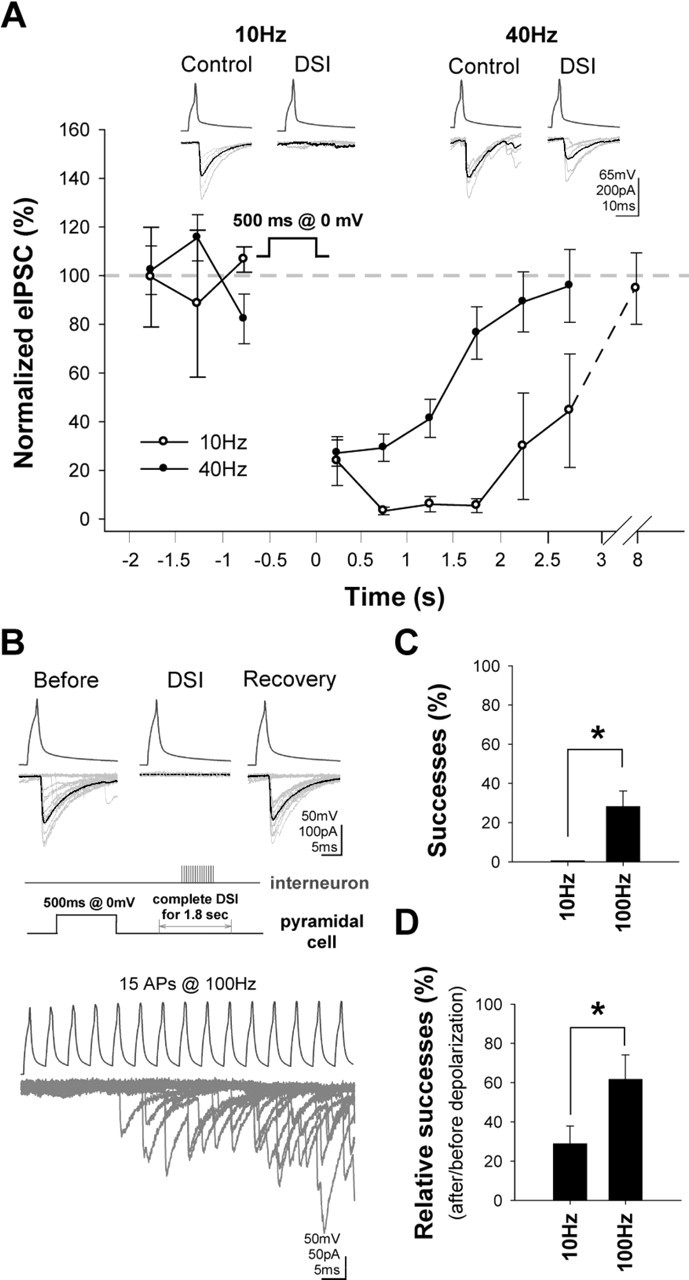
Basket cell firing rates modulate CB1-mediated inhibition of GABA release induced by the endogenous mobilization of cannabinoids. A, Depolarization of the postsynaptic cell evoked strong DSI when the presynaptic firing was 10 Hz. However, the same depolarization evoked a significantly smaller and shorter-lasting DSI when the interneuron firing was 40 Hz. The plot shows summary data from n = 6 pairs (insets, example traces; the averaged eIPSCs include failures). B, C, Burst-like trains of 15 action potentials at 100 Hz were able to evoke eIPSCs during DSI that was complete (i.e., 100% failure rate) with 10 Hz firing. Twelve traces are superimposed in B. Summary data are shown in C from n = 4 pairs. D, Summary data of relative success rates (during vs preceding DSI) observed with trains of 15 action potentials either at 10 or 100 Hz, in n = 8 pairs. Errors bars represent SEM.
N-type Ca2+ channels mediate the activity-dependent recovery of transmission
GABA release from CB1-receptor-expressing, CCK+ basket cells depends on presynaptic, ω-conotoxin-sensitive, N-type Ca2+ channels (Wilson et al., 2001). As shown in Figure 1, D and E, the “recovered” GABAergic transmission in the presence of WIN at a 40 Hz presynaptic firing rate was abolished by ω-conotoxin GVIA (250 nm; n = 4), but it was not affected by the L-type Ca2+ channel blocker nifedipine (10 μm; n = 4). These data demonstrate that the same type of presynaptic Ca2+ channel is responsible for GABA release from CCK+ basket cells in the absence and presence of the exogenous CB1 receptor agonist.
Magnitude of DSI is a function of interneuronal firing rates
Subsequently, we used the DSI paradigm (Llano et al., 1991; Pitler and Alger, 1992) to determine whether interneuronal firing modulates CB1-receptor-mediated inhibition of GABA release under conditions in which the CB1 receptors are activated by the endogenous ligand. In n = 6 pairs, when the transmission was tested with 10 Hz presynaptic firing rate, 500-ms-long depolarizations to 0 mV in the postsynaptic cells evoked strong DSI (Fig. 2A). However, when the interneuronal firing rate was increased to 40 Hz (in the same n = 6 pairs), identical postsynaptic depolarizations caused significantly smaller and shorter-lasting DSI [note that CB1 receptor antagonist AM251 abolished DSI; n = 4 (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material)]. Therefore, these data show that presynaptic activity can modulate CB1-receptor-mediated inhibition of GABA release after both exogenous application and endogenous mobilization of cannabinoids.
Short bursts of action potentials can overcome even complete DSI
In the previous experiments, we used tonic firing in the presynaptic cells to probe the activity-dependent changes in the efficacy of CB1 receptors in inhibiting GABA release. However, interneurons do not typically fire in a sustained, tonic manner in vivo. Therefore, in two sets of experiments, we tested whether short bursts of high-frequency action potential discharges in the presynaptic cells decrease the CB1-receptor-mediated inhibition of GABA release during DSI. In the first experiment, we used pairs (n = 4; pairs different from those in the experiments related to Fig. 2A) in which, when tested with 10 Hz firing, the DSI was complete for a prolonged period (for 2.2 ± 0.3 s). During this period of complete DSI, we evoked a brief train of 15 action potentials at 100 Hz. As shown in Figure 2, B and C, despite the 100% failure rate during the complete DSI at 10 Hz, successful transmission appeared during the high-frequency train even after a few (6.7 ± 1.2) action potentials (note that the bottom trace in Fig. 2B shows superimposed responses from several trials). These data show that complete DSI can be overcome with even a short burst of high-frequency firing by the CCK-positive basket cells.
In the second set of experiments, we used pairs that were not preselected for displaying 100% DSI at 10 Hz firing (i.e., all recorded pairs, regardless of the magnitude of DSI at 10 Hz, were examined; n = 8). In addition, in these experiments, the relative probability of successes (i.e., percent successes during versus before DSI) were compared at 10 and 100 Hz (in both cases, 15 action potentials were delivered in a train). As shown in Figure 2D, the relative probability of successes was 23.8 ± 9.0% with the 10 Hz train, which increased to 61.7 ± 12.5% with the 100 Hz train, again indicating that the magnitude of DSI is strongly dependent on the presynaptic firing frequency.
Discussion
Interneuronal firing rate as a major determinant of the efficacy of presynaptic cannabinoid actions
Based on the present paired recording data, we conclude that cannabinoid-dependent inhibition of GABA release is significantly modulated by activity in both the postsynaptic (e.g., depolarization and firing of the postsynaptic cell during DSI) and the presynaptic neurons. There have been several indications in the literature that suggested that presynaptic firing frequency may modulate cannabinoid effects on GABA release. First, at the most general level, the context dependency of some cannabinoid actions on behavior is well established. For example, users report widely different effects of marijuana, depending on expectations of the experience, social environment, and emotional state (Iversen, 2000). Second, certain pharmacological agents, such as the K+ channel blocker 4-AP, have been reported to modulate DSI through a presynaptic action (Varma et al., 2002). Because 4-AP not only suppressed DSI but also induced synchronized burst firing in hippocampal slices, the hitherto unproven hypothesis was proposed that the repetitively discharging interneurons may have played a key role in the 4-AP-induced suppression of DSI (Varma et al., 2002). Third, membrane depolarization is known to relieve G-protein-mediated inhibition of voltage-gated Ca2+ channels (Bean, 1989; Boland and Bean, 1993; Ikeda, 1996), suggesting that higher presynaptic firing rates may be able to counteract and relieve CB1-receptor-induced depression of GABA release from basket cell terminals. However, N-type Ca2+ channels in cultured hippocampal neurons showed weaker relief from G-protein inhibition during action potential trains compared with P/Q-type channels (Brody and Yue, 2000). Finally, a recent study showed that, at synapses between CCK-positive, mossy-fiber-associated interneurons and pyramidal cells in the CA3 region, a strong increase in the presynaptic interneuronal firing relieved the tonic silencing of these synapses by persistent CB1 receptor activity (Losonczy et al., 2004). However, it was not clear whether CB1-expressing interneurons outside the CA3 region can also overcome complete CB1-induced suppression of GABA release by increased presynaptic discharge rates.
Although these previous behavioral, pharmacological, and physiological data were consistent with the hypothesis that interneuronal firing rates may modulate CB1-mediated inhibition of GABA release, the present results are the first to conclusively demonstrate that the efficacy of cannabinoid control of GABA release, including DSI-related processes, is strongly influenced by the activity levels in the presynaptic interneurons. The ability of presynaptic activity to overcome even complete inhibition of GABA release by CB1 receptors likely involves the voltage-dependent removal of G-protein-mediated inhibition of N-type Ca2+ channels (Bean, 1989; Boland and Bean, 1993; Ikeda, 1996), perhaps together with the summation of subthreshold presynaptic Ca2+ rises in the presence of CB1 agonists (Brown et al., 2004). The present data show that presynaptic CB1 receptors are similar to GABAB receptors in this regard, because the efficacy of GABAB receptors can also be significantly regulated by presynaptic discharge rates (Davies and Collinridge, 1993; Brenowitz et al., 1998; Brody and Yue, 2000). The effect of the elevated firing rate on synaptic transmission in WIN, but not in the absence of WIN (Fig. 1B,C), may indicate that presynaptic firing modulates specific processes within the CB1-mediated pathway, although roles for nonspecific factors cannot be excluded (e.g., upregulation of GABA release by high firing rate and depletion of the releasable vesicle pool).
Functional implications
Recent in vivo recordings revealed that CA1 interneuronal subtypes, including CCK-positive basket cells, show highly characteristic, brain state- and behavior-specific action potential discharges (Klausberger et al., 2003, 2005). Therefore, a functional corollary of our findings is the prediction, to be tested in future studies, that the degree of cannabinoid-mediated inhibition of GABA release is strongly dependent on the behavioral state of the animal. The exact in vivo pattern of firing by CCK-positive cells is only beginning to be described precisely. A recent in vivo study (Klausberger et al., 2005) showed that CCK-positive basket cells, similar to the previously studied other interneuronal subtypes in the CA1 (Klausberger et al., 2003), displayed significant changes in firing probabilities during different phases of theta cycles and between theta-ripple oscillations. Although the CCK-positive basket cells recorded in the Klausberger et al. (2005) study fired at a mean rate of 10 Hz during theta under anesthesia, the discharge rate of these cells is likely to be higher under anesthesia-free conditions. Indeed, presumed CCK-expressing interneurons (identified on the basis of their characteristic phase relationships during theta and ripple oscillations) recorded in nonanesthetized rats did show occasional high-frequency action potential bursts during theta waves [e.g., in Klausberger et al. (2005), the first burst in their Fig. 7B exhibits an ∼100 Hz discharge, similar to that used in our Fig. 2B,C]. These in vivo data indicate that it is likely that CCK-positive basket cells, particularly in freely behaving, nonanesthetized animals, exhibit a wide dynamic range of action potential firing that include both the lower (10 Hz) and higher (20 to 100 Hz) frequencies that are sufficient to modulate cannabinoid-mediated inhibition of GABA release. It is interesting to speculate that the opposing influence of pyramidal cell and interneuronal action potential discharges on DSI may lead to a certain normalization of cannabinoid-mediated suppression of GABA release in brain states in which pyramidal cell and CCK-positive basket cell populations are coactivated, because, in such situations, increases in DSI attributable to enhanced pyramidal cell bursts would be expected to be counteracted by the DSI-decreasing effects of the augmented interneuronal discharges.
Footnotes
This work was supported by National Institutes of Health Grant NS 38580 (I.S.) and Deutsche Forschungsgemeinschaft Grant NE1185/1-1 (A.N.). We thank R. Zhu for expert technical assistance and Dr. Allyson Howard for advice.
References
- Bean PB (1989). Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature 340:153–156. [DOI] [PubMed] [Google Scholar]
- Boland LM, Bean PB (1993). Modulation of N-type calcium channels in bullfrog sympathetic neurons by luteinizing hormone-release hormone: kinetics and voltage-dependence. J Neurosci 13:516–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz S, David J, Trussell L (1998). Enhancement of synaptic efficacy by presynaptic GABAB receptors. Neuron 20:135–141. [DOI] [PubMed] [Google Scholar]
- Brody DL, Yue DT (2000). Relief of G-protein inhibition of calcium channels and short-term synaptic facilitation in cultured hippocampal neurons. J Neurosci 20:889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SP, Safo PK, Regehr WG (2004). Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci 24:5623–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CH, Collinridge GL (1993). The physiological regulation of synaptic inhibition by GABAB receptors in rat hippocampus. J Physiol (Lond) 472:245–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P (2005). Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci 8:1319–1328. [DOI] [PubMed] [Google Scholar]
- Ikeda SR (1996). Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature 380:255–258. [DOI] [PubMed] [Google Scholar]
- Iversen LL (2000). In: The science of marijuana pp. 78–79. New York: Oxford UP.
- Katona I, Sperlagh B, Sik A, Kofalvi A, Vizi ES, Mackie K, Freund TF (1999). Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19:4544–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Magill PJ, Marton LF, Roberts JD, Cobden PM, Buzsaki G, Somogyi P (2003). Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature 421:844–848. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Marton LF, O’Neill J, Huck JHJ, Dalezios Y, Fuentealba P, Yee Suen W, Papp E, Kaneko T, Watanabe M, Csicsvari J, Somogyi P (2005). Complementary roles of cholecystokinin- and parvalbumin-expressing GABAergic neurons in hippocampal network oscillations. J Neurosci 25:9782–9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001). Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727. [DOI] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A (1991). Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron 6:565–574. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Biro AA, Nusser Z (2004). Persistently active cannabinoid receptors mute a subpopulation of hippocampal interneurons. Proc Natl Acad Sci USA 101:1362–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M (2001). Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29:729–738. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Matsui M, Fukudome Y, Shosaku J, Tsubokawa H, Taketo MM, Manabe T, Kano M (2003). Postsynaptic M1 and M3 receptors are responsible for the muscarinic enhancement of retrograde endocannabinoid signalling in the hippocampus. Eur J Neurosci 18:109–116. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE (1992). Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci 12:4122–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE (2001). Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. In: J Neurosci 21:1-5 pp. RC188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma N, Brager D, Morishita W, Lenz RA, London B, Alger B (2002). Presynaptic factors in the regulation of DSI expression in hippocampus. Neuropharmacology 43:550–562. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001). Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410:588–592. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA (2001). Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31:453–462. [DOI] [PubMed] [Google Scholar]