

Abstract
The development of ordered connections or “maps” within the nervous system is a common feature of sensory systems and is crucial for their normal function. NMDA receptors are known to play a key role in the formation of these maps; however, the intracellular signaling pathways that mediate the effects of glutamate are poorly understood. Here, we demonstrate that SynGAP, a synaptic Ras GTPase activating protein, is essential for the anatomical development of whisker-related patterns in the developing somatosensory pathways in rodent forebrain. Mice lacking SynGAP show only partial segregation of barreloids in the thalamus, and thalamocortical axons segregate into rows but do not form whisker-related patches. In cortex, layer 4 cells do not aggregate to form barrels. In Syngap+/− animals, barreloids develop normally, and thalamocortical afferents segregate in layer 4, but cell segregation is retarded. SynGAP is not necessary for the development of whisker-related patterns in the brainstem. Immunoelectron microscopy for SynGAP from layer 4 revealed a postsynaptic localization with labeling in developing postsynaptic densities (PSDs). Biochemically, SynGAP associates with the PSD in a PSD-95-independent manner, and Psd-95−/− animals develop normal barrels. These data demonstrate an essential role for SynGAP signaling in the activity-dependent development of whisker-related maps selectively in forebrain structures indicating that the intracellular pathways by which NMDA receptor activation mediates map formation differ between brain regions and developmental stage.
Keywords: SynGAP, barrels, NMDA receptors, somatosensory, development, PSD-95
Introduction
Sensory cortices are organized into topographic maps whereby the pattern of the peripheral receptors is faithfully recapitulated onto the cortical surface via thalamocortical afferents (TCAs). Within these topographic maps, structural and functional specializations occur, such as the whisker-related modules, the “barrels,” of the rodent somatosensory cortex (Woolsey and Van der Loos, 1970; Killackey and Belford, 1979). Initially, TCAs overlap tangentially in layer 4 and subsequently segregate into whisker-related clusters (Rebsam et al., 2002) in a process that is modulated by presynaptic serotonin signaling (Gaspar et al., 2003). Subsequently, cortical neurons form cellular aggregates around the TCA clusters (Woolsey and Van der Loos, 1970) in a process that is regulated by glutamate neurotransmission (Erzurumlu and Kind, 2001; Kind and Neumann, 2001). Mice lacking the metabotropic glutamate receptor 5 (mGluR5) or cortical NMDA receptors (NMDARs) fail to form barrels despite at least partial segregation of TCAs (Iwasato et al., 2000; Hannan et al., 2001). Similarly, activity-dependent changes in neuronal phenotype in response to altered sensory activity are dependent on NMDAR activation (Schlaggar et al., 1993; Fox et al., 1996; Datwani et al., 2002).
The characterization of the postsynaptic density (PSD) and NMDA receptor complex has provided a framework for identifying candidate proteins regulated by glutamate receptors that may be involved in the cellular processes mediating barrel formation (Husi et al., 2000; Walikonis et al., 2000). One such protein is SynGAP, a synaptic GTPase activating protein that regulates small G-proteins (Ras, Rab, and Rap) (Chen et al., 1998; Kim et al., 1998; Krapivinsky et al., 2004; Tomoda et al., 2004). SynGAP binds to the NMDA receptor via its association with membrane associated guanylate kinases (MAGUKs), including PSD-95 and synapse-associated protein 102 (SAP-102) and negatively regulates the extracellular-regulated kinase (ERK) signaling cascade (Komiyama et al., 2002). ERK regulates numerous forms of plasticity, including the critical period plasticity in rodent visual cortex, long-term potentiation (LTP), and learning and memory (Di Cristo et al., 2001; Adams and Sweatt, 2002). It also regulates cytoskeletal rearrangements, synaptogenesis, dendritic morphology, and cellular migration (Ho et al., 2001; Adams and Sweatt, 2002). Thus, SynGAP may represent a key link from NMDARs to downstream pathways regulating barrel development.
A single gene encodes SynGAP, although the mRNA can be spliced to give several distinct proteins, the functions of which are not clear (Li et al., 2001). It is expressed throughout the cortex, hippocampus, and thalamus of postnatal mice, suggesting a role in developmental plasticity (Porter et al., 2005). SynGAP homozygous null mutant mice die perinatally (Komiyama et al., 2002; Kim et al., 2003; Vazquez et al., 2004), although some live to postnatal day 5 (P5) to P7 (Kim et al., 2003); heterozygotes are viable. The brains of Syngap−/− mice appear grossly normal, and the cause of death is not clear. Syngap+/− mice show a significant reduction in LTP) and a shift in the plasticity frequency function (Komiyama et al., 2002; Kim et al., 2003) and defects in spatial learning (Komiyama et al., 2002). Cultured hippocampal neurons lacking SynGAP show precocious spine and synapse formation, and spines are significantly larger than normal (Vazquez et al., 2004).
Materials and Methods
Breeding and genotyping of transgenic mice.
Syngap heterozygous mating pairs on the MF1 background were used to derive wild-type (WT), heterozygous, and homozygous pups for experimental analysis. PCR genotyping from purified genomic DNA was performed as described previously (Komiyama et al., 2002). All animals were treated in accordance with the UK Animal Scientific Procedures Act (1986).
Biochemistry.
Mice were killed either by cervical dislocation or decapitation, and S1 cortices dissected from P0, P4, P7, P14, P21, and adult mice were immediately frozen on dry ice and stored at −70°C. For developmental analysis, barrel cortices were homogenized in lysis buffer (50 mm HEPES, pH 7.5, 1% Triton X-100, 50 mm NaCl containing protease inhibitors, phosphatase inhibitor cocktails I and II) (P2850 and P5276; Sigma, Poole, UK). Protein concentrations were determined by Bradford assays, and immunoblot analysis was performed according to the methods of Kind et al. (1994). Briefly, 10 μg of protein was loaded per lane of each age on a 7 or 10% polyacrylamide gel with a 4% stacking gel. The proteins were then transferred to nitrocellulose membranes, which were then stained with amido black to confirm equal loading of protein. The blots were then incubated in primary antibody [calcium calmodulin-dependent protein kinase II (CaMKII), 1:1000 (Promega, Madison, WI); PSD-95, 1:20,000 (Upstate Biotechnology, Lake Placid, NY); pan-SynGAP, 1:4000 (Affinity BioReagents, Golden, CO); SynGAPα, 1:2000 (Upstate Biotechnology); SAP-102, 1:5000 (Santa Cruz Biotechnology, Santa Cruz, CA; Chemicon, Temecula, CA; Alomone Labs, Jerusalem, Israel; or Synaptic Systems, Gottingen, Germany); NR2A, 1:2000 (Chemicon); NR1, 1:10,000 (Santa Cruz Biotechnology)] overnight at room temperature before being placed in secondary antibodies (anti-mouse IgG 1:10,000, anti-rabbit IgG 1:25,000, anti-goat IgG 1:50,000; Sigma) coupled to HRP for 1–2 h. Proteins were visualized using ECL reagents (Amersham Biosciences, Piscataway, NJ) and XAR Kodak (Rochester, NY) autoradiographic film.
Synaptosome and PSD fractionation.
Synaptosome preparations were prepared according to the methods of Dunkley et al. (1986). Briefly, S1 cortex was homogenized in a 320 mm sucrose solution (pH 7.4, containing 1 mm EDTA and 5 mm Tris) and then poured onto Percoll gradients (3 ml layers of 24, 10, and 3% Percoll) that were centrifuged for 12 min at 15,000 rpm at 4°C. Synaptosomes were removed from between the 24 and 10% Percoll layers and spun for 30 min at 13,000 rpm in ice-cold 320 mm sucrose solution. The resulting synaptosome pellet was then resuspended and centrifuged in cold minus Ca2+ Krebs buffer (containing NaCl, KCl, MgSO4 glucose, Na2HPO4·12H20, and HEPES) at 13,000 rpm for 10 min, twice in succession. Synaptosomes were treated with lysis buffer [50 mm HEPES, pH 7.5, 1% Triton X-100, 50 mm NaCl containing protease inhibitors, phosphatase inhibitor cocktails I and II (Sigma P2850 and P5276)]. The PSD fraction was then pelleted by two successive centrifugations at 36,800 × g for 45 min.
Tissue preparation for histology.
Mice were anesthetized with an overdose of sodium pentobarbital (Euthanal; 200 mg/kg, i.p.) and were perfused with PB followed by 4% (w/v) paraformaldehyde in 0.1 m phosphate buffer. The brains were removed, fixed overnight in 4% paraformaldehyde, and cryoprotected overnight in 30% (w/v) sucrose. The brains were sectioned on a freezing microtome either coronally or tangentially to the pial surface at 48 μm. For tangential sections, cortices were dissected from the thalamus, and the hippocampus and striatum were removed. The cortex was then flattened on the freezing microtome stage with the pial surface facing up.
Histology.
5-Bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) staining was performed as described previously (Porter et al., 2005). At least three animals at each age from at least two separate litters were used for X-Gal staining. Thionin (Nissl) and cytochrome oxidase (CO) staining was performed as described previously (Hannan et al., 2001). For 5-HT and serotonin transporter (5-HTT) immunohistochemistry, sections were incubated overnight in rat anti-mouse 5-HT (1:10 to 1:20; Harlan Sprague Dawley, Indianapolis, IN) or rabbit anti-mouse 5-HTT (1:2000; Calbiochem, La Jolla, CA), respectively, diluted in PBS containing 0.2–0.5% Triton X-100 or DMEM containing 5% fetal calf serum and 0.2–0.5% Triton X-100. Visualization of 5-HT was then performed as described previously (Vitalis et al., 2002). Sections reacted for calretinin (1:3000; Swant, Bellinzona, Switzerland) and cAMP-dependent protein kinase A regulatory subunit IIβ (PKARIIβ) (1:600; Santa Cruz Biotechnology, Santa Cruz, CA) were incubated overnight in antibody diluted in DMEM containing 5% fetal calf serum and 0.2–0.5% Triton X-100. Visualization was performed using a Vectastain ABC kit (Vector Laboratories, Burlingame, CA). To reveal barrels, flattened sections were mounted on slides, dried overnight, and stained with cresyl violet acetate (0.5%). In both cases, sections were dehydrated into xylene and coverslipped. For propidium iodide (PI) staining, free-floating sections were collected serially in PBS and incubated for 30 min in a solution containing 1:1000 propidium iodide (Invitrogen, San Diego, CA). Sections were rinsed in PBS, mounted on gelatin-coated slides, and coverslipped in PBS-glycerol (3:1; w/v). To reveal SynGAP protein localization, 48 μm frozen sections were incubated free-floating overnight at room temperature in a 1:200 to 1:600 dilution of rabbit anti-mouse pan-SynGAP antibodies (1:4000; Affinity BioReagents) in DMEM containing 5% fetal calf serum and 0.2% Triton X-100. Signal was then amplified by incubation in biotinylated goat anti-rabbit secondary antibody and streptavidin-coupled HRP.
Measurements.
All analyses were performed blind to genotype. No difference in cortical thickness or neocortical area was observed in mice between P5 and P7; therefore, animals at these ages were combined for analysis. Cortical thickness measurements in posterior medial barrel subfield (PMBSF) were obtained from Nissl-stained 48 μm coronal sections equivalent to between −1.82 and −1.94 mm posterior and between 2.8 and 3.2 mm lateral to bregma (Franklin and Paxinos, 1997). Additional cortical thickness measurements in the anterior snout region were obtained from Nissl-stained 48 μm coronal sections equivalent to between −0.94 and −1.06 mm posterior and between 3.0 and 3.4 mm lateral to bregma. In all cases, adjacent sections stained with 5-HTT were used to confirm the location within PMBSF or the anterior snout region. Layer 4 thickness and layer 5/6 thickness measurements were obtained in identical sections stained with anti-PKARIIβ and anti-calretinin antibodies, respectively. All area measurements were obtained from tangential sections stained with anti-5-HTT or anti-5HT antibodies. Images were analyzed at a final magnification of 40× (S1 cortex area), 80× (PMBSF area), and 160× (C1 barrel area, cortical thickness and cortical layer thickness measurements) using a Leica DMLB microscope and the Leica DMLB Image Manager version 4.0 program. All linear and area measurements were performed using the Image Tool for Windows version 3.0 software (University of Texas Health Science Centre at San Antonio, San Antonio, TX). Each image measurement was calibrated using a 1 mm graticule (Graticules, Tonbridge, Kent).
Cell counts.
Sections were viewed with a Leica confocal microscope. Three to four adjacent sections containing the barrel-field representation were analyzed for each animal. From these sections, the position of individual barrels were determined, and a series of confocal images of B3 and its neighboring barrels were taken with 7 μm intervals using the 10 and 20× objectives. Morphometric analysis was performed with the Leica software (TCNST). For each series of optical images, the section containing the clearer representation of the barrel of interest was used to start the analysis. All analysis was performed blind to genotype. On this selected section, three rectangles along the B2–B4 axis, one containing the B2–B3 wall, one the B3 hollow, and one the B3–B4 wall were drawn to calculate the density of propidium-stained nuclei in each rectangle. Within each animal, rectangle size remained constant but varied slightly between animals to ensure that its outer limits were restricted to the structure (barrel wall or hollow) of interest. Each rectangle was 60–70 × 80 μm, and in all cases, the rectangle was restricted to the structure (i.e., barrel wall or hollow) being measured. Counts were then normalized to the area of the rectangle being measured. Measurements were done on the selected section and on the two adjacent sections (upper and lower). From these data, the average density of propidium-stained nuclei in the walls and hollow of the chosen barrel was calculated. Measures are means of average densities ± SEM (P8, wild type, n = 8; Syngap+/−, n = 8). Average differences in nuclear densities taken from walls and hollows of the wild-type and Syngap+/− groups were statistically different (Student’s t test; *p < 0.01).
Electron microscopy.
Animals for electron microscopy were perfused as above, except that 0.1% glutaraldehyde was included in the fixative. Vibratome sections, 50-μm-thick, were placed in 1:200 dilution of rabbit anti-mouse SynGAP overnight at 4°C in the absence of detergent and reacted for DAB histochemistry as described above. They were then postfixed in 1% osmium tetroxide in 0.1 m phosphate buffer for 45 min. After dehydration through an ascending series of ethanol solutions and propylene oxide, all sections were embedded on glass slides in Durcupan resin. Regions of cortex (∼1 × 1 mm) to be used for assessment were then cut out using a scalpel and glued onto a resin block for sectioning. Ultrathin sections (∼70 nm) were cut and collected on formvar-coated grids (Agar Scientific, Stansted, UK), stained with uranyl acetate and lead citrate in a LKB Ultrostainer, and then assessed in a Philips CM12 transmission electron microscope. Negatives taken in the microscope were scanned onto an Apple Macintosh G5 computer using an Epson 4870 Photo flatbed scanner at 1200 dpi, before being prepared for presentation in Adobe Photoshop (Adobe Systems, San Jose, CA).
Analysis of gene expression using real-time reverse transcription-PCR.
The barrel cortex was dissected from mice between P0 and adult, frozen on dry ice, and stored at −70°C. Total RNA was extracted using an RNeasy mini-kit (Qiagen) and an RNase-Free DNase set (Qiagen). Total RNA was run on 0.8% agarose gel to ensure that RNA was not degraded (28S ribosomal band was well defined and double the intensity of 18S ribosomal band) or the sample was discarded. First-strand cDNA synthesis was performed as described by Barnett et al. (1998). Real-time reverse transcription (RT)-PCR was performed using MJ Research DNA Engine Opticon and Quantitect SYBR Green PCR kit (Qiagen). In each PCR, 1 μl of cDNA was combined with gene-specific primers (0.5 μm) and 12.5 μl of QuantiTect SYBR Green PCR Master Mix to a total volume of 25 μl. To compare expression levels at different developmental stages, a dilution series of control cDNA was made and assayed in each Opticon run. The dilution series was used from cDNA of the developmental stage predicted to give the highest expression of the gene product being amplified. Other controls performed in each run were RT and water blanks.
At the end of each run, melting curve analysis was performed between 60 and 90°C, and single melting peak demonstrated specific product. OpticonMonitor analysis software (version 1.01) was used to compare amplification in experimental samples during the log-linear phase to the standard curve from the dilution series of control cDNA. Comparisons were displayed as histograms. 18S rRNA and glyceraldehyde-3-phosphate dehydrogenase were used as a loading control, and each bar was normalized to the level of 18S rRNA expression. Primer sets used were pan-Syngap-F 5′-CGAAGTGCTGACCATGAC-3′, pan-Syngap-R 5′-CGGCTGTTGTCCTTGTTG-3′, 18S-F 5′-GTGGAGCGATTTGT-CTGGTT-3′, and 18S-R 5′-CAAGCTTATGACCCGCACTT-3′.
Analysis of SynGAP splice variant expression.
Ventral posterior medial nucleus (VpM) and layer IV of S1 cortex were dissected from P8 C57BL/6 mice in PBS and immediately frozen on dry ice. Total RNA was extracted using an RNeasy mini kit (Qiagen) and dissolved in RNase-free dH2O. RNA quality was verified by visualization on an agarose gel. cDNA synthesis was performed as done by Barnett et al. (1998), and PCR was performed with primers that detect all known Syngap 3′ isoforms (Li et al., 2001; Kim et al., 1998). The resulting PCR products were electrophoretically separated on an agarose gel, extracted, and ligated into P-GEM-T Easy Vector (Promega) and transformed into JM109 competent cells. Plasmid DNA was extracted using a Quiagen mini-prep kit, and the insert was sequenced by MWG (Ebersberg, Germany).
Results
SynGAP is necessary for barrel formation
Previous reports have demonstrated that Syngap−/− mice die within 48 h of birth (Komiyama et al., 2002; Vazquez et al., 2004), although one report showed mice living as late as P7. We observed a propensity for Syngap−/− mice to die when part of large litters, however, when competition between littermates is reduced, either as a result of naturally small litters or deliberate culling of littermates, we have been able to maintain homozygous knockout animals to 1 week of age.
To determine whether SynGAP plays a role in the general development and lamination of the cortex, we examined the expression of several layer-specific markers including 5-HTT, PKARIIβ, and calretinin in the barrel field in P5–P7 Syngap−/−, Syngap+/−, and Syngap+/+ mice (Fig. 1). Qualitatively, no difference was seen in the laminar expression patterns of these proteins in Syngap+/− or Syngap−/− mice relative to littermate control animals. Quantitatively we examined the following: (1) cortical thickness in coronal Nissl sections through both PMBSF and the anterior snout whiskers region; (2) areas of neocortex, S1, and PMBSF in flattened sections labeled with 5-HTT; (3) radial thickness of TCA terminals in coronal sections labeled with 5-HTT in sections through both PMBSF and the anterior snout whiskers region; (4) radial thickness of layers 1–4 and 5–6 in calretinin-labeled sections through both PMBSF and the anterior snout whiskers region; and (5) area of B1–B3, C1–C3, and D1–D3 barrels in Syngap+/+ and Syngap+/− animals (Figs. 1m, 2f) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). No significant difference was present between WT and heterozygous animals indicating that the cortex develops normally in these mice. In comparison with Syngap+/+ and Syngap+/− animals, Syngap−/− mice demonstrate a small reduction in cortical thickness in PMBSF and the anterior snout region. The decrease in cortical thickness affected all layers. These results (summarized in supplemental Table 1, available at www.jneurosci.org as supplemental material) are in good agreement with the reduction in body weight and brain size reported previously (Kim et al., 2003;Vasquez et al., 2004).
Figure 1.

Normal cortical lamination in Syngap+/− and Syngap−/− mice. Coronal sections through S1 cortex of P6/7 Syngap+/+ (a, d, g, j), Syngap+/− (b, e, h, k), and Syngap−/− (c, f, i, l) mice stained for Nissl substance (a–c), calretinin (d–f), 5-HTT (g–i), and PKA RIIβ (j–l). No qualitative difference in pattern of staining is visible between genotypes with the exception of the lack of segregation of TCAs seen in the Syngap−/− animals. Quantitative analysis of cortical thickness, radial thickness of TCA terminals, layer 5/6 thickness, and layer 1–4 thickness was also calculated (m). In all cases, there was no significant difference between Syngap+/+ and Syngap+/− animals. There was a significant decrease in Syngap−/− compared with Syngap+/+ and Syngap+/− animals in all parameters measured except 5-HTT terminal zone thickness in PMBSF. A complete numerical account of these data is presented in supplemental Table 1 (available at www.jneurosci.org as supplemental material). Scale bar, 250 μm. Error bars represent SE.
Figure 2.

Lack of barrel formation in Syngap−/− mice. Nissl staining of flattened sections through layer 4 of P6.5 Syngap+/+ (a) and Syngap−/− (b) mice showing a complete absence of cellular aggregation in Syngap−/− mice. Flattened sections through layer 4 of Syngap+/+ (c), Syngap+/− (d), and Syngap−/− (e) mice were immunostained for 5-HT to reveal the distribution of TCAs. Clear segregation of primary visual (V1), somatosensory (S1), and auditory (A1) as well as secondary somatosensory (S2) cortical areas can be seen in all three genotypes indicating no general defect in TCA pathfinding in Syngap mutants. Within S1, the representation of different body regions (PMBSF and the anterior snout, lower lip, forepaw, and hindpaw representations) are also clearly defined (arrows). However, within the anterior snout, no TCA segregation is visible and, within PMBSF, TCAs can be seen segregating into rows, but patches corresponding to individual whiskers fail to form. Scale bar (in e): a, b, 1 mm; c–e, 800 μm. Quantification of neocortical area, area of S1, and PMBSF in Syngap+/+, Syngap+/−, and Syngap−/− animals revealed a small but significant decrease in neocortical area and area of PMBSF in Syngap−/− animals. The area of S1 was reduced in Syngap−/− animals, although this decrease was not significant. No significant difference between Syngap+/+ and Syngap+/− was seen in any these measurements. In addition, there was no significant difference in the area of individual TCA patches of PMBSF barrels in Syngap+/+ compared with Syngap+/− mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Error bars represent SE.
To determine whether SynGAP plays a role in the development of the primary somatosensory cortex, we examined the distribution of TCAs and soma of layer 4 neurons in Syngap+/+ (n = 4 for both TCAs and cell distribution) and Syngap−/− (n = 5) mice. Nissl staining in flattened sections through layer 4 of P6/7 Syngap−/− mice demonstrates a complete loss of cellular segregation into barrels in layer 4 (Fig. 2b). Supplemental Figure 2 (available at www.jneurosci.org as supplemental material) shows three adjacent sections through layer 4 of another Syngap−/− mouse. No barrels are visible in any section. Wild-type littermate control animals demonstrate a normal cellular aggregation in layer 4 (Fig. 2a). To elucidate the locus of the defect in Syngap−/− mice, we examined the state of TCA segregation using 5-HT immunohistochemistry in Syngap−/−, Syngap+/−, and Syngap+/+ mice (Figs. 1g–i, 2c–e). Serotonin immunoreactivity in coronal sections through the posteromedial barrel subfield of P6/P7 animals revealed TCAs were restricted to layer 4 in all three genotypes (Fig. 1g–i). In tangential sections through layer 4 of Syngap−/− animals (Fig. 2e), however, TCAs segregated into rows but not into individual whisker-related patches, indicating that while TCAs terminate correctly in the radial dimension but fail to segregate into a barrel-like pattern. In contrast, wild-type and Syngap+/− littermates showed normal segregation of TCAs in layer 4 (Fig. 2c,d). A small decrease in the size of S1 or PMBSF was seen in Syngap−/− animals compared with WT and Syngap+/− animals; this decrease was small and was not significant in the former (Fig. 2f) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Therefore, decrease in area of S1 and/or PMBSF cannot account for the loss of segregation seen in Syngap−/− animals. Interestingly, despite the reduced tangential TCA segregation in S1, the segregation of TCAs between cortical areas appears normal (i.e., V1, S1, S2, and A1 are all easily identifiable). Even subregions of S1 such as the PMBSF and the anterior snout, lower lip, forepaw, and hindpaw representations are all identifiable, indicating that the effects in S1 do not arise from a general defect in TCA pathfinding.
The complete lack of barrels, despite partial segregation of TCAs, suggested a role for cortically expressed SynGAP in barrel formation. To gain additional insight into the role of SynGAP, we examined the Syngap heterozygotes in more detail (Fig. 3). P8 Syngap+/− mice (Fig. 3b,d) demonstrated reduced segregation in PMBSF compared with age-matched wild-type mice (Fig. 3a,c). To quantify the change in segregation, tangential sections through layer 4 from eight wild-type and eight Syngap+/− mice were labeled with PI and labeled nuclei were visualized using the confocal microscope. All eight Syngap+/− mice showed reduced segregation compared with wild-type controls. In 7 μm optical sections, PI-labeled nuclei were counted in both the barrel wall and barrel hollow of barrel B3 (Fig. 3e). The mean number of cells in the wall of the B3 barrel was 32.7 ± 2.8 per 500 μm2 in Syngap+/+ and was significantly reduced to 26.6 ± 0.13 in Syngap+/− mice (Student’s t test; p < 0.01). Analysis of cell density in the barrel hollows revealed a significant increase in the Syngap+/− (23.8 ± 1.7) compared with Syngap+/+ (21.1 ± 2.8) mice (p < 0.01). In Syngap+/+, the ratio of PI-labeled nuclei in the barrel wall to barrel hollow was 1.73 ± 0.16. In contrast, this ratio was significantly reduced in Syngap+/− mice to 1.25 ± 0.13 (p < 0.01) (Fig. 3f). The decrease in wall-to-hollow ratio reflects an increase in the number of neurons in the barrel hollow and a decrease in the number in the barrel wall (Fig. 3f). No difference in the overall number of barrel neurons was observed. This reduction in barrel segregation in Syngap+/− mice is not attributable to a delay in barrel formation, because it is visible at all ages examined (data not shown). Furthermore, the decrease in cortical cell segregation did not result from a decrease in TCA segregation, because TCA patch formation appears normal in Syngap+/− animals (Fig. 2d). To address this issue quantitatively, we measured the area of PMBSF and individual TCA patches identified with 5-HTT immunoreactivity in normal compared with WT animals (Fig. 2f). (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). PMBSF size and TCA patch sizes were not significantly different between Syngap+/+ and Syngap+/− animals indicating normal TCA patch segregation.
Figure 3.
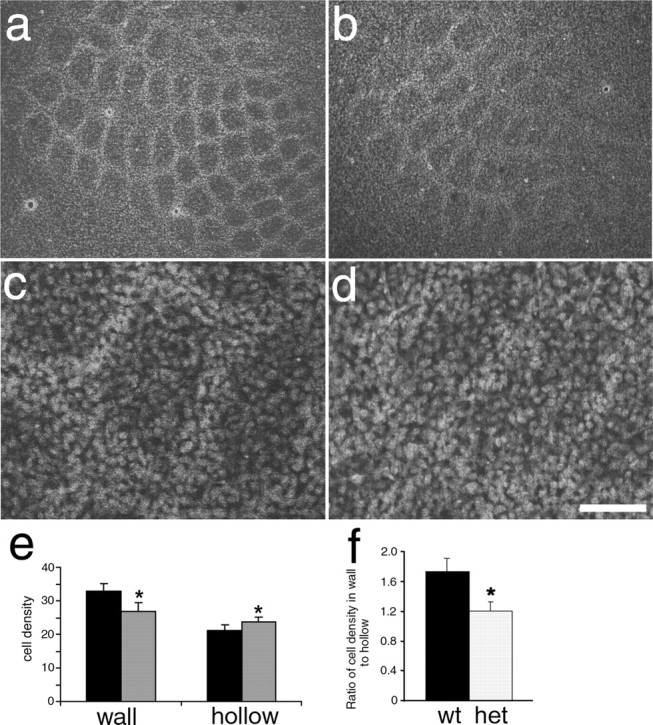
Reduced barrel segregation in Syngap+/− mice. Propidium iodide staining showing neuronal distribution in flattened sections from WT (a, c) and Syngap+/− (b, d) mice showing a clear reduction in the ratio of cells in the barrel wall to barrel hollow in Syngap+/− mice. Cell counts revealed a significant decrease (p < 0.01) in the density of barrel wall neurons and a significant increase (p < 0.01) in the density of barrel hollow neurons in Syngap+/− (gray bars) compared with Syngap+/+ (black bars) animals (e). The barrel wall to hollow ratio is significantly reduced (p < 0.01) from 1.8 in WT mice to 1.2 in Syngap+/− mice (f). The reduction in this ratio appears to be caused by a change in the distribution of cells rather than an overall change in the number of cells (f). Scale bar (in d): a, b, 250 μm; c, d, 70 μm. Error bars represent SE. het, Heterozygotes.
SynGAP is needed for barreloid but not barrelette formation
In the trigeminal pathway, the segregation into whisker-specific modules also occurs within the brainstem and thalamus where they are referred to as barrelettes and barreloids, respectively. The incomplete segregation of TCAs in Syngap−/− mice raised the possibility that SynGAP plays a role in the segregation of barreloids in the VpM of the thalamus. CO histochemistry in wild-type mice showed clear segregation of barreloids at both P4 and P7 (Fig. 4a and d, respectively). In contrast, Syngap−/− showed reduced segregation at P4 and P7 (Fig. 4c and f, respectively). Patches of CO label are visible in the dorsolateral region of VpM corresponding to the large whisker representations; however, no segregation is visible in the regions receiving input from whiskers on the anterior snout. In P7 Syngap+/− mice (Fig. 4e), barreloid segregation was indistinguishable from Syngap+/+ animals. At P4, barreloids were visible in the Syngap+/− animals; however, segregation does not appear as complete as in WT mice (Fig. 4b and a, respectively). These data indicate that reduced levels of SynGAP may result in a delay in barreloid formation. No delay in TCA segregation in layer 4 has been observed (data not shown). In contrast to the developmental pattern in the barreloids, CO staining revealed normal formation of barrelettes in both the principal nucleus (Fig. 4g,h) of the trigeminal complex (PrV) and the subnucleus interpolaris (SpI) (Fig. 4i,j) of P4 Syngap+/− (Fig. 4g,i) and Syngap−/− (Fig. 4h,j) mice. The normal formation of whisker-related structures in these brainstem nuclei is in good agreement with the lack of SynGAP expression in the developing trigeminal nuclei (see below).
Figure 4.

Reduced barreloid segregation in Syngap−/− mice. Cytochrome oxidase staining in VpM of thalamus to reveal barreloids in Syngap+/+ (a, d), Syngap+/− (b, e), and Syngap−/− (c, f) mice at P4 (a–c) and P7 (d–f). Barreloids can be clearly seen in all genotypes at both P3/4 and P7; however, segregation is clearly reduced in Syngap−/− mice (n = 5) at both ages relative to Syngap+/+ (n = 8) and Syngap+/− (n = 12) mice, especially in the anterior snout representations. Coronal section through the brainstem trigeminal complex stained for cytochrome oxidase in P3/4 control (g, i; n = 10) and Syngap−/− (h, j; n = 6) mice showing normal barrelette formation in PrV (g, h) and SpI (i, j). Scale bars: (in f) a–f, 350 μm; (in j) g, h, 225 μm; (in j) i, j, 250 μm.
The smaller body weight and cortical size of Syngap−/− animals raises the possibility that the lack of barrels in these animals results from general ill health or defects in brain development. We find this possibility very unlikely for several reasons. First, general ill health does not prevent TCA or barrel segregation (Vongdokmai, 1980). For example, mice lacking trkB show reduced body and brain weight and, similar to Syngap−/− mice, die during the first postnatal week. Despite their general ill health, we have shown that TCAs segregate normally in layer 4, and barrels are indistinguishable from wild-type controls (Vitalis et al., 2002). Second, barrelettes form normally in the trigeminal brainstem nuclei of Syngap−/− mice where SynGAP is not expressed strongly, suggesting the lack or decrease in barrel or barreloid segregation is specific to the lack of SynGAP. Third, cortical development appears normal in all features, apart from TCA segregation and barrel formation in S1. We observe no general problem with TCA pathfinding; TCAs stop in layer 4 of S1 and also form their typical segregation into sensory areas. Even within S1, the segregation of body regions occurs normally as the representation of the forepaw, hindpaw, lower jaw, anterior snout, and PMBSF can all be clearly demarcated (Fig. 2). The only defects observed are within PMBSF and anterior snout regions of S1. Fourth, levels of cytochrome oxidase activity appear normal in VpM and cortex in Syngap−/− animals, again suggesting no general defect in activity levels in cortex (Fig. 4) (data not shown). Finally, Syngap+/− animals show clear deficits in barrel development despite being indistinguishable from the WT littermates in general health and other aspects of brain development examined.
Postnatal development of SynGAP expression in the somatosensory pathway
The analysis of the mutant mice indicates a temporal and spatial requirement for SynGAP in the development of the trigeminal system. To identify the cells expressing Syngap, we examined the spatiotemporal expression profile of Syngap in Syngap+/− animals, taking advantage of the presence of the insertion of the gene encoding β-galactosidase (β-gal) into the Syngap locus (Komiyama et al., 2002). It should be noted that because there is an internal ribosomal entry site upstream of the β-galactosidase gene, translation of the protein would not be effected by regulatory sequences in the 3′ and 5′ untranslated regions of the mRNA. Hence, X-Gal expression is more likely to reflect the expression profile of Syngap mRNA rather than SynGAP protein. To validate the LacZ reporter, we examined the levels of Syngap mRNA using real-time RT-PCR with primers specific for the conserved GAP domain (supplemental Fig. 3b, available at www.jneurosci.org as supplemental material). These results were consistent with the X-Gal histochemistry as Syngap mRNA levels peaked during the second postnatal week in the barrel cortex before being dramatically reduced in the adult. In addition, because lacZ does not contain any SynGAP peptide sequences, including those for localization to dendrites, staining will accurately report on the cells in which SynGAP is normally transcribed and not the localization of SynGAP protein. Hence, X-Gal staining appears as one or several cytoplasmic inclusions and was not seen in dendrites or axons.
In barrel cortex, X-Gal staining clearly reveals a regionalized pattern of Syngap expression throughout development (Fig. 5) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). At P0, Syngap expression was located throughout the dorsal thalamus with high levels in both the dorsal lateral geniculate nucleus and ventrobasal complex (VB) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). No X-Gal staining was observed in the ventral thalamus indicating a selective expression in projection nuclei of the thalamus. X-Gal staining remained high throughout the dorsal thalamus at P4 (Fig. 5c), including within VB, where it increased dramatically by P8 (Fig. 5d). Staining remained high until P14 before decreasing at P21 (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). No staining was seen in any region of the adult thalamus.
Figure 5.

SynGAP expression in the developing cortex and thalamus. Histochemistry for β-galactosidase to reveal the expression profile of Syngap in the S1 (a, b) and thalamus (c, d) at P4 (a, c) and P8 (b, d). For a complete developmental series, see supplemental Figure 2 (available at www.jneurosci.org as supplemental material). In cortex, β-gal is first expressed at P0 in layer 4 of the developing cortical plate. By P4, staining can be seen throughout the supragranular layers as well as layer 4 and the upper region of layer 5. It is also present in a thin strip of cells located at the bottom of the cortical plate (a). By P8, staining can be seen throughout the cortical plate, and the barrels are clearly distinguished in layer 4 (c). By adulthood, staining has been dramatically reduced and can only be seen at the layer 4/5 border and in layer 1. A similar developmental profile can be seen in the VB and throughout the dorsal thalamus with strong staining visible at P4 (c) and P8 (d) and reduced expression in adults. Immunohistochemical localization of SynGAP protein at P7 (e–g) is in good agreement with β-gal localization. Low-power images show high levels of SynGAP in the cortex (C), hippocampus (H), and amygdaloid complex (A) with lower levels in the VB of the thalamus. In S1 (f), SynGAP levels are highest in layer 4, and the supragranular layers and barrels can be clearly seen. Expression is just starting to appear in the infragranular layers, although a dense band of label can be seen at the layer 4/5 border. The immunohistochemical localization therefore is in good agreement with the large increase in X-Gal staining between P4 and P7. In thalamus, SynGAP is expressed throughout the VB. Electron microscopy analysis of SynGAP expression in layer 4 at P14 reveals a postsynaptic localization for SynGAP (h–j). DAB reaction product can be clearly seen as dark labeling in the PSD (asterisks) opposing presynaptic terminals containing synaptic vesicles. In h, three PSDs in tandem on the shaft of a dendrite are clearly visible. Higher magnification of the middle synapse in h shows clear amalgamation of presynaptic vesicles abutting the SynGAP-positive PSD. Clear presynaptic vesicles are also seen in j. Reaction product was never seen in axons or axon terminals. Scale bar (in j): a, b, 200 μm; c, d, 400 μm; e, 775 μm; f, 150 μm; g, 275 μm; h, 250 nm; i, 175 nm; j, 100 nm.
Throughout cortical development, X-Gal expression appeared in a pattern consistent with a neuronal localization. It was not found in the white matter, suggesting it is not present in oligodendrocytes. Furthermore, no X-Gal-positive cells were visible in layer 1, and no double-labeled cells were seen when sections were stained with the astrocyte marker GFAP, strongly suggesting it is not present in astrocytes (data not shown). At P0 in the somatosensory cortex, X-Gal expression appeared as a thin dense band at the bottom of the cortical plate (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). The ventricular zone, subventricular zone, intermediate zone, and upper layers of the cortical plate showed little or no staining. At P4, Syngap expression was clearly present through the granular and supragranular layers of the cortex (Fig. 5b). Little staining was observed in the infragranular layers, although a thin band of cells can be seen at the bottom of layer 6. During the second postnatal week, X-Gal staining was present throughout the cortical plate with highest levels in layer 4, where clear staining was seen in the barrel walls (Fig. 5a). By P35, the density of labeled cells had decreased and in the adult two thin bands of label remained in upper layer 2 and at the layer 4/5 boundary (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Immunohistochemical distribution of SynGAP protein in S1 cortex and VpM was in good agreement with the results obtained for X-Gal staining (Fig. 5e–g). At P7 (Fig. 5e,f), SynGAP is expressed throughout S1 with highest staining in the layer 4 and the supragranular layers. Patchy label representing barrels is clearly present in layer 4 corresponding to high expression in barrels (Fig. 5f). SynGAP is also expressed in the barreloids in good agreement with its role in barreloid segregation (Fig. 5g).
To determine the subcellular localization of SynGAP, we performed immunoelectron microscopy on layer 4 from P14 barrel cortex (Fig. 5h–j). SynGAP immunoreactivity can be clearly seen in postsynaptic densities abutting presynaptic terminals containing synaptic vesicles. SynGAP was also seen throughout dendritic shafts; however, a precise subcellular localization was prevented because of the large amount of DAB reaction product. Reaction product was never seen in the axons or presynaptic terminals. A postsynaptic localization for SynGAP in cortex is in good agreement with previous findings from several groups showing SynGAP selectively associates with PSD in cultured hippocampal neurons. To further examine the likely localization of SynGAP during barrel development, we examined the expression of Syngap splice variants during barrel cortex development. Syngap is a heteromeric mRNA consisting of three known N-terminal (a, b, c) and seven known C-terminal (α1, 2, β1–4, γ) splice variants (Li et al., 2001). SynGAPα1 is the isoform of SynGAP originally characterized by Kim et al. (1998) and Chen et al. (1998). In cultured cerebellar neurons, SynGAPα2 was shown to localize to axons (Tomoda et al., 2004). To determine whether SynGAPα2 was present in developing VpM and hence could localize to TCAs, we examined the expression of the mRNA encoding the SynGAP isoforms in layer 4 and VpM of P8 mice. Using primers designed to amplify all known 3′ isoforms of the Syngap gene, we used PCR to amplify, clone, and sequence Syngap mRNAs from layer 4 cells and VpM. Of the 17 Syngap cDNAs cloned from VpM, none encoded sequence for Syngapα2, indicating that the axon-associated form of Syngap is not present in the VpM cells during barrel development (data not shown).
SynGAP interactions with PSD-95 and H-Ras
The family of MAGUK proteins that interact with the NMDAR include PSD-95, PSD-93/Chapsyn-110, and SAP102. Each contains three PDZ domains, of which the first two bind to the C termini of NR2 subunits. The third PDZ domain of PSD-95 and SAP-102 binds to the C terminus of SynGAP. To explore whether PSD-95 was the specific MAGUK protein responsible for regulating SynGAP, we examine the barrel phenotype of adult (n = 3) and P7 (n = 7) Psd-95−/− mutant mice (Migaud et al., 1998). There was no abnormality detected (Fig. 6c). The lack of any discernible alteration in barrel pattern in the Psd-95−/− animals raised the possibility that SynGAP may be associating with the PSD in a PSD-95-independent manner. To examine this possibility, we isolated PSDs from barrel cortex taken from wild-type and Psd-95−/− animals at P7 (Fig. 7b). A 135 kDa band corresponding to the SynGAP protein was clearly visible in both synaptosomes and PSD fractions of wild-type and Psd-95−/− animals. We also examined the developmental profile of PSD component proteins in S1 cortex homogenates and in synaptosomal fractions from P7 S1 cortex (Fig. 7a). SynGAP, NR1, and SAP-102 were all present in neonatal homogenates of barrel cortex and increased their expression levels into adulthood. In contrast, NR2A was barely detectable in homogenates of barrel cortex at P7, increased dramatically by P14 and continued to increase gradually into adulthood. PSD-95 is expressed throughout the first postnatal week and then increases dramatically from P14 to adulthood. This increase in PSD-95 expression was so dramatic that two separate concentrations of primary antibody were needed to clearly analyze the low and high end of the developmental expression. PSD-95 was highly enriched in synaptosomes at P7; however, the presence of PSD-95 was not necessary for SynGAP association with the PSD. These data indicate that PSD-95 is not the key MAGUK for mediating SynGAP function during barrel formation; instead, this function may reside in either SAP102, other MAGUKs, or via MAGUK-independent interactions.
Figure 6.

Normal barrel formation in mice lacking PSD-95 and H-Ras. Nissl staining in flattened sections through layer 4 of WT (a), H-Ras (b), PSD-95 (c), and SynGAP (d) mutant mice.
Figure 7.
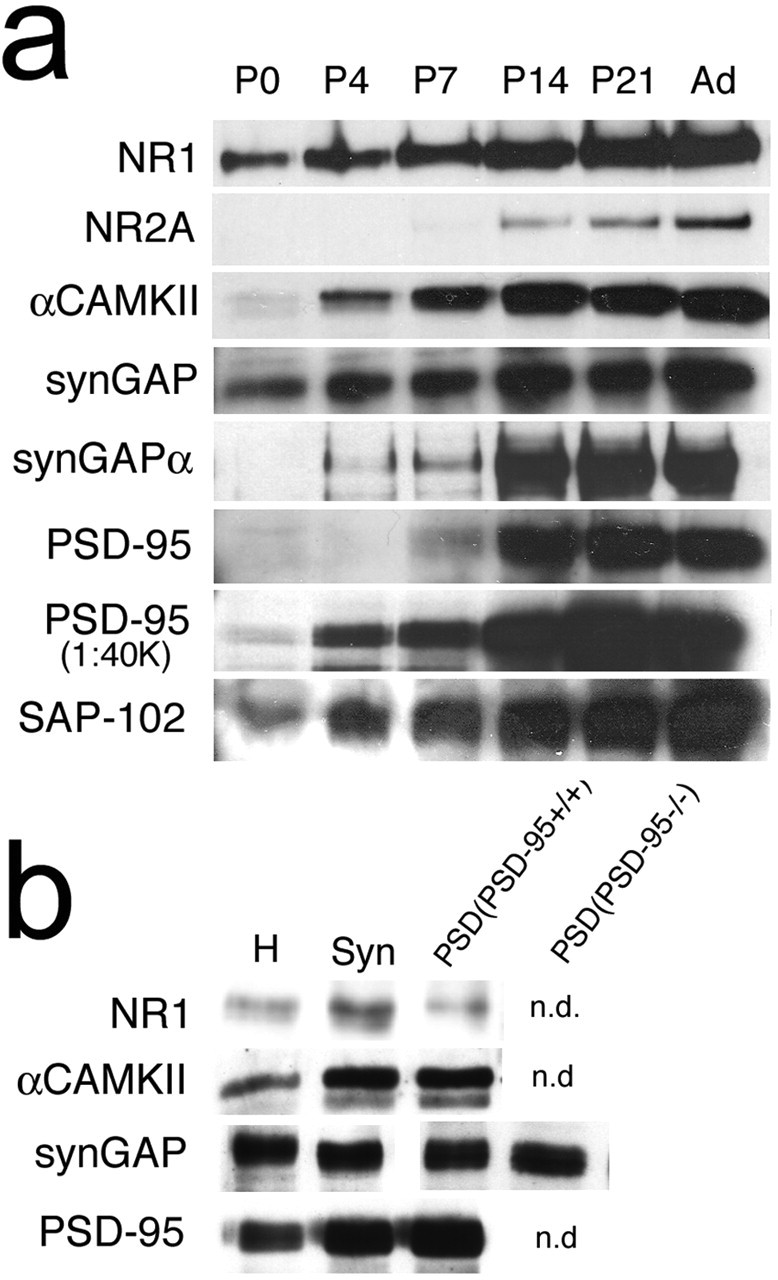
SynGAP still associates with the PSD in Psd-95−/− mice. Western blotting for PSD components in homogenates of S1 cortex reveals a dramatic increase in PSD components during the first postnatal week (a). b, Western blotting for NR1, CaMKII, PSD-95, and SynGAP (Syn) in homogenates, synaptosomes, and PSDs isolate from S1 of P7 WT mice as well as SynGAP expression in the PSD of P7 Psd-95−/− mice. n.d., Not done.
Our results examining the Syngap splice variants expressed during cortical development are in good agreement with these biochemical findings. SynGAPα1 is the C-terminal splice variant that contains the QTRV sequence necessary for binding to the PDZ domain of PSD-95. Interestingly, of the 13 clones analyzed from layer 4 of barrel cortex, Syngapα1 was not found. One of 13 clones encoded a protein that differed by only two amino acids from SynGAPα1 and contained the coding region for the QTRV sequence. It also coded for the peptide sequence recognized by the SynGAPα1-specific antibody. To determine the relative abundance of SynGAPα1 protein in the developing barrel cortex, we compared the developmental profile of SynGAPα1 to all SynGAPs using a pan-SynGAP antibody, the epitope of which lies in the GAP domain (Fig. 7a). Both pan-SynGAP and SynGAPα1 levels increase dramatically during the second postnatal week; however, unlike pan-SynGAP, very little SynGAPα1 is present during the first postnatal week. These data strongly suggest that the principal isoforms of SynGAP present during barrel formation are not those that bind directly to the MAGUKs.
In vitro studies show that SynGAP hydrolyzes H-Ras-GTP to H-Ras-GDP (Chen et al., 1998). If H-Ras is the key effector for SynGAP, then H-Ras mutants may show a barrel phenotype. We detected no overt phenotype in the barrel cortex of either adult (n = 3) (Fig. 6b) or P7 (n = 5) H-Ras homozygous null mice. The absence of an in vivo effector role for H-Ras in the cortex is consistent with previous data in the hippocampus (Komiyama et al., 2002). The presence of other Ras isoforms in the barrel cortex may provide the physiological substrate for SynGAP.
Discussion
NMDA receptors play a critical role in the development of the somatosensory cortex (Schlaggar et al., 1993; Fox et al., 1996; Iwasato et al., 2000; Datwani et al., 2002). The intracellular pathways through which NMDARs signal to initiate these morphological changes are not clear. We show that SynGAP, a component of the mature NMDA receptor complex (Chen et al., 1998; Kim et al., 1998, 2003; Komiyama et al., 2002), is expressed in the developing PSD and regulates S1 cortical development. Mice lacking SynGAP show an absence of barrels and only partial formation of barreloids in VpM. Thalamocortical axons segregate into rows, but individual whisker-related clusters do not form. SynGAP+/− animals also show reduced barrel segregation but normal clustering of TCA and normal barreloid formation. Barrel development is normal in Psd-95−/− animals. SynGAP maintains its association with the PSD in P7 S1 cortex in the absence of PSD-95. These data demonstrate a crucial role for SynGAP in the organization of sensory maps selectively in forebrain structures. They also highlight the heterogeneity of intracellular pathways used by different brain regions at different developmental stages.
SynGAP in barreloid and barrel formation
SynGAP was initially identified as a Ras-GAP (Chen et al., 1998; Kim et al., 1998), the activity of which is regulated by NMDA receptor stimulation in hippocampus (Komiyama et al., 2002; Oh et al., 2004). Our findings suggest that SynGAP is also downstream of NMDA receptor activation during trigeminal pathway development. SynGAP associates with the PSD in developing S1. Furthemore, the phenotype resulting from the loss of NMDA receptors or SynGAP are very similar. In cortex, NMDA receptors and SynGAP are necessary for barrel formation but not for the development of cortical layers or cell type (Messersmith et al., 1997; Iwasato et al., 2000). In Syngap−/− mice, cortical lamination, radial distribution of calretinin-positive interneurons, distribution of PKARIIβ, and radial termination of TCAs are similar to WT mice. Barreloid formation is also dependent on NMDARs (Iwasato et al., 1997) and SynGAP. In contrast, we found no role for SynGAP in barrelette formation, although NMDARs are required (Li et al., 1994; Iwasato et al., 1997). These observations suggest that NMDA receptor-dependent development uses different signaling proteins in different neuronal populations to achieve very similar cellular outcomes (e.g., axon segregation). These findings are also in agreement with previous studies showing that mGluR5 signaling via PLC-β1 is critical in barrel formation (Hannan et al., 2001) but not for barreloid and barrelette formation (our unpublished observations).
The finding that TCAs in Syngap−/− mice form rows but not individual patches raises the possibility that the cortical defect in Syngap−/− mice is secondary to thalamic defects. However, two main findings indicate a cortical role for SynGAP during barrel formation in addition to its role in the thalamus. First, a significant reduction in barrel segregation was seen in Syngap+/− animals despite normal segregation of TCAs into whisker-related patches. Second, in Syngap−/− mice, TCAs segregate into rows, but no barrels form around these rows. In contrast, in WT mice, the soma of layer 4 neurons cluster around the row of TCAs that forms after cauterization of row C follicles to form one large barrel (Van der Loos and Woolsey, 1973). If barrel formation is independent of cortically expressed SynGAP, a large barrel would be expected to form around the rows of TCAs in Syngap−/− mice; this result was never observed.
It is not yet clear whether the incomplete segregation of TCAs into rows results from the loss of SynGAP in the cortex or thalamus. In CxNR1−/− mice, TCAs form rudimentary patches, and there is a significant decrease in TCA complexity within layer 4 (Lee et al., 2005), indicating that cortically derived signals regulate TCA elaboration independently of axon segregation. NMDA receptors similarly regulate axon dynamics in Xenopus tectum (Ruthazer et al., 2003). NMDA receptors may be regulating axon branch dynamics via SynGAP-regulated release of a retrograde signal. Alternatively, the loss of TCA segregation in Syngap+/− animals could arise from a cell-autonomous defect in the thalamic neurons that prevents terminal segregation in the cortex. Distinguishing between these two possibilities awaits examination of mice with a cortex-specific Syngap deletion.
Association of SynGAP with the PSD
SynGAP has been demonstrated to associate with the PSD via PSD-95 (Chen et al., 1998; Kim et al., 1998). However, we have shown that Psd-95−/− mice develop normal barrels, and SynGAP remains associated with the developing PSD in these mice. The apparent discrepancy between these findings may be resolved by the recent finding that in adult hippocampus, SynGAP is preferentially associated with NR2B receptors via an interaction with SAP-102 (Kim et al., 2005). It is also consistent with previous studies examining the developmental and spatial expression profiles of these PSD components (Porter et al., 2005). During development, NMDA receptors are predominantly NR2B containing and associated with SAP-102; NR2A and PSD-95 are expressed later in development and may displace NR2B/SAP-102 complexes from the PSD (Shi et al., 1997; Sans et al., 2000; Yoshii et al., 2003; Van Zundert et al., 2004). In S1, we found high levels of both PSD-95 and SAP-102 in the developing PSD, and their increase correlates well with the rapid increase in synapse formation in rodent S1 (Micheva and Beaulieu, 1996; White et al., 1997; Spires et al., 2004). However, PSD-95 levels increase more dramatically with age and therefore more closely parallel the NR2A development expression profile. These findings suggest that, during barrel development, SynGAP may be associating with NR2B-containing receptors via SAP-102 and not PSD-95.
Alternatively, SynGAPs association with the PSD in developing S1 cortex may be independent of MAGUKs. Analysis of the C-terminal isoforms of SynGAP in layer 4 revealed a preponderance of SynGAP splice variants that do not contain the MAGUK-binding consensus sequence (Li et al., 2001). Thus, in developing S1, SynGAP may be associating with the PSD in a MAGUK-independent manner as has been described previously (Vazquez et al., 2004), possibly via its Plecktrin homology (PH) or C2 domains.
Pathways downstream of SynGAP
SynGAP has been shown to regulate the activation of ERK-mitogen-activated protein kinase (MAPK) through its role as a Ras-GAP (Komiyama et al., 2002). Furthermore, the Ras-ERK-MAPK pathway has been demonstrated to control numerous developmental processes, including synaptic plasticity, cell proliferation, survival, migration, and differentiation (Gille and Downward, 1999; Di Cristo et al., 2001; Adams and Sweatt, 2002; Sweatt, 2004). These findings indicate that SynGAP maybe regulating barrel formation by regulating ERK activation and are in good agreement with previous work demonstrating a role for ERK-MAPK in visual cortical development as well as in NMDAR-dependent LTP in visual cortex (Di Cristo et al., 2001) and hippocampus (Winder et al., 1999) and performance on spatial learning tasks (Bozon et al., 2003). These results strongly indicate a conservation of signaling pathways for various forms of plasticity in a variety of brain areas at various developmental ages.
We have shown previously that mGluR5 signaling through PLC-β1 is crucial for barrel formation (Hannan et al., 2001). Because mGluR activation is also capable of regulating ERK activation (Choe and Wang, 2001; Berkeley and Levey, 2003; Gallagher et al., 2004), it is possible that ERK may be a common downstream target of multiple signaling pathways initiated from mGluR5 and NMDA receptors during cortical neuronal development. Interestingly, mGluR5−/− mice also show segregation of TCAs into rows, similar to the Syngap−/− mice, raising the possibility that mGluR5 may regulate SynGAP activity, possibly via PLC-β1 stimulated Ca2+ release from endoplasmic reticulum and subsequent CaMKII activity. A similar integrative role for ERK has been proposed previously (Watabe et al., 2000; Adams and Sweatt, 2002). This could explain why loss of any of the pathways leading to ERK regulation results in a disruption of barrel development.
It is also possible that SynGAP may be regulating pathways that are Ras dependent but ERK independent. In support of this possibility Syngap+/− mice showed defects in LTP induction using pairing protocols and 100 Hz stimulation (Komiyama et al., 2002), two forms of LTP induction that are ERK independent in wild-type slices (Winder et al., 1999; Watabe et al., 2000). Furthermore, Ras can regulate the activity of PI3-kinase (Sanna et al., 2002), which plays a crucial role in certain types of synaptic plasticity (Kelly and Lynch, 2000; Sanna et al., 2002; Opazo et al., 2003). Alternatively, SynGAP may be a GTPase for other non-RAS small G-proteins. For example, SynGAP has recently been shown to regulate Rab-5 activity (Tomoda et al., 2004) and Rab-5 has been shown to regulate the actin cytoskeleton during the formation of “circular ruffles” and three-dimensional migration (Lanzetti et al., 2004).
The mechanisms by which activity and, more specifically, glutamate neurotransmission, mediates early cortical development and map formation will be crucial to understand normal brain development and the cellular events that underlie many forms of mental retardation and possibly provide treatments for adult neurodegenerative diseases. We have shown that SynGAP plays a crucial role in the anatomical development of maps in the primary somatosensory cortex of mice and hence may be crucial in establishing the complex neuronal circuitry that mediates normal cortical function.
Footnotes
*M.W.B., R.F.W., T.V., and K.P. contributed equally to this work.
This work was supported by the Wellcome Trust (P.C.K.), the Wellcome Trust Genes to Cognition Programme (S.G.N.G.), and the Medical Research Council UK (P.C.K.).
References
- Adams JP, Sweatt JD (2002). Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol 42:135–163. [DOI] [PubMed] [Google Scholar]
- Barnett MW, Old RW, Jones EA (1998). Neural induction and patterning by fibroblast growth factor, notochord and somite tissue in Xenopus. Dev Growth Differ 40:47–57. [DOI] [PubMed] [Google Scholar]
- Berkeley JL, Levey AI (2003). Cell-specific extracellular signal-regulated kinase activation by multiple G protein-coupled receptor families in hippocampus. Mol Pharmacol 63:128–135. [DOI] [PubMed] [Google Scholar]
- Bozon B, Kelly A, Josselyn SA, Silva AJ, Davis S, Laroche S (2003). MAPK, CREB and zif268 are all required for the consolidation of recognition memory. Philos Trans R Soc Lond B Biol Sci 358:805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB (1998). A synaptic Ras-GTPase activating protein (p135 synGAP) inhibited by CaM kinase II. Neuron 20:895–904. [DOI] [PubMed] [Google Scholar]
- Choe ES, Wang JQ (2001). Group I metabotropic glutamate receptor activation increases phosphorylation of cAMP response element-binding protein, Elk-1, and extracellular signal-regulated kinases in rat dorsal striatum. Brain Res Mol Brain Res 94:75–84. [DOI] [PubMed] [Google Scholar]
- Datwani A, Iwasato T, Itohara S, Erzurumlu RS (2002). NMDA receptor-dependent pattern transfer from afferents to postsynaptic cells and dendritic differentiation in the barrel cortex. Mol Cell Neurosci 21:477–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristo G, Berardi N, Cancedda L, Pizzorusso T, Putignano E, Ratto GM, Maffei L (2001). Requirement of ERK activation for visual cortical plasticity. Science 292:2337–2340. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, Heath JW, Kidd GJ, Rostas JA (1986). A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res 372:115 129. [DOI] [PubMed] [Google Scholar]
- Erzurumlu RS, Kind PC (2001). Neural activity: sculptor of “barrels” in the neocortex. Trends Neurosci 24:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K, Schlaggar BL, Glazewski S, O’Leary DD (1996). Glutamate receptor blockade at cortical synapses disrupts development of thalamocortical and columnar organization in somatosensory cortex. Proc Natl Acad Sci USA 93:5584–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G (1997). In: The mouse brain in stereotaxic coordinates San Diego: Academic.
- Gallagher SM, Daly CA, Bear MF, Huber KM (2004). Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci 24:4859–4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar P, Cases O, Maroteaux L (2003). The developmental role of serotonin: news from mouse molecular genetics. Nat Rev Neurosci 4:1002–1012. [DOI] [PubMed] [Google Scholar]
- Gille H, Downward J (1999). Multiple ras effector pathways contribute to G(1) cell cycle progression. J Biol Chem 274:22033–22040. [DOI] [PubMed] [Google Scholar]
- Hannan AJ, Blakemore C, Katsnelson A, Vitalis T, Huber KM, Bear M, Roder J, Kim D, Shin HS, Kind PC (2001). Phospholipase C-β1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat Neurosci 4:282–288. [DOI] [PubMed] [Google Scholar]
- Ho W, Uniyal S, Meakin SO, Morris VL, Chan BM (2001). A differential role of extracellular signal-regulated kinase in stimulated PC12 pheochromocytoma cell movement. Exp Cell Res 263:254–264. [DOI] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG (2000). Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci 3:661–669. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Erzurumlu RS, Huerta PT, Chen DF, Sasaoka T, Ulupinar E, Tonegawa S (1997). NMDA receptor-dependent refinement of somatotopic maps. Neuron 19:1201–1210. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S (2000). Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 406:726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, Lynch MA (2000). Long-term potentiation in dentate gyrus of the rat is inhibited by the phosphoinositide 3-kinase inhibitor, wortmannin. Neuropharm 39:643–651. [DOI] [PubMed] [Google Scholar]
- Killackey HP, Belford GR (1979). The formation of afferent patterns in the somatosensory cortex of the neonatal rat. J Comp Neurol 183:285–304. [DOI] [PubMed] [Google Scholar]
- Kim JH, Liao D, Lau LF, Huganir RL (1998). SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 20:683–691. [DOI] [PubMed] [Google Scholar]
- Kim JH, Lee HK, Takamiya K, Huganir RL (2003). The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J Neurosci 23:1119–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M (2005). Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron 46:745–760. [DOI] [PubMed] [Google Scholar]
- Kind PC, Neumann PE (2001). Plasticity: downstream of glutamate. Trends Neurosci 24:553–555. [DOI] [PubMed] [Google Scholar]
- Kind P, Blakemore C, Fryer H, Hockfield S (1994). Identification of proteins down-regulated during the postnatal development of the cat visual cortex. Cereb Cortex 4:361–375. [DOI] [PubMed] [Google Scholar]
- Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJ, O’Carroll CM, Martin SJ, Morris RG, O’Dell TJ, Grant SG (2002). SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci 22:9721–9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Medina I, Krapivinsky L, Gapon S, Clapham DE (2004). SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron 43:563–574. [DOI] [PubMed] [Google Scholar]
- Lanzetti L, Palamidessi A, Areces L, Scita G, Di Fiore PP (2004). Rab5 is a signalling GTPase involved in actin remodelling by receptor tyrosine kinases. Nature 429:309–314. [DOI] [PubMed] [Google Scholar]
- Lee LJ, Iwasato T, Itohara S, Erzurumlu RS (2005). Exuberant thalamocortical axon arborization in cortex-specific NMDAR1 knockout mice. J Comp Neurol 485:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Okano A, Tian QB, Nakayama K, Furihata T, Nawa H, Suzuki T (2001). Characterization of a novel synGAP isoform, synGAP-beta. J Biol Chem 276:21417–21424. [DOI] [PubMed] [Google Scholar]
- Li Y, Erzurumlu RS, Chen C, Jhaveri S, Tonegawa S (1994). Whisker-related neuronal patterns fail to develop in the trigeminal brainstem nuclei of NMDAR1 knockout mice. Cell 76:427–437. [DOI] [PubMed] [Google Scholar]
- Messersmith EK, Feller MB, Zhang H, Shatz CJ (1997). Migration of neocortical neurons in the absence of functional NMDA receptors. Mol Cell Neurosci 9:347–357. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Beaulieu C (1996). Quantitative aspects of synaptogenesis in the rat barrel field cortex with special reference to GABA circuitry. J Comp Neurol 373:340–354. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O’Dell TJ, Grant SG (1998). Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396:433–439. [DOI] [PubMed] [Google Scholar]
- Oh JS, Manzerra P, Kennedy MB (2004). Regulation of the neuron-specific Ras GTPase-activating protein, synGAP, by Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 279:17980–17988. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SG, O’Dell TJ (2003). Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci 23:3679–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter K, Komiyama NH, Vitalis T, Kind PC, Grant SGN (2005). Differential expression of two NMDA receptor interacting proteins, PSD-95 and SynGAP during mouse development. EJN 21:351–362. [DOI] [PubMed] [Google Scholar]
- Rebsam A, Seif I, Gaspar P (2002). Refinement of thalamocortical arbors and emergence of barrel domains in the primary somatosensory cortex: a study of normal and monoamine oxidase a knock-out mice. J Neurosci 22:8541–8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthazer ES, Akerman CJ, Cline HT (2003). Control of axon branch dynamics by correlated activity in vivo. Science 301:66–70. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W (2002). Phosphatidylinositol 3-kinase is required for the expression but not the induction or maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci 22:3359–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J Jr, Hell JW, Wenthold RJ (2000). A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci 20:1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaggar BL, Fox K, O’Leary DD (1993). Postsynaptic control of synaptic plasticity in developing somatosensory cortex. Nature 364:623–626. [DOI] [PubMed] [Google Scholar]
- Shi J, Aamodt SM, Constantine-Paton M (1997). Temporal correlations between functional and molecular changes in NMDA receptors and GABA neurotransmission in the superior colliculus. J Neurosci 17:6264–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Molnar Z, Kind PC, Cordery PM, Upton AL, Blakemore C, Hannan AJ (2004). Activity-dependent regulation of synapse and dendritic spine morphology in developing barrel cortex requires phospholipase C-β1 signalling. Cereb Cortex 15:385–393. [DOI] [PubMed] [Google Scholar]
- Sweatt JD (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14:311–317. [DOI] [PubMed] [Google Scholar]
- Tomoda T, Kim JH, Zhan C, Hatten ME (2004). Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev 18:541–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Loos H, Woolsey TA (1973). Somatosensory cortex: structural alterations following early injury to sense organs. Science 179:395–398. [DOI] [PubMed] [Google Scholar]
- Van Zundert B, Yoshii A, Constantine-Paton M (2004). Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal. Trends Neurosci 27:428–437. [DOI] [PubMed] [Google Scholar]
- Vazquez LE, Chen HJ, Sokolova I, Knuesel I, Kennedy MB (2004). synGAP regulates spine formation. J Neurosci 24:8862–8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis T, Cases O, Gillies K, Hanoun N, Hamon M, Seif I, Gaspar P, Kind PC, Price DJ (2002). Interactions between TrkB-signalling and serotonin excess in the developing murine somatosensory cortex: a role in tangential and radial organisation of thalamocortical axons. J Neurosci 22:4987–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vongdokmai R (1980). Effect of protein malnutrition on development of mouse cortical barrels. J Comp Neurol 191:283–294. [DOI] [PubMed] [Google Scholar]
- Walikonis RS, Jensen ON, Mann M, Provance DW Jr, Mercer JA, Kennedy MB (2000). Identification of proteins in the postsynaptic density fraction by mass spectrometry. J Neurosci 20:4069–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe AM, Zaki PA, O’Dell TJ (2000). Coactivation of β-adrenergic and cholinergic receptors enhances the induction of long-term potentiation and synergistically activates mitogen-activated protein kinase in the hippocampal CA1 region. J Neurosci 20:5924–5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EL, Weinfeld L, Lev DL (1997). A survey of morphogenesis during the early postnatal period in PMBSF barrels of mouse SmI cortex with emphasis on barrel D4. J Comp Neurol Somatosens Mot Res 14:34–55. [DOI] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER (1999). ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron 24:715–726. [DOI] [PubMed] [Google Scholar]
- Woolsey DH, Van der Loos H (1970). The structural organization of layer 4 in the somatosensory region (SI) of the mouse cerebral cortex. Brain Res 17:205–242. [DOI] [PubMed] [Google Scholar]
- Yoshii A, Sheng MH, Constantine-Paton M (2003). Eye opening induces a rapid dendritic localization of PSD-95 in central visual neurons. Proc Natl Acad Sci USA 100:1334–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]