

Abstract
Major depressive disorder (MDD) is clinically heterogeneous with prevalence rates twice as high in women as in men. There are many possible sources of heterogeneity in MDD most of which are not measured in a sufficiently comparable way across study samples. Here, we assess genetic heterogeneity based on two fundamental measures, between-cohort and between-sex heterogeneity. First, we used genome-wide association study (GWAS) summary statistics to investigate between-cohort genetic heterogeneity using the 29 research cohorts of the Psychiatric Genomics Consortium (PGC; N cases = 16,823, N controls = 25,632) and found that some of the cohort heterogeneity can be attributed to ascertainment differences (such as recruitment of cases from hospital vs community sources). Second, we evaluated between-sex genetic heterogeneity using GWAS summary statistics from the PGC, Kaiser Permanente GERA, UK Biobank and the Danish iPSYCH studies but did not find convincing evidence for genetic differences between the sexes as the genetic correlation between the sexes was not significantly different from one (0.90 s.e. 0.06; pH0:rg=1= 0.096). We conclude that there is no evidence that the heterogeneity between MDD data sets and between sexes reflects genetic heterogeneity. Larger sample sizes with detailed phenotypic records and genomic data remain the key to overcome heterogeneity inherent in assessment of MDD.
Keywords: MDD, depression, genetic heterogeneity, sex differences, LD score regression
INTRODUCTION
Major Depressive Disorder (MDD) is a common debilitating disorder with lifetime risk of ~15% (R. C. Kessler & Bromet, 2013; Lohoff, 2010). Genetic factors contribute to etiology of MDD with heritability estimated to be ~37% (Kendler, Gatz, Gardner, & Pedersen, 2006; Sullivan, Neale, & Kendler, 2000) of which about one-third is tracked by common-genetic variants (Cross-Disorder Group of the Psychiatric Genomics et al., 2013; Wray et al., 2018). Non-genetic factors also contribute and environmental risk factors include childhood psychological trauma (Chapman et al., 2004; Heim, Newport, Mletzko, Miller, & Nemeroff, 2008; Vythilingam et al., 2002), social isolation (Bruce & Hoff, 1994), and medical conditions, such as cardiovascular disease (Fiedorowicz, 2014; Fraguas Jr et al., 2007; Huffman, Celano, Beach, Motiwala, & Januzzi, 2013). Most complex disorders are considered to be heterogeneous at clinical presentation. For MDD, heterogeneity is inherent in the diagnostic framework since diagnosis is achieved through different combinations of endorsements of at least five out of nine criteria in the context of depressed mood for most of the day every day for two weeks (Diagnostic and Statistical Manual of Mental Disorders (DSM) criteria). Heterogeneity in symptom profiles between individuals reflects not only the symptoms endorsed, but for some criteria (those assessing sleep, weight/appetite and psychomotor function) the endorsement can reflect either increase or decrease (or both). It is plausible that these clinical differences reflect different biological pathways. The lack of a biological “gold standard” definition in psychiatric illness is well recognised (Kapur, Phillips, & Insel, 2012), and a key question for the field is whether genetic heterogeneity underpins phenotypic heterogeneity (Fanous & Kendler, 2005), and if genome-wide genetic data can support analyses that demonstrate genetic heterogeneity (Han et al., 2016). Here, we assess genetic heterogeneity based on two fundamental measures available to us, between-cohort and between-sex heterogeneity. While non-biological factors (such as ascertainment strategy) could contribute to both between-cohort and between-sex heterogeneity, evidence for between-sex heterogeneity may reflect, at least in part, biological differences.
Prevalence rates of MDD in women that are double those of men are consistently reported in epidemiological studies, with lifetime risk approximately 0.2 for females and 0.1 for males (Ronald C. Kessler, 2003) Women tend to have younger age of onset, greater comorbidity with panic and other anxiety disorders, whereas men exhibit stronger comorbidity with alcohol dependence or abuse (Schuch, Roest, Nolen, Penninx, & de Jonge, 2014). Attempts to link the epidemiological differences to biological differences have been less consistent. Some twin studies reported significantly higher heritability in females (0.42, 95% CI=0.36–0.47) than males (0.29, 95% CI=0.19–0.38), and with genetic correlation significantly different from 1 (rg~0.60, 95% CI=0.31–0.99) (Kendler et al., 2006). Other studies failed to find differences between sexes (Fernandez-Pujals et al., 2015). Drawing strong conclusions may be confounded by reporting biases as males are more likely to under-report their symptoms when compared to females (Hunt, Auriemma, & Cashaw, 2003; Thornicroft et al., 2017).
We use genome-wide association study (GWAS) summary statistics data to investigate genetic heterogeneity of MDD. We study between-cohort genetic heterogeneity using data from the 29 independent studies that comprise the wave 2 PGC-MDD study (PGC29 (Wray et al., 2018)). We also investigate genetic heterogeneity by sex using GWAS summary statistics from PGC29 and three other large data sets. We evaluate between-cohort and between-sex genetic heterogeneity estimates of SNP-heritabilities and genetic correlations. These estimates of genetic parameters, calculated from genome-wide data, provide single statistic summaries of the data. Specifically, differences in SNP-heritability estimates between samples could imply real differences in the relative magnitude of genetic risk effect sizes between samples or could reflect biases due to ascertainment characteristics of the sample. In contrast, an estimate of a genetic correlation less than one may reflect differences in the relative ordering of genetic risk effects between samples. It is possible for SNP-heritabilities to differ between samples but the genetic correlations to be one.
MATERIALS & METHODS
Between-cohort heterogeneity
We investigate heterogeneity between cohorts from the PGC Working Group for MDD (PGC-MDD) (Major Depressive Disorder Working Group of the Psychiatric et al., 2013), which comprises 29 cohorts (PGC29, 10 from wave 1 (Major Depressive Disorder Working Group of the Psychiatric et al., 2013) and 19 from wave 2 (Wray et al., 2018)), totalling 16,815 cases (68% female) and 25,485 controls (51% female) (Table 1, Supplementary Table 1). Cohorts represent individual studies in which cases and controls were imputed together to the 1000 Genomes reference panel (Genomes Project et al., 2010) from a common set of SNPs that had been processed through a common quality control (QC) pipeline (Wray et al., 2018). For the majority of cohorts (but not all), cases and controls were collected by the same research group and were genotyped together on the same genotyping array. All 29 case cohorts passed a structured methodological review by MDD assessment experts (DF Levinson and KS Kendler). Cases were required to meet international consensus criteria (DSM-IV, International Statistical Classification of Diseases (ICD)-9, or ICD-10) (American Psychiatric Association, 1994; World Health Organization, 1978, 1992) for a lifetime diagnosis of MDD established using structured diagnostic instruments from assessments by trained interviewers, clinician-administered checklists, or medical record review. Nonetheless, there were differences in ascertainment across cohorts (Supplementary Table 1). For example the RADIANT cohort (rad3) (C. M. Lewis et al., 2010) recruited cases of clinically assessed recurrent MDD, which being more severe have lower lifetime risk ~5% (McGuffin, Katz, Watkins, & Rutherford, 1996), compared to community samples such as the QIMR cohorts (qi3c, qi6c, qio2) assessed by self-report interview and with lifetime risk ~24% (Mosing et al., 2009). To capture heterogeneity due to ascertainment, we coded the 29 cohorts as identified in community, psychiatric outpatient, psychiatric inpatients, or mixed in-/out-patient settings (Supplementary Table 1).
Table 1. Description of GWAS data sets for between-sex heterogeneity analyses.
| Data Set | Cases | Controls | Female cases |
Female controls |
Male cases |
Male controls |
Number of Cohortsa |
|---|---|---|---|---|---|---|---|
| PGC29 | 16,823 | 25,632 | 11,438 | 12,463 | 5,377 | 13,022 | 29b |
| GERA | 7,162 | 38,287 | 5,152 | 20,650 | 2,010 | 17,637 | 1 |
| UKB | 113,769 | 208,801 | 73,292 | 99,385 | 40,477 | 109,426 | 1 |
| iPSYCH | 18,577 | 17,637 | 12,690 | 8,534 | 5,887 | 9,103 | 1 |
| Total | 156,331 | 290,357 | 102,572 | 141,032 | 53,751 | 149,188 | 32 |
: Cohort is defined as the cases and controls with genome-wide genotypes imputed from the same set of SNPs that have passed through a common quality control pipeline. Mostly, cohort reflects a case-control sample collected by a PGC principal investigator.
: cohorts ranged in size from 246 to 3760 cases plus controls.
Between-sex heterogeneity
We investigate between sex heterogeneity using four large MDD data sets (Table 1). In addition to PGC29, we used the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort (Banda et al., 2015) (where electronic medical records from the Kaiser Permanente healthcare system were used to identify cases as individuals being treated clinically for MDD, and controls had no recorded treatment for any psychiatric disorder), the Danish iPSYCH cohort (where national hospital records identified cases as those ever treated clinically for MDD and controls as those who have not), and the volunteer UK Biobank (Bycroft et al., 2018; Lane et al., 2016) (UKB) study. UKB cases were those with either recorded ICD10 codes for MDD (F32, F33) or self-report for seeking treatment for nerves, anxiety or depression; for detailed description of the “broad depression” definition see reference (Howard et al., 2018)). Exclusions for both cases and controls were those with recorded schizophrenia, bipolar or mental retardation diagnoses or prescriptions associated with these disorders. Additional exclusions for controls included those with recorded anxiety, phobic or autistic spectrum disorders. In all studies, cases and controls were unrelated. GWAS summary statistics for each cohort used the same methods as for PGC29.
Statistical methods
We use GWAS summary statistics and linkage disequilbrium (LD) score analysis (LDSC) (B. K. Bulik-Sullivan et al., 2015) to estimate the total proportion of variance in liability attributable to SNPs genome-wide (i.e., SNP-heritability). Bivariate LDSC was used to estimate the genetic correlation tagged by genome-wide SNPs (rg) between two traits. LDSC has been applied widely to GWAS summary statistics of psychiatric (Anttila et al., 2018) and other disorders (B. Bulik-Sullivan et al., 2015), and results have been shown to agree well with estimates made from full individual-level genotype and phenotype data using linear mixed model analysis (e.g., GREML (Yang et al., 2010)), as long as the LD reference sample is drawn from a population that appropriately reflects the samples contributing the GWAS summary statistics(Yang et al., 2015). A key advantage of LDSC is the minimal computational requirements compared to methods that use individual level data, and the ability to differentiate between genomic inflation due to polygenicity and due to population stratification. Disadvantages of LDSC are that standard errors (s.e.) of estimates can be (about 50%) higher compared to when estimates are based on full data, particularly for rg estimates (Ni, Moser, Schizophrenia Working Group of the Psychiatric Genomics, Wray, & Lee, 2018).
SNP-heritability is estimated on the observed binary scale , but these estimates depend on the proportion of cases in the sample (P) and so are not easily comparable across cohorts. Hence, for improved interpretability and comparison across studies, is transformed to the liability scale (Lee, Wray, Goddard, & Visscher, 2011) based on normal distribution theory, given an assumed lifetime risk of disease in the population(K):
| [1] |
where z is the height of the standard normal density function when truncated at proportion K. However, this transformation assumes that controls are screened. Peyrot et al (2016) (Peyrot, Boomsma, Penninx, & Wray, 2016) showed that when the proportion of controls that are unscreened is u, then transformation should be
| [2] |
which reduces to equation [1] when all controls are screened, u = 0. When diseases are uncommon, assuming controls are screened when they are not makes little impact(Peyrot et al., 2016). However, for very common disorders, such as MDD, the difference is not trivial. For example, for K = 0.15, = 0.15, P = 0.5, then = 0.18 when controls are screened and 0.24 when unscreened. The rg estimates are robust to P, K and u, since these factors contribute to both numerator and denominator of the correlation (which is defined as the estimate of the additive genetic covariance divided by the product of the square root of the SNP-heritabilities for the two traits). Hence rg estimates are robust to ascertainment practices and approximately the same where estimated on the case-control observed scale or liability scales (B. Bulik-Sullivan et al., 2015). If the same genetic effects contribute to disease risk between sexes or between cohorts then rg is expected to be 1.
It was not possible to compare of each PGC29 cohort, because the per-cohort estimates had high s.e. (e.g. a cohort of 500 cases and 500 controls would be expected to produce with standard error of at minimum 0.38 (Visscher et al., 2014)). Instead we estimated the attributed to a cohort by evaluating its contribution to estimates calculated from 500 random samplings of cohorts drawn from the 29 PGC29 cohorts. In each sampling, we randomly selected cohorts until the total sample size was ≥ 5000, then used the GWAS summary statistics meta-analysed (weighted by s.e.) in LDSC to estimate assuming lifetime risk of K = 0.15, and assuming controls are screened (equation [1]). To determine the contribution to the estimate from each cohort we fitted a linear model with estimated as the dependent variable regressed on indicator variables set as 1 if the cohort contributed to the estimate (was included in the random sampling), and 0 otherwise.
RESULTS
Between-cohort heterogeneity within PGC29
We estimated in 500 random samplings of the cohorts from PGC29. From a linear regression of on indicator variables set as 1 if the cohort contributed to the estimate and 0 if it was not, we estimated an effect size deviation per cohort (y-axis Figure 1). Fifteen of the 29 cohorts had deviations different from zero (p < 0.05/29). We found that the cohorts nes1 (combined sample of the Netherlands Study of Depression and Anxiety and the Netherlands Twin Registry) (Boomsma et al., 2008; Penninx et al., 2008) and gep3 (GenPod/NEWMEDS) (G. Lewis et al., 2011) contributed most to variation in estimates of , and explain 0.14 and 0.16, respectively, of the variance in estimates across the 500 samplings. Samplings that included cohort nes1 had the highest average estimates of , while samplings including gep3 had the lowest average estimates. These differences are in line with expectations based on screening strategies for controls (Supplementary Table 1). The nes1 cohort used super-screened controls (Boomsma et al., 2008), such that controls never scored higher than 0.65 on a general factor score for anxious depression (mean =0, SD=0.7) derived from a combined measure of neuroticism, anxiety, and depressive symptoms assessed via longitudinal questionnaires over 15 years. In contrast, the gep3 cohort was a case-only research cohort which was matched to independently collected and genotyped controls (hence particularly stringent QC is needed to combine the genotype data of the contributing cases and controls). In fact, gep3 is one of seven cohorts for which controls were unscreened for MDD (Figure 1), but only one other cohort used independently genotyped controls (STAR*D, coded as stm2); together the seven cohorts have lower mean beta-values, but not significantly so (p=0.055). The trend in these results might be explained by recognising that SNP-heritability is first estimated on the observed binary case-control scale and then transformed to the liability scale Indeed, we find that increasing sample prevalence (P in equation 1) is significantly associated with the estimated (p=0.00057), but not sex ratio (p=0.72). The application of the standard transformation (equation [1]), as we have done, assumes screened controls and could generate an under-estimate of the SNP-heritability if controls were in fact unscreened. Similarly, super-screening of controls could generate an over-estimate of the true Hence, we expect that the standard transformation would generate an overestimate for the nes1 cohort (super-screened controls) and an underestimate for cohorts with unscreened controls, consistent with our results.
Figure 1. Cohort deviation estimates from the linear regression of estimates (from each of the 500 samplings of cohorts) on cohort indicator variables set at 1 if the cohort was included in the sampling that generated the and 0 otherwise.
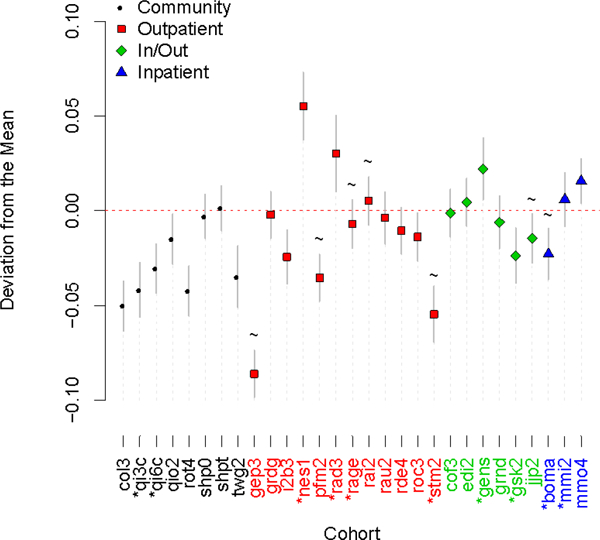
In each sampling, cohorts were selected at random until the total case/control sample size exceeded 5000. Cohort GWAS results were meta-analysed and these results passed into LDscore. was estimated using the equation 1 transformation (K =0.15) which assumes controls are screened. estimates of samplings were highest, on average, when cohort nes1 was included and lowest, on average, when cohort gep3 was included. Wave 1 cohorts have an asterisk by their name and cohorts that have unscreened controls are marked by a tilde. Continuous lines around data-points are 95% confidence Intervals. For explanation of cohort names see Supplementary Table 1.
Next, we investigated if estimates differed based on the research protocol to ascertain cases. For the same proportion of cases and controls in the GWAS sample, we would expect the to be higher for a clinically ascertained cohort than a community ascertained cohort, further we would expect the transformation based on K = 0. 15 (equation [1]) to overestimate when the true K is lower (clinical cohort) and underestimate when the true K is higher (community cohort). There is evidence to support this hypothesis (Figure 1). We found significant difference between the mean estimates of community (−0.027, s.e. 0.007) vs non-community cohorts (−0.08 s.e. 0.006) (with non-community comprising the three in- and out-patient categories), using a one-sided, two-sample t-test assuming unequal variance (p=0.028) (Supplementary Table 4). The difference became more significant (p=0.015) when the cohorts we had a priori reason to exclude, namely nes1 and gep3, based on discussions above were removed.
Between-sex heterogeneity
Using the four large data sets (Table 1) we investigate sex-specific heterogeneity. We used bivariate LDSC to estimate the rg between all pairs of the two sexes by four data sets, but the standard errors were high (Supplementary Table 2). rg involving the GERA_M data set were not estimable, because of the negative/zero of used in the denominator of the rg estimate. The meta-analysis of 12 male-female rg estimates was 0.74 (s.e. 0.043; pH0:rg=1= 1.9 ×10−9). At face value this estimate implies genetic factors are only partially shared between the sexes. However, this interpretation should be considered with caution when benchmarked by the meta-analysis of 6 female-female rg estimates of 0.71 (s.e. 0.051; pH0:rg=1= 1.0 × 10−8) and the meta-analysis of 3 male-male rg estimates of 0.75 (s.e. 0.13; pH0:rg=1= 0.05) Hence, the between-sex estimate of rg being significantly different from zero likely reflects the general heterogeneity between the data sets rather than being sex-specific. Indeed, the between-sex rg estimated from the meta-analysis of the GWAS summary statistics of the 4 data sets was 0.90 (s.e. 0.06; pH0:rg=1= 0.096), implying no differences in genetic risk factors between the sexes.
Next, we investigated sex-specific estimates of using LDSC (Table 2, Supplementary Table 3) to determine if there is evidence for a greater genetic contribution to MDD risk in females then males. We have power to detect differences of the order of 2*(s.e. of male estimate + s.e. of female estimate). Initially, in the transformation of the estimate to the liability scale (equation [1]) we assumed K = 0.20 for females and K =0.10 for males (Table 2), consistent with literature reports that MDD is twice as common in females as males (Weissman, Leaf, Holzer, Myers, & Tischler, 1984). The estimates were smaller for males (range −0.02 to 0.15) than for females (range 0.15 to 0.23), but given the magnitude of the standard errors, none of the sex differences were significantly different for any individual data set. Meta-analysis of the estimates of the four data sets did lead to estimates that were significantly different (Meta-4 in Table 2; 0.12 in males vs. 0.19 in females, p=0.0016). However, estimated from the meta-analysed GWAS results of the 4 data sets showed only nominal significance for the difference between males and females (0.10 vs 0.12, p = 0.041; Table 2 GWAS-Meta). This discrepancy can be explained, in part, by the fact that the meta-analysed GWAS summary statistics include association signal from between data-sets as well as within-data sets, and/or may be diluted by between data set heterogeneity. To investigate this discrepancy, we also meta-analysed the six values estimated from the genetic covariance between pairs of same-sex data sets in bivariate LDSC analysis. Since the traits are (presumed to be) the same, the genetic covariance is also an estimate of genetic variance (Supplementary Table 3; Table 2 Meta-6). This again showed lower mean estimates for males with a significant difference between the sexes (0.09 in males vs 0.12 in females, p=0.0070). For completeness, a meta-analysis from all 10 of the estimates is provided (Table 2 Meta-10); this uses the same data sets as the GWAS-Meta, but the latter uses all the information jointly rather than pairwise.
Table 2. Estimates of from LDSC applied to sex-specific GWAS summary statistics.
| Female (se) | Males v1 (se) | Males v2 (se) | P-value v1 |
P-value v2 |
|
|---|---|---|---|---|---|
| K | 0.2 | 0.1 | 0.2 | ||
| u | 0 | 0 | 0.1 | ||
| PGC29 | 0.20 (0.03) | 0.07 (0.04) | 0.09 (0.05) | 0.60 | 0.68 |
| GERA | 0.15 (0.04) | −0.02 (0.05) | −0.03 (0.07) | 0.55 | 0.57 |
| UKB | 0.18 (0.02) | 0.15 (0.02) | 0.20 (0.03) | 0.90 | 0.93 |
| iPSYCH | 0.23 (0.03) | 0.15 (0.04) | 0.20 (0.05) | 0.77 | 0.91 |
| Meta-4 | 0.19 (0.01) | 0.12 (0.02) | 0.16 (0.02) | 1.6×10−3 | 0.23 |
| Meta-6 | 0.12 (0.01) | 0.09 (0.01) | 0.11 (0.01) | 7.0×10−3 | 0.58 |
| Meta-10 | 0.14 (0.01) | 0.10 (0.01) | 0.12 (0.01) | 8.6×10−5 | 0.28 |
| GWAS-Meta | 0.12 (0.01) | 0.10 (0.01) | 0.12 (0.01) | 4.1×10−2 | 0.92 |
estimates are presented on the liability scale achieved through transformation of the LDSC estimate accounting for the case prevalence in the sample (P), the lifetime risk (K) of the disorder, and the proportion of cases in the control sample (u), equation [1]. Meta-4: meta-analysis of the estimates for the 4 data sets (PGC29,GERA,UKB, iPSYCH). Meta-6: meta-analysis of the 6 estimates derived from the genetic covariance estimates from bivariate LDSC between the 6 possible same-sex data-set pairwise combinations. Meta-10: meta-analysis based on all estimates contributing to Meta-4 and Meta-6. GWAS-Meta: estimated from the GWAS summary statistics of the 4 data sets. Versions v1 and v2 differ by K and u values; v2 hypothesis that the lifetime risk of MDD is the same in men and women but that more cases go unreported in men and hence could be included in a screened control set.
For completeness, we also estimated X-chromosome SNP-heritability from the meta-analysed cohorts for males and females separately. However, the standard errors of the estimates were large relative to the estimates (=0.0025 (se=0.06); =0.0005 (se=0.03), which meant estimation of the rg between them was not meaningful.
Before drawing strong conclusions from these results, it is important to recognise that the estimates of depend on the choice of the lifetime risk estimates (K in equations [1] and [2]) (Figure 2). The point estimates are more similar if the same lifetime risk is assumed between the sexes, but it is difficult to justify such an assumption, because it is not, at face value, supported by epidemiological data. However, since depression maybe under-reported in males (Martin, Neighbors, & Griffith, 2013; Thornicroft et al., 2017), for illustration purposes we could assume the true lifetime risk of MDD is the same between the sexes (K =0.20), but that through under-reporting the controls are contaminated by 0.10 of cases (Equation [2], u=0.1). Under these assumptions, the estimates are not significantly different between the sexes for any data set (Figure 2, Table 2)
Figure 2. Impact of choice of lifetime risk on estimate of
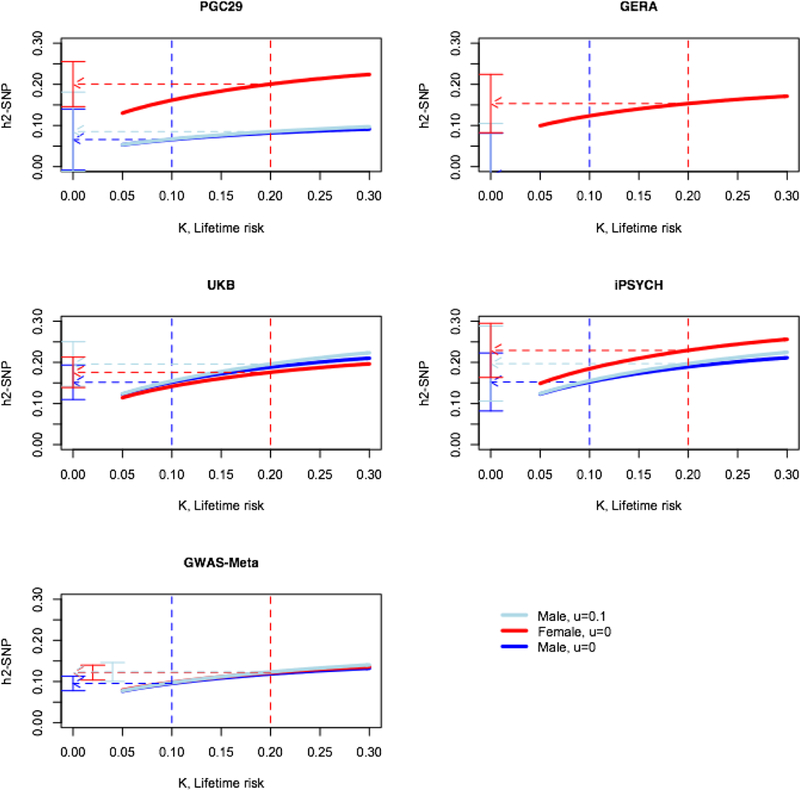
The graphs shows on the liability scale from equation [2], u (proportion of controls that are unrecognised cases). The blue/red dashed lines are positioned at the lifetime risk for males/females. The flat ended bars show the 95% confidence intervals of the estimates at the chosen lifetime risk.
DISCUSSION
Heterogeneity in MDD is often discussed, but hard to investigate. In a novel set of analyses, we explored the heterogeneity of MDD using genetic data. The first set of analyses contrasted 29 PGC cohorts, by estimating their average contribution to estimates of from repeated random samplings of cohorts selected into GWAS meta-analyses. While we found notable differences between cohorts in the contribution estimates (Figure 1), these differences could be explained, at least partly, via knowledge of cohort ascertainment practices: higher contributions for cohorts ascertained in clinical compared to community settings (Figure 1, p=0.028), higher contribution from a sample known to use super-screened controls (nes1), and a trend towards lower contributions from samples that used unscreened controls. One conclusion is that known cohort information about case ascertainment status could be included usefully in analysis methods to increase power. A framework for such an analysis has been proposed (Zaitlen et al., 2012), but in practice the necessary parameters relating to cohort specific risks are usually unknown. In the seven samples contributing to the published PGC meta-analysis (PGC29, GERA, iPSYCH, UK Biobank, deCode, Generation Scotland, 23andMe) (Wray et al., 2018), estimates ranged from 0.09 to 0.25 and the weighted mean rg for all pairwise combinations was 0.76 (s.e. = 0.03), which is significantly different from one. The cohorts had different recruitment strategies with ascertainment ranging from self-report to national hospital records. Moreover, even within the wave 1 PGC-MDD research cohorts endorsement proportions of the nine DSMIV criteria showed considerable heterogeneity including between cohorts that had similar clinical ascertainment strategies(Major Depressive Disorder Working Group of the Psychiatric et al., 2013). For example, endorsement rates of 56%, 27% and 10% were recorded for the criterion symptom 4b, hypersomnia nearly every day, for different early onset (< 30 years) recurrent MDD samples(Major Depressive Disorder Working Group of the Psychiatric et al., 2013). Despite the heterogeneity, out-of-sample prediction demonstrated that the self-reported 23andMe GWAS results explained variance in clinically ascertained cohorts with high significance (Wray et al., 2018). Sample size remains the driving force for genetic discovery in MDD. Ideally, larger sample sizes should be accompanied by collection of detailed, consistent, and longitudinal phenotypic data to enable more precise case and control definitions.
We also investigated between-sex genetic heterogeneity. Our sex-specific analyses found significantly smaller for males than females, a trend replicated in all four data sets, and hence was highly significant in the meta-analysis of the four cohort estimates (Table 2 Meta-4). However, a set of follow-up analyses leads to a somewhat different conclusion. Since the estimates generated from the meta-analysis of the summary statistics of the four data sets was only nominally significantly different (Table 2, GWAS-Meta, males vs. females: 0.10 s.e. 0.01 vs. 0.12 s.e. 0.01, p= 0.041), it could be argued that heterogeneity between data sets (as reported in our between cohort analyses above) removes a signal in sex differences, a point of view supported by the lower estimates for both sexes. However, it is noteworthy that for the largest sample, with data collected under standardised protocols (UKB), the point estimates were similar (Table 2). Moreover, we recognised that the comparisons of between the sexes depended on the choice of their respective lifetime risks (Figure 2). For baseline analyses we used lifetime risk estimates of K = 0.20 for females and K = 0.10 for males, consistent with a 2:1 risk for females vs. males (Weissman et al., 1984), with higher K values generating higher estimates. One explanation for a lower lifetime risk for males could be higher rates of under-reporting (Martin et al., 2013; Thornicroft et al., 2017). We calculated in males assuming the same lifetime risk as females, but with incomplete screening of controls. Such a hypothetical scenario generated similar estimates of between the sexes (Figure 2, Table 2). The last piece of evidence supporting no sex differences in genetic factors contributing to depression is the high estimated rg between the sexes (0.90 s.e. 0.06; pH0:rg=1= 0.096).
In summary, our analyses demonstrate between-cohort genetic heterogeneity, but this can be explained, at least in part, by known factors such as case/control ascertainment. Investigation of between sex heterogeneity provided no convincing evidence to support genetic differences between the sexes. A robust conclusion is simply that large sample sizes will overcome sample heterogeneity as demonstrated in the latest major depression GWAS meta-analyses (Howard et al., 2018; Wray et al., 2018). Based on differences in lifetime disease risk and differences in heritability, while assuming a similar number of contributing risk loci, we previously estimated that sample sizes for GWAS need to be five times bigger for MDD than for schizophrenia (SCZ) (Wray et al., 2012). On the one hand, heterogeneity between samples may push this estimate higher. On the other hand, the heterogeneity may already account for the higher prevalence and lower heritability. The PGC GWAS meta-analysis for MDD/major depression based on 135K cases (Wray et al., 2018) identified 44 independent significant loci. This compares to 145 independent loci for SCZ from 41K cases (Pardiñas et al., 2018), hence requiring 3.4 times as many cases for major depression compared to SCZ per genome-wide significant locus. However, the relationship between sample size and variant discovery is not linear (Wray et al., 2018) and so observing the sample size ratios for discovery will be of interest as sample sizes increase. Very large MDD case-control samples will allow novel methods to be applied to assess evidence for genetic subsets. Larger data sets are likely to lead to the development of new methods to assess genetic heterogeneity (Han et al., 2016). There is a growing interest in machine learning methods (Libbrecht & Noble, 2015) as a strategy to identify phenotypically relevant genetic subsets, but cohort heterogeneity must diminish their utility, making large electronic health or biobank samples collected and genotyped in a uniform way of most value.
Supplementary Material
Acknowledgements
We acknowledge funding from the Australian National Health & Medical Research Council (1078901, 1113400, 1087889). The PGC has received major funding from the US National Institute of Mental Health and the US National Institute of Drug Abuse (U01 MH109528 and U01 MH1095320). A full list of funding is provided in ref(Wray et al., 2018). UK Biobank: this research has been conducted using the UK Biobank 593 Resource (URLs), including applications #4844 and #6818. The Genetic Epidemiology Research on Adult Health and Aging (GERA) study was supported by grant RC2 AG036607 from the National Institute of Health, grants from Robert Wood Johnson Foundation, the Ellison Medical Foundation, the Wayne and Gladys Valley Foundation and Kaiser Permanente. The authors thank the Kaiser Permanente Medical Care Plan, Northern California Region (KPNC) members who have generously agreed to participate in the Kaiser Permanente Research Program on Genes, Environment and Health (RPGEH).
References
- American Psychiatric Association. (1994). Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition ed.). Washington, DC: American Psychiatric Association. [Google Scholar]
- Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, Duncan L, . . . Neale BM (2018). Analysis of shared heritability in common disorders of the brain. Science, 360(6395). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda Y, Kvale MN, Hoffmann TJ, Hesselson SE, Ranatunga D, Tang H, . . . Risch N (2015). Characterizing Race/Ethnicity and Genetic Ancestry for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics, 200(4), 1285–1295. doi: 10.1534/genetics.115.178616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomsma DI, Willemsen G, Sullivan PF, Heutink P, Meijer P, Sondervan D, . . . Penninx BW (2008). Genome-wide association of major depression: description of samples for the GAIN Major Depressive Disorder Study: NTR and NESDA biobank projects. Eur J Hum Genet, 16(3), 335–342. doi: 10.1038/sj.ejhg.5201979 [DOI] [PubMed] [Google Scholar]
- Bruce ML, & Hoff RA (1994). Social and physical health risk factors for first-onset major depressive disorder in a community sample. Social Psychiatry and Psychiatric Epidemiology, 29(4), 165–171. doi: 10.1007/BF00802013 [DOI] [PubMed] [Google Scholar]
- Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, . . . Neale BM (2015). An atlas of genetic correlations across human diseases and traits. Nat Genet, 47(11), 1236–1241. doi: 10.1038/ng.3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics, C., . . . Neale BM (2015). LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet, 47(3), 291–295. doi: 10.1038/ng.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, . . . Marchini J (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature, 562(7726), 203–209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman DP, Whitfield CL, Felitti VJ, Dube SR, Edwards VJ, & Anda RF (2004). Adverse childhood experiences and the risk of depressive disorders in adulthood. J Affect Disord, 82(2), 217–225. doi: 10.1016/j.jad.2003.12.013 [DOI] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics, C., Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, . . . International Inflammatory Bowel Disease Genetics, C. (2013). Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet, 45(9), 984–994. doi: 10.1038/ng.2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanous AH, & Kendler KS (2005). Genetic heterogeneity, modifier genes, and quantitative phenotypes in psychiatric illness: searching for a framework. Mol Psychiatry, 10(1), 6–13. doi: 10.1038/sj.mp.4001571 [DOI] [PubMed] [Google Scholar]
- Fernandez-Pujals AM, Adams MJ, Thomson P, McKechanie AG, Blackwood DH, Smith BH, . . . McIntosh AM (2015). Epidemiology and Heritability of Major Depressive Disorder, Stratified by Age of Onset, Sex, and Illness Course in Generation Scotland: Scottish Family Health Study (GS:SFHS). PLoS One, 10(11), e0142197. doi: 10.1371/journal.pone.0142197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedorowicz JG (2014). Depression and Cardiovascular Disease: An Update on How Course of Illness May Influence Risk. Current psychiatry reports, 16(10), 492–492. doi: 10.1007/s11920-014-0492-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraguas R Jr, Iosifescu DV, Alpert J, Wisniewski SR, Barkin JL, Trivedi MH, . . . Fava M (2007). Major Depressive Disorder and Comorbid Cardiac Disease: Is There a Depressive Subtype With Greater Cardiovascular Morbidity? Results From the STAR*D Study. Psychosomatics, 48(5), 418–425. doi:doi: 10.1176/appi.psy.48.5.418 [DOI] [PubMed] [Google Scholar]
- Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, . . . McVean GA (2010). A map of human genome variation from population-scale sequencing. Nature, 467(7319), 1061–1073. doi: 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Pouget JG, Slowikowski K, Stahl E, Lee CH, Diogo D, . . . Raychaudhuri S (2016). A method to decipher pleiotropy by detecting underlying heterogeneity driven by hidden subgroups applied to autoimmune and neuropsychiatric diseases. Nat Genet, 48(7), 803–810. doi: 10.1038/ng.3572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim C, Newport DJ, Mletzko T, Miller AH, & Nemeroff CB (2008). The link between childhood trauma and depression: Insights from HPA axis studies in humans. Psychoneuroendocrinology, 33(6), 693–710. doi:doi: 10.1016/j.psyneuen.2008.03.008 [DOI] [PubMed] [Google Scholar]
- Howard DM, Adams MJ, Shirali M, Clarke T-K, Marioni RE, Davies G, . . . McIntosh AM (2018). Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nature Communications, 9(1), 1470. doi: 10.1038/s41467-018-03819-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman JC, Celano CM, Beach SR, Motiwala SR, & Januzzi JL (2013). Depression and cardiac disease: epidemiology, mechanisms, and diagnosis. Cardiovasc Psychiatry Neurol, 2013, 695925. doi: 10.1155/2013/695925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt M, Auriemma J, & Cashaw AC (2003). Self-report bias and underreporting of depression on the BDI-II. J Pers Assess, 80(1), 26–30. doi: 10.1207/s15327752jpa8001_10 [DOI] [PubMed] [Google Scholar]
- Kapur S, Phillips AG, & Insel TR (2012). Why has it taken so long for biological psychiatry to develop clinical tests and what to do about it? Mol Psychiatry, 17(12), 1174–1179. doi: 10.1038/mp.2012.105 [DOI] [PubMed] [Google Scholar]
- Kendler KS, Gatz M, Gardner CO, & Pedersen NL (2006). A Swedish national twin study of lifetime major depression. Am J Psychiatry, 163(1), 109–114. doi: 10.1176/appi.ajp.163.1.109 [DOI] [PubMed] [Google Scholar]
- Kessler RC (2003). Epidemiology of women and depression. Journal of Affective Disorders, 74(1), 5–13. doi:doi: 10.1016/S0165-0327(02)00426-3 [DOI] [PubMed] [Google Scholar]
- Kessler RC, & Bromet EJ (2013). The epidemiology of depression across cultures. Annu Rev Public Health, 34, 119–138. doi: 10.1146/annurev-publhealth-031912-114409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JM, Vlasac I, Anderson SG, Kyle SD, Dixon WG, Bechtold DA, . . . Saxena R (2016). Genome-wide association analysis identifies novel loci for chronotype in 100,420 individuals from the UK Biobank. Nat Commun, 7, 10889. doi: 10.1038/ncomms10889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Wray NR, Goddard ME, & Visscher PM (2011). Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet, 88(3), 294–305. doi: 10.1016/j.ajhg.2011.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CM, Ng MY, Butler AW, Cohen-Woods S, Uher R, Pirlo K, . . . McGuffin P (2010). Genome-wide association study of major recurrent depression in the U.K. population. Am J Psychiatry, 167(8), 949–957. doi: 10.1176/appi.ajp.2010.09091380 [DOI] [PubMed] [Google Scholar]
- Lewis G, Mulligan J, Wiles N, Cowen P, Craddock N, Ikeda M, . . . Peters TJ (2011). Polymorphism of the 5-HT transporter and response to antidepressants: randomised controlled trial. Br J Psychiatry, 198(6), 464–471. doi: 10.1192/bjp.bp.110.082727 [DOI] [PubMed] [Google Scholar]
- Libbrecht MW, & Noble WS (2015). Machine learning applications in genetics and genomics. Nat Rev Genet, 16(6), 321–332. doi: 10.1038/nrg3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohoff FW (2010). Overview of the Genetics of Major Depressive Disorder. Current psychiatry reports, 12(6), 539–546. doi: 10.1007/s11920-010-0150-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major Depressive Disorder Working Group of the Psychiatric, G. C., Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, . . . Sullivan PF (2013). A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry, 18(4), 497–511. doi: 10.1038/mp.2012.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LA, Neighbors HW, & Griffith DM (2013). The experience of symptoms of depression in men vs women: analysis of the National Comorbidity Survey Replication. JAMA Psychiatry, 70(10), 1100–1106. doi: 10.1001/jamapsychiatry.2013.1985 [DOI] [PubMed] [Google Scholar]
- McGuffin P, Katz R, Watkins S, & Rutherford J (1996). A hospital-based twin register of the heritability of DSM-IV unipolar depression. Arch Gen Psychiatry, 53, 129–136. [DOI] [PubMed] [Google Scholar]
- Mosing MA, Gordon SD, Medland SE, Statham DJ, Nelson EC, Heath AC, . . . Wray NR (2009). Genetic and environmental influences on the co-morbidity between depression, panic disorder, agoraphobia, and social phobia: a twin study. Depress Anxiety, 26(11), 1004–1011. doi: 10.1002/da.20611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni G, Moser G, Schizophrenia Working Group of the Psychiatric Genomics, C., Wray NR, & Lee SH (2018). Estimation of Genetic Correlation via Linkage Disequilibrium Score Regression and Genomic Restricted Maximum Likelihood. American journal of human genetics, 102(6), 1185–1194. doi: 10.1016/j.ajhg.2018.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardiñas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, . . . Walters JTR (2018). Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nature Genetics, 50(3), 381–389. doi: 10.1038/s41588-018-0059-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penninx BW, Beekman AT, Smit JH, Zitman FG, Nolen WA, Spinhoven P, . . . Consortium NR (2008). The Netherlands Study of Depression and Anxiety (NESDA): rationale, objectives and methods. Int J Methods Psychiatr Res, 17(3), 121–140. doi: 10.1002/mpr.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrot WJ, Boomsma DI, Penninx BW, & Wray NR (2016). Disease and Polygenic Architecture: Avoid Trio Design and Appropriately Account for Unscreened Control Subjects for Common Disease. Am J Hum Genet, 98(2), 382–391. doi: 10.1016/j.ajhg.2015.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuch JJ, Roest AM, Nolen WA, Penninx BW, & de Jonge P (2014). Gender differences in major depressive disorder: results from the Netherlands study of depression and anxiety. J Affect Disord, 156, 156–163. doi: 10.1016/j.jad.2013.12.011 [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Neale MC, & Kendler KS (2000). Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry, 157(10), 1552–1562. doi: 10.1176/appi.ajp.157.10.1552 [DOI] [PubMed] [Google Scholar]
- Thornicroft G, Chatterji S, Evans-Lacko S, Gruber M, Sampson N, Aguilar-Gaxiola S, . . . Kessler RC (2017). Undertreatment of people with major depressive disorder in 21 countries. Br J Psychiatry, 210(2), 119–124. doi: 10.1192/bjp.bp.116.188078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Hemani G, Vinkhuyzen AAE, Chen GB, Lee SH, Wray NR, . . . Yang J (2014). Statistical Power to Detect Genetic (Co)Variance of Complex Traits Using SNP Data in Unrelated Samples. Plos Genetics, 10(4). doi:ARTN e1004269 10.1371/journal.pgen.1004269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vythilingam M, Heim C, Newport J, Miller AH, Anderson E, Bronen R, . . . Bremner JD (2002). Childhood trauma associated with smaller hippocampal volume in women with major depression. Am J Psychiatry, 159(12), 2072–2080. doi: 10.1176/appi.ajp.159.12.2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman MM, Leaf PJ, Holzer CE 3rd, Myers JK, & Tischler GL (1984). The epidemiology of depression. An update on sex differences in rates. J Affect Disord, 7(3–4), 179–188. [DOI] [PubMed] [Google Scholar]
- World Health Organization. (1978). International Classification of Diseases (9th revised ed.). Geneva: World Health Organization. [Google Scholar]
- World Health Organization. (1992). International Classification of Diseases (10th revised ed.). Geneva: World Health Organization. [Google Scholar]
- Wray NR, Pergadia ML, Blackwood DH, Penninx BW, Gordon SD, Nyholt DR, . . . Sullivan PF (2012). Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry, 17(1), 36–48. doi: 10.1038/mp.2010.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, . . . Major Depressive Disorder Working Group of the Psychiatric Genomics, C. (2018). Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet, 50(5), 668–681. doi: 10.1038/s41588-018-0090-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AA, Nolte IM, . . . Visscher PM (2015). Genome-wide genetic homogeneity between sexes and populations for human height and body mass index. Hum Mol Genet, 24(25), 7445–7449. doi: 10.1093/hmg/ddv443 [DOI] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, . . . Visscher PM (2010). Common SNPs explain a large proportion of the heritability for human height. Nat Genet, 42(7), 565–569. doi: 10.1038/ng.608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitlen N, Lindstrom S, Pasaniuc B, Cornelis M, Genovese G, Pollack S, . . . Price AL (2012). Informed conditioning on clinical covariates increases power in case-control association studies. PLoS Genet, 8(11), e1003032. doi: 10.1371/journal.pgen.1003032 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.