

Abstract
Cancer patients have been treated with various types of therapies, including conventional strategies like chemo‐, radio‐, and targeted therapy, as well as immunotherapy like checkpoint inhibitors, vaccine and cell therapy etc. Among the therapeutic alternatives, T‐cell therapy like CAR‐T (Chimeric Antigen Receptor Engineered T cell) and TCR‐T (T Cell Receptor Engineered T cell), has emerged as the most promising therapeutics due to its impressive clinical efficacy. However, there are many challenges and obstacles, such as immunosuppressive tumor microenvironment, manufacturing complexity, and poor infiltration of engrafted cells, etc still, need to be overcome for further treatment with different forms of cancer. Recently, the antitumor activities of CAR‐T and TCR‐T cells have shown great improvement with the utilization of CRISPR/Cas9 gene editing technology. Thus, the genome editing system could be a powerful genetic tool to use for manipulating T cells and enhancing the efficacy of cell immunotherapy. This review focuses on pros and cons of various gene delivery methods, challenges, and safety issues of CRISPR/Cas9 gene editing application in T‐cell‐based immunotherapy.
Keywords: CRISPR, gene editing, immunotherapy
1. INTRODUCTION
1.1. Cell immunotherapy and the challenges it faces
With precise targeting and impressive efficacy, CAR‐T (Chimeric Antigen Receptor Engineered T cell) and TCR‐T (T Cell Receptor Engineered T cell) cell therapies have become powerful and innovative therapeutic modalities for cancer patients. CARs are recombinant receptors that redirect the T‐cell activity towards target cells expressing specific surface antigen, independent of the classic peptide/MHC‐TCR recognition patterns. While TCR‐T cells are directed to recognize tumor‐specific peptide epitopes‐generated from inside the cells with the dependence on MHC molecules.
The first‐generation of CAR consists of the binding moiety from a monoclonal antibody fused to the constant regions of a TCR,1, 2 and later this design was modified to use a single‐chain Fv fragment (scFv) of an antibody linked with CD3 zeta or FcγRIIIA γ signaling chain (Figure 1).3, 4 Such engineered T cells specifically lysed target cells and produced cytokines.5, 6, 7, 8 However, clinical studies showed its limited antitumor efficacies (Table 1), probably owing to the short persistence of CAR‐T cells in vivo.9, 10 The second‐generation of CAR (Figure 1) provided a costimulatory signal in combination with the primary activation signal.11, 12 These CAR‐T cells have a higher level of cytokine production, improved persistence in vivo and potent clinical activities (Table 1),13, 14 enabling FDA's first two approvals of CAR‐T therapies in 2017. The third‐generation of CAR contained two costimulatory domains combined with an activation domain (Figure 1), which suggested an enhancement of antitumor response compared to the second‐generation CAR‐T cells (Table 1).15, 16 A clinical trial comparing the antitumor efficacy between the second‐ and the third‐generation CAR‐T cells is currently underway (NCT01853631). The fourth generation of CAR‐T cells (Table 1), engineered based on the backbone of the second‐generation CAR, were equipped with an inducible expression cassette to produce a transgenic cytokine, for example, IL‐12, IL‐18, upon the engagement of CAR to the specific tumor target (Figure 1).17
Figure 1.
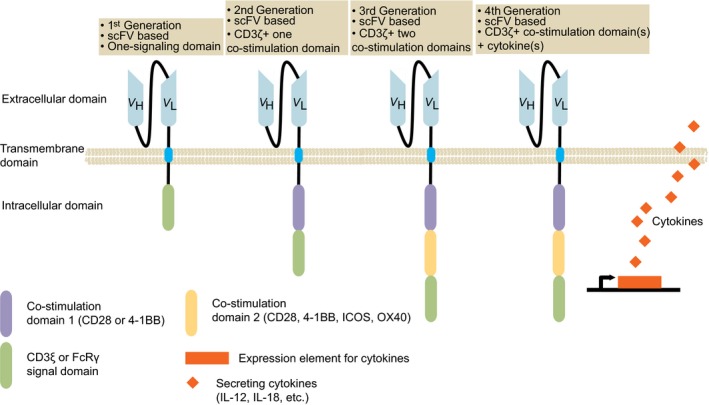
Different generations of CAR. The basic design of CAR is composed of an extracellular binding domain (usually a scFv), a hinge, transmembrane domain and one to three intracellular domains. The fourth‐generation‐CAR‐T cells are engineered to deliver a transgenic payload, such as proinflammatory cytokines, released upon engagement of CAR with its target. CAR, chimeric antigen receptor; scFv, single‐chain variable fragment
Table 1.
Different generations of CAR‐T cells
| Generation | First generation | Second generation | Third generation | Fourth generation |
|---|---|---|---|---|
| Composition of CAR | scFv, hinge, Fc receptor γ or CD3 ζ signaling chain | scFv, hinge, transmembrane region, one costimulatory domain, CD3 ζ | scFv, hinge, transmembrane region, two costimulatory domains, CD3 ζ | scFv, hinge, transmembrane region, one or two costimulatory domains, CD3 ζ, transgenic payload (cytokines, antibodies, enzymes, etc) |
| Preclinical studies | Effective in vitro and in mouse models | Superior antitumor effects, improved persistence, and higher level of cytokine production than first‐generation CAR | Compared to second‐generation CAR‐T, showed enhanced antitumor potency in some studies | Improved the effector function of CAR‐T cells in suppressive tumor microenvironment of solid tumor; provided a safety switch |
| Summary of clinical results | Limited antitumor efficacy due to short persistence in vivo | Most often used in clinic, significant antitumor response in various hematological malignancies | Used in a few clinical studies and most of them are still ongoing. Limited data suggested good antitumor efficacies and modest toxicity in B cell malignancies | Clinical studies for hematological malignancies and solid tumors are still in the early stage |
| Reference | 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 | 11, 12, 13, 14 | 15, 16 | 17 |
Abbreviations: CAR, chimeric antigen receptor; scFv, single‐chain variable fragment.
CAR‐T and TCR‐T cell therapies are showing promising results for cancer treatment, especially for targeting the B‐cell lineage‐restricted CD19 molecule expressed on B‐cell leukemias and lymphomas with CD19‐specific CAR‐T cells.13, 18, 19, 20 However, challenges including poor persistence, long manufacturing time, and limited infiltration of engineered T cells into immunosuppressive environment, still remain to be addressed. Through disrupting TCR and HLA genes, knocking out checkpoint inhibitory molecules, etc, CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas9 (CRISPR‐Associated protein) technology holds enormous promise to enhance T‐cell functionality and improve drug efficacy.
1.2. CRISPR‐Cas9 genome editing system: a genetic tool to enhance T‐cell functionality
Cas9 functions as a RNA‐dependent endonuclease and can be directed to the DNA target sites under the limitation of the protospacer adjacent motif (PAM), guided by a chimeric single‐guide RNA (sgRNA).21 Cas9/sgRNA system could target any DNA sequence of interest by changing sgRNA guide sequence, and cut DNA to cause double‐strand breaks (DSBs).22 These DSBs are repaired by error‐prone nonhomologous end joining (NHEJ) or precise homology‐directed repair (HDR) pathways. NHEJ leads to insertions or deletions (indel) of target gene and makes gene knock‐out possible. HDR uses assisted recombination of DNA donor templates to reconstruct cleaved DNA with precise repair, which could be used to knock‐in desired DNA.
Compared to other genome editing strategies such as transcription activator‐like effector nucleases (TALENs), and zinc‐finger nucleases (ZFNs), CRISPR/Cas9 genome editing is more rapid, cost‐effective, and it has been applied widely in plants and animals with its easier feasibility. Differences among the three types of genome editing systems have been discussed in numbers of review papers.23, 24, 25, 26
2. APPLICATIONS OF CRISPR/CAS9 TECHNOLOGY IN T‐CELL THERAPY
Although commercial products of CAR‐T have been successfully lunched,20, 27 including Kymriah and Yescarta from Novatis and Gilead/Kite, respectively, there is still much room to improve the existing T‐cell therapy. We summarized the recent progress about how CRISPR/Cas9 system could be harnessed to produce advanced CAR‐T cell products, with lower cost, reduced risk of causing malignancies, improved antitumor activities and efficacies (Table 2).
Table 2.
Applications of CRISPR/Cas9 technology in T‐cell therapy
| Application | Generate off‐the‐shelf CAR‐T | Knock‐in CAR or TCR | Knock‐out checkpoint molecules | Generation of CAR‐T cells expressing exogenous cytokines |
|---|---|---|---|---|
| Summary | TCR, B2M and PD‐1 molecules were eliminated simultaneously to enhance the antitumor activity. Other genes such as CTLA‐4 and Fas were also disrupted together with TCR and B2M | CAR or TCR cassette is knocked into endogenous TCR gene locus to mitigate GvHD | PD‐1, CTLA‐4, and LAG‐3 genes were knocked out separately or in combination | Beneficial cell cytokines (‐IL‐12, IL‐15, IL‐18, IL‐17, etc) can be knocked in designed gene locus |
| Advantages | Cheaper and faster, more potent | Avoid random integration; uniform CAR expression | Higher efficacy, less side effects, durable | More natural, less side effects |
| Disadvantages | The elimination of HLA‐class I could increase the attack from NK cells | Low knock‐in efficiency | Potential off‐target effects | Limited knock‐in efficiency |
| References | 28, 29, 30, 31 | 33, 34 | 35, 36, 37, 40, 41, 42, 45 | 46, 47, 48, 49, 50 |
Abbreviations: CRISPR/Cas9, clustered, regularly interspaced, short palindromic repeats/ CRISPR‐associated protein 9. CAR, chimeric antigen receptor; TCR, T cell receptor; PD‐1, programmed cell death protein 1; CTLA‐4, cytotoxic T‐lymphocyte–associated antigen 4; GvHD, graft‐vs‐host disease; LAG‐3, lymphocyte activation gene 3; IL, interleukin.
2.1. Generation of off‐the‐shelf CAR‐T cells
Current CAR‐T therapy mostly focused on autologous T cells owing to the limitation of intrinsic MHC restriction. To shorten the manufacture cycle and lower the cost of CAR‐T cell products, the concept of off‐the‐shelf CAR‐T cells was emerged (Figure 2). Endogenous TCR on allogeneic T cells were eliminated by ZFN and TALEN to avoid graft‐vs‐host disease (GVHD), and HLA molecules were disrupted to prevent a rejection from recipient's immune system.28, 29, 30 It has been reported that universal CAR‐T cells with TCRα chain and CD52 gene disrupted by TALEN were infused to two infants with relapsed refractory CD19+ ALL,31 demonstrating the therapeutic potential of gene‐editing technology.
Figure 2.
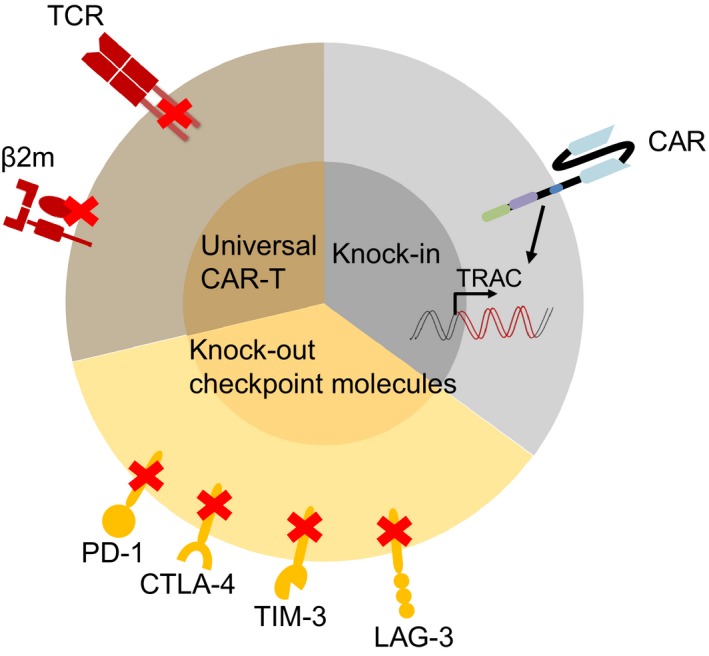
Applications of CRISPR/Cas9 technology in CAR‐T cell therapy. Pie diagram shows that three main aspects of CRISPR/Cas9 system can be applied in CAR‐T cell therapy: to generate universal CAR‐T cell products by disrupting endogenous TCR and MHC molecules, knock‐in CAR at a designed gene locus such as TCR locus to avoid random integration, and knock‐out checkpoint inhibitors to improve antitumor activities. CRISPR/Cas9, clustered regularly interspaced short palindromic repeats/ CRISPR‐associated protein 9; CAR, chimeric antigen receptor; TCR, T‐cell receptor
Compared to ZEN and TALEN technologies, CRISPR/Cas9 gene editing system holds greater promise due to its simplicity and high effectiveness to increase the efficacy of therapeutic agents or work as standalone therapeutics. TRAC and B2M genes have been knocked out simultaneously by CRISPR/Cas9 to generate universal CAR‐T cells.32 Besides, to improve antitumor activity, multiplex genomic editing of CAR‐T cells by CRISPR/Cas9 has been reported.33, 34 This one‐shot CRISPR system has shown to improve gene targeting efficiency and facilitate the manufacture of universal CAR‐T cells deficient in CD3 and HLA‐class I.33
Nevertheless, there is a potential issue when β2M and TCR gene loci are eliminated to prevent allo‐rejection. The elimination of HLA‐class I of T cell could increase the attack from NK cell due to its “missing self” phenotype,35 which should be taken into consideration for the future therapy.
2.2. Mitigation of malignancy risk by knocking in CAR or TCR at a designed gene locus
To avoid oncogenic transformation and transcriptional silencing caused by random integration of CAR into genome by lentivirus infection, knocking in CAR at designed gene locus via homologous recombination (HR) has been achieved (Figure 2). Schumann et al36 conducted a targeted nucleotide replacement in CXCR4 and PD‐1 (PDCD1) gene loci by electroporating Cas9:sgRNA ribonucleoproteins (Cas9 RNPs) with homology‐directed repair template oligonucleotides, establishing applications of Cas9 RNP technology for genome engineering in human T cells. Eyquem et al showed that human T cells were electroporated with Cas9 mRNA and sgRNA to specifically insert a CD19‐specific CAR into TRAC locus, which resulted in not only uniform CAR expression but also enhanced T‐cell potency.37 These results indicate site‐specific knocking‐in a CAR may provide a safer and potent T‐cell product.
In addition to CAR knock‐in, a TCR that recognizes NY‐ESO‐1 tumor antigen has also been knocked into endogenous TCR gene locus, giving rise to specific recognition of tumor antigens and productive antitumor cell responses.38 Such precise knock‐in of CAR or TCR into a specific gene locus by CRISPR/Cas9 system leads to the enhancement of antitumor responses and brings additional clinical benefits to the patients engrafted with engineered T cells.
2.3. Knocking out of inhibitory checkpoint molecules to improve antitumor activity
To conquer the inhibitory effects of immune checkpoints in human T cells, and protect normal cells from being disrupted by checkpoint inhibitors in a nonspecific manner, PD‐1 gene in T cells has been abolished by CRISPR/Cas9 to enhance the cytotoxicity against tumor target cells (Figure 2).39, 40 Hu et al reported that PD‐1 gene was eliminated in anti‐CD133 CAR‐T cells by nucleofection of CRISPR/Cas9 plasmids,41 producing enhanced cytotoxicity of CAR‐T cells and inhibition of tumor growth. Interestingly, it has been reported that blockade of PD‐1, LAG‐3 (Lymphocyte Activation Gene 3) or CTLA‐4 led to a compensatory upregulation of the other checkpoint pathways,42 which means combinatorial blockade strategies should be applied in practice. This conclusion is in accordance with Tanvetyanon's review that combinatorial blockade of PD‐1 and CTLA‐4 may produce a higher antitumor response than PD‐1 blockade alone in patients.43 The clinical potential of combined disruption of PD‐144, 45 and LAG‐346 has also been explored in the CAR‐T cell therapy.
Except knock‐out of checkpoint molecules, an alternative way is to coexpress the PD‐1‐blocking scFv with CAR, which has improved the antitumor activities of CAR‐T cells.47, 48 CRISPR/Cas9 has also been used in TCR‐T cells to disrupt PD‐1 gene.49 The clinical trial with the strategy to disrupt endogenous TCR and PD‐1 gene is ongoing (NCT03399448).
2.4. Generation of CAR‐T cells expressing exogenous cytokines for efficacy improvement
In addition to TCR engagement (Signal 1) and costimulatory signaling (Signal 2), cytokines play essential roles in regulating T‐cell function. Constitutive expression of IL‐12 in CAR‐T cells to destroy antigen‐loss cancer cells,50 and improve antitumor efficiency51 has been reported. IL‐15, which is functionally associated with T‐cell memory, was coexpressed with anti‐CD19 CAR to develop long‐term persistence of CAR‐T cells.52 IL‐18 was also expressed in CAR‐T cells to augment antitumor effects against melanoma.53 IL‐7 and CCL19 have been expressed together with anti‐CD20 CAR‐T to treat preestablished solid tumors.54
Most of the cytokines discussed above including IL‐12, IL‐15, IL‐18, and IL‐7 are overexpressed through gamaretrovirus or lentivirus. Their expression is artificially regulated and may lead to side effects such as T‐cell exhaustion caused by higher secretion of cytokines. A better strategy would be to drive the expression of these cytokines under the control of an endogenous promoter through knocking in by CRISPR/Cas9 at a designated gene locus such as TRAC.
3. DIFFERENT METHODS OF GENE DELIVERY SYSTEMS FOR CRISPR/CAS9‐BASED CELL IMMUNOTHERAPY
An efficient gene delivery system is critical for the success of CAR and TCR cell therapies and the efficacy of gene editing. There are several major delivery systems (Table 3). Gamaretro‐, lenti‐virus‐based gene delivery, and transposon systems are used to stably express CAR/TCR in T cells, while adenoviruses (AdV), adeno‐associated viruses (AAV), electroporation and nanocarriers are used to deliver CRISPR/Cas9 for transient expression (Figure 3). A new delivery system called cell squeezing, recently emerges to deliver a wide range of compounds including DNA, RNA, and protein.55, 56, 57
Table 3.
Different methods of delivery systems for CRISPR/Cas9‐based cell immunotherapy
| Forms of payload | Capacity | Advantages | ||
|---|---|---|---|---|
| Viral delivery system | Infection | AdV | 7.5‐37 kb | 1) Broad infectivity; 2) High titers; 3) Large cloning capacity; 4) Transient expression without integration |
| AAV | 5 kb | 1) Relatively broad host spectrum; 2) Low immunogenicity; 3) Transient gene expression | ||
| Gammaretroviral & lentiviral vectors | 8‐9 kb | 1) Stable gene expression; 2) Lentivirus infects dividing and nondividing cells | ||
| Nonviral delivery system | Electroporation | Minigene/minicircle (Transposon) | More than 100 kb | 1) Lower immunogenicity; 2) Efficient stable genome modification; 3) Reduced cost |
| DNA | More than 100 kb | 1) Easy to operate; 2) Lower cost; 3) scalable manufacturing | ||
| mRNA | Flexible | 1) Higher efficiency; 2) Rapid expression; 3) Reduce off‐target effect | ||
| RNP | Flexible | 1) Lower off‐target; 2) Lower cellular toxicity | ||
| Nano‐carrier | RNA, DNA, or protein | Flexible | 1) Flexible payload sizes and formats; 2) Low immunogenicity; 3) Transient or stable gene expression | |
| Squeeze | Different bioactive materials, including small molecules, polysaccharides, siRNA, proteins, carbon nanotubes and quantum dots | Flexible | 1) Diverse range of payloads; 2) Higher efficiently; 3) Unchanged expression profiles; 4) High throughput; 5) Improved safety |
| Disadvantages | Reference | Clinical trials |
|---|---|---|
| 1) High immunogenicity | 55, 56, 57 | None |
| 1) Limited cargo size; 2) Difficult to produce pure viral stock | 58, 59 | Projected by Editas Medicine in LCA10 patients |
| 1) Genotoxicity; 2) Random integration | 60, 61, 62 |
CAR‐T (gammaretroviral): NCT01583686, NCT02744287 (lentiviral): NCT02935543, NCT03274219, NCT03166878 |
| 1) Cellular toxicity (Transposon); 2) Random integration | 63, 64, 65 | NCT03389035, NCT00968760, NCT01497184, NCT01362452, NCT01653717, NCT02529813 |
| 1) Reduced cell viability; 2) Spontaneous vector integration | 28, 29, 37, 40 | CRISPR: NCT03057912, NCT03164135, NCT03655678, NCT03545815, NCT03081715, NCT03398967, NCT03690011, NCT02863913, NCT02867345, NCT02867332, NCT02793856, NCT03044743 |
| 1) Medium cost; 2) Fast degradation | NCT03399448, NCT03166878 | |
| 1) High cost; 2) Fast degradation | None for CRISPR/Cas9 | |
| Requires special expertise | 66, 67, 68, 69, 70 | NCT02340156, NCT03020017, NCT01455389 |
| Requires special facilities | 51, 52, 53, 71 | None |
AdV, adenovirus; AAV, adenovirus associated virus; RNP, ribonucleoprotein.
LCA10, TYPE 10 leber's congenital amaurosis; CAR, chimeric antigen receptor; CRISPR, clustered, regularly interspaced, short palindromic repeats.
Figure 3.
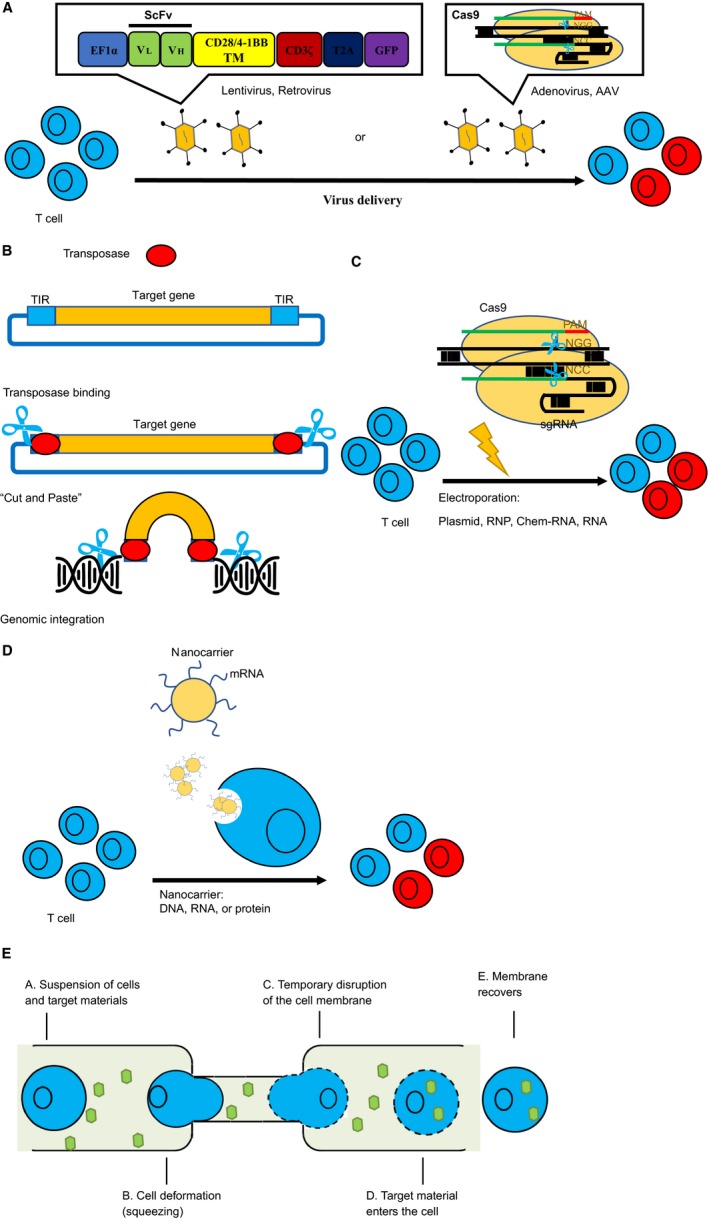
Different methods of delivery systems for CRISPR/Cas9 based cell immunotherapy. A, Viral delivery of CAR or Cas9/sgRNA into T cells. CAR genes are delivered by lentivirus or retrovirus and integrate into the genome for stable expression, while cas9 and sgRNA are often transiently expressed by AdV or AAV. B, Transposition mechanisms of DNA transposon. DNA transposon contains a target gene in the middle, flanked by TIRs. Transposase binds to the TIRs, and mobilizes the transposon for integration into the target genome via a cut‐and‐paste mechanism. TIR: terminal inverted repeats. C, Cas9 and sgRNA are delivered by electroporation in the form of plasmid, RNA, chem‐RNA, or RNP. D, Delivery of DNA, RNA, or protein into target cells via nanoparticles. E, Five main steps of delivery via CellSqueeze technology. scFv, single‐chain variable fragment; Cas9, CRISPR associated protein 9; AAV, adenovirus associated virus; RNP, ribonucleoprotein
3.1. Virus‐mediated gene delivery
Most commonly used viral vectors are derived from AdV, AAV, gammaretrovirus or lentivirus (Figure 3A).58 AdV‐derived vectors are able to infect a broad range of nondividing or dividing vertebrate cells and have many advantages (Table 3).59 Using AdV‐based CRISPR/cas9 system, Cheng et al demonstrated that gene editing in the mouse liver was highly efficient, specific, and persisted long in vivo while the expression of Cas9 protein was transient.60 Nevertheless, AdV has strong immunogenicity in nature, which raises safety concerns for clinical uses.61 AAV vectors have broad spectrum of target cell types and low immunogenicity, with its capacity less than 4.8 kb (Table 3). Using a dual‐vector system with SpCas9 (~4.1 kb) and sgRNA expressing separately, Swiech et al demonstrated effective editing of single or multiple genes in the mouse brain.62 On the other hand, Ran et al identified that smaller Cas9 ortholog, SaCas9 (~3.3 kb) with similar efficiency as SpCas9 allowed the delivery of SaCas9 and its sgRNA in a single AAV vector.63
Gammaretrovirus and lentivirus belong to the retroviral family and possess the intrinsic capability to integrate into the host genome, allowing long‐term and stable transgene expression (Table 3). Lentiviruses are able to infect both dividing and nondividing cells, while gammaretroviruses have been shown to preferentially infect dividing cells.64 CAR‐T cells were generated with 50%‐80% transduction efficiencies using replication‐deficient gammaretroviral or lentiviral vectors,65, 66 which are the main gene delivery systems in manufacturing CAR‐T cells for clinical usage. A major concern for retroviral and lentiviral vectors is the random insertion of transgene into chromosomes, posing risks of oncogenesis, although no apparent oncogenic consequences have been observed from existing clinical practice yet.
3.2. Transposon
Besides the viral vector delivery systems described above, Transposon has emerged as a new potential delivery tool for transferring genes of interest (Figure 3B). The vector system of DNA transposon comprises a transposon containing a gene of interest flanked by terminal inverted repeats (TIRs), and a transposase that binds to TIRs.67 These nonviral vector integration systems, such as PiggyBac (PB) and Sleeping Beauty (SB), also showed advantages for gene delivery (Table 3).
Transposon system was exploited in clinical trials for cancer immunotherapy. Human T cells genetically modified by the SB transposon/transposase system to express a CD19‐specific CAR were evaluated in 2016.68 The clinical results demonstrated that these SB platform‐engineered CAR‐T cells were safe, and further supported the clinical development of this nonviral gene therapy approach. Clinical trials at MD Anderson Cancer (Table 3) have shown that these transposon‐engineered CAR‐T products work robustly and feasibly.69
3.3. Electroporation
Cas9 mRNAs or proteins were electroporated into CAR‐T cells (Figure 3C) to knock‐out specific genes like TCR,33 CTLA‐4, and PD‐1 gene.32, 33, 44 Ren et al accomplished a versatile system for rapidly generating multiplex genome‐edited CAR‐T cells, by lentiviral infection of one‐shot CAR vector with multiple sgRNAs and electroporation of Cas9 mRNA.33 Other labs developed a protocol for combined Cas9 RNP‐mediated gene editing and lentiviral transduction to generate PD‐1 deficient anti‐CD19 CAR T cells.44 Hu et al explored a simplified protocol for generating PD‐1 deficient CD133‐specific CAR T cells, by nucleofecting plasmids of CRISPR/Cas9 system to disrupt PD‐1 gene and piggyBac transposon system for CAR gene expression. This convenient method avoids manufacturing of RNAs or proteins and reduces the processing of T‐cell modification.41
3.4. Nanocarriers
Nanoparticles are emerging synthetic delivery systems with favorable characteristics (Table 3).70, 71 The flexible design of nanoparticles allows to carry versatile types of cargoes or a combination of multiple components (Figure 3D). A novel nanocarrier CRISPR‐gold, designed to deliver three components simultaneously, was able to internalize by primary cells and stem cells via endocytosis.72 Moreover, administration of CRISPR‐gold carrying Cas9 RNP and donor DNA to correct the Dystrophin gene mutation in MDX mice, resulted in the gene repair and restored the protein expression.
Selective targeting of specific cell subgroups is another feature that can be achieved via nanocarriers. In the application of Stephan lab, plasmids encoding CD19‐targeted CAR gene flanked with piggyBac system were coencapsulated in such nanoparticles conjugated with CD3‐targeting antibodies. The strategy showed robust CAR production inside the T cells both in vitro and in vivo.73 This system could also favor transient expression of transgenes by delivering mRNA, as Moffett et al described as “hit‐and‐run programming.”74
The key benefit of using nanoparticle over electroporation is high viability and expansion capability of manipulated cells, allowing its broader applications.
3.5. Microfluidics‐based CellSqueeze
Although gene delivery by electroporation has been wide‐used, it was found by genome‐wide approach that electroporation treatment may disrupt the expression profiles of key functional transcripts and lead to the perturbation of cytokine secretion. A microfluidic delivery system called cell squeezing was recently used for compound delivery. Its mechanism of action is based on mechanical membrane disruption (Figure 3E), which has minimal effects on transcriptional responses and will not modulate T‐cell activity.75 The CellSqueeze technology from SQZ Biotechnologies Co. (Table 3) is able to introduce a wide range of compounds into varieties of cell types like immune cells, embryonic stem cells etc.55, 56, 57
Compared with other delivery systems described above, CellSqueeze technology could achieve high delivery efficacy without adversely affecting cell viability and expression profiles.
4. CHALLENGES AND THE FUTURE DIRECTIONS OF THE APPLICATION OF CRISPR/CAS9 TECHNOLOGY
4.1. An increased risk of tumor malignancy
CRISPR‐Cas9 technology has made gene editing simpler and faster than ever. However, a study points out this popular gene‐editing tool could inadvertently cause cancer. It found that Cas9 RNP delivery triggers a p53‐dependent DNA damage response that suppresses gene correction.76
4.2. Failure of genome editing caused by the immunogenicity elicited from anti‐Cas9 responses
A recent study showed that the most widely used forms of CRISPR could be an immunogen in humans. Instead of modifying the genome while used therapeutically, the editing tool could trigger an adaptive immunity to Cas9 proteins, raising considerable concerns for the future CRISPR clinical trials.77, 78
4.3. An increased risk of off‐target mutagenesis
The off‐target mutation, which may cause genomic instability and disrupt the functionality of other normal genes, is still the major concern of CRISPR/Cas9 system in biomedical and clinical application. Although the targeting specificity of Cas9 is believed to be tightly controlled by the guide sequence of sgRNA and the presence of PAM, potential off‐target cleavage activity could still occur with even three to five base pair mismatches in the PAM‐distal part of the sgRNA‐guiding sequence.79 Moreover, Cas9 stays in the cells for a period of time after treatment, which increases the incidence of DNA being cut in the wrong place.
4.4. Inefficient delivery systems for CRISPR/Cas9
There are several delivery systems for CRISPR/Cas9 as described previously, but the delivery efficiency of each system is still not satisfying, especially for in vivo application. In addition, large DNA fragment knock‐in and multiplex genome editing are generally hard to achieve.
5. CONCLUSIONS AND FUTURE DIRECTIONS
CRISPR/Cas9 has provided a simple, cheap, and fast way to manipulate genomes. CRISPR‐edited CAR‐T and TCR‐T cells hold out great hope and potential for the next generation cancer immunotherapy, particularly for the treatment of solid tumors. Considering the safety issues related to CRISPR/Cas9 system, mutated Cas9 including Cas9 nickase,80 truncated sgRNA,81 or other more accurate nucleases with longer PAM sequences are explored to reduce off‐target effect. As to improve delivery efficiency of CRISPR, new delivery platforms like nanocarrier and CellSqueeze technology, have recently emerged. With rapid improvement in the field of gene therapy, CRISPR/Cas9 genome editing system is expected to have broader therapeutic applications in cancer immunotherapies.
ACKNOWLEDGMENTS
The authors are grateful to Dr. Kun Ma (BGI‐Shenzhen) for his helpful suggestions and helping us revise our manuscript. We also thank our lab members Yijian Li and Renpeng Ding for their assistance. Thank Science, Technology and Innovation Commission of Shenzhen Municipality, China, for its funding (grant number: JCYJ20170817150015170).
Gao Q, Dong X, Xu Q, et al. Therapeutic potential of CRISPR/Cas9 gene editing in engineered T‐cell therapy. Cancer Med. 2019;8:4254–4264. 10.1002/cam4.2257
REFERENCES
- 1. Kuwana Y, Asakura Y, Utsunomiya N, et al. Expression of chimeric receptor composed of immunoglobulin‐derived V regions and T‐cell receptor‐derived C regions. Biochem Biophys Res Comm. 1987;149(3):960‐968. [DOI] [PubMed] [Google Scholar]
- 2. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin‐T‐cell receptor chimeric molecules as functional receptors with antibody‐type specificity. Proc Natl Acad Sci USA. 1989;86(24):10024‐10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody‐binding domains and the gamma or zeta subunits of the immunoglobulin and T‐cell receptors. Proc Natl Acad Sci USA. 1993;90(2):720‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brocker T, Peter A, Traunecker A, Karjalainen K. New simplified molecular design for functional T cell receptor. Eur J Immunol. 1993;23(7):1435‐1439. [DOI] [PubMed] [Google Scholar]
- 5. Hwu P, Shafer GE, Treisman J, et al. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J Exp Med. 1993;178(1):361‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stancovski I, Schindler DG, Waks T, Yarden Y, Sela M, Eshhar Z. Targeting of T lymphocytes to Neu/HER2‐expressing cells using chimeric single chain Fv receptors. J Immunol. 1993;151(11):6577‐6582. [PubMed] [Google Scholar]
- 7. Moritz D, Wels W, Mattern J, Groner B. Cytotoxic T lymphocytes with a grafted recognition specificity for ERBB2‐expressing tumor cells. Proc Natl Acad Sci USA. 1994;91(10):4318‐4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hwu P, Yang JC, Cowherd R, et al. In vivo antitumor activity of T cells redirected with chimeric antibody/T‐cell receptor genes. Can Res. 1995;55(15):3369‐3373. [PubMed] [Google Scholar]
- 9. Lamers C, Sleijfer S, Vulto AG, et al. Treatment of metastatic renal cell carcinoma with autologous T‐lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20‐22. [DOI] [PubMed] [Google Scholar]
- 10. Lamers C, Sleijfer S, van Steenbergen S, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR‐engineered T cells: clinical evaluation and management of on‐target toxicity. Mol Ther. 2013;21(4):904‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Finney HM, Lawson A, Bebbington CR, Weir A. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161:2791‐2797. [PubMed] [Google Scholar]
- 12. Golubovskaya V, Berahovich R, Xu QM, et al. GITR domain inside CAR co‐stimulates activity of CAR‐T cells against cancer. Front Biosci. 2018;23:2245‐2254. [DOI] [PubMed] [Google Scholar]
- 13. Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee DW, Kochenderfer JN, Stetler‐Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose‐escalation trial. Lancet. 2015;385(9967):517‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilkie S, Picco G, Foster J, et al. Retargeting of human T cells to tumor‐associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180(7):4901‐4909. [DOI] [PubMed] [Google Scholar]
- 16. Guedan S, Posey AD, Shaw C, et al. T cell persistence through ICOS and 4–1BB costimulation. JCI insight. 2018;3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145‐1154. [DOI] [PubMed] [Google Scholar]
- 18. Brentjens RJ, Riviere I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19‐targeted T cells in patients with relapsed or chemotherapy refractory B‐cell leukemias. Blood. 2011;118(18):4817‐4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kochenderfer JN, Dudley ME, Feldman SA, et al. B‐cell depletion and remissions of malignancy along with cytokine‐associated toxicity in a clinical trial of anti‐CD19 chimeric‐antigen‐receptor–transduced T cells. Blood. 2012;119(12):2709‐2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor‐modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nishimasu H, Ran F, Hsu P, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156(5):935‐949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(8):167‐170. [DOI] [PubMed] [Google Scholar]
- 23. Ren J, Zhao Y. Advancing chimeric antigen receptor T cell therapy with CRISPR/Cas9. Protein cell. 2017;8(9):634‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mollanoori H, Shahraki H, Rahmati Y, Teimourian S. CRISPR/Cas9 and CAR‐T cell, collaboration of two revolutionary technologies in cancer immunotherapy, an instruction for successful cancer treatment. Hum Immunol. 2018;79(12):876–882. [DOI] [PubMed] [Google Scholar]
- 25. Xia A‐L, He Q‐F, Wang J‐C, et al. Applications and advances of CRISPR‐Cas9 in cancer immunotherapy. J Med Genet. 2018;56(1):4–9. [DOI] [PubMed] [Google Scholar]
- 26. Singh N, Shi J, June CH, Ruella M. Genome‐editing technologies in adoptive T cell immunotherapy for cancer. Curr Hematol Malig Rep. 2017;12(6):522‐529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garfall AL, Maus MV, Hwang W‐T, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med. 2015;373(11):1040‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Torikai H, Reik A, Liu P‐Q, et al. A foundation for universal T‐cell based immunotherapy: T cells engineered to express a CD19‐specific chimeric‐antigen‐receptor and eliminate expression of endogenous TCR. Blood. 2012;119(24):5697‐5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Provasi E, Genovese P, Lombardo A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012;18(5):807‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Poirot L, Philip B, Schiffer‐Mannioui C, et al. Multiplex genome‐edited T‐cell manufacturing platform for "Off‐the‐Shelf" adoptive T‐cell immunotherapies. Can Res. 2015;75(18):3853‐3864. [DOI] [PubMed] [Google Scholar]
- 31. Waseem Qasim HZ, Samarasinghe S, Adams S, Persis A. molecular remission of infant b‐cell after infusion of universal TALEN gene‐edited CAR T cells. Sci Transl Med. 2017. [DOI] [PubMed] [Google Scholar]
- 32. Liu X, Zhang Y, Cheng C, et al. CRISPR‐Cas9‐mediated multiplex gene editing in CAR‐T cells. Cell Res. 2017;27(1):154‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ren J, Zhang X, Liu X, et al. A versatile system for rapid multiplex genome‐edited CAR‐T cell generation. Oncotarget. 2017;8(10):17002‐17011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23(9):2255‐2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCreedy BJ, Senyukov VV, Nguyen KT. Off the shelf T cell therapies for hematologic malignancies. Best Pract Res Clin Haematol. 2018;31(2):166‐175. [DOI] [PubMed] [Google Scholar]
- 36. Schumann K, Lin S, Boyer E, et al. Generation of knock‐in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci USA. 2015;112(33):10437‐10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eyquem J, Mansilla‐Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roth TL, Puig‐Saus C, Yu R, et al. Reprogramming human T cell function and specificity with non‐viral genome targeting. Nature. 2018;559:405‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Su S, Hu B, Shao J, et al. CRISPR‐Cas9 mediated efficient PD‐1 disruption on human primary T cells from cancer patients. Sci Rep. 2016;6:20070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang C, Peng Y, Hublitz P, Zhang H, Dong T. Genetic abrogation of immune checkpoints in antigen‐specific cytotoxic T‐lymphocyte as a potential alternative to blockade immunotherapy. Sci Rep. 2018;8(1):5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu B, Zou Y, Zhang L, et al. Nucleofection with plasmid DNA for CRISPR/Cas9‐mediated inactivation of programmed cell death protein 1 in CD133‐specific CAR T cells. Hum Gene Ther. 2018;30(4):446‐458. [DOI] [PubMed] [Google Scholar]
- 42. Huang RY, Francois A, McGray AR, Miliotto A, Odunsi K. Compensatory upregulation of PD‐1, LAG‐3, and CTLA‐4 limits the efficacy of single‐agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology. 2017;6(1):e1249561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanvetyanon T, Gray JE, Antonia SJ. PD‐1 checkpoint blockade alone or combined PD‐1 and CTLA‐4 blockade as immunotherapy for lung cancer? Expert Opin Biol Ther. 2017;17(3):305‐312. [DOI] [PubMed] [Google Scholar]
- 44. Rupp LJ, Schumann K, Roybal KT, et al. CRISPR/Cas9‐mediated PD‐1 disruption enhances anti‐tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jiangtao Ren XL, Chongyun F, Shuguang J. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Y, Zhang X, Cheng C, et al. CRISPR‐Cas9 mediated LAG‐3 disruption in CAR‐T cells. Front Med. 2017;11(4):554‐562. [DOI] [PubMed] [Google Scholar]
- 47. Li SI, Siriwon N, Zhang X, et al. Enhanced cancer immunotherapy by chimeric antigen receptor‐modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res. 2017;23(22):6982‐6992. [DOI] [PubMed] [Google Scholar]
- 48. Rafiq S, Yeku OO, Jackson HJ, et al. Targeted delivery of a PD‐1‐blocking scFv by CAR‐T cells enhances anti‐tumor efficacy in vivo. Nat Biotechnol. 2018;36(9):847‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Blank C, Brown I, Peterson AC, et al. PD‐L1/B7H‐1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Can Res. 2004;64:1140‐1145. [DOI] [PubMed] [Google Scholar]
- 50. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL‐12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen‐independent macrophage response on tumor cells that have shut down tumor antigen expression. Can Res. 2011;71(17):5697‐5706. [DOI] [PubMed] [Google Scholar]
- 51. Pegram HJ, Lee JC, Hayman EG, et al. Tumor‐targeted T cells modified to secrete IL‐12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119(18):4133‐4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hurton LV, Singh H, Najjar AM, et al. Tethered IL‐15 augments antitumor activity and promotes a stem‐cell memory subset in tumor‐specific T cells. PNAS. 2016;113(48):E7788‐E7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hu B, Ren J, Luo Y, et al. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL‐18. Cell Rep. 2017;20(13):3025‐3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL‐7 and CCL19 expression in CAR‐T cells improves immune cell infiltration and CAR‐T cell survival in the tumor. Nat Biotechnol. 2018;36(4):346‐351. [DOI] [PubMed] [Google Scholar]
- 55. Adamo A, Sharei A, Adamo L, Lee B, Mao S, Jensen KF. Microfluidics‐based assessment of cell deformability. Anal Chem. 2012;84(15):6438‐6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sharei A, Zoldan J, Adamo A, et al. A vector‐free microfluidic platform for intracellular delivery. Proc Natl Acad Sci USA. 2013;110(6):2082‐2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sharei A, Poceviciute R, Jackson EL, et al. Plasma membrane recovery kinetics of a microfluidic intracellular delivery platform. Integr Biol. 2014;6(4):470‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lundstrom K. Viral Vectors in Gene Therapy. Diseases. 2018;6(2):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wold WS, Toth K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr Gene Ther. 2013;13(6):421‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cheng R, Peng J, Yan Y, et al. Efficient gene editing in adult mouse livers via adenoviral delivery of CRISPR/Cas9. FEBS Lett. 2014;588(21):3954‐3958. [DOI] [PubMed] [Google Scholar]
- 61. Raper SE, Chirmule N, Lee FS, et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80(1–2):148‐158. [DOI] [PubMed] [Google Scholar]
- 62. Swiech L, Heidenreich M, Banerjee A, et al. In vivo interrogation of gene function in the mammalian brain using CRISPR‐Cas9. Nat Biotechnol. 2015;33(1):102‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ran FA, Cong LE, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yamashita M, Emerman M. Retroviral infection of non‐dividing cells: old and new perspectives. Virology. 2006;344(1):88‐93. [DOI] [PubMed] [Google Scholar]
- 65. Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B‐cell maturation antigen is a promising target for adoptive T‐cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048‐2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kochenderfer JN, Feldman SA, Zhao Y, et al. Construction and preclinical evaluation of an anti‐CD19 chimeric antigen receptor. J Immunother. 2009;32(7):689‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huang CR, Burns KH, Boeke JD. Active transposition in genomes. Annu Rev Genet. 2012;46:651‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kebriaei P, Singh H, Huls MH, et al. Phase I trials using Sleeping Beauty to generate CD19‐specific CAR T cells. J Clin Investig. 2016;126(9):3363‐3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kebriaei P, Izsvak Z, Narayanavari SA, Singh H, Ivics Z. Gene therapy with the sleeping beauty transposon system. TIG. 2017;33(11):852‐870. [DOI] [PubMed] [Google Scholar]
- 70. Riley MK, Vermerris W. Recent advances in nanomaterials for gene delivery—a review. Nanomaterials. 2017;7(5):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sau S, Alsaab HO, Bhise K, Alzhrani R, Nabil G, Iyer AK. Multifunctional nanoparticles for cancer immunotherapy: A groundbreaking approach for reprogramming malfunctioned tumor environment. J Control Release. 2018;274:24‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee K, Conboy M, Park HM, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology‐directed DNA repair. Nat Biomed Eng. 2017;1:889‐901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Smith TT, Stephan SB, Moffett HF, et al. In situ programming of leukaemia‐specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol. 2017;12(8):813‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Moffett HF, Coon ME, Radtke S, et al. Hit‐and‐run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat Commun. 2017;8(1):389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. DiTommaso T, Cole JM, Cassereau L, et al. Cell engineering with microfluidic squeezing preserves functionality of primary immune cells in vivo. Proc Natl Acad Sci USA. 2018;115(46):E10907‐E10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR‐Cas9 genome editing induces a p53‐mediated DNA damage response. Nat Med. 2018;24(7):927‐930. [DOI] [PubMed] [Google Scholar]
- 77. Li L, He ZY, Wei XW, Gao GP, Wei YQ. Challenges in CRISPR/CAS9 delivery: potential roles of nonviral vectors. Hum Gene Ther. 2015;26(7):452‐462. [DOI] [PubMed] [Google Scholar]
- 78. Charlesworth CT, Deshpande PS, Dever DP, et al. Identification of Pre‐Existing Adaptive Immunity to Cas9 Proteins in Humans. bioRxiv. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH. Off‐target effects in crispr/cas9‐mediated genome engineering. Mol Ther Nucleic acids. 2015;4:e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ran F, Hsu P, Lin C‐Y, et al. Double nicking by RNA‐guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380‐1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR‐Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]