

Bluetongue virus (BTV) is responsible of the arthropod-borne disease bluetongue (BT) transmitted to ruminants by blood-feeding midges. In this report, we found that BTV, through its nonstructural protein NS3 (BTV-NS3), interacts with BRAF, a key component of the MAPK/ERK pathway. In response to growth factors, this pathway promotes cell survival and increases protein translation. We showed that BTV-NS3 enhances the MAPK/ERK pathway, and this activation is BRAF dependent. Treatment of MAPK/ERK pathway with the pharmacologic inhibitor U0126 impairs viral replication, suggesting that BTV manipulates this pathway for its own benefit. Our results illustrate, at the molecular level, how a single virulence factor has evolved to target a cellular function to increase its viral replication.
KEYWORDS: MAP kinases, Reoviridae, interferons, molecular virus-host interactions, virulence factors
ABSTRACT
Bluetongue virus (BTV) is an arbovirus transmitted by blood-feeding midges to a wide range of wild and domestic ruminants. In this report, we showed that BTV, through its nonstructural protein NS3 (BTV-NS3), is able to activate the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway, as assessed by phosphorylation levels of ERK1/2 and the translation initiation factor eukaryotic translation initiation factor 4E (eIF4E). By combining immunoprecipitation of BTV-NS3 and mass spectrometry analysis from both BTV-infected and NS3-transfected cells, we identified the serine/threonine-protein kinase B-Raf (BRAF), a crucial player in the MAPK/ERK pathway, as a new cellular interactor of BTV-NS3. BRAF silencing led to a significant decrease in the MAPK/ERK activation by BTV, supporting a model wherein BTV-NS3 interacts with BRAF to activate this signaling cascade. This positive regulation acts independently of the role of BTV-NS3 in counteracting the induction of the alpha/beta interferon response. Furthermore, the intrinsic ability of BTV-NS3 to bind BRAF and activate the MAPK/ERK pathway is conserved throughout multiple serotypes/strains but appears to be specific to BTV compared to other members of Orbivirus genus. Inhibition of MAPK/ERK pathway with U0126 reduced viral titers, suggesting that BTV manipulates this pathway for its own replication. Altogether, our data provide molecular mechanisms that unravel a new essential function of NS3 during BTV infection.
IMPORTANCE Bluetongue virus (BTV) is responsible of the arthropod-borne disease bluetongue (BT) transmitted to ruminants by blood-feeding midges. In this report, we found that BTV, through its nonstructural protein NS3 (BTV-NS3), interacts with BRAF, a key component of the MAPK/ERK pathway. In response to growth factors, this pathway promotes cell survival and increases protein translation. We showed that BTV-NS3 enhances the MAPK/ERK pathway, and this activation is BRAF dependent. Treatment of MAPK/ERK pathway with the pharmacologic inhibitor U0126 impairs viral replication, suggesting that BTV manipulates this pathway for its own benefit. Our results illustrate, at the molecular level, how a single virulence factor has evolved to target a cellular function to increase its viral replication.
INTRODUCTION
Bluetongue virus (BTV) is the etiologic agent of the arthropod-borne disease bluetongue (BT) transmitted to ruminants by blood-feeding midges of the genus Culicoides. It belongs to the Orbivirus genus within the Reoviridae family, with 27 serotypes currently identified (1) and at least 6 putative new serotypes (2–7). BTV infects a broad spectrum of wild and domestic ruminants even if sheep are the most sensitive species to the disease. During the 20th century, BTV was principally circumscribed to tropical and subtropical geographical areas (8). In 2006, BTV serotype 8 (BTV-8, strain 2006) emerged in Northern Europe (9) from which it rapidly spread to Central and Western Europe, causing significant economic losses (mortality, morbidity, reduced production, and restrictions in the trade of ruminants). Despite the fact that a high vaccination coverage has been achieved across many European countries, allowing the control of the BT disease, BTV outbreaks are still a major concern for the World Organization for Animal Health (OIE), particularly in Europe (1). Clinical signs include hemorrhagic fever, ulcer in the oral cavity and upper gastrointestinal tract, necrosis of the skeletal and cardiac muscle, and edema of the lungs (10). These variabilities in its host range and clinical manifestations are due to several factors related both to the infected hosts and the viral serotypes and strains.
The BTV genome is composed of 10 double-stranded RNA (dsRNA) segments encoding seven structural (VP1 to VP7) and five, or possibly six, nonstructural (NS1 to NS4, NS3A, and possibly NS5) proteins (11–13). The BT virion is an icosahedral particle organized as a triple-layered capsid. Viral genomic segments are associated with replication complexes containing VP1 (RNA-dependent RNA polymerase), VP4 (capping enzyme, including methyltransferase), and VP6 (RNA-dependent ATPase and helicase) and enclosed by VP3 (subcore) and VP7 (core) (14). Cell attachment and viral entry involve the two structural proteins of the outer capsid VP5 and the most variable of BTV protein VP2 representing the main target of neutralizing antibodies and determines the serotype specificity (15, 16). Nonstructural proteins contribute to the control of BTV replication (17), viral protein synthesis (18), maturation, and export from infected cells (19–23). Initially described for NS4, NS3 has also been shown to counteract the innate immune response and in particular the type I interferon (IFN-α/β) pathway (13, 24, 25).
NS3 is encoded by the segment 10 and expressed as two isoforms, NS3 and NS3A, the latter being translated from an second in-frame start codon by which the first N-terminal 13 amino acids residues are lacking (26). NS3 proteins are glycoproteins that promote viral release either through its viroporin activity (20) or by budding. The latter implies interactions between NS3 and outer capsid VP2/VP5 proteins (27), and cellular proteins involved in the pathway of endosomal sorting complexes required for transport (ESCRT; TSG101 and NEDD4-like ubiquitin ligase) and the calpactin light chain p11 (23, 28, 29). Altogether, these reports provide molecular basis of the multifunctional role of BTV-NS3 as a virulence factor and determinant of pathogenesis, as also illustrated by other in vivo studies using BTV monoreassortants and NS3/NS3A knockout mutants (30–32).
Many viruses can modulate and hijack signaling pathways related to the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway for more efficient replication (33). In response to extracellular stimuli such as growth factors, several downstream components of the MAPK/ERK pathway, including RAS, RAF, MEK1/2, and ERK1/2, are successively activated. Then, ERK1/2 directly or indirectly regulates not only transcription factors (e.g., Elk1) involved in cell proliferation, differentiation, and survival (34, 35) but also cellular factors that control mRNA translation such as eukaryotic initiation factor 4E (eIF4E) (36). In 2010, Mortola and Larsen were the first to show the modulation, as activation, of the MAPK/ERK pathway by BTV (37). In contrast to this finding, other studies demonstrated that the phosphorylation of ERK1/2 was reduced (38) or unchanged (39) after BTV infection. The discrepancy of these studies may be due to different virus infection kinetics or differences in viral serotype/strain and/or cell line used. In addition, the molecular mechanisms underlying the potential modulation of this pathway and its possible contribution to the BTV pathogenesis remain to be established. Altogether, these data led us to investigate the functional impact of BTV on the MAPK/ERK signaling pathway. Using different strategies, we provided unprecedented evidence for the crucial role of BTV-NS3 in the activation of the MAPK/ERK pathway by BTV, as well as the importance of this positive regulation in BTV replication. We demonstrate that this modulation involves a key MAPK component, BRAF, and that this activation acts independently of the role of BTV-NS3 in counteracting the induction of the IFN-α/β response.
(This article was submitted to an online preprint archive [40].)
RESULTS
BTV-NS3 activates the MAPK/ERK signaling pathway.
To address the question of BTV modulation with the MAPK/ERK pathway, we used a trans-reporter gene assay that measures Elk1 activation by ERK1/2. This reporting system allows quick detection of the MAPK/ERK activation and provides not only qualitative but also quantitative comparison between different conditions. In this system, Elk1 transcription factor is fused to the DNA binding domain of Gal4 (Gal4-DB) and leads to the expression of the firefly luciferase reporter gene downstream of a promoter sequence containing a Gal4 binding site. Upon stimulation with a growth factor like EGF, Elk1 was activated, as assessed by a 5-fold increase of luciferase activity compared to unstimulated HEK-293T cells (Fig. 1A). Cells infected with BTV at different multiplicities of infection (MOIs) showed a very strong enhancement in this cellular pathway, even in the absence of epidermal growth factor (EGF) stimulation (Fig. 1A), and in an MOI-dependent manner. Moreover, the inability of a UV-inactivated BTV to activate the luciferase reporter gene indicated that the induction of MAPK/ERK was dependent on viral replication or de novo viral protein expression. To determine whether viral protein(s) could be involved in the MAPK/ERK activation, we tested separately all the BTV proteins in our reporter assay. As shown in Fig. 1B, only BTV-NS3 is able to strongly activate the MAPK/ERK pathway both in the presence and in the absence of EGF stimulation. In parallel, the expression levels of all BTV proteins have been verified by immunoblotting (Fig. 1C). Most of BTV proteins were robustly expressed with few exceptions like NS4 and NS5, which were barely detectable. In order to confirm the impact of NS3 on the MAPK/ERK pathway taking account of the expression profile of all BTV proteins, we tested different amounts of expression vector encoding BTV-NS3 (300, 100, 30, 10, and 3 ng) using the same luciferase reporting system. As shown in Fig. 1D, BTV-NS3 is able to activate the MAPK/ERK pathway even with small amounts of plasmid (up to 10 ng). In conclusion, the activation of the MAPK/ERK pathway by BTV notably involves its NS3 viral protein.
FIG 1.

Activation of the MAPK/ERK pathway by BTV-NS3. (A) HEK-293T cells were transfected with pFA2-Elk1 to express Elk1 transcription factor fused to the DNA binding domain of Gal4, pGal4-UAS-Luc, which contains the firefly luciferase reporter gene downstream of a promoter sequence containing Gal4 binding site, and pRL-CMV, which drives Renilla luciferase expression constitutively. At 12 h after transfection, the cells were serum starved, and 6 h later EGF was added at a final concentration of 400 ng/ml. At the time of EGF stimulation, the cells were also infected with WT BTV at the indicated MOIs or UV inactivated BTV (MOI = 0.05). After 24 h, the relative luciferase activity was determined. ND, not determined. (B) Same experiments as in panel A but, in addition to these three plasmids, the cells were cotransfected with 300 ng of expression vectors encoding BTV ORFs in fusion with the 3×FLAG tag. At 12 h after transfection, the cells were serum starved, and 6 h later EGF was added at a final concentration of 400 ng/ml. After 24 h, the relative luciferase activity was determined. All experiments were achieved in triplicate, and the data represent means ± the SD. (C) HEK-293T cells were transfected with 300 ng of expression vectors encoding BTV ORFs in fusion with the 3×FLAG tag. At 12 h after transfection, the cells were serum starved for 24 h. The cells were then lysed, and the expression levels of 3×FLAG-tagged BTV proteins were detected by anti-3×FLAG immunoblotting. (D) As described in panels A and B, HEK-293T cells were cotransfected with pFA2-Elk1, pGal4-UAS-Luc, pRL-CMV, and different amounts (300, 100, 30, 10, and 3 ng) of an expression vector encoding 3×FLAG-tagged BTV-NS3, BTV-VP3 (300 ng), or the corresponding empty vector pCI-neo-3×FLAG (300 ng). At 12 h after transfection, the cells were serum starved, and 6 h later EGF was added at a final concentration of 400 ng/ml. After 24 h, the relative luciferase activity was determined. All experiments were achieved in triplicate, and data represent means ± the SD. *, P < 0.05; **, P < 0.005; ***, P < 0.0005 (differences observed with the corresponding control vector pCI-neo-3×FLAG). ns, nonsignificant differences. In parallel, the indicated 3×FLAG-tagged BTV proteins were detected by anti-3×FLAG immunoblotting. Sizes are shown in kilodaltons (kDa).
BTV-NS3 interacts with BRAF.
As a first approach to understand at molecular level how BTV-NS3 activates the MAPK/ERK pathway, we undertook proteomic analyses after immunoprecipitation of NS3 from either BTV-infected or NS3-transfected HEK-293T cells. The whole purification protocol and liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses are presented in Materials and Methods. These analyses revealed that BTV-NS3 copurified with BRAF in both BTV-infected and NS3-transfected cells (with Mascot scores of 64 and 104, respectively), whereas BRAF was not detected in the control conditions (mock-infected or empty pCI-neo-3×FLAG-transfected cells). The identified peptides corresponding to BRAF are listed in Table 1 .
TABLE 1.
Identified peptides corresponding to BRAF
| BRAF sequence | Positions (according to BRAF) | Ion score (by search engine)a
|
|
|---|---|---|---|
| 3×FLAG-tagged NS3 | BTV infection | ||
| QTAQGMDYLHAK | 559–570 | 6 | 31 |
| LLFQGFR | 254–260 | 16 | 28 |
| IGDFGLATVK | 592–601 | 56 | 38 |
| SSSAPNVHINTIEPVNIDDLIR | 363–384 | 33 | NA |
| DQIIFMVGR | 663–671 | 29 | NA |
| LDALQQR | 89–95 | 39 | NA |
| GYLSPDLSK | 672–680 | 17 | NA |
With a percolator 1% FDR threshold. NA, not applicable.
BRAF, together with ARAF and CRAF, are members of the RAF kinase family that play a central role in regulating the MAPK/ERK signaling pathway. Therefore, binding to BRAF represents a potential molecular mechanism underlying this manipulation that we decided to investigate. To validate this interaction, HEK-293T cells were infected with BTV (MOI = 0.1), and BTV-NS3 was immunoprecipitated 24 h later using the same purification protocol mentioned above. As shown in Fig. 2A, BTV-NS3 was able to copurify with BRAF, confirming the mass spectrometry analysis. Then, full-length BTV-NS3 (NS3FL), NS3A, and different fragments of NS3 (Fig. 2B) were tested for their ability to interact with endogenous BRAF. To do so, glutathione S-transferase (GST)-tagged NS3FL or the indicated fragments were expressed in HEK-293T cells and purified 48 h later with glutathione-Sepharose beads. As expected, endogenous BRAF copurified with NS3FL (Fig. 2C). In contrast to the indicated fragments, NS3A was also able to bind BRAF, suggesting (Fig. 2C) that the first N-terminal 13 amino acids residues are not essential for this interaction.
FIG 2.

Both BTV-NS3 and -NS3A interact with BRAF and activate the MAPK/ERK pathway. (A) HEK-293T cells were infected with BTV (MOI = 0.1), and NS3 was purified with a specific antibody 24 h after infection. BTV-NS3 and endogenous BRAF were detected in both immunoprecipitated proteins (left panel) and total cell lysates (right panel). (B) Schematic representation of BTV-NS3 protein. Deletion fragments of BTV-NS3 were designed. Transmembrane (T) and extracellular domains (E) are indicated. (C) HEK-293T cells were transfected with expression vectors encoding GST alone or fused to the full-length NS3 protein (NS3FL), NS3A, or the indicated fragments and then tested for interaction with endogenous BRAF. Total cell lysates were prepared 48 h posttransfection (cell lysate; middle panel), and copurifications of endogenous BRAF were assayed by pulldown using glutathione-Sepharose beads (pulldown; upper panel). GST-tagged NS3FL, NS3A, and fragments were detected by immunoblotting with anti-GST antibody (pulldown; lower panel), while endogenous BRAF was detected with a specific antibody. (D) As described in Fig. 1, HEK-293T cells were transfected with pFA2-Elk1, pGal4-UAS-Luc, and pRL-CMV, the expression vectors encoding 3×FLAG-tagged NS3FL, NS3A, or fragments, as indicated. At 12 h after transfection, the cells were serum starved, and 6 h later EGF was added at a final concentration of 400 ng/ml. After 24 h, the relative luciferase activity was determined. All experiments were achieved in triplicate, and data represent means ± the SD. Sizes are shown in kilodaltons (kDa).
Using the same luciferase assay, we showed that both full-length BTV-NS3 and -NS3A are able to enhance Elk1 activation (Fig. 2D). The indicated fragments of NS3 were unable to do so, consistently with the previous pulldown assays (Fig. 2C). Altogether, these results demonstrate that the full-length BTV-NS3 and its NS3A isoform interact with BRAF and activate the MAPK/ERK signaling pathway.
Phosphorylation of ERK1/2 and eIF4E is stimulated by BTV infection and in cells expressing BTV-NS3.
To further decipher the impact of BTV-NS3 on the MAPK/ERK signaling pathway, we measured the activation of ERK1/2 and the translation initiation factor eIF4E, the latter being indirectly regulated by ERK1/2 and considered a crucial factor that control mRNA translation (41). We compared the phosphorylation kinetics of ERK1/2 and eIF4E in HEK-293T cells infected by BTV (Fig. 3A) and in cells expressing BTV-NS3 (Fig. 3B). HEK-293T cells were infected with BTV (MOI = 0.01) and 24 h later, cells were serum starved for 12 h before being stimulated with EGF. Phosphorylation levels of ERK1/2, determined at 10 min, 30 min, 120 min, 6 h, and 24 h after stimulation, were markedly and reproducibly higher after BTV infection compared to the mock-infected control (Fig. 3A). Interestingly, we observed that BTV also induced ERK1/2 phosphorylation even in the absence of EGF stimulation. This is reminiscent to what was observed for Elk1 activation (Fig. 1A), showing a significant activation of the MAPK/ERK pathway by BTV. In contrast to ERK1/2, the phosphorylation level of eIF4E in BTV-infected cells was increased only at later time points after EGF stimulation, whereas p-eIF4E decreased in the mock condition (Fig. 3A).
FIG 3.

Stimulation of ERK1/2 and eIF4E phosphorylations by BTV infection or BTV-NS3 expression. HEK-293T cells were transfected with an expression vector encoding 3×FLAG-tagged BTV-NS3 or the corresponding empty vector pCI-neo-3×FLAG (B). After 18 h, the cells were serum starved and, where indicated, infected with WT BTV (MOI = 0.01) (A). After 12 h, the cells were stimulated with 400 ng/ml of EGF. Phosphorylations of ERK1/2 and eIF4E were measured at 10 min, 30 min, 2 h, 6 h, and 24 h after EGF stimulation (A and B). BTV infection was confirmed by anti-NS3 immunoblotting (A, lower panel), and the expression of 3×FLAG-tagged BTV-NS3 in transfected cells was detected by anti-3×FLAG immunoblotting (B, lower panel). Densitometric analysis of the gels were performed for p-ERK, ERK1/2, peIF4E, and eIF4E, and the graphs represent the ratio of phosphorylated proteins to total proteins. Data presented are representative of at least three independent experiments. Sizes are shown in kilodaltons (kDa).
In parallel to the BTV infectious context, cells were transfected with 3×FLAG-tagged BTV-NS3 or a control plasmid. After 24 h, cells were serum starved, and the phosphorylation levels of ERK1/2 and eIF4E were measured at the same time points post-EGF stimulation as before. Like BTV infection, similar phosphorylation kinetics of ERK1/2 and eIF4E were observed for BTV-NS3 expression alone (Fig. 3B). In conclusion, the phosphorylation kinetics of ERK1/2 and eIF4E confirm that either BTV infection or transient expression of BTV-NS3 can both activate the MAPK/ERK pathway.
BRAF intracellular localization is modified by BTV-NS3.
Although HEK-293T cells are highly efficient for transfection and support BTV replication, we aimed to complement our analysis by carrying out similar experiments with a cell line derived from a host naturally infected by BTV. To do so, we measured phosphorylation levels of ERK1/2 in a bovine kidney cell line (MDBK). MDBK cells were serum starved and infected with BTV at different MOIs for 24 h. As shown in Fig. 4A, BTV increased phosphorylation levels of ERK1/2 in an MOI-dependent manner, which is consistent to what was observed for Elk1 activation in HEK-293T cells infected with BTV (Fig. 1A).
FIG 4.

BRAF subcellular localization in the BTV infectious context. (A) MDBK cells were serum starved and infected with BTV at the indicated MOIs for 24 h. Phosphorylated ERK1/2, total ERK1/2, and BTV-NS3 were detected by Western blot analysis. Sizes are shown in kilodaltons (kDa). (B) MDBK cells were serum starved and infected with BTV (MOI = 0.01). After 24 h, the cells were fixed with 4% PFA and labeled with Hoechst 33258 dye to stain the nuclei and with specific antibodies for BRAF and BTV-NS3. Intracellular localization of Hoechst-stained nuclei (blue), endogenous BRAF (green), and BTV-NS3 (red) was visualized by fluorescence microscopy (×63 magnification). Scale bars, 20 μm.
To determine the potential consequences of NS3-BRAF interaction on their own subcellular localizations, we carried out fluorescence microscopy in MDBK cells. Cells were serum starved and infected with BTV (MOI = 0.01). Then, BTV-NS3 and BRAF localizations were analyzed at 24 h postinfection by fluorescence microscopy (Fig. 4B). First, in mock-infected cells, BRAF is present throughout the cytosol with a sparse punctate distribution. As expected, in cells infected by BTV, NS3 is localized in specific cytoplasmic structures evocative of the Golgi apparatus but also at the plasma membrane. Interestingly, we also found that the subcellular distribution of BTV-NS3 matched the relocalization of BRAF in BTV-infected cells. It should also be noted that similar distribution patterns were also observed in HeLa cells infected with BTV, where NS3 matched the relocalization of BRAF in structures evocative of the Golgi apparatus (data not shown). These results demonstrated that BTV-NS3 alters the localization of BRAF, which may contribute to the BTV-activated MAPK/ERK pathway.
U0126 inhibitor blocks the activation of MAPK/ERK by BTV and alters viral replication.
To demonstrate that enhancement of Elk1 activation by BTV-NS3 is completely dependent on ERK1/2 activation, HEK-293T cells were transfected with 3×FLAG-tagged BTV-NS3 or a control plasmid and 24 h later treated with 20 μM of MEK1/2 inhibitor U0126 (Fig. 5A). The U0126 molecule targets MEK1/2 that are directly activated by BRAF proteins (42). As shown in Fig. 5A, NS3-induced Elk1 activation was completely inhibited by U0126. To confirm these results, we measured the phosphorylation levels of ERK1/2 and eIF4E in HEK-293T cells treated with different concentrations of U0126 (20, 10, 5, and 2.5 μM) before infection with BTV (Fig. 5B). As observed for Elk1 activation, U0126 efficiently blocked the phosphorylation of ERK1/2 after BTV infection whatever the concentration of U0126 used. In contrast to ERK1/2, the phosphorylation of eIF4E was only inhibited at 20 and 10 μM U0126. Interestingly, the presence of U0126 at the same concentrations (20 and 10 μM) also prevented the expression of BTV-NS3 and -VP5. To test whether this inhibitor could have an antiviral effect on BTV replication, HEK-293T cells were treated with MEK1/2 inhibitor U0126 and infected with BTV (MOI = 0.01). As shown in Fig. 5C, HEK-293T cells treated with 20 and 10 μM U0126 exhibited significant lower viral titers compared to the dimethyl sulfoxide control. It should also be noted that cell viability is not affected at 10, 5, and 2.5 μM U0126 (data not shown). Altogether, these results suggest that BTV manipulates the MAPK/ERK signaling pathway to increase replication efficiency.
FIG 5.

Effects of U0126 inhibitor on BTV-induced MAPK/ERK pathway. (A) As described in Fig. 1, HEK-293T cells were transfected with pFA2-Elk1, pGal4-UAS-Luc, and pRL-CMV to determine the activation level of the MAPK/ERK signaling pathway. In addition to these three plasmids, the cells were cotransfected with an expression vector encoding 3×FLAG-tagged BTV-NS3 or the corresponding empty vector pCI-neo-3×FLAG. At 12 h after transfection, the cells were serum starved, and 6 h later the cells were treated with a 20 μM concentration of the MEK1/2-specific inhibitor U0126 where indicated. After 24 h, the relative luciferase activity was determined. All experiments were achieved in triplicate, and data represent means ± the SD. (B) HEK-293T cells were serum starved and infected with BTV (MOI = 0.01). At the time of infection, the cells were also treated with different concentrations of U0126 inhibitor, as indicated. After 24 h, phosphorylations of ERK1/2 and eIF4E were measured, and BTV infection was confirmed by anti-NS3 and -VP5 immunoblotting. Sizes are shown in kilodaltons (kDa). (C) HEK-293T cells were serum starved and infected with BTV (MOI = 0.01). At the time of infection, the cells were also treated with different concentrations of U0126 inhibitor, as indicated. After 24 h, the supernatants were harvested and titrated by determining the 50% tissue culture infective doses (TCID50)/ml. The experiment was performed in triplicates, and data represent means ± the SD. *, P < 0.05.
BRAF silencing impairs BTV-activated MAPK/ERK pathway.
To further confirm the experiments with the MAPK/ERK pharmacologic inhibitor U0126, we used a gene silencing approach targeting BRAF. HEK-293T cells were transfected with BRAF-specific or control nonspecific small interfering RNA (siRNA) before infection with BTV (Fig. 6A and C) or transfection with 3×FLAG-tagged BTV-NS3 (Fig. 6B and D). In both cases, the reduction of BRAF expression led to a significant decrease of the MAPK/ERK activation, as assessed by luciferase reporter gene assays and anti-pERK/1/2 immunoblotting. These results support a model where BTV-NS3 interaction with BRAF enhances MAPK/ERK pathway activation.
FIG 6.

BRAF silencing impairs activation of MAPK/ERK pathway by BTV. (A to D) HEK-293T cells were transfected with nonspecific or a BRAF-specific siRNA. One day later, the cells were either infected with BTV (MOI = 0.01) (A and C) or transfected to express 3×FLAG-tagged BTV-NS3 (B and D), as described in Fig. 1. After 24 h, the relative luciferase activity was determined (A and B), and cell lysates were analyzed by immunoblotting with antibodies against the indicated proteins (C and D). (A and B) All experiments were achieved in triplicate, and data represent means ± the SD. ***, P < 0.0005 (C and D). Sizes are shown in kilodaltons (kDa).
NS3 interaction with BRAF and activation of the MAPK/ERK signaling pathway are characteristic of BTV.
To address the question of the specificity of the BRAF interaction, we first compared NS3 proteins from three serotypes (BTV1, -8, and -27) in their ability to interact with endogenous BRAF. GST-tagged NS3 from BTV1, -8, and -27 were expressed in HEK-293T cells and purified 48 h later with glutathione-Sepharose beads. As shown in Fig. 7A, NS3 proteins from BTV1, -8, and -27 have similar binding capacities for BRAF. As a consequence, BTV1-NS3 and BTV27-NS3 were also able to enhance the MAPK/ERK pathway, although the activation by BTV8-NS3 was stronger compared to NS3 proteins from BTV1 and 27 (Fig. 7B). Then, NS3 proteins from other members of Orbivirus genus, such as epizootic hemorrhagic disease virus (EHDV), African horse sickness virus (AHSV), and equine encephalosis virus (EEV) were also tested for binding to endogenous BRAF. Only BTV-NS3 was able to copurify with BRAF, demonstrating the specificity of this interaction (Fig. 7C). Moreover, only BTV-NS3 highly activates MAPK/ERK pathway, in contrast to EHDV-NS3, AHSV-NS3, and EEV-NS3 (Fig. 7D). Thus, the interaction with BRAF and the activation of the MAPK/ERK pathway are unique to BTV-NS3.
FIG 7.

Comparative analysis of MAPK/ERK pathway for NS3 proteins from different orbiviruses. (A and C) HEK-293T cells were transfected with expression vectors encoding GST alone or fused to BTV8-NS3, BTV1-NS3, BTV27-NS3, EHDV-NS3, AHSV-NS3, or EEV-NS3 as indicated and tested for interaction with endogenous BRAF. Total cell lysates were prepared at 48 h posttransfection (cell lysate; middle panel), and copurifications of endogenous BRAF were assayed by pulldown using glutathione-Sepharose beads (pulldown; upper panel). Sizes are shown in kilodaltons (kDa). (B and D) As described in Fig. 1, HEK-293T cells were transfected with pFA2-Elk1, pGal4-UAS-Luc, and pRL-CMV. In addition to these three plasmids, cells were cotransfected with an expression vector encoding 3×FLAG-tagged BTV8-NS3, BTV1-NS3, BTV27-NS3, EHDV-NS3, AHSV-NS3, EEV-NS3, or the corresponding empty vector pCI-neo-3×FLAG, as indicated. At 12 h after transfection, the cells were serum starved for 24 h, and the relative luciferase activity was then determined. The experiment was performed in triplicate, and data represent means ± the SD. ***, Differences observed between BTV8-NS3, BTV1-NS3, or BTV27-NS3 and the corresponding control vector pCI-neo-3×FLAG were statistically significant (P < 0.0005); **, differences observed between BTV-NS3 and the corresponding control vector pCI-neo-3×FLAG were statistically significant (P < 0.005); ns, nonsignificant differences between EHDV-NS3, AHSV-NS3, or EEV-NS3 and the corresponding control vector pCI-neo-3×FLAG.
BTV-NS3 inhibits the induction of IFN-α/β independently of MAPK/ERK signaling.
Our team has demonstrated the major role of BTV-NS3 in counteracting the induction of the type I interferon (IFN-α/β) response (24). Interestingly, it has been reported that the activation of the MAPK/ERK pathway could be associated with the inhibition of IFN-α/β synthesis (43). Therefore, we sought to determine whether the activation of the MAPK/ERK pathway by BTV-NS3 is required for an efficient control of the IFN-α/β response. Using a luciferase gene reporter assay, we tested NS3FL, NS3A, and its fragments for their capacity to inhibit an IFN-β-specific promoter downstream of a stimulation with a constitutively active form of RIG-I (NΔRIG-I for N-terminal CARDs of RIG-I). As shown in Fig. 8A, both NS3FL and NS3A fully blocked the IFN-β promoter activity. Moreover, while NS31–182, NS3118–229, and NS3Δ118–182 were not able to activate Elk1, as previously shown in Fig. 2C, these fragments are partially, but significantly, able to block the IFN-β promoter activity (Fig. 8A). As a complementary approach, we used the MEK1/2 inhibitor U0126 and measured its impact on the capacity of BTV-NS3 to inhibit the IFN-β promoter activity. As shown in Fig. 8B, the U0126 molecule was unable to prevent the antagonist function of BTV-NS3 on the induction of IFN-α/β and thus to rescue a significant activation of the IFN-β promoter. In conclusion, our data demonstrate that the activation of MAPK/ERK by BTV-NS3 does not contribute to its antagonist activity on the IFN-α/β synthesis.
FIG 8.
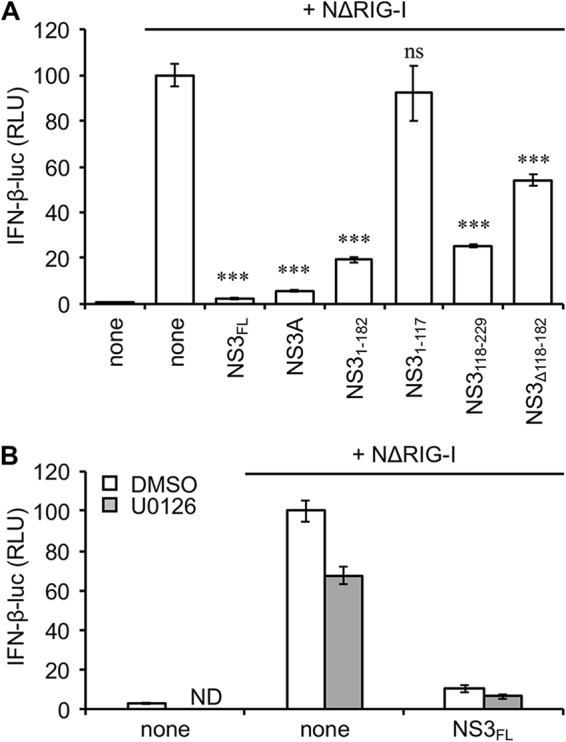
Activation of MAPK/ERK pathway is not related to the inhibition of IFN-α/β signaling by BTV-NS3. (A) HEK-293T cells were cotransfected with IFN-β-pGL3 plasmid that contains the firefly luciferase reporter gene downstream of an IFN-β-specific promoter sequence, pRL-CMV reference plasmid, and pCI-neo-3×FLAG expression vectors encoding 3×FLAG alone or fused to NΔRIG-I (N-terminal CARDs of RIG-I) and BTV-NS3FL, BTV-NS3A, or fragments. After 48 h, the relative luciferase activity was determined. ***, Differences observed between NS3FL, NS3A, NS31–182, NS3118–229, or NS3Δ118–182 and the corresponding control vector pCI-neo-3×FLAG were statistically significant P < 0.0005; ns, nonsignificant differences between NS31–117 and the corresponding control vector pCI-neo-3×FLAG. (B) Same experiments as in panel A but, at 24 h posttransfection, the cells were serum starved for 6 h and then treated with 20 μM U0126, as indicated. After 24 h, the relative luciferase activity was determined. All experiments were performed in triplicates, and data represent means ± the SD. ND, not determined.
DISCUSSION
As obligate intracellular parasites, viruses have evolved multiples strategies to hijack their host cellular machineries and control them to survive, replicate, and spread. The MAPK/ERK pathway contains one of the most highly conserved family of serine/threonine kinases from yeast to humans, which regulates a multiplicity of cellular processes, including cell survival, proliferation, and differentiation, as well as immune and inflammatory responses. Therefore, many viruses have been shown to modulate this pathway for their own benefit (33). Considering that its aberrant activation represents an important step toward carcinogenesis, the modulation of the MAPK/ERK pathway has been initially described for DNA tumor viruses and oncogenic retroviruses. However, nononcogenic RNA viruses are also able to activate this signaling cascade even if the molecular mechanisms underlying this manipulation, in particular in terms of protein-protein interactions, often remain a pending question.
In this report, we show that BTV activates the MAPK/ERK pathway, as assessed by Elk1 transactivation and phosphorylation levels of ERK1/2 and eIF4E, which is reminiscent of the findings of Mortola and Larsen (37), but we also give the first example of a BTV protein that contributes to this positive regulation. We further demonstrate that both BTV infection and NS3 expression alone also activate the MAPK/ERK pathway in the absence of external stimuli. Moreover, our data provide a molecular basis for this activity through the identification of BRAF as a new interactor of BTV-NS3, together with the fact that BRAF silencing impairs BTV-activated MAPK/ERK signaling. Intriguingly, two other studies have shown that BTV does not activate the MAPK/ERK pathway (38, 39). However, these findings do not necessarily contradict our current data. The apparent discrepancy does not appear to depend on factors related to the virus used, as we have demonstrated that NS3 proteins from at least three serotypes of BTV have similar abilities to bind BRAF and activate the MAPK/ERK pathway. In addition, we have confirmed this activation in both human and bovine cells. One possible explanation could be that, in contrast to these previous studies, all of our experiments were performed under starvation conditions, which is likely to be essential since the MAPK/ERK pathway is activated in response to growth factors.
In contrast to BTV, NS3 proteins from EHDV, AHSV, and EEV are unable to activate the MAPK/ERK pathway, suggesting that BTV-NS3 is likely to be functionally distinct from other Orbivirus NS3 proteins. Although BTV-NS3 shares several domains with EHDV-NS3, AHSV-NS3, and EEV-NS3 (e.g., the amphipathic helix at the N terminus, the late-domain motifs, and the extracellular and two transmembrane domains) (23, 44), these proteins are genetically different. Indeed, the NS3 proteins of AHSV and EEV only share ∼30% of sequence homology with BTV-NS3, whereas the protein sequence is more conserved between BTV-NS3 and EHDV-NS3 (57% of sequence homology), which is consistent with the fact that both BTV and EHDV are transmitted to ruminants. Although we have currently no explanation for this apparent specificity of BTV, these differences in protein sequence could account for their capacity or incapacity to activate the MAPK/ERK pathway. Nevertheless, further investigations will be needed to understand how the MAPK activation could provide an advantage for BTV at molecular/cellular level in comparison to other orbiviruses.
Considering that ERK1/2 regulate more than 160 downstream target factors (45), activation of the MAPK/ERK pathway by BTV could have several consequences on host cell biology. It triggers both NF-κB, c-Jun, and STAT1 transcription factors (46, 47), leading to an increased expression of inflammatory factors, in particular cytokines and chemokines, that participate in immunity and inflammation, such as interleukin-6 (IL-6) and IL-8 (48–50). Although the expression of cytokines and chemokines is critical for developing an efficient antiviral response, their excessive production could also lead to deleterious inflammation by causing tissue damage and therefore contribute to disease pathogenesis. Indeed, both in vivo and in vitro studies have shown that BTV-infected cells secrete numerous proinflammatory cytokines, including IL-6 and IL-8 (51–57). A consequence of such production could be an aggravation of the endothelial injury associated with an increased vascular permeability, as already observed in severe cases of BT disease (10, 58). Altogether, our report provides instrumental data to further investigate in vivo whether BTV-NS3 interaction with BRAF enhances MAPK/ERK activation above normal level in infected cells, possibly contributing to a deregulation of the blood vessels permeability to promote BTV replication and spreading.
Besides its effects on immunity and inflammation, activation of the MAPK/ERK pathway has considerable consequences on viral replication, as assessed by experiments using MEK1/2 inhibitor U0126. Indeed, it is now well documented that MAPK/ERK pathway inhibition with U0126 highly alters the replication of several viruses such as influenza virus, Junin virus, herpes simplex virus 1, astrovirus, Borna disease virus, coronavirus, human parainfluenza virus type 3, and porcine epidemic diarrhea virus (59–66). Similarly, we show in this report that MAPK/ERK pathway inhibition by U0126 prevents BTV protein expression in infected cells, resulting in a reduced viral titer. It should also be noted that the inhibition of the MAPK/ERK pathway, as assessed by Elk1 transactivation and phosphorylation levels of ERK1/2, was more efficient in cells treated with U0126 than in those transfected with the BRAF siRNA (see Fig. 5A compared to Fig. 6B and see Fig. 5B compared to Fig. 6C). These differences could be due to the fact that BRAF expression was not fully blocked following gene silencing (Fig. 6C and D), and this would explain why NS3 expression, as well as the viral titers, are not affected in cells transfected with the BRAF siRNA (data not shown). Our findings could correlate with data obtained with other members of the Reoviridae family. Indeed, activation of the EGF signaling pathway has been correlated with increased reovirus replication and spread through the regulation of multiple steps of the infectious life cycle, including viral uncoating and disassembly, viral protein translation, and the generation of viral progeny (67–69). However, and in contrast to reovirus (70), we showed that the activation of the MAPK/ERK pathway by BTV-NS3 does not contribute to its ability to inhibit the induction of IFN-α/β. Rhesus and group A rotaviruses also activate the MAPK/ERK pathway to facilitate viral replication and viral uncoating, respectively (71, 72). A possible link between MAPK/ERK pathway and BTV protein expression could be related to the downstream targets of this pathway, which are essential factors of the cellular translational machinery, like eIF4E.
eIF4E is believed to be the least abundant of all initiation factors. Therefore, it can be considered an excellent target to regulate protein synthesis. Phosphorylation of eIF4E is mediated by MAPK interacting kinase 1 (MNK1), itself mainly activated by ERK1/2 (36). However, MNK1 has also other downstream targets, including eukaryotic initiation factor 4G (eIF4G). Once activated, eIF4E interacts with the cap structure and brings translation initiation factors together with the small ribosomal subunit via the scaffold protein eIF4G to initiate cap-dependent mRNA translation (73). Since BTV mRNAs are capped like their host counterparts (74, 75), it may be assumed that BTV increases the phosphorylation level of eIF4E, thereby stimulating cap-dependent translation, to promote its own viral protein synthesis within infected cells.
Our findings support a model wherein BTV-NS3 interacts with BRAF to activate MAPK/ERK pathway. To the best of our knowledge, this is the first report demonstrating both BRAF as a target of a viral protein and this interaction as an important step toward a viral manipulation of the MAPK/ERK pathway. We also demonstrate that BTV infection leads to a relocalization of BRAF either at the cell membrane or the Golgi apparatus. Initially described to be mainly regulated at the plasma membrane (76, 77), it has now been clearly established that the control of MAPK/ERK pathway can also occur in the Golgi apparatus (78). Thus, these specific localizations could contribute to the aggregation of NS3-BRAF complexes to enhance MAPK/ERK signaling. Further analyses with confocal microscopy will be needed to confirm the colocalization between NS3 and BRAF, and additional biochemical investigations are still required to decipher how this viral protein activates BRAF and the downstream events of this pathway. Altogether, NS3 interactions with BRAF represents a potential target for the development of antiviral molecules against BTV.
MATERIALS AND METHODS
Cell lines and viral infections.
HEK-293T and MDBK cells were maintained in Dulbecco modified Eagle medium (Gibco-Invitrogen) containing 10% fetal bovine serum, penicillin, and streptomycin at 37°C and 5% CO2. BTV8 wild-type (WT) strain was amplified and titrated on BSR-T7. Inactivated virus was prepared by exposing live virus to 254-nm UV light, as previously described (79). Serum-free medium was used as an inoculum for BTV-infected cells. BTV infection was analyzed at the indicated time points by fluorescence microscopy or Western blotting with specific NS3 (kindly provided by Frederick Arnaud) (80) and VP5 antibodies (monoclonal antibody 10AE12; Ingenasa).
Plasmid DNA constructs.
Open reading frame (ORF)-encoding sequences from the BTV8 WT strain (isolated in the French Ardennes in 2006 [81]), the BTV1 WT strain (isolated in France in 2008 [82]), the BTV27 WT strain (isolated in Corsica in 2014 [83]), the EHDV6 WT strain (isolated in Reunion Island in 2009 [84]), the AHSV4 WT strain (isolated in Morocco in 1990 [85]), and the EEV3 WT strain (isolated in South Africa in 1974 [86]) were amplified by reverse transcription-PCR (RT-PCR; Roche) from purified infected-cell RNAs. Amplification was performed using ORF-specific primers flanked with the Gateway cloning sites 5′-GGGGACAACTTTGTACAAAAAAGTTGGC and 5′-GGGGACAACTTTGTACAAGAAAGTTGG. PCR products were cloned by in vitro recombination into pDONR207 (Gateway System; Invitrogen). ORF coding sequences were subsequently transferred by in vitro recombination from pDONR207 into different Gateway-compatible destination vectors (see below) according to the manufacturer’s recommendations (LR cloning reaction; Invitrogen). In mammalian cells, GST tag and 3×FLAG tag fusions were achieved using pDEST27 (Invitrogen) and pCI-neo-3×FLAG vector, respectively (87). An expression vector pNRIG-I carrying genes for the constitutively active N-terminal CARDs of RIG-I (NRIG-I) has been used to stimulate the luciferase reporter gene downstream of an IFN-β-specific promoter sequence, as previously described (88).
Luciferase reporter gene assay.
HEK-293T were plated in 24-well plates (5 × 105 cells per well). One day later, cells were transfected with either pFA2-Elk1 (0.3 μg/well; PathDetect signal transduction pathway trans-reporting system from Stratagene [catalog number 219005]) and pGal4-UAS-Luc plasmids (0.3 μg/well; provided by Yves Jacob) or IFN-β-pGL3 (0.3 μg/well; Stratagene), together with the pRL-CMV reference plasmid (0.03 μg/well; Promega). Cells were simultaneously cotransfected with 0.3 μg/well of the empty pCI-neo-3×FLAG expression vector or encoding viral proteins as specified. At 12 h after transfection, the cells were serum starved for 6 h and then stimulated with EGF (Sigma) at 400 ng/ml. When specified, at the time of EGF stimulation, the cells were infected by BTV at the indicated MOIa. After 24 h, the cells were lysed, and both firefly and Renilla luciferase activities in the lysate were determined using the Bright-Glo and Renilla-Glo luciferase assay systems (Promega), respectively. The reporter activity was calculated as the ratio of firefly luciferase activity to the reference Renilla luciferase activity. All graphs show mean values and include error bars indicating the standard deviations (SD).
Statistical analyses.
P values were determined using an unpaired two-tailed Student t test. Differences were considered significant if the P value was <0.05 (*), <0.005 (**), or <0.0005 (***).
Coaffinity purification experiments.
To perform coaffinity purification experiments coupled to mass spectrometry analyses, HEK-293T cells were either infected with BTV8 WT strain or transfected with pCI-neo-3×FLAG expression vectors encoding 3×FLAG alone or fused to BTV-NS3FL. Briefly, 2 × 106 HEK-293T cells were dispensed in each well of a 6-well plate (three wells per condition) and, after 24 h, infected (MOI = 0.1) or transfected with 1 μg of each plasmid DNA (JetPRIME; Polyplus). At 18 h postinfection or at 24 h posttransfection, the cells were washed in phosphate-buffered saline (PBS) and then resuspended in lysis buffer (20 mm MOPS [morpholinepropanesulfonic acid]-KOH [pH 7.4], 120 mm of KCl, 0.5% IGEPAL, 2 mM β-mercaptoethanol), supplemented with Complete protease inhibitor cocktail (Roche). Cell lysates were incubated on ice for 20 min and then clarified by centrifugation at 14,000 × g for 10 min. Protein extracts were incubated for 3 h on a spinning wheel at 4°C with 40 μl of protein G-Sepharose beads (Roche) and 2.5 μg of the specific BTV-NS3 antibody. Beads were then washed with ice-cold lysis buffer three times for 5 min on a spinning wheel.
For other coaffinity purification experiments, ORFs encoding NS3 or fragments were transferred from pDONR207 to pDEST27 expression vector (Invitrogen) to achieve GST fusion. HEK-293T cells were transfected with 300 ng of each plasmid DNA per well. At 2 days posttransfection, the cells were collected in PBS, incubated on ice in lysis buffer for 20 min, and clarified by centrifugation at 14,000 × g for 10 min. For pulldown analysis, protein extracts were incubated for 2 h at 4°C with 30 μl of glutathione-Sepharose beads (Amersham Biosciences) to purify GST-tagged proteins. Beads were then washed with ice-cold lysis buffer three times for 5 min, and proteins were recovered by boiling in denaturing loading buffer (Invitrogen).
LC-MS/MS analyses.
Coimmunoprecipitation beads from two independent biological replicates were eluted in Laemmli buffer and run on a 4 to 12% acrylamide gel (Invitrogen), and proteins were stained with Coomassie blue (Bio-Rad). The experiment was reproduced one time to obtain two independent biological replicates of the total experiment. For both experiments, three gel plugs were cut for each condition. Plugs were reduced with 10 mM dithiothreitol, alkylated with 55 mM iodoacetamide, and incubated with 20 μl of 25 mM NH4HCO3 containing 12.5 μg/ml sequencing-grade trypsin (Promega, France) overnight at 37°C. The resulting peptides were sequentially extracted from the gel with 30% acetonitrile, 0.1% formic acid, and 70% acetonitrile. Digests were pooled (three per condition) according to the different experimental conditions. Peptide mixtures were analyzed by using a Q-Exactive Plus coupled to a Nano-LC Proxeon 1000 (both from Thermo Scientific). Peptides were separated by chromatography according to the following parameters: Acclaim PepMap100 C18 precolumn (2 cm, 75 μm [inner diameter], 3 μm, 100 Å), Pepmap-RSLC Proxeon C18 column (50 cm, 75 μm [inner diameter], 2 μm, 100 Å), 300 nl/min flow rate, and a 98-min gradient from 95% solvent A (water, 0.1% formic acid) to 35% solvent B (100% acetonitrile, 0.1% formic acid). Peptides were analyzed in the Orbitrap cell, at a resolution of 70,000, with a mass range of m/z 375 to 1,500. Fragments were obtained by higher-energy collisional dissociation activation with a collisional energy of 28%. MS/MS data were acquired in the Orbitrap cell in a Top20 mode, at a resolution of 17,500. For the identification step, all MS and MS/MS data were processed using Proteome Discoverer software (Thermo Scientific, version 2.2) and with the Mascot search engine (Matrix Science, version 5.1). The mass tolerance was set to 6 ppm for precursor ions and 0.02 Da for fragments. The following modifications were allowed: oxidation (M), phosphorylation (ST), acetylation (N-term of protein), and carbamidomethylation (C). The Swiss-Prot database (02/17) with the Homo sapiens taxonomy and a database including all of the viral proteins encoded by BTV were used in parallel. Peptide identifications were validated using a 1% false discovery rate (FDR) threshold calculated with the Percolator algorithm. Proteins with at least two unique peptides were considered. Identified proteins were considered potential partners of BTV-NS3 if no identifications were reported in the control condition (mock-infected or empty pCI-neo-3×FLAG-transfected cells).
Western blot analysis.
Purified complexes and protein extracts were resolved by SDS-PAGE on 4 to 12% NuPAGE Bis-Tris gels with MOPS running buffer and transferred to a nitrocellulose membrane (Invitrogen). Proteins were detected using standard immunoblotting techniques. 3×FLAG- and GST-tagged proteins were detected with a mouse monoclonal horseradish peroxidase (HRP)-conjugated anti-3×FLAG antibody (M2; Sigma-Aldrich) and a rabbit polyclonal anti-GST antibody (Sigma-Aldrich), respectively. Specific antibodies (all from Cell Signaling) were used to detect endogenous BRAF (clone D9T6S), phospho-ERK1/2 (clone-E10), ERK1/2, phospho-eIF4E (Ser209), and eIF4E. Secondary anti-mouse and anti-rabbit HRP-conjugated antibodies were purchased from Invitrogen. Densitometric analysis of the gels was performed using the ImageJ program.
Immunofluorescence assays.
Plates (24-well; Ibidi μ-plates, BioValley) were seeded with MDBK cells (1 × 105 cells per well). One day later, cells were serum starved and then infected with BTV (MOI = 0.01). At 18 h postinfection, the cells were washed three times with PBS, incubated with a 4% paraformaldehyde (PFA) solution (Electron Microscopy Sciences) for 30 min at room temperature, and then treated with PBS-glycine (0.1 M) and PBS-Triton (0.5%) for 5 min/each at room temperature to quench and permeabilize the cells, respectively. The cells were washed three times with PBS and incubated for 1 h with a PBS–1% bovine serum albumin (BSA) blocking solution. Finally, the cells were incubated with specific BRAF (Sc-5284; Santa Cruz) and NS3 antibodies for 2 h at room temperature, followed by incubation for 1 h at room temperature in a PBS–1% BSA solution containing Hoechst 33258 dye and secondary antibodies (anti-rabbit/A11035 and anti-mouse/A11029; Thermo Fisher). Preparations were visualized using an Axio Observer Z1 fluorescence inverted microscope (Zeiss). Each experiment was repeated at least three times.
MAPK inhibitors and BRAF silencing.
When specified, the cells were treated with MEK1/2 specific inhibitor U0126 at the indicated final concentrations (Promega). Transfections with siRNAs were performed using JetPRIME (Polyplus) according to the manufacturer’s instructions. Control nonspecific and BRAF-specific siRNAs (SMARTpool; ON-TARGETplus human BRAF SiRNA) were purchased from Dharmacon and used at a final concentration of 50 nM.
ACKNOWLEDGMENTS
This study was supported by Laboratoire d’Excellence Integrative Biology of Emerging Infectious Diseases grant ANR-10-LABX-62-IBEID. The LC-MS/MS equipment was funded by the Region Ile-de-France (SESAME), the Paris-Diderot University (ARS), and the CNRS. C.K. is supported by a Ph.D. fellowship from ANSES. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank Frederick Arnaud, Marc Therrien, Pierre-Olivier Vidalain, Maxime Ratinier, Virginie Doceul, and all members of UMR 1161 Virology for fruitful discussions. We thank Yves Jacob and Frederick Arnaud for providing the pGal4-UAS-Luc plasmid and the NS3 antibody, respectively.
REFERENCES
- 1.Belbis G, Zientara S, Bréard E, Sailleau C, Caignard G, Vitour D, Attoui H. 2017. Bluetongue virus: from BTV-1 to BTV-27 Adv Virus Res 99:161–167. [DOI] [PubMed] [Google Scholar]
- 2.Bumbarov V, Golender N, Erster O, Khinich Y. 2016. Detection and isolation of bluetongue virus from commercial vaccine batches. Vaccine 34:3317–3323. doi: 10.1016/j.vaccine.2016.03.097. [DOI] [PubMed] [Google Scholar]
- 3.Lorusso A, Sghaier S, Di Domenico M, Barbria ME, Zaccaria G, Megdich A, Portanti O, Seliman IB, Spedicato M, Pizzurro F, Carmine I, Teodori L, Mahjoub M, Mangone I, Leone A, Hammami S, Marcacci M, Savini G. 2018. Analysis of bluetongue serotype 3 spread in Tunisia and discovery of a novel strain related to the bluetongue virus isolated from a commercial sheep pox vaccine. Infect Genet Evol 59:63–71. doi: 10.1016/j.meegid.2018.01.025. [DOI] [PubMed] [Google Scholar]
- 4.Maan S, Maan NS, Belaganahalli MN, Rao PP, Singh KP, Hemadri D, Putty K, Kumar A, Batra K, Krishnajyothi Y, Chandel BS, Reddy GH, Nomikou K, Reddy YN, Attoui H, Hegde NR, Mertens P. 2015. Full-genome sequencing as a basis for molecular epidemiology studies of bluetongue virus in India. PLoS One 10:e0131257. doi: 10.1371/journal.pone.0131257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marcacci M, Sant S, Mangone I, Goria M, Dondo A, Zoppi S, van Gennip RGP, Radaelli MC, Cammà C, van Rijn PA, Savini G, Lorusso A. 2018. One after the other: a novel bluetongue virus strain related to Toggenburg virus detected in the Piedmont region (North-western Italy), extends the panel of novel atypical BTV strains. Transbound Emerg Dis 65:370–374. doi: 10.1111/tbed.12822. [DOI] [PubMed] [Google Scholar]
- 6.Savini G, Puggioni G, Meloni G, Marcacci M, Di Domenico M, Rocchigiani AM, Spedicato M, Oggiano A, Manunta D, Teodori L, Leone A, Portanti O, Cito F, Conte A, Orsini M, Cammà C, Calistri P, Giovannini A, Lorusso A. 2017. Novel putative bluetongue virus in healthy goats from Sardinia, Italy. Infect Genet Evol 51:108–117. doi: 10.1016/j.meegid.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 7.Sun EC, Huang LP, Xu QY, Wang HX, Xue XM, Lu P, Li WJ, Liu W, Bu ZG, Wu DL. 2016. Emergence of a novel bluetongue virus serotype, China 2014. Transbound Emerg Dis 63:585–589. doi: 10.1111/tbed.12560. [DOI] [PubMed] [Google Scholar]
- 8.Mellor PS, Carpenter S, Harrup L, Baylis M, Mertens P. 2008. Bluetongue in Europe and the Mediterranean basin: history of occurrence prior to 2006. Prev Vet Med 87:4–20. doi: 10.1016/j.prevetmed.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Toussaint JF, Sailleau C, Mast J, Houdart P, Czaplicki G, Demeestere L, VandenBussche F, van Dessel W, Goris N, Bréard E, Bounaadja T, Etienne T, Zientara S, De Clercq K. 2007. Bluetongue in Belgium, 2006. Emerg Infect Dis 13:614–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maclachlan NJ, Drew CP, Darpel KE, Worwa G. 2009. The pathology and pathogenesis of bluetongue. J Comp Pathol 141:1–16. doi: 10.1016/j.jcpa.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Stewart M, Hardy A, Barry G, Pinto RM, Caporale M, Melzi E, Hughes J, Taggart A, Janowicz A, Varela M, Ratinier M, Palmarini M. 2015. Characterization of a second open reading frame in genome segment 10 of bluetongue virus. J Gen Virol 96:3280–3293. doi: 10.1099/jgv.0.000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belhouchet M, Mohd Jaafar F, Firth AE, Grimes JM, Mertens PPC, Attoui H. 2011. Detection of a fourth orbivirus nonstructural protein. PLoS One 6:e25697. doi: 10.1371/journal.pone.0025697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ratinier M, Caporale M, Golder M, Franzoni G, Allan K, Nunes SF, Armezzani A, Bayoumy A, Rixon F, Shaw A, Palmarini M. 2011. Identification and characterization of a novel nonstructural protein of bluetongue virus. PLoS Pathog 7:e1002477. doi: 10.1371/journal.ppat.1002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel A, Roy P. 2014. The molecular biology of bluetongue virus replication. Virus Res 182:5–20. doi: 10.1016/j.virusres.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huismans H, Erasmus BJ. 1981. Identification of the serotype-specific and group-specific antigens of bluetongue virus. Onderstepoort J Vet Res 48:51–58. [PubMed] [Google Scholar]
- 16.Kahlon J, Sugiyama K, Roy P. 1983. Molecular basis of bluetongue virus neutralization. J Virol 48:627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kar AK, Bhattacharya B, Roy P. 2007. Bluetongue virus RNA binding protein NS2 is a modulator of viral replication and assembly. BMC Mol Biol 8:4. doi: 10.1186/1471-2199-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyce M, Celma CCP, Roy P. 2012. Bluetongue virus nonstructural protein 1 is a positive regulator of viral protein synthesis. Virol J 9:178. doi: 10.1186/1743-422X-9-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forzan M, Wirblich C, Roy P. 2004. A capsid protein of nonenveloped bluetongue virus exhibits membrane fusion activity. Proc Natl Acad Sci U S A 101:2100–2105. doi: 10.1073/pnas.0306448101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han Z, Harty RN. 2004. The NS3 protein of bluetongue virus exhibits viroporin-like properties. J Biol Chem 279:43092–43097. doi: 10.1074/jbc.M403663200. [DOI] [PubMed] [Google Scholar]
- 21.Hyatt AD, Zhao Y, Roy P. 1993. Release of bluetongue virus-like particles from insect cells is mediated by BTV nonstructural protein NS3/NS3A. Virology 193:592–603. doi: 10.1006/viro.1993.1167. [DOI] [PubMed] [Google Scholar]
- 22.Owens RJ, Limn C, Roy P. 2004. Role of an arbovirus nonstructural protein in cellular pathogenesis and virus release. J Virol 78:6649–6656. doi: 10.1128/JVI.78.12.6649-6656.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wirblich C, Bhattacharya B, Roy P. 2006. Nonstructural protein 3 of bluetongue virus assists virus release by recruiting ESCRT-I protein Tsg101. J Virol 80:460–473. doi: 10.1128/JVI.80.1.460-473.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chauveau E, Doceul V, Lara E, Breard E, Sailleau C, Vidalain P-O, Meurs EF, Dabo S, Schwartz-Cornil I, Zientara S, Vitour D. 2013. NS3 of bluetongue virus interferes with the induction of type I interferon. J Virol 87:8241–8246. doi: 10.1128/JVI.00678-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ratinier M, Shaw AE, Barry G, Gu Q, Gialleonardo LD, Janowicz A, Varela M, Randall RE, Caporale M, Palmarini M. 2016. Bluetongue virus NS4 protein is an interferon antagonist and a determinant of virus virulence. J Virol 90:5427–5439. doi: 10.1128/JVI.00422-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy P, Marshall JJA, French TJ. 1990. Structure of the bluetongue virus genome and its encoded proteins, p 43–87. In Roy P, Gorman BM (ed), Bluetongue viruses. Springer, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 27.Bhattacharya B, Roy P. 2008. Bluetongue virus outer capsid protein VP5 interacts with membrane lipid rafts via a SNARE domain. J Virol 82:10600–10612. doi: 10.1128/JVI.01274-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beaton AR, Rodriguez J, Reddy YK, Roy P. 2002. The membrane trafficking protein calpactin forms a complex with bluetongue virus protein NS3 and mediates virus release. Proc Natl Acad Sci U S A 99:13154–13159. doi: 10.1073/pnas.192432299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Celma CCP, Roy P. 2009. A viral nonstructural protein regulates bluetongue virus trafficking and release. J Virol 83:6806–6816. doi: 10.1128/JVI.00263-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janowicz A, Caporale M, Shaw A, Gulletta S, Gialleonardo LD, Ratinier M, Palmarini M. 2015. Multiple genome segments determine virulence of bluetongue virus serotype 8. J Virol 89:5238–5249. doi: 10.1128/JVI.00395-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feenstra F, van Gennip RGP, Maris-Veldhuis M, Verheij E, van Rijn PA. 2014. Bluetongue virus without NS3/NS3a expression is not virulent and protects against virulent bluetongue virus challenge. J Gen Virol 95:2019–2029. doi: 10.1099/vir.0.065615-0. [DOI] [PubMed] [Google Scholar]
- 32.Ftaich N, Ciancia C, Viarouge C, Barry G, Ratinier M, van Rijn PA, Breard E, Vitour D, Zientara S, Palmarini M, Terzian C, Arnaud F. 2015. Turnover rate of NS3 proteins modulates bluetongue virus replication kinetics in a host-specific manner. J Virol 89:10467–10481. doi: 10.1128/JVI.01541-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonjardim CA. 2017. Viral exploitation of the MEK/ERK pathway: a tale of vaccinia virus and other viruses. Virology 507:267–275. doi: 10.1016/j.virol.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Buday L, Downward J. 1993. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 73:611–620. doi: 10.1016/0092-8674(93)90146-H. [DOI] [PubMed] [Google Scholar]
- 35.Lavoie JN, L’Allemain G, Brunet A, Müller R, Pouysségur J. 1996. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem 271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- 36.Pyronnet S. 2000. Phosphorylation of the cap-binding protein eIF4E by the MAPK-activated protein kinase Mnk1. Biochem Pharmacol 60:1237–1243. doi: 10.1016/S0006-2952(00)00429-9. [DOI] [PubMed] [Google Scholar]
- 37.Mortola E, Larsen A. 2010. Bluetongue virus infection: Activation of the MAP kinase-dependent pathway is required for apoptosis. Res Vet Sci 89:460–464. doi: 10.1016/j.rvsc.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 38.Mohl B-P, Emmott E, Roy P. 2017. Phosphoproteomic analysis reveals the importance of kinase regulation during orbivirus infection. Mol Cell Proteomics 16:1990–2005. doi: 10.1074/mcp.M117.067355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lv S, Xu Q-Y, Sun E-C, Zhang J-K, Wu D-L. 2016. Dissection and integration of the autophagy signaling network initiated by bluetongue virus infection: crucial candidates ERK1/2, Akt and AMPK. Sci Rep 6:23130. doi: 10.1038/srep23130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kundlacz C, Pourcelot M, Fablet A, Amaral Da Silva Moraes R, Léger T, Morlet B, Viarouge C, Sailleau C, Turpaud M, Gorlier A, Breard E, Lecollinet S, van Rijn PA, Zientara S, Vitour D, Caignard G. 2019. Novel function of bluetongue virus NS3 protein in regulation of the MAPK/ERK signaling pathway. bioRxiv 10.1101/562421. [DOI] [PMC free article] [PubMed]
- 41.Sonenberg N. 2008. eIF4E, the mRNA cap-binding protein: from basic discovery to translational research. Biochem Cell Biol 86:178–183. doi: 10.1139/O08-034. [DOI] [PubMed] [Google Scholar]
- 42.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EWT, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. 2007. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Battcock SM, Collier TW, Zu D, Hirasawa K. 2006. Negative regulation of the alpha interferon-induced antiviral response by the Ras/Raf/MEK pathway. J Virol 80:4422–4430. doi: 10.1128/JVI.80.9.4422-4430.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huismans H, van Staden V, Fick WC, van Niekerk M, Meiring TL. 2004. A comparison of different orbivirus proteins that could affect virulence and pathogenesis. Vet Ital 40(4):417–425. [PubMed] [Google Scholar]
- 45.Li L, Zhao G-D, Shi Z, Qi L-L, Zhou L-Y, Fu Z-X. 2016. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol Lett 12:3045–3050. doi: 10.3892/ol.2016.5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakano H, Shindo M, Sakon S, Nishinaka S, Mihara M, Yagita H, Okumura K. 1998. Differential regulation of IκB kinase α and β by two upstream kinases, NF-κB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc Natl Acad Sci U S A 95:3537–3542. doi: 10.1073/pnas.95.7.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. 2004. JAK/STAT, Raf/MEK/ERK, PI3K/Akt, and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 18:189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- 48.Clemente MG, Patton JT, Anders RA, Yolken RH, Schwarz KB. 2015. Rotavirus infects human biliary epithelial cells and stimulates secretion of cytokines IL-6 and IL-8 via MAPK pathway. Biomed Res 2015:1. doi: 10.1155/2015/697238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alcorn MJ, Booth JL, Coggeshall KM, Metcalf JP. 2001. Adenovirus type 7 induces interleukin-8 production via activation of extracellular regulated kinase 1/2. J Virol 75:6450–6459. doi: 10.1128/JVI.75.14.6450-6459.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Booth JL, Coggeshall KM, Gordon BE, Metcalf JP. 2004. Adenovirus type 7 induces interleukin-8 in a lung slice model and requires activation of Erk. J Virol 78:4156–4164. doi: 10.1128/jvi.78.8.4156-4164.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drew CP, Heller MC, Mayo C, Watson JL, MacLachlan NJ. 2010. Bluetongue virus infection activates bovine monocyte-derived macrophages and pulmonary artery endothelial cells. Vet Immunol Immunopathol 136:292–296. doi: 10.1016/j.vetimm.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeMaula CD, Leutenegger CM, Bonneau KR, MacLachlan NJ. 2002. The role of endothelial cell-derived inflammatory and vasoactive mediators in the pathogenesis of bluetongue. Virology 296:330–337. doi: 10.1006/viro.2002.1476. [DOI] [PubMed] [Google Scholar]
- 53.Hemati B, Contreras V, Urien C, Bonneau M, Takamatsu H-H, Mertens PPC, Bréard E, Sailleau C, Zientara S, Schwartz-Cornil I. 2009. Bluetongue virus targets conventional dendritic cells in skin lymph. J Virol 83:8789–8799. doi: 10.1128/JVI.00626-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schwartz-Cornil I, Mertens PPC, Contreras V, Hemati B, Pascale F, Bréard E, Mellor PS, MacLachlan NJ, Zientara S. 2008. Bluetongue virus: virology, pathogenesis, and immunity. Vet Res 39:1. [DOI] [PubMed] [Google Scholar]
- 55.Dhanasekaran S, Vignesh AR, Raj GD, Reddy YKM, Raja A, Tirumurugaan KG. 2013. Comparative analysis of innate immune response following in vitro stimulation of sheep and goat peripheral blood mononuclear cells with bluetongue virus: serotype 23. Vet Res Commun 37:319–327. doi: 10.1007/s11259-013-9579-5. [DOI] [PubMed] [Google Scholar]
- 56.Marín-López A, Bermúdez R, Calvo-Pinilla E, Moreno S, Brun A, Ortego J. 2016. Pathological characterization of IFNAR–/– mice infected with bluetongue virus serotype 4. Int J Biol Sci 12:1448–1460. doi: 10.7150/ijbs.14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruscanu S, Jouneau L, Urien C, Bourge M, Lecardonnel J, Moroldo M, Loup B, Dalod M, Elhmouzi-Younes J, Bevilacqua C, Hope J, Vitour D, Zientara S, Meyer G, Schwartz-Cornil I. 2013. Dendritic cell subtypes from lymph nodes and blood show contrasted gene expression programs upon bluetongue virus infection. J Virol 87:9333–9343. doi: 10.1128/JVI.00631-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sánchez-Cordón PJ, Pedrera M, Risalde MA, Molina V, Rodríguez-Sánchez B, Núñez A, Sánchez-Vizcaíno JM, Gómez P, Villamandos JC. 2013. Potential role of proinflammatory cytokines in the pathogenetic mechanisms of vascular lesions in goats naturally infected with bluetongue virus serotype 1. Transbound Emerg Dis 60:252–262. doi: 10.1111/j.1865-1682.2012.01343.x. [DOI] [PubMed] [Google Scholar]
- 59.Cai Y, Liu Y, Zhang X. 2007. Suppression of coronavirus replication by inhibition of the MEK signaling pathway. J Virol 81:446–456. doi: 10.1128/JVI.01705-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Colao I, Pennisi R, Venuti A, Nygårdas M, Heikkilä O, Hukkanen V, Sciortino MT. 2017. The ERK-1 function is required for HSV-1-mediated G1/S progression in HEP-2 cells and contributes to virus growth. Sci Rep 7:9176. doi: 10.1038/s41598-017-09529-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ludwig S, Wolff T, Ehrhardt C, Wurzer WJ, Reinhardt J, Planz O, Pleschka S. 2004. MEK inhibition impairs influenza B virus propagation without emergence of resistant variants. FEBS Lett 561:37–43. doi: 10.1016/S0014-5793(04)00108-5. [DOI] [PubMed] [Google Scholar]
- 62.Moser LA, Schultz-Cherry S. 2008. Suppression of astrovirus replication by an ERK1/2 inhibitor. J Virol 82:7475–7482. doi: 10.1128/JVI.02193-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Planz O, Pleschka S, Ludwig S. 2001. MEK-specific inhibitor U0126 blocks spread of Borna disease virus in cultured cells. J Virol 75:4871–4877. doi: 10.1128/JVI.75.10.4871-4877.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rodríguez ME, Brunetti JE, Wachsman MB, Scolaro LA, Castilla V. 2014. Raf/MEK/ERK pathway activation is required for Junín virus replication. J Gen Virol 95:799–805. doi: 10.1099/vir.0.061242-0. [DOI] [PubMed] [Google Scholar]
- 65.Kim Y, Lee C. 2015. Extracellular signal-regulated kinase (ERK) activation is required for porcine epidemic diarrhea virus replication. Virology 484:181–193. doi: 10.1016/j.virol.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Caignard G, Komarova AV, Bouraï M, Mourez T, Jacob Y, Jones LM, Rozenberg F, Vabret A, Freymuth F, Tangy F, Vidalain P-O. 2009. Differential regulation of type I interferon and epidermal growth factor pathways by a human respirovirus virulence factor. PLoS Pathog 5:e1000587. doi: 10.1371/journal.ppat.1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gong J, Mita MM. 2014. Activated Ras signaling pathways and reovirus oncolysis: an update on the mechanism of preferential reovirus replication in cancer cells. Front Oncol 4:167. doi: 10.3389/fonc.2014.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Phillips MB, Stuart JD, Rodríguez Stewart RM, Berry JT, Mainou BA, Boehme KW. 2018. Current understanding of reovirus oncolysis mechanisms. Oncolytic Virother 7:53–63. doi: 10.2147/OV.S143808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bourhill T, Mori Y, Rancourt DE, Shmulevitz M, Johnston RN. 2018. Going (reo)viral: factors promoting successful reoviral oncolytic infection. Viruses 10:421. doi: 10.3390/v10080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shmulevitz M, Pan L-Z, Garant K, Pan D, Lee P. 2010. Oncogenic Ras promotes reovirus spread by suppressing IFN-β production through negative regulation of RIG-I signaling. Cancer Res 70:4912–4921. doi: 10.1158/0008-5472.CAN-09-4676. [DOI] [PubMed] [Google Scholar]
- 71.Lobeck I, Donnelly B, Dupree P, Mahe MM, McNeal M, Mohanty SK, Tiao G. 2016. Rhesus rotavirus VP6 regulates ERK-dependent calcium influx in cholangiocytes. Virology 499:185–195. doi: 10.1016/j.virol.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Soliman M, Seo J-Y, Kim D-S, Kim J-Y, Park J-G, Alfajaro MM, Baek Y-B, Cho E-H, Kwon J, Choi J-S, Kang M-I, Park S-I, Cho K-O. 2018. Activation of PI3K, Akt, and ERK during early rotavirus infection leads to V-ATPase-dependent endosomal acidification required for uncoating. PLoS Pathog 14 doi: 10.1371/journal.ppat.1006820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Richter JD, Sonenberg N. 2005. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433:477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 74.Ramadevi N, Roy P. 1998. Bluetongue virus core protein VP4 has nucleoside triphosphate phosphohydrolase activity. J Gen Virol 79:2475–2480. doi: 10.1099/0022-1317-79-10-2475. [DOI] [PubMed] [Google Scholar]
- 75.Ramadevi N, Burroughs NJ, Mertens PPC, Jones IM, Roy P. 1998. Capping and methylation of mRNA by purified recombinant VP4 protein of bluetongue virus. Proc Natl Acad Sci U S A 95:13537–13542. doi: 10.1073/pnas.95.23.13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stokoe D, Macdonald SG, Cadwallader K, Symons M, Hancock JF. 1994. Activation of Raf as a result of recruitment to the plasma membrane. Science 264:1463–1467. doi: 10.1126/science.7811320. [DOI] [PubMed] [Google Scholar]
- 77.Leevers SJ, Paterson HF, Marshall CJ. 1994. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature 369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- 78.Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, Johnson RL, Cox AD, Philips MR. 2002. Ras signaling on the endoplasmic reticulum and the Golgi. Nat Cell Biol 4:343–350. doi: 10.1038/ncb783. [DOI] [PubMed] [Google Scholar]
- 79.Chauveau E, Doceul V, Lara E, Adam M, Breard E, Sailleau C, Viarouge C, Desprat A, Meyer G, Schwartz-Cornil I, Ruscanu S, Charley B, Zientara S, Vitour D. 2012. Sensing and control of bluetongue virus infection in epithelial cells via RIG-I and MDA5 helicases. J Virol 86:11789–11799. doi: 10.1128/JVI.00430-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shaw AE, Veronesi E, Maurin G, Ftaich N, Guiguen F, Rixon F, Ratinier M, Mertens P, Carpenter S, Palmarini M, Terzian C, Arnaud F. 2012. Drosophila melanogaster as a model organism for bluetongue virus replication and tropism. J Virol 86:9015–9024. doi: 10.1128/JVI.00131-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Le Gal MC, Dufour B, Geoffroy E, Zanella G, Moutou F, Millemann Y, Rieffel JN, Pouilly F. 2008. Bluetongue virus serotype 8 in the Ardennes in 2007. Vet Rec 163:668. doi: 10.1136/vr.163.22.668-b. [DOI] [PubMed] [Google Scholar]
- 82.Nomikou K, Hughes J, Wash R, Kellam P, Breard E, Zientara S, Palmarini M, Biek R, Mertens P. 2015. Widespread reassortment shapes the evolution and epidemiology of bluetongue virus following European invasion. PLoS Pathog 11:e1005056. doi: 10.1371/journal.ppat.1005056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zientara S, Sailleau C, Viarouge C, Höper D, Beer M, Jenckel M, Hoffmann B, Romey A, Bakkali-Kassimi L, Fablet A, Vitour D, Bréard E. 2014. Novel bluetongue virus in goats, Corsica, France, 2014. Emerg Infect Dis 20:2123–2125. doi: 10.3201/eid2012.140924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sailleau C, Zanella G, Breard E, Viarouge C, Desprat A, Vitour D, Adam M, Lasne L, Martrenchar A, Bakkali-Kassimi L, Costes L, Zientara S. 2012. Co-circulation of bluetongue and epizootic haemorrhagic disease viruses in cattle in Reunion Island. Vet Microbiol 155:191–197. doi: 10.1016/j.vetmic.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 85.Zientara S, Sailleau C, Moulay S, Cruciere C. 1994. Diagnosis of the African horse sickness virus serotype 4 by a one-tube, one manipulation RT-PCR reaction from infected organs. J Virol Methods 46:179–188. doi: 10.1016/0166-0934(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 86.Erasmus BJ, Boshoff ST, Pieterse LM. 1978. The isolation and characterisation of equine encephalosis and serologically related orbiviruses from horses, p 447–450. In Bryans JT, Gerber H (ed), Proceedings of the Fourth International Conference on Equine Infectious Diseases. Veterinary Publications Inc., Princeton, NJ. [Google Scholar]
- 87.Mendoza J-A, Jacob Y, Cassonnet P, Favre M. 2006. Human papillomavirus type 5 E6 oncoprotein represses the transforming growth factor beta signaling pathway by binding to SMAD3. J Virol 80:12420–12424. doi: 10.1128/JVI.02576-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vitour D, Dabo S, Pour MA, Vilasco M, Vidalain P-O, Jacob Y, Mezel-Lemoine M, Paz S, Arguello M, Lin R, Tangy F, Hiscott J, Meurs EF. 2009. Polo-like kinase 1 (PLK1) regulates interferon (IFN) induction by MAVS. J Biol Chem 284:21797–21809. doi: 10.1074/jbc.M109.018275. [DOI] [PMC free article] [PubMed] [Google Scholar]