

The pathogenic potential of HTLV-1 is linked to the indispensable multifaceted functions of the viral regulatory proteins Tax and HBZ, encoded by the sense and antisense viral transcripts, respectively. The interaction between Tax and the SWI/SNF family of chromatin remodeling complexes has been associated with HTLV-1 transcriptional activation. To date, the relationship between the SWI/SNF chromatin remodeling family and HBZ, the only viral protein that is consistently expressed in infected cells and ATL cells, has not been elucidated. Here, we have characterized the biological significance of the SWI/SNF family in regard to viral transcriptional repression by HBZ. This is important because it provides a better understanding of the function and role of HBZ in downregulating viral transcription and, hence, its contribution to viral latency and persistence in vivo, a process that may ultimately lead to the development of ATL.
KEYWORDS: BRG1, HBZ, HTLV-1, PBAF, SWI/SNF, Tax, leukemia
ABSTRACT
The human T-cell leukemia virus type 1 (HTLV-1) regulatory proteins Tax and HBZ play indispensable roles in regulating viral and cellular gene expression. BRG1, the ATPase subunit of the SWI/SNF chromatin remodeling complex, has been demonstrated to be essential not only for Tax transactivation but also for viral replication. We sought to investigate the physical interaction between HBZ and BRG1 and to determine the effect of these interactions on Tax-mediated long terminal repeat (LTR) activation. We reveal that HTLV-1 cell lines and adult T-cell leukemia (ATL) cells harbor high levels of BRG1. Using glutathione S-transferase (GST) pulldown and coimmunoprecipitation assays, we have demonstrated physical interactions between BRG1 and HBZ and characterized the protein domains involved. Moreover, we have identified the PBAF signature subunits BAF200 and BAF180 as novel interaction partners of HBZ, suggesting that the PBAF complex may be required for HTLV-1 transcriptional repression by HBZ. Additionally, we found that BRG1 expression translocates HBZ into distinct nuclear foci. We show that HBZ substantially represses HTLV-1 LTR activation by Tax/BRG1. Interestingly, we found that Tax stabilizes the expression of exogenous and endogenous BRG1 and that HBZ reverses this effect. Finally, using a chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR) assay, we illustrate that HBZ facilitates the downregulation of HTLV-1 transcription by deregulating the recruitment of SWI/SNF complexes to the promoter. Overall, we conclude that SWI/SNF complexes, in addition to other cellular transcription factors, are involved in HBZ-mediated suppression of HTLV-1 viral gene expression.
IMPORTANCE The pathogenic potential of HTLV-1 is linked to the indispensable multifaceted functions of the viral regulatory proteins Tax and HBZ, encoded by the sense and antisense viral transcripts, respectively. The interaction between Tax and the SWI/SNF family of chromatin remodeling complexes has been associated with HTLV-1 transcriptional activation. To date, the relationship between the SWI/SNF chromatin remodeling family and HBZ, the only viral protein that is consistently expressed in infected cells and ATL cells, has not been elucidated. Here, we have characterized the biological significance of the SWI/SNF family in regard to viral transcriptional repression by HBZ. This is important because it provides a better understanding of the function and role of HBZ in downregulating viral transcription and, hence, its contribution to viral latency and persistence in vivo, a process that may ultimately lead to the development of ATL.
INTRODUCTION
Human T-cell leukemia virus type 1 (HTLV-1) is the first human retrovirus discovered in the early 1980s and belongs to the Deltaretrovirus family (1–4). HTLV-1 infects at least 5 million to 10 million people worldwide (5), with 6.6% of males and 2.1% of females eventually developing an aggressive and fatal CD4+ T-cell malignancy termed adult T-cell leukemia (ATL) after many years of clinically latent infection (2, 6–8). HTLV-1 infection can also cause a chronic neurodegenerative disorder called tropical spastic paraparesis/HTLV-1-associated myelopathy (HAM/TSP) in another 2% to 3% of infected individuals (2, 9–11). Other inflammatory diseases, such as uveitis, polymyositis, and alveolitis, as well as infective dermatitis and some types of skin lesions, have been associated with HTLV-1 infection (12). Treatment options are limited, and HTLV-1 vaccines that could prevent infection and, hence, ATL and HAM/TSP development in infected populations are not available. A high proviral load is a major risk factor for the development of ATL and HAM/TSP (13, 14). The cooperation between the viral oncoproteins Tax and HBZ plays a crucial role in the high HTLV-I proviral load in carriers (15–20).
HTLV-1 gene expression is determined by the viral regulatory proteins Tax and HBZ, which play key, sometimes opposing, roles in regulating viral and cellular gene expression. Tax is expressed from the 5′ long terminal repeat (LTR) using the sense strand of the viral genome, while HBZ is expressed from the 3′ LTR using the antisense strand of the HTLV-1 genome. Tax is a powerful transactivator of viral gene expression and is recruited to the viral promoter as part of a complex with the host cellular transcription factors of the ATF/CREB family (21–25). These complexes promote local nucleosome modification via histone acetylation at the HTLV-1 transcription start site, stimulating viral gene expression (24–27). The production of viral proteins in infected cells, in particular Tax, targets them for immune destruction (19, 28–30). Persistence of HTLV-1 in the host is ensured by HBZ, which downregulates Tax activity by competing for binding to the cellular transcription factors of the ATF/CREB family (15, 17, 19, 31, 32). Such activity suppresses HTLV-1 replication and abolishes the expression of Tax and other viral genes, allowing infected cells to evade immune surveillance and persist in the host (33–35).
In addition to regulating viral transcription via the CREB/CBP pathway, previous studies revealed that SWI/SNF chromatin remodeling complexes are critical for Tax transactivation and viral replication (36). The SWI/SNF complexes are classified into two major classes: one is BRG/hBRM-associated factor (BAF) complexes, and the other is polybromo-associated BAF (PBAF) complexes. The BAF complex can contain either of two closely related catalytic ATPase subunits, Brahma (BRM) or BRM-related gene 1 (BRG1), while the PBAF complex contains only BRG1 (37). These complexes share a high degree of similarity and can be distinguished only by the presence of specific subunits, BAF250A/B in the case of the BAF complex or BAF180 and BAF200 in the case of the PBAF complex (38–41).
BRG1 has been reported to possess both tumor-suppressive and oncogenic activities, depending on the type of cancer. For instance, BRG1 has been shown to be essential for the proliferation and survival of acute myeloid leukemic cells, as leukemic cells lacking BRG1 rapidly undergo cell cycle arrest and apoptosis, indicating the role of BRG1 in cell cycle regulation and cancer promotion (42, 43). In pancreatic cancer, BRG1 has been reported to play opposing roles in the development of different precancerous lesions that lead to pancreatic cancer in a stage-specific manner. In the pancreatic intraepithelial neoplasia (PanIN) stage that precedes neoplastic transformation, BRG1 functions as a tumor suppressor to prevent dedifferentiation of pancreatic duct cells (PDCs) and, hence, attenuates tumor initiation. In contrast, once pancreatic ductal adenocarcinoma (PDA) develops, BRG1 drives PDA tumorigenesis by inducing an epithelial-to-mesenchymal transition (44). In malignant melanoma and breast cancer, enhanced BRG1 expression is correlated with tumorigenesis and poor patient survival (45–47).
In the context of HTLV-1 gene expression, BRG1 has been shown to be essential for optimal transcriptional activation of the HTLV-1 LTR by Tax (36, 48). Tax and BRG1 have been shown to be recruited to the viral promoter together with the components of the basal transcription machinery (polymerase II [Pol II] and CBP/p300), all of which are required for transcription initiation. This was previously shown by chromatin immunoprecipitation (ChIP) and viral particle production assays in HEK293T cells transfected with the HTLV-1 infectious clone ACH.WT (36). BRG1 coelutes with Tax and has been shown to be required for efficient nucleosome removal and optimal Tax transactivation (36). This suggests that both SWI/SNF and p300/CREB are involved in Tax-mediated activation of transcription. Furthermore, the knockdown of the signature subunits unique to either the BAF complex (BAF250) or the PBAF complex (BAF180) suggested that the SWI/SNF complex PBAF may be responsible for optimal HTLV-1 transcriptional activation by Tax and viral production (36).
Moreover, Tax interacts with multiple other SWI/SNF components, including BAF53, BAF57, and BAF155, as shown by mass spectrophotometry and immunoprecipitation (IP) (48). Given that these subunits have been shown to enhance the chromatin remodeling process (49), these interactions suggest that Tax may affect SWI/SNF complex function at multiple levels. In addition, recent research has demonstrated that binding sites for BRG1 upstream and downstream of the proviral integration sites have opposite effects on Tax expression. The presence of a BRG1 site upstream of the provirus is associated with transcriptional silencing of Tax expression, while a BRG1 site downstream is associated with Tax expression (50). Collectively, these studies indicate that SWI/SNF complexes play a role in HTLV-1 viral gene expression.
Interestingly, our previous studies using the yeast two-hybrid system revealed a possible interaction between HBZ and BRG1 (our unpublished data). This finding suggests that HBZ/BRG1 interactions, together with those involving Tax, may play a role in the regulation of HTLV-1 gene expression. In this study, we report that HBZ binds BRG1 and downregulates HTLV-1 LTR activation mediated by Tax/BRG1. Interestingly, we show that HBZ also interacts with BAF200 and BAF180, members of PBAF chromatin remodeling complexes of the SWI/SNF family, and can modulate the occupancy of PBAF complexes at the HTLV-1 promoter. Specifically, HBZ displaces BRG1 and BAF200 from the viral promoter and conversely recruits BAF180 to the viral promoter. This study presents further insights into the mechanisms involved in HBZ-mediated repression of Tax-dependent transactivation of the HTLV-1 LTR via the SWI/SNF family.
RESULTS
The HTLV-1 HBZ antisense protein interacts with BRG1 in mammalian cells and in vitro.
Data from previous studies point to the correlation between suppressed and elevated expression levels of BRG1 and the development of various human cancers (51, 52). However, to date, the expression of BRG1 in ATL patient cells or in HTLV-1/2 carrier cells has not been defined. Therefore, we first wanted to gain insight into the level of BRG1 expression in these cell types. To this end, we performed immunoblot analysis of BRG1 expression levels in nuclear extracts derived from HTLV-1-positive cell lines (C91 and MT2), an HTLV-2-positive cell line (MO), ATL patient cell lines (ATL-CR and ATL-TH), and HTLV-1-negative T-cell lymphoma cells (Jurkat) (Fig. 1A). This analysis indicates that BRG1 was abundantly expressed in HTLV-1-positive cell lines and ATL cell lines, compared to the HTLV-2-positive cell line or an uninfected cell line. Even though the number of HTLV-infected cell lines tested is small, these findings appear compatible with previous reports showing that BRG1 is overexpressed in different cancer types, such as gastric carcinomas (53), liver hepatocellular carcinoma (HCC), kidney renal clear cell carcinoma, and breast cancer (45, 54).
FIG 1.

HTLV-1 HBZ antisense protein interacts with BRG1. (A) BRG1 expression levels in HTLV-1/2-infected T cells and uninfected cells. Nuclear fractions from HTLV-1-positive cell lines (C91 and MT2), an HTLV-2-positive cell line (MO), ATL patient cell lines (ATL-CR and ATL-TH), and HTLV-1-negative T-cell lymphoma cells (Jurkat) were isolated using a nuclear and cytoplasmic extraction kit from Pierce (NE-PER). The expression of BRG1 was detected using anti-BRG1. Anti-HDAC1 was used as a loading control. Anti-CYPA was used as an indicator of the integrity of the nuclear fractions. The bottom panel represents the relative optical density (ROD) of BRG1 expression normalized against HDAC1. (B and C) HBZ interacts with endogenous (B) and exogenous (C) BRG1 in mammalian cells. HEK293T cells were transfected with 5 μg of FLAG-HBZ or an empty vector alone (B) or together with 6 μg of a MYC-tagged expression vector for BRG1 (C). Transfected cells were lysed after 24 h, and coimmunoprecipitations were performed using anti-FLAG M2 beads overnight. Interactions were analyzed by Western blotting using anti-FLAG, anti-BRG1, and anti-MYC antibodies, as indicated. IP, immunoprecipitation; WCL, whole-cell lysate. (D) Direct interaction between HBZ and BRG1 in vitro. GST pulldown assays were performed by incubating purified BRG1 protein (6 μg) with GST resins precoated with either GST-HBZ or GST proteins. Bound proteins were eluted twice (E1 and E2), and interactions were analyzed by immunoblotting using anti-BRG1 (top) and Coomassie blue staining (bottom). The BRG1 input corresponds to 40% of the total BRG1 loaded for each pulldown. M indicates the protein molecular weight marker.
To verify our previous finding from the yeast two-hybrid study that shows the possible interaction between HBZ and BRG1, we sought to confirm the interaction between HBZ and BRG1 in mammalian cells. To this end, we cotransfected HEK293T cells with an expression plasmid encoding FLAG-HBZ or FLAG alone (Fig. 1B) or together with an expression plasmid encoding MYC-BRG1 (Fig. 1C). Cellular lysates were subjected to coimmunoprecipitation (co-IP) using anti-FLAG M2 beads, and the resultant interactions were analyzed by immunoblotting using anti-FLAG, anti-BRG1, or anti-MYC antibodies. Our findings confirm that HBZ specifically interacts with endogenous (Fig. 1B) and exogenous (Fig. 1C) BRG1.
We next investigated whether HBZ interacts with BRG1 directly. To this end, glutathione S-transferase (GST) pulldown assays were carried out with GST or GST-HBZ and purified BRG1 proteins. Our results illustrate that GST-HBZ binds purified BRG1, indicating a direct physical interaction between these two proteins in vitro (Fig. 1D).
HBZ selectively interacts with PBAF complexes in mammalian cells.
We next sought to extend our analysis to determine which BRG1-associated complex (BAF or PBAF) interacts with HBZ and hence plays a role in HBZ-mediated suppression of HTLV-1 transcription. BAF and PBAF complexes share most subunits but can be distinguished by the presence of BAF250A in the BAF complex and BAF200 and BAF180 exclusively in the PBAF complex (37, 40). To investigate this, the cellular lysates from HEK293T cells transfected with FLAG-tagged HBZ constructs were subjected to co-IP assays using anti-FLAG M2 beads. Western blot analyses of precipitated complexes show that HBZ does not bind endogenous BAF250, the specific subunit of the BAF complex (Fig. 2A), and instead binds to endogenous BAF200 and BAF180, the specific subunits of the PBAF complex (Fig. 2B and C). These findings suggest that HBZ/PBAF may play a role in the downregulation of viral transcription by HBZ.
FIG 2.

HBZ interacts with the PBAF complex in mammalian cells. Shown are data from immunoblot analysis of the interaction between HBZ and endogenous BAF250 (BAF complex) (A) or endogenous BAF200 (B) and BAF180 (C) (PBAF complex). HEK293T cells were transfected with 7 μg of FLAG-HBZ. Transfected cells were lysed after 24 h, and co-IPs were performed using anti-FLAG M2 beads overnight. Precipitates were analyzed by Western blotting using anti-FLAG, anti-BAF250, anti-BAF200, and anti-BAF180, as indicated. IP, immunoprecipitation; WCL, whole-cell lysate.
The N terminus of HBZ is involved in the interaction with BRG1.
Based on the demonstrated physical interaction between HBZ and BRG1 in mammalian cells and directly in vitro, we sought to determine the protein domains in HBZ that mediate its interaction with BRG1. HBZ contains three key interaction domains, namely, an activation domain (AD), a central domain (CD), and a basic ZIP (bZIP) domain (Fig. 3A) (55). These domains are responsible for its interactions with numerous cellular factors that are involved in the regulation of viral transcription as well as cellular signaling pathways that may contribute to the development of ATL (56). To ascertain the domain of HBZ that binds BRG1, HEK293T cells were cotransfected with MYC-BRG1 together with FLAG-HBZ deletions (Δ-AD, Δ-CD, Δ-bZIP). Lysates were immunoprecipitated using anti-FLAG M2 beads. Western blot analyses of precipitated complexes show a strong interaction between MYC-BRG1 and FLAG-HBZ-Δ-CD, indicating that this domain is not involved in the interaction between these two proteins (Fig. 3B). An interaction (although weaker) could also be observed between MYC-BRG1 and FLAG-HBZ-Δ-bZIP, while no interaction was observed using FLAG-HBZ-Δ-AD. Overall, these findings suggest that the N-terminal activation domain of HBZ may be involved in the interaction with BRG1.
FIG 3.

Identification of domains in HBZ and BRG1 involved in their interaction in mammalian cells. (A) Schematic diagram of HBZ functional domains and deletion mutants used in co-IP experiments. HBZ deletions are as follows: Δ-AD (lacking the N-terminal activation domain), Δ-CD (lacking the central domain), and Δ-bZIP (lacking the C-terminal leucine zipper domain [59]). WT, wild type. (B) The N terminus of HBZ is involved in the interaction with BRG1. HEK293T cells were cotransfected with 6 μg of MYC-BRG1, alone or together with 5 μg of each of the three FLAG-tagged expression HBZ deletion vectors, as indicated. Transfected cells were lysed after 24 h, and co-IPs were performed using an anti-FLAG M2 beads overnight. Interactions were analyzed by immunoblotting using anti-FLAG and anti-MYC, as indicated. (C) Schematic diagram of BRG1 functional domains and deletion mutants used in co-IP experiments. Deletions are as follows: Δ-N-T (lacking the N-terminal domain), Δ-ATPase (lacking the ATPase domain), and Δ-Bromo domain (lacking the bromodomain). (D to F) Multiple domains of BRG1 may be involved in the interaction with HBZ. HEK293T cells were cotransfected with 5 μg of FLAG-HBZ, alone or together with 6 μg of MYC-tagged expression vectors for BRG1 Δ-N-T (D), Δ-ATPase (E), or Δ-Bromo (F). Transfected cells were lysed after 24 h, and co-IPs were performed using anti-FLAG M2 beads overnight. Precipitates were analyzed by Western blotting using anti-FLAG and anti-MYC, as indicated. IP, immunoprecipitation; WCL, whole-cell lysate.
Multiple domains of BRG1 may be involved in the interaction with HBZ.
We also sought to ascertain the protein domains in BRG1 that are responsible for binding HBZ. BRG1 contains conserved N-terminal, ATPase, and C-terminal regions necessary for its function as a coregulator of transcription (57). To evaluate their contribution to the interaction with HBZ, we generated three expression constructs, lacking the N-terminal region (Δ-N-T,1–710), the ATPase domain (Δ-ATPase,701–1405), and the C-terminal bromodomain (Δ-Bromo,1463–1593), using site-directed mutagenesis techniques (Fig. 3C). Plasmids containing these deletions were cotransfected together with a FLAG-HBZ expression plasmid into HEK293T cells, and lysates were immunoprecipitated using anti-FLAG M2 beads. Western blot analyses of precipitated complexes failed to identify a discrete region in BRG1 required to mediate the interaction with HBZ, as all BRG1-deleted proteins showed positive interactions (Fig. 3D to F). These findings suggest two possibilities. The first is that that several regions of the BRG1 protein may be involved in the interaction with HBZ in mammalian cells. This could possibly explain why missing one of BRG1 functional regions does not affect HBZ and BRG1 binding. These findings could also suggest that the interaction between HBZ and BRG1 may occur via regions bordering the deleted region spanning amino acids 1405 to 1463 located between Δ-ATPase,701–1405 and Δ-Bromo,1463–1593 or in the region spanning amino acid residues 1593 to 647 bordering the Δ-Bromo domain.
HBZ colocalizes with BRG1 in distinct nuclear foci.
Based on the physical interaction studies (Fig. 1), we sought to explore the possibility that HBZ colocalizes with BRG1 in mammalian cells, which may impact the functions of both proteins. To this end, HeLa cells were transfected with expression vectors encoding green fluorescent protein (GFP)-HBZ and MYC-BRG1 individually or together. Exogenous BRG1 was detected using an anti-BRG1 antibody, followed by Alexa Fluor 594-conjugated goat anti-mouse IgG1 antibody (red), and nuclei were stained using 4′,6-diamidino-2-phenylindole (DAPI) (blue). As shown in previous reports, GFP-HBZ was observed in speckle-like structures in the nuclear compartment (Fig. 4A) (58, 59). BRG1 localized in the nucleus in distinct foci (Fig. 4B) (60, 61). Examination of the colocalization of GFP-HBZ and MYC-BRG1 in cotransfected HeLa cells shows that the overexpression of BRG1 changes the nuclear distribution pattern of HBZ (Fig. 4C). HBZ partially colocalizes with BRG1 in distinct nuclear foci, which we suspect to be nucleolar foci, but this needs to be confirmed. These results are consistent with our previous data showing an interaction between HBZ and BRG1 in mammalian cells and in vitro.
FIG 4.

Subcellular colocalization of HBZ and BRG1. Immunofluorescence was performed on HeLa cells that were transfected with an expression vector for either green florescent protein (GFP)-HBZ (A), MYC-BRG1 (B), or GFP-HBZ and MYC-BRG1 (C). At 24 h posttransfection, the cells were fixed and stained for BRG1 using anti-BRG1, followed by Alexa Fluor 594-conjugated goat anti-mouse IgG1 antibody (red). Nuclei were labeled with DAPI (blue), and slides were mounted using ProLong gold antifade reagent. Images were obtained using a Zeiss AxioImager MI fluorescence microscope and an AxioCam HR microscope camera.
BRG1 and HBZ cooperate in the downregulation of Tax-mediated HTLV-1 LTR transactivation.
Previous studies have reported that HBZ plays a crucial role in the downregulation of Tax-dependent viral transcription by directly interacting with CREB and p300/CBP (62). In this study, we demonstrate that HBZ directly interacts with BRG1, which is essential for LTR transcriptional activation by Tax (36). On this basis, we wished to evaluate the impact of BRG1 on the ability of HBZ to repress Tax-mediated LTR activation using overexpression and knockdown approaches. We overexpressed BRG1 in HEK293T cells in the presence of Tax and increasing concentrations of HBZ, as indicated (Fig. 5A). Our results showed that the addition of BRG1 enhances Tax-mediated LTR activation. This observation is consistent with previous results by Easley et al. demonstrating the positive regulatory effects of BRG1 on Tax-activated transcription (36). We also observed that HBZ potently inhibits BRG1/Tax-mediated activation of the LTR in a dose-dependent manner (Fig. 5A). This suggests that HBZ can counteract Tax/BRG1 activation of viral transcription, possibly by competing with Tax for BRG1 binding. However, this effect coincided with a significant reduction in BRG1 expression levels in the presence of HBZ compared to the levels observed in the presence of Tax alone. Remarkably, the stability of BRG1 expression in the presence of Tax expression was counteracted by HBZ in a dose-dependent manner (Fig. 5A). To further investigate the effect of Tax on BRG1 expression, we overexpressed increasing amounts of His-Tax in HEK293T and Jurkat cells in the presence MYC-BRG1 (Fig. 5B and D) or in the absence of MYC-BRG1 (Fig. 5C and E). Western blotting and reverse transcription-PCR (RT-PCR) were used to determine the effect of Tax on BRG1 expression at the protein and RNA levels, respectively. Our data show that increasing levels of Tax expression stabilize both exogenous and endogenous BRG1 expression in HEK293T (Fig. 5B and C) and in Jurkat (Fig. 5D and E) cells. We also show that Tax has no effect on BRG1 expression at the transcriptional level (Fig. 5F), indicating that Tax possibly stabilizes BRG1 expression only at the protein level. Collectively, our results support the hypothesis that HBZ negatively modulates Tax-dependent viral transcription via the SWI/SNF pathway and suggest that BRG1 can function as either a coactivator or a corepressor of HTLV-1 transcription, depending on its interaction with the viral oncoprotein Tax or HBZ.
FIG 5.

BRG1 overexpression enhances the repressive effects of HBZ on Tax-dependent viral transcription. (A) Luciferase assays were performed on lysates from HEK293T cells cotransfected with 250 ng of HTLV-1-LTR-Luc and 25 ng of HIS-pCAGGS-Tax, alone or together with 2 μg MYC-BRG1 with or without increasing concentrations of FLAG-HBZ (0.2, 0.4, and 0.8 μg). Luciferase activity was measured at 24 h posttransfection. LTR-dependent luciferase activity was normalized to the protein concentration in lysates, and the means from three independent experiments are expressed as fold activation. Error bars represent standard deviations (SD) calculated in three independent experiments. * indicates significance at a P value of <0.05. The expressions of HBZ and BRG1 were analyzed by immunoblotting using anti-BRG1 and anti-FLAG. Tubulin was probed as a loading control. NS, not significant. (B and D) Immunoblot analyses were performed on lysates from HEK293T cells and Jurkat cells cotransfected with 4 μg of an expression vector for MYC-BRG1, together with increasing concentrations of HIS-pCAGGS-Tax (0.025, 0.05, 0.1, and 0.2 μg [B] or 1 and 2 μg [D]). Transfected cells were lysed after 24 h, and the expression levels of Tax and BRG1 were analyzed by Western blotting, using anti-His, anti-MYC, and antitubulin antibodies, as indicated. The bottom of panel B represents the relative optical density (ROD) of BRG1 expression normalized against tubulin. (C and E) Immunoblot analysis was performed on lysates from HEK293T cells and Jurkat cells transfected with increasing concentrations of HIS-pCAGGS-Tax (0.025, 0.05, 0.1, or 0.2 μg [C] or 1 and 2 μg [E]) alone. Transfected cells were lysed after 24 h, and the expression levels of Tax and endogenous BRG1 were analyzed by Western blotting, using anti-His and anti-BRG1. Tubulin was probed as a loading control. The bottom of panel C represents the relative optical density of BRG1 expression normalized against tubulin. (F) RT-PCR analyses were performed on RNA extracts from HEK293T cells cotransfected with increasing concentrations of HIS-pCAGGS-Tax (0, 0.5, and 1 μg). RNA was extracted from transfected cells after 24 h, and the BRG1 mRNA levels were determined using real-time RT-PCR. Beta-actin was used as a housekeeping gene for normalization. Reported values are the averages of data from three independent experiments, and the error bars represent SD from the triplicates. The expressions of Tax and BRG1 were analyzed by immunoblotting, using anti-BRG1 and anti-His. Tubulin was probed as a loading control.
To further substantiate the effect of HBZ/BRG1 on HTLV-1 LTR activation, we knocked down BRG1 expression in HeLa and Jurkat cells, both of which contain an integrated luciferase reporter gene under the transcriptional control of the HTLV-1 LTR. Cells were transfected with short hairpin RNA (shRNA) to knock down BRG1 or an shRNA negative control together with expression vectors encoding His-Tax, His-Tax plus FLAG-HBZ, or FLAG-HBZ alone. Using this approach, we consistently observed that BRG1 knockdown in HeLa-HTLV-1-LTR-Luc (Fig. 6A) and Jurkat-U3-LTR-HTLV-1-Luc (Fig. 6C) cells negatively modulates Tax-dependent promoter activity. This was in agreement with a previous study reporting the positive impact of BRG1 on Tax-dependent viral transcription (36). Moreover, BRG1 knockdown significantly limited HBZ-dependent suppression of Tax-dependent promoter activation in both cell lines (Fig. 6B and D). This suggests that the presence of BRG1 plays a role in HBZ-mediated suppression of Tax-dependent viral transcription. Furthermore, we found that BRG1 knockdown resulted in a reduction of basal promoter activity and impeded the negative impact of HBZ on the basal transcriptional activation of the viral promoter in Jurkat-U3-LTR-HTLV-1-Luc cells (Fig. 6E and F). Collectively, our observations suggest that BRG1 may be involved in HBZ-mediated suppression of basal and Tax-dependent viral transcription.
FIG 6.

Impact of BRG1 knockdown on HBZ inhibition of basal and Tax-dependent viral LTR transcription. (A) Effect of BRG1 knockdown on Tax-dependent viral promoter activity. HeLa-HTLV-1-LTR-Luc cells were cotransfected with 25 ng of HIS-pCAGGS-Tax, alone or together with 4 μg shRNA against BRG1 (ShBRG1). Luciferase activity was measured at 72 h posttransfection. LTR-dependent luciferase activity was normalized to the protein concentration in lysates and is expressed as fold activation. Error bars represent SD calculated from three independent experiments. * indicates significance at a P value of <0.05. (B) Effect of BRG1 knockdown on the ability of HBZ to suppress Tax-dependent viral promoter activity. HeLa-HTLV-1-LTR-Luc cells were cotransfected with 25 ng of HIS-pCAGGS-Tax together with 2 μg FLAG-HBZ plus shRNA against BRG1. Luciferase assays were performed as described above for panel A. (C) Effect of BRG1 knockdown on Tax-dependent viral promoter activity in Jurkat-U3-LTR-HTLV-1-Luc cells. Luciferase assays were performed on lysates from Jurkat-U3-LTR-HTLV-1-Luc cells cotransfected with 25 ng of HIS-pCAGGS-Tax, alone or together with 4 μg shRNA against BRG1 or the shRNA negative control. Luciferase activity was measured at 48 h posttransfection. LTR-dependent luciferase activity was normalized to the protein concentration in the lysates, and the means of data from three independent experiments are expressed as fold activation. The P value indicates no significance (NS). (D) Effect of BRG1 knockdown on the ability of HBZ to suppress Tax-dependent viral promoter activity. Luciferase assays were performed on lysates from Jurkat-U3-LTR-HTLV-1-Luc cells cotransfected with either 4 μg shRNA against BRG1 or the shRNA negative control together with 25 ng of HIS-pCAGGS-Tax, alone or together 1 μg FLAG-HBZ. Luciferase assays were performed as described above for panel C. (E) Effect of BRG1 knockdown on basal LTR activation. Luciferase assays were carried out on lysates from Jurkat-U3-LTR-HTLV-1-Luc cells cotransfected with either 4 μg shRNA against BRG1 or the shRNA negative control. Luciferase assays were performed as described above for panel C. (F) Effect of BRG1 knockdown on the ability of HBZ to suppress basal activation of the viral promoter. Luciferase assays were performed on lysates from Jurkat-U3-LTR-HTLV-1-Luc cells cotransfected with either 4 μg shRNA against BRG1 or the shRNA negative control together with 1 μg FLAG-HBZ. Luciferase assays were performed as described above for panel C. (G) Immunoblot analysis of BRG1 knockdown was performed on Jurkat-U3-LTR-HTLV-1-Luc cells. Cells were transfected with 4 μg of shRNA against BRG1 plasmids (lane 2) and the shRNA negative control (lane 1).
HBZ alters the recruitment of SWI/SNF subunits to the HTLV-1 promoter.
An important question raised by our findings is how HBZ can downregulate viral transcription via the SWI/SNF pathway. Previous studies report that Tax recruits BRG1 to the HTLV-1 promoter (48, 63). Moreover, HBZ has been shown to downregulate the viral promoter by interacting with basal transcription factors and disturbing their recruitment to the viral promoter (32, 62, 64). In this study, we observed that HTLV-1-positive cells and ATL cells harbor particularly high levels of BRG1 protein, and we demonstrate that HBZ interacts not only with BRG1 in mammalian cells but also with BAF180 and BAF200, the specific subunits of the PBAF complex. Based on these observations, we hypothesized that HBZ may downregulate Tax-dependent viral promoter transcription by affecting the recruitment of BRG1 and/or other subunits of the SWI/SNF complex to the viral promoter. To investigate this hypothesis, Jurkat-U3-LTR-HTLV-1-Luc cells were cotransfected with either an empty vector (control) or His-Tax, alone or together with FLAG-HBZ expression plasmids. About 1 × 106 cells under each condition were assayed at 24 h posttransfection by luciferase assays to confirm that HBZ significantly reduced Tax transactivation of the integrated promoter as expected. Another 3 × 106 cells under each condition were subjected to chromatin immunoprecipitation (ChIP) to analyze the effect of HBZ on the recruitment of endogenous BRG1, BAF200, and BAF180 to the viral promoter. The protein-DNA complexes were immunoprecipitated by incubating chromatin fragments with magnetic beads, precoated with an antibody directed against BRG1, BAF200, and BAF180 proteins. Twenty-four hours after ChIP, cross-links were reversed, and the HTLV-1 promoter was amplified using quantitative PCR (qPCR). Data were quantified by comparing the signal from the coimmunoprecipitated DNA to that of the input DNA in each experiment and are presented as percentages of the DNA input (Fig. 7A). As shown previously, transfection of these cells with the Tax expression vector strongly increases BRG1 enrichment at the viral promoter (Fig. 7A). We also observed a slight increase in the enrichment of BAF200 and BAF180 at the viral promoter, under the same conditions, which all coincided with an increase in HTLV-1 LTR luciferase activity (Fig. 7B). Interestingly, cotransfection with HBZ reduces the levels of BRG1 and BAF200 at the LTR to about 50% at the promoter. Unexpectedly, we consistently observed a 2- to 3-fold increase in BAF180 enrichment at the viral promoter under the same conditions (Fig. 7A). Together, these findings reveal that HBZ facilitates the downregulation of HTLV-1 transcription by regulating the recruitment of the SWI/SNF complex to the promoter (Fig. 7B). Moreover, these experiments highlight the opposite impact of HBZ on the recruitment of BAF200 and BAF180 to the viral promoter, despite belonging to the same complex. This raised the possibility that these subunits may have distinct, but complementary, functions in the regulation of Tax-dependent viral promoter transcription in the presence of HBZ.
FIG 7.
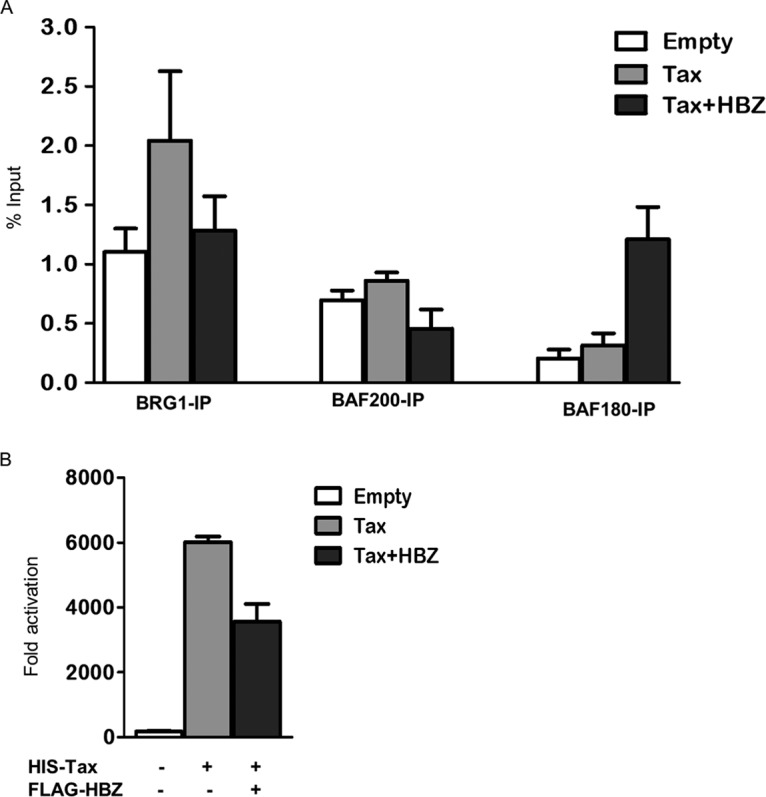
HBZ displaces BRG1 and BAF200 from the HTLV-1 promoter and recruits BAF180. (A) ChIP analyses were performed on Jurkat-U3-LTR-HTLV-1-Luc cells cotransfected with either an empty vector or HIS-pCAGGS-Tax, alone or together with FLAG-HBZ. At 24 h posttransfection, genomic DNA was fixed with 1% formaldehyde and sheared using a Bioruptor instrument. Sheared chromatin was diluted and immunoprecipitated using anti-BRG1, anti-BAF180, and anti-BAF200 antibodies, with an IgG isotype control. The immunoprecipitated material was quantified by qPCR. Results were normalized to inputs and are expressed as a percentage of the DNA input. (B) Analysis of LTR luciferase activity in cells used in ChIP experiments. Error bars represent SD calculated from three independent experiments.
DISCUSSION
The SWI/SNF chromatin remodeling complexes were demonstrated to function as either transcriptional activators or repressors (65–67). In the context of HTLV-1 gene expression, BRG1, the ATPase subunit of the SWI/SNF complex, has been shown to be essential for optimal transcriptional activation of the HTLV-1 LTR by Tax (36, 48). In the present study, we investigated physical interactions between HBZ and BRG1 and the biological significance of these interactions for basal and Tax-mediated viral gene expression.
Alterations of BRG1 expression and its role in tumorigenesis and tumor suppression have been demonstrated in various human cancers (52, 68–70). Here, we have illustrated that BRG1 is highly expressed in HTLV-1-infected cell lines and ATL cell lines, suggesting that elevated BRG1 expression may contribute to the phenotype of these cells. Our findings appear compatible with previously reported evidence showing that BRG1 is overexpressed in other cancer types, including, but not limited to, gastric carcinomas (53), liver hepatocellular carcinoma (HCC), kidney renal clear cell carcinoma, and breast cancer (45, 54). Several studies have presented that BRG1 is required for tumor cell proliferation. For example, BRG1 has been shown to maintain acute myeloid leukemia (AML) cell proliferation by stimulating MYC gene expression (42, 43). Overexpression of BRG1 in HCC, malignant melanoma, and breast cancer has been demonstrated to promote proliferation and invasion (45, 47, 71). Based on these reports and others, we speculate that BRG1 expression may contribute to the phenotype of ATL cells. However, more experiments will be required to test this hypothesis.
In previous studies using the yeast two-hybrid system, we identified BRG1 as a possible HBZ-interacting candidate. In this study, using coimmunoprecipitation and GST pulldown assays, we confirmed that HBZ interacts with BRG1 in mammalian cells and in vitro. This finding is of particular interest not only because BRG1 is involved in Tax-mediated HTLV-1 transcription (36) but also for the reason that HBZ and BRG1 individually govern distinct pathways to direct multiple cell biological processes, including cell differentiation, proliferation, apoptosis, tumorigenesis, and cancer progression (42, 72–76). We show that the N-terminal activation domain (AD) of HBZ is responsible for the interaction with BRG1. This domain is important for HBZ function because it accommodates two LXXLL-like motifs that have been shown to bind to the KIX domain of CBP/p300 (62). These complexes are well-known transcription coactivators involved in a variety of cellular functions, including proliferation, cell cycle regulation, apoptosis, and differentiation (77). The formation of the HBZ-CBP/p300 complex inhibits the ability of Tax-CREB to recruit p300/CBP to the viral promoter, which results in the reduction of viral transcription (62). In ATL cells, HBZ-p300/CBP complexes were illustrated to repress p53 (tumor suppressor) function (78). Interestingly, BRG1 also interacts with p300/CBP, and BRG1 and CBP cooperate to constrain p53 activity and to permit cancer cell proliferation (78). BRG1-CBP was also found in a complex with an activated form of the transforming growth factor β (TGF-β) pathway effector Smad2 (79). The LXXLL motifs are also required for HBZ-binding Smad2/3 to activate TGF-β/Smad signaling and to induce Foxp3 expression (80), which is critical for the suppressive function of regulatory T cells (Tregs) (81). The AD of HBZ was also shown to interact with interferon regulatory factor 1 (IRF-1), a known tumor suppressor that is downregulated in leukemia (82, 83). HBZ/IRF-1 interaction reduced both IRF-1 DNA binding and transcriptional activity, resulting in reduced numbers of cells undergoing apoptosis (84). These reports and our results suggest that the BRG1/AD-HBZ interaction might form complexes with multiple cellular factors that may influence leukemogenesis.
BRG1 contains conserved N-terminal, ATPase, and C-terminal regions necessary for its function as a coregulator of transcription (57). Unfortunately, we were unable to define a unique region in BRG1 that is required for its interaction with HBZ, as all BRG1-deleted proteins retained the ability to interact with HBZ. Hence, we suggest that multiple regions of BRG1 might be involved in the interaction. To verify this, multiple-site deletions should be generated in BRG1 and tested for interaction with HBZ.
BRG1 is associated with two related SWI/SNF complexes, BAF and PBAF. These complexes share most subunits but can be distinguished by the presence of BAF250A in the BAF complex and BAF200 and BAF180 exclusively in the PBAF complex (37, 40). We revealed that HBZ interacts with endogenous BAF200 and BAF180, PBAF signature subunits (37, 40). These SWI/SNF subunits have also been shown to interact with the HIV transactivator Tat, which is required for Tat-mediated activation of the HIV-1 promoter (63). Furthermore, BAF180 is required for HTLV-1 replication, as the virus does not replicate in a BAF180 mutant cell line, indicating that BAF180 may play a role in viral replication (36). The interaction between HBZ and BAF180 may regulate viral replication, but this requires further investigation. Moreover, BAF180 was shown to negatively regulate interleukin-10 (IL-10) transcription (85). As IL-10 was shown to contribute to the proliferation of HTLV-1-infected cells (86), it is possible that the HBZ/BAF180 interaction may influence the negative effect of BAF180 on IL-10 and hence contribute to HTLV-1 leukemogenesis. BAF200 depletion was also reported to result in a concomitant destabilization of the PBAF complex (40). Thus, the interaction between HBZ and BAF200 may disrupt the stability of the PBAF complex, which may lead to either activation or repression of PBAF-regulated genes in favor of the HTLV-1 life cycle. Moreover, BAF200 regulates the expression of the interferon alpha (IFN-α)-inducible gene IFITM1 in HeLa cells (40). Based on these studies, together with our results, we hypothesize that the HBZ/PBAF complex may play a role in the HTLV-1 life cycle and pathogenesis.
While BRG1 has a euchromatin nucleoplasmic distribution with several prominent foci that localize in the nucleolus (60, 61), HBZ is a predominately nucleus-localizing protein with a speckled pattern associated with heterochromatin (15, 55, 58). Using immunofluorescence (IF) studies, we observed that BRG1 expression changes the localization pattern of HBZ. BRG1 expression partially recruits HBZ into distinct nuclear foci in HeLa cells. This pattern of localization was observed previously in COS cells transfected with HBZ constructs lacking the activation domain and bZIP domain (55). Considering that the colocalization of HBZ and BRG1 detected in this study is specific and is not simply a result of their coexpression in HeLa cells, and given that we demonstrated that BRG1 interacts with HBZ via the AD, we hypothesize that this interaction may promote the localization of HBZ to distinct nuclear foci.
A previous study has shown that BRG1 is required for Tax-mediated activation of HTLV-1 viral gene expression (36). Here, the determination of the functional significance of the HBZ/BRG1 interaction in the suppression of HTLV-1 promoter activity using knockdown and overexpression studies revealed that BRG1 is required for HBZ’s optimal suppression of basal and Tax transactivation of the HTLV-1 LTR. These data suggest that HBZ cooperates with BRG1 and core transcriptional factors, including CREB and CBP/P300, to inhibit Tax-mediated activation of the HTLV LTR.
Interestingly, we consistently observed enhanced levels of BRG1 expression in the presence of Tax alone compared to those observed in cells expressing Tax and HBZ. We speculate that this may contribute to transcriptional repression in the case of HBZ and transcriptional activation in the case of Tax. Immunoblotting and RT-PCR data indicate that the effect of Tax on BRG1 is not likely to occur at the transcriptional level but occurs chiefly through posttranscriptional mechanisms. It has been reported that BRG1 degradation is mediated by a proteasome-dependent pathway (87). It has also been demonstrated that after Tax transfection, BRG1 exclusively colocalizes with Tax in the nucleus (88). Thus, it is possible that the interaction between BRG1 and Tax in the nucleus protects BRG1 from proteasomal degradation. This mechanism may facilitate BRG1 stability, which, in turn, may affect the ability of Tax to activate viral transcription via the SWI/SNF pathway. However, our data showing elevated levels of BRG1 expression in the presence of Tax are inconsistent with observations (although data not shown) made in other studies which suggest lower levels of BRG1 expression in the presence of Tax (36, 89). The reason for this discrepancy is unclear. Having clearly demonstrated the interaction between HBZ and BRG1, we hypothesize that the reduction of BRG1 stability in Tax/HBZ-expressing cells may be the result of competition between HBZ and Tax to bind BRG1. This may release BRG1 from the protective zone and affect its degradation. It is also possible that HBZ forms a complex with BRG1/Tax and impedes the ability of Tax to stabilize BRG1 and, hence, inhibit Tax/BRG1-mediated HTLV-1 LTR activation.
An important question raised by our findings is how HBZ can downregulate viral transcription via the SWI/SNF pathway. Using ChIP-qPCR studies and luciferase reporter assays, we observed that Tax actively promoted BRG1 recruitment to an integrated HTLV-1 LTR. These results are in agreement with previous reports which show that BRG1 interacts with Tax physically and functionally assists Tax-mediated LTR activation (36, 48). Under the same conditions, we also show that BAF200 and BAF180 are slightly recruited to the viral promoter, which suggests that they may be required for optimal LTR activation by Tax via the SWI/SNF pathway. Interestingly, we show that the coexpression of HBZ with Tax displaced about 50% of BRG1, which correlates with the repressive effect of HBZ on Tax-dependent LTR activation. This observation supports the hypothesis that HBZ limits the association of BRG1 and the viral promoter in Tax-expressing cells. Strikingly, under the same conditions, we observed that HBZ potently recruits BAF180, while it displaces BAF200, despite the fact that both subunits belong to the same complex. This suggests that BAF200 and BAF180 may temporarily play opposing roles in coregulating viral LTR activation depending on their stimulation by the viral oncoprotein Tax or HBZ.
One explanation for the different enrichment scenarios for the PBAF complex subunits at the viral promoter could relate to the interaction between these subunits and the Tax and HBZ proteins. Previously, we showed that HBZ interacts with BAF200 and BAF180, whereas no interactions were reported between Tax and these subunits. It is known that BAF200 is required for PBAF complex stability in vivo and that disrupting BAF200 results in the dissociation of the PBAF complex subunit BAF180 (40). Therefore, we speculate that HBZ/BAF200 may disrupt the assembly of BAF180 into the PBAF complex, releasing BAF108 subunits which could then be recruited to the LTR by HBZ. However, further experiments would be required to confirm this hypothesis.
Given that previous studies have shown that the interaction between Tax and BRG1 causes their recruitment to the viral promoter and also recruits the basal transcriptional machinery to activate viral transcription (36, 48), it is possible that the displacement of BRG1 by HBZ is the molecular mechanism behind HBZ-mediated downregulation of HTLV-1 transcription. Multiple lines of evidence support this suggestion. This study demonstrates that HBZ diminishes the stability of BRG1 induced by Tax. This correlates with a significant reduction of BRG1/Tax-mediated LTR activation. BRG1 degradation was shown in previous studies to enhance its removal from chromatin and inhibit persistent chromatin remodeling in SWI/SNF-regulated genes (87, 90). We show that HBZ expression displaces 50% of BRG1 from the promoter. This effect coincides with a significant reduction in LTR activation, indicating that BRG1 displacement may serve as the primary mechanism for the downregulation of HTLV-1 promoter activity. This is consistent with our previous finding showing that BRG1 knockdown induces the suppression of basal and Tax-mediated HTLV-1 LTR activation. Moreover, a previous study showed that HBZ expression displaced more than 50% of Tax at the HTLV-1 promoter despite the fact that HBZ does not interact with Tax (32). It was suggested that this displacement occurs via HBZ interacting with CREB/Tax complexes. Since we have revealed here that HBZ interacts with BRG1 and that this reduces promoter activity, correlating with the displacement of BRG1, it is likely that the drop in the level of Tax at the promoter after HBZ transfection in the study by Lemasson et al. is also associated with the BRG1 displacement that we demonstrated in our study. Overall, our data indicate that BRG1 may have a dual function in HTLV-1 transcription: first as a coactivator that binds to Tax to stimulate LTR transcription and second as a corepressor that binds to HBZ to promote efficient inhibition of viral LTR activation.
In conclusion, the results presented here demonstrate that HBZ represses basal and Tax-dependent HTLV-1 transcription via SWI/SNF pathways. They also place BRG1 and SWI/SNF complexes on the list of proteins that are positively and negatively regulated by Tax and HBZ to influence viral silencing and foster viral latency and pathogenesis.
MATERIALS AND METHODS
Cell culture.
HEK293T and HeLa cells (ATCC) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco). HeLa-LTR-HTLV-1-Luc clone 1 cells (generated in our laboratory) were maintained in DMEM supplemented with 10% FBS and 400 μg/ml of G418 (GE Healthcare). Jurkat T cells, HTLV-infected cells, and ATL cells (C91, ATL-TH, ATL-CR, MT2, and MO) were maintained in RPMI 1640 medium (Gibco) supplemented with 10% FBS. Jurkat-U3-LTR-HTLV-1-Luc cells were a kind gift from Andrea K. Thoma-Kress, Friedrich Alexander University of Erlangen-Nürnberg (91), and were maintained in RPMI 1640 medium supplemented with 10% FBS and 0.25 μg/ml of puromycin antibiotic (Gibco). Cells were cultured under standard tissue culture conditions.
shRNA and plasmid constructs.
(i) shRNA. BRG1 knockdown was performed using a sure silencing shRNA plasmid GFP kit (human SMARCA4, catalogue no. 336311; Qiagen).
(ii) Plasmids.
The expression plasmids encoding GST, GST-HBZ, FLAG-Δ-AD, FLAG-HBZ-Δ-CD, and FLAG-Δ-bZIP were previously described (92). GFP-HBZ was a kind gift from Jean-Michel Mesnard, Université de Montpellier, France. The MYC-BRG1 plasmid was a kind gift from Kristen Kroll, Washington University School of Medicine, and was previously described (93). The MYC-BRG1 mutants MYC-Δ-N-T, MYC-Δ-ATPase, and MYC-Δ-Bromo were generated using a Phusion site-directed mutagenesis kit (Thermo Fisher Scientific), using primers at the indicated positions and the MYC-BRG1 plasmid as a template. Primer sequences for deletion were as follows: 5′-GCC AAG CAA GAT GTC GAT GAT-3′ and 5′-CTT CAC CCA ATT CAA GTC CTC TTC-3′ for MYC-BRG1-Δ-N-T, 5′-CGG CAG AAG AAA TCA TCA G-3′ and 5′-CTC AGA GAC GTC ATC GCT GTC-3′ for MYC-BRG1-Δ-ATPase, and 5′-GTC AAA GTG AAG ATC AAG CTT GGC-3′ and 5′-CTT CTT CAT CTT CTT GGT GAG GTT-3′ for MYC-BRG1-Δ-Bromo. HTLV-1-LTR-Luc and HIS-pCAGGS-Tax were previously described (94).
Transfections. HEK293T cells were transiently transfected using Lipofectamine 2000 transfection reagents (Thermo Fisher Scientific), according to the manufacturer’s guidelines. Transfections of HeLa cells and HeLa-LTR-HTLV-1-Luc clone 1 cells were performed using TurboFect transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s manual. Jurkat cell and Jurkat-U3-LTR-HTLV-1-Luc cell transfections were performed by electroporation, using Amaxa cell line Nucleofector V kit (Lonza) program X-001, according to the manufacturer’s instructions for nucleofection of suspension cell lines.
Immunoblotting and antibodies.
Cellular lysates were resolved by 4 to 12% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) using 1× morpholineethanesulfonic acid (MES)-SDS running buffer or 1× Tris-acetate-SDS running buffer (Invitrogen). Proteins were transferred to iBlot2 nitrocellulose regular stacks, using an iBlot2 gel transfer device (Invitrogen), for 7 to 10 min. Membranes were blocked in 4% nonfat dry milk (NFDM) power in Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBST) for 45 min. Thereafter, membranes were incubated with the indicated primary antibodies diluted in 4% NFDM–TBST and incubated overnight at 4°C with gentle shaking. The membranes were washed three times for 5 min in TBST and incubated with a 1:5,000 dilution of the horseradish peroxidase (HRP)-conjugated secondary antibody diluted in 4% NFDM–TBST for 40 min at room temperature. Thereafter, the membranes were washed three times for 5 min in 0.1% TBST. Protein bands were detected by using Amersham ECL Western blotting detection reagents (GE Healthcare Life Sciences) and visualized on Fuji medical X-ray film.
The antibodies used in immunoblotting, immunofluorescence, coimmunoprecipitation, and chromatin immunoprecipitation procedures were as follows: anti-FLAG (catalogue no. F7425; Sigma-Aldrich), anti-α-tubulin (catalogue no. ab7291; Abcam), anti-MYC (catalogue no. 46-0603; Novex), anti-cyclophilin A (anti-CYPA) (catalogue no. sc-133494; Santa Cruz), anti-histone deacetylase 1 (HDAC1) (catalogue no. sc-7872; Santa Cruz), anti-BRG1 (H-10) (catalogue no. sc-374197; Santa Cruz), anti-BAF180 (catalogue no. ABE70; Merck Millipore), anti-6×His (catalogue no. 631212; Clontech), anti-BAF200 (E-3) (catalogue no. sc-166117i; Santa Cruz), anti-BAF250 (catalogue no. sc-32761; Santa Cruz), and Alexa Fluor 594-conjugated goat anti-rabbit IgG (Invitrogen).
Coimmunoprecipitation.
HEK293T cells were seeded to 90% confluence in 60-mm-diameter dishes and transfected with relevant amounts of expression plasmids as indicated for the individual experiments. At 24 h posttransfection, cells were washed with phosphate-buffered saline (PBS) and lysed at 4°C for 30 min in 300 μl of ice-cold immunoprecipitation lysis buffer (1× TBS, 5 mM EDTA, 1% Triton X-100, 50 mM NaCl) supplemented with 1% SDS and protease inhibitors. Lysates were centrifuged at 4°C for 5 min at 14,000 rpm and incubated with anti-FLAG M2 beads (Sigma) overnight at 4°C with gentle rotation. The beads were washed in lysis buffer, and proteins were eluted in 2× SDS loading buffer and 100 mM dithiothreitol (DTT). Eluted proteins were boiled for 5 min and analyzed by immunoblotting using anti-FLAG, anti-BRG1, anti-MYC, anti-BAF180, anti-ARID2, and anti-ARID1A antibodies.
GST protein expression and purification.
Recombinant GST and GST-HBZ were produced in Escherichia coli BL2. This protocol was adopted from a protocol obtained from Jean-Marie Peloponnese, Jr. (CNRS, Montpellier, France). In summation, transfected E. coli BL2 cells were grown to an optical density at 600 nm (OD600) of 0.5 to 0.7 at 37°C and induced with 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG; Sigma) for 3 to 5 h at 37°C. GST was optimally induced with 300 μM IPTG at an OD600 of 0.6 to 0.8 for 1 h at 37°C. One-liter cultures were harvested, resuspended in 30 ml lysis buffer (25 mM phenylmethylsulfonyl fluoride [PMSF] and 1 mg/ml lysozyme in 1× ice-cold PBS), and freeze/thawed three times. Lysates were sonicated four times for 30 s at 70% power. A total of 300 μl of Triton X-100 was added to the lysates, and the mixture was incubated for 60 min on a rotator at 4°C and clarified by centrifugation at 1,200 rpm for 10 min at 4°C. Thereafter, the supernatant was incubated with 2 ml of washed glutathione-Sepharose 4 fast flow beads (GE Healthcare) at 4°C with rotation overnight. After extensive washing, cleared GST fusion proteins were eluted in 1 ml elution buffer (10 mM reduced glutathione and 50 mM Tris-HCl [pH 8]). GST-eluted protein purity was determined by SDS-polyacrylamide gel electrophoresis, followed by Coomassie blue staining.
GST pulldown assays.
Pulldown assays for GST and GST-HBZ fusion proteins were performed by incubating 100 μl of GST-HBZ and GST with 50 μl of glutathione-Sepharose 4 fast flow beads (GE Healthcare) in a final volume of 800 μl GST binding buffer (0.5% Triton X-100 in PBS plus propidium iodide [PI]) and rotating the mixture overnight at 4°C. The beads were centrifuged, washed twice with GST binding buffer, and resuspended in 1 ml binding buffer. Six micrograms of purified human BRG1 protein (catalogue no. ab198137; Abcam) was incubated with GST-bound beads overnight at 4°C, and following washing, bound proteins were eluted in 40 μl GST elution buffer (50 mM Tris-HCl [pH 8.0] containing 10 mM reduced glutathione). Interactions were analyzed by immunoblotting using an anti-BRG1 antibody and Coomassie brilliant blue staining.
Chromatin immunoprecipitation-qPCR.
ChIP was performed using the high-cell-number chromatin immunoprecipitation kit (catalogue no. C01010063; Diagenode), according to the manufacturer’s instructions, with minor modifications. In brief, approximately 1.8 × 106 Jurkat-U3-LTR-HTLV-1-Luc cells were electroporated with 18 μg plasmid DNA (9 μg HIS-pCAGGS-Tax, alone or together with 9 μg FLAG-HBZ). Cells were harvested 24 h later for luciferase assays and ChIP analyses. A total of 3 × 106 cells per IP were cross-linked with 1% formaldehyde (Sigma) for 15 min at room temperature and stopped by the addition of glycine at a final concentration of 125 mM. Following washing with ice‐cold PBS, nuclei were isolated using ice‐cold lysis buffer L1 for 10 min, followed by one wash with ice‐cold lysis buffer L2 for 10 min and centrifugation. The pellet was resuspended in 250 μl shearing buffer supplemented with a protease inhibitor and sonicated using a Bioruptor instrument (Diagenode) for 30 cycles, 30 s on and 30 s off on high frequency, such that it will give between 100- and 400-bp fragments. Samples were centrifuged to pellet debris, and aliquots of 50 μl of sheared chromatin were taken out of each sample for DNA analysis in a 1% agarose gel. A total of 200 μl of sheared chromatin was diluted in 800 μl of C1 buffer supplemented with a protease inhibitor. Immunoprecipitation was conducted by incubating 5 μg of anti-ARID2x, anti-ARID1Ax, or anti-BRG1x with 900 μl of diluted chromatin overnight at 4°C with rotation. Washed and purified DNAs were subjected to qPCR using a QuantiNova SYBR green PCR kit (Qiagen) according to the manufacturer’s instructions. The primers directed against the HTLV-1 promoter region were as follows: 5′‐TTC CGA GAA ACA GAA GTC TG‐3′; and 5′-GTG AGG GGT TGT CGT CA‐3′. The enrichment of protein in the target DNA fragment under each condition was compared to the percent DNA quantity in the input sample. The percent input was calculated as % input = efficiency[(CTinput−6.644)−CTsamples)] × 100, where the efficiency was the mean PCR efficiency for all reactions within the primer set, calculated using the LinRegPCR program (http://www.hartfaalcentrum.nl/index.php?main=files&fileName=LinRegPCR.zip&sub=LinRegPCR), and CT is the threshold cycle. Amplification was carried out on an Applied Biosystems 7300 instrument, according to the manufacturer’s instructions.
Real-time PCR.
RT-qPCR was performed on RNA templates extracted from HEK293T cells transfected with increasing concentrations of HIS-pCAGGS-Tax to investigate its effect on endogenous BRG1 mRNA levels. The extraction of RNA was carried out using an RNeasy minikit (Qiagen), according to the manufacturer’s instructions, with minor modifications. Equal amounts of RNA were subjected to RT-qPCR using a QuantiNova SYBR green RT-PCR kit (Thermo Fisher Scientific) with specific primers for BRG1 and an actin internal control (Qiagen): 5′‐GTCGACAACGGCTCCGGC‐3′ and 5′-GGTGTGGCAGATTTTCT‐3′ for actin and 5′-GCTCATGGCTGAAGATGAGG-3′ and 5′-CAGGCGTCTGTCCTTCTGC-3′ for BRG1. Relative RNA expression was calculated using the following calculation: efficiency(CTinternal control−CTsamples). RT-qPCR amplification was carried out on an Applied Biosystems 7300 instrument, according to the manufacturer’s instructions.
Immunofluorescence.
HeLa cells were seeded to 60% confluence onto two-well chamber slides (Thermo Fisher Scientific) and cotransfected with the indicated plasmids. Cells were washed twice with PBS at 24 h posttransfection and fixed with 4% paraformaldehyde for 10 min at room temperature. Cells were permeabilized with PBS–0.5% Triton X-100 for 5 min followed by washing buffer (PBS–0.05% Triton X-100) and blocked in 1% Invitrogen blocking reagent plus 10% goat serum at room temperature for 1 h. The cells were then incubated with appropriate primary antibodies (diluted in IF blocking buffer) overnight at 4°C. The primary antibodies were then cleared by washing the slides three times with PBS, followed by incubation with the appropriate fluorophore-labeled secondary antibody diluted in IF blocking buffer in a dark humid chamber at room temperature for 2 h. Following washing, the nuclei were stained with DAPI (Sigma), and slides were mounted using Prolong gold antifade (Invitrogen) and analyzed by using a Zeiss MI AxioImager fluorescence microscope and an AxioCam HR camera.
Luciferase assay.
A luciferase assay was performed using the dual-luciferase assay system (catalogue no. E1501; Promega), according to the manufacturer’s specifications. Luciferase activity results were normalized to the protein concentration quantified by using a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. Results were expressed as fold changes relative to values for cells transfected with the controls.
ACKNOWLEDGMENTS
This work was supported by the Ministry of Higher Education, Kingdom of Saudi Arabia.
We thank Andrea K. Thoma-Kress, Friedrich Alexander University of Erlangen-Nürnberg, for providing the Jurkat-U3-LTR-HTLV-1-Luc cell line and Jean-Michel Mesnard, Université de Montpellier, France, for providing the GFP-HBZ plasmid. We thank Kristen Kroll, Washington University School of Medicine, for providing the MYC-BRG1 plasmid and Jonathan Dean, University College Dublin National Virus Reference Laboratory, for helping with sequencing analysis.
REFERENCES
- 1.Gallo RC. 2005. History of the discoveries of the first human retroviruses: HTLV-1 and HTLV-2. Oncogene 24:5926–5930. doi: 10.1038/sj.onc.1208980. [DOI] [PubMed] [Google Scholar]
- 2.Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. 1980. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A 77:7415–7419. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poiesz BJ, Ruscetti FW, Mier JW, Woods AM, Gallo RC. 1980. T-cell lines established from human T-lymphocytic neoplasias by direct response to T-cell growth factor. Proc Natl Acad Sci U S A 77:6815–6819. doi: 10.1073/pnas.77.11.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshida M. 2005. Discovery of HTLV-1, the first human retrovirus, its unique regulatory mechanisms, and insights into pathogenesis. Oncogene 24:5931–5937. doi: 10.1038/sj.onc.1208981. [DOI] [PubMed] [Google Scholar]
- 5.Gessain A, Cassar O. 2012. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol 3:388. doi: 10.3389/fmicb.2012.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshida M, Miyoshi I, Hinuma Y. 1982. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci U S A 79:2031–2035. doi: 10.1073/pnas.79.6.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahieux R, Gessain A. 2003. HTLV-1 and associated adult T-cell leukemia/lymphoma. Rev Clin Exp Hematol 7:336–361. [PubMed] [Google Scholar]
- 8.Takatsuki K. 2005. Discovery of adult T-cell leukemia. Retrovirology 2:16. doi: 10.1186/1742-4690-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gessain A, Barin F, Vernant JC, Gout O, Maurs L, Calender A, de Thé G. 1985. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet ii:407–410. [DOI] [PubMed] [Google Scholar]
- 10.Osame M, Usuku K, Izumo S, Ijichi N, Amitani H, Igata A, Matsumoto M, Tara M. 1986. HTLV-I associated myelopathy, a new clinical entity. Lancet i:1031–1032. [DOI] [PubMed] [Google Scholar]
- 11.Levin MC, Jacobson S. 1997. HTLV-I associated myelopathy/tropical spastic paraparesis (HAM/TSP): a chronic progressive neurologic disease associated with immunologically mediated damage to the central nervous system. J Neurovirol 3:126–140. doi: 10.3109/13550289709015802. [DOI] [PubMed] [Google Scholar]
- 12.Bhigjee AI, Hlela C. 2014. HTLV-1 infection and disease with special reference to the dermatological manifestations. OA Dermatology 2:1. [Google Scholar]
- 13.Mangano A, Costantini P, Altamirano N, De Stefano G, Aulicino P, Sen L. 2011. Persistent high HTLV-1 proviral load in a patient with complete remission of adult T-cell leukemia/lymphoma (ATL). Retrovirology 8:A44. doi: 10.1186/1742-4690-8-S1-A44. [DOI] [Google Scholar]
- 14.Akbarin MM, Rahimi H, Hassannia T, Shoja Razavi G, Sabet F, Shirdel A. 2013. Comparison of HTLV-I proviral load in adult T cell leukemia/lymphoma (ATL), HTLV-I-associated myelopathy (HAM-TSP) and healthy carriers. Iran J Basic Med Sci 16:208–212. [PMC free article] [PubMed] [Google Scholar]
- 15.Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM. 2002. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol 76:12813–12822. doi: 10.1128/jvi.76.24.12813-12822.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Georges SA, Kraus WL, Luger K, Nyborg JK, Laybourn PJ. 2002. p300-mediated Tax transactivation from recombinant chromatin: histone tail deletion mimics coactivator function. Mol Cell Biol 22:127–137. doi: 10.1128/mcb.22.1.127-137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Satou Y, Yasunaga J, Yoshida M, Matsuoka M. 2006. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci U S A 103:720–725. doi: 10.1073/pnas.0507631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Afonso PV, Mekaouche M, Mortreux F, Toulza F, Moriceau A, Wattel E, Gessain A, Bangham CR, Dubreuil G, Plumelle Y, Hermine O, Estaquier J, Mahieux R. 2010. Highly active antiretroviral treatment against STLV-1 infection combining reverse transcriptase and HDAC inhibitors. Blood 116:3802–3808. doi: 10.1182/blood-2010-02-270751. [DOI] [PubMed] [Google Scholar]
- 19.Lever AML, Jeang K-T, Berkhout B. 2010. Recent advances in human retroviruses: principles of replication and pathogenesis. Advances in retroviral research. World Scientific, Hackensack, NJ. [Google Scholar]
- 20.Barbeau B, Mesnard JM. 2011. Making sense out of antisense transcription in human T-cell lymphotropic viruses (HTLVs). Viruses 3:456–468. doi: 10.3390/v3050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwok RP, Laurance ME, Lundblad JR, Goldman PS, Shih H, Connor LM, Marriott SJ, Goodman RH. 1996. Control of cAMP-regulated enhancers by the viral transactivator Tax through CREB and the co-activator CBP. Nature 380:642–646. doi: 10.1038/380642a0. [DOI] [PubMed] [Google Scholar]
- 22.Yao J, Wigdahl B. 2000. Human T cell lymphotropic virus type I genomic expression and impact on intracellular signaling pathways during neurodegenerative disease and leukemia. Front Biosci 5:D138–D168. doi: 10.2741/Yao. [DOI] [PubMed] [Google Scholar]
- 23.Wessner R, Tillmann-Bogush M, Wigdahl B. 1995. Characterization of a glial cell-specific DNA-protein complex formed with the human T cell lymphotropic virus type I (HTLV-I) enhancer. J Neurovirol 1:62–77. doi: 10.3109/13550289509111011. [DOI] [PubMed] [Google Scholar]
- 24.Nyborg JK, Dynan WS, Chen IS, Wachsman W. 1988. Binding of host-cell factors to DNA sequences in the long terminal repeat of human T-cell leukemia virus type I: implications for viral gene expression. Proc Natl Acad Sci U S A 85:1457–1461. doi: 10.1073/pnas.85.5.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Orden K, Yan JP, Ulloa A, Nyborg JK. 1999. Binding of the human T-cell leukemia virus Tax protein to the coactivator CBP interferes with CBP-mediated transcriptional control. Oncogene 18:3766–3772. doi: 10.1038/sj.onc.1202703. [DOI] [PubMed] [Google Scholar]
- 26.Hansen JC, Wolffe AP. 1992. Influence of chromatin folding on transcription initiation and elongation by RNA polymerase III. Biochemistry 31:7977–7988. doi: 10.1021/bi00149a032. [DOI] [PubMed] [Google Scholar]
- 27.Lemasson I, Polakowski NJ, Laybourn PJ, Nyborg JK. 2002. Transcription factor binding and histone modifications on the integrated proviral promoter in human T-cell leukemia virus-I-infected T-cells. J Biol Chem 277:49459–49465. doi: 10.1074/jbc.M209566200. [DOI] [PubMed] [Google Scholar]
- 28.Kannagi M, Harada S, Maruyama I, Inoko H, Igarashi H, Kuwashima G, Sato S, Morita M, Kidokoro M, Sugimoto M, Funahashi S, Osame M, Shida H. 1991. Predominant recognition of human T cell leukemia virus type I (HTLV-I) pX gene products by human CD8+ cytotoxic T cells directed against HTLV-I-infected cells. Int Immunol 3:761–767. doi: 10.1093/intimm/3.8.761. [DOI] [PubMed] [Google Scholar]
- 29.Jacobson S, Shida H, McFarlin DE, Fauci AS, Koenig S. 1990. Circulating CD8+ cytotoxic T lymphocytes specific for HTLV-I pX in patients with HTLV-I associated neurological disease. Nature 348:245–248. doi: 10.1038/348245a0. [DOI] [PubMed] [Google Scholar]
- 30.Mendez E, Kawanishi T, Clemens K, Siomi H, Soldan SS, Calabresi P, Brady J, Jacobson S. 1997. Astrocyte-specific expression of human T-cell lymphotropic virus type 1 (HTLV-1) Tax: induction of tumor necrosis factor alpha and susceptibility to lysis by CD8+ HTLV-1-specific cytotoxic T cells. J Virol 71:9143–9149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnold J, Yamamoto B, Li M, Phipps AJ, Younis I, Lairmore MD, Green PL. 2006. Enhancement of infectivity and persistence in vivo by HBZ, a natural antisense coded protein of HTLV-1. Blood 107:3976–3982. doi: 10.1182/blood-2005-11-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemasson I, Lewis MR, Polakowski N, Hivin P, Cavanagh MH, Thebault S, Barbeau B, Nyborg JK, Mesnard JM. 2007. Human T-cell leukemia virus type 1 (HTLV-1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV-1 transcription. J Virol 81:1543–1553. doi: 10.1128/JVI.00480-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landry S, Halin M, Lefort S, Audet B, Vaquero C, Mesnard JM, Barbeau B. 2007. Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology 4:71. doi: 10.1186/1742-4690-4-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Landry S, Halin M, Vargas A, Lemasson I, Mesnard JM, Barbeau B. 2009. Upregulation of human T-cell leukemia virus type 1 antisense transcription by the viral Tax protein. J Virol 83:2048–2054. doi: 10.1128/JVI.01264-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M, Kesic M, Yin H, Yu L, Green PL. 2009. Kinetic analysis of human T-cell leukemia virus type 1 gene expression in cell culture and infected animals. J Virol 83:3788–3797. doi: 10.1128/JVI.02315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Easley R, Carpio L, Guendel I, Klase Z, Choi S, Kehn-Hall K, Brady JN, Kashanchi F. 2010. Human T-lymphotropic virus type 1 transcription and chromatin-remodeling complexes. J Virol 84:4755–4768. doi: 10.1128/JVI.00851-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohrmann L, Verrijzer CP. 2005. Composition and functional specificity of SWI2/SNF2 class chromatin remodeling complexes. Biochim Biophys Acta 1681:59–73. doi: 10.1016/j.bbaexp.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Côté J, Xue Y, Zhou S, Khavari PA, Biggar SR, Muchardt C, Kalpana GV, Goff SP, Yaniv M, Workman JL, Crabtree GR. 1996. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J 15:5370–5382. doi: 10.1002/j.1460-2075.1996.tb00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Nagl NG, Wilsker D, Van Scoy M, Pacchione S, Yaciuk P, Dallas PB, Moran E. 2004. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem J 383:319–325. doi: 10.1042/BJ20040524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A, Parsons R, Zhao K, Wang W. 2005. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev 19:1662–1667. doi: 10.1101/gad.1323805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia W, Nagase S, Montia AG, Kalachikov SM, Keniry M, Su T, Memeo L, Hibshoosh H, Parsons R. 2008. BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res 68:1667–1674. doi: 10.1158/0008-5472.CAN-07-5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, Campbell AE, Kawaoka S, Shareef S, Zhu Z, Kendall J, Muhar M, Haslinger C, Yu M, Roeder RG, Wigler MH, Blobel GA, Zuber J, Spector DL, Young RA, Vakoc CR. 2013. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev 27:2648–2662. doi: 10.1101/gad.232710.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buscarlet M, Krasteva V, Ho L, Simon C, Hebert J, Wilhelm B, Crabtree GR, Sauvageau G, Thibault P, Lessard JA. 2014. Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood 123:1720–1728. doi: 10.1182/blood-2013-02-483495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roy N, Malik S, Villanueva KE, Urano A, Lu X, Von Figura G, Seeley ES, Dawson DW, Collisson EA, Hebrok M. 2015. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev 29:658–671. doi: 10.1101/gad.256628.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bai J, Mei P, Zhang C, Chen F, Li C, Pan Z, Liu H, Zheng J. 2013. BRG1 is a prognostic marker and potential therapeutic target in human breast cancer. PLoS One 8:e59772. doi: 10.1371/journal.pone.0059772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Do SI, Yoon G, Kim HS, Kim K, Lee H, Do IG, Kim DH, Chae SW, Sohn JH. 2016. Increased Brahma-related gene 1 expression predicts distant metastasis and shorter survival in patients with invasive ductal carcinoma of the breast. Anticancer Res 36:4873–4882. doi: 10.21873/anticanres.11051. [DOI] [PubMed] [Google Scholar]
- 47.Lin H, Wong RP, Martinka M, Li G. 2010. BRG1 expression is increased in human cutaneous melanoma. Br J Dermatol 163:502–510. doi: 10.1111/j.1365-2133.2010.09851.x. [DOI] [PubMed] [Google Scholar]
- 48.Wu K, Bottazzi ME, de la Fuente C, Deng L, Gitlin SD, Maddukuri A, Dadgar S, Li H, Vertes A, Pumfery A, Kashanchi F. 2004. Protein profile of tax-associated complexes. J Biol Chem 279:495–508. doi: 10.1074/jbc.M310069200. [DOI] [PubMed] [Google Scholar]
- 49.Phelan ML, Sif S, Narlikar GJ, Kingston RE. 1999. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell 3:247–253. doi: 10.1016/S1097-2765(00)80315-9. [DOI] [PubMed] [Google Scholar]
- 50.Melamed A, Laydon DJ, Gillet NA, Tanaka Y, Taylor GP, Bangham CR. 2013. Genome-wide determinants of proviral targeting, clonal abundance and expression in natural HTLV-1 infection. PLoS Pathog 9:e1003271. doi: 10.1371/journal.ppat.1003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manelyte L. 2017. Chromatin remodelers, their implication in cancer and therapeutic potential. J Rare Dis Res Treat 2:34–40. doi: 10.29245/2572-9411/2017/3.1108. [DOI] [Google Scholar]
- 52.Wu Q, Lian JB, Stein JL, Stein GS, Nickerson JA, Imbalzano AN. 2017. The BRG1 ATPase of human SWI/SNF chromatin remodeling enzymes as a driver of cancer. Epigenomics 9:919–931. doi: 10.2217/epi-2017-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sentani K, Oue N, Kondo H, Kuraoka K, Motoshita J, Ito R, Yokozaki H, Yasui W. 2001. Increased expression but not genetic alteration of BRG1, a component of the SWI/SNF complex, is associated with the advanced stage of human gastric carcinomas. Pathobiology 69:315–320. doi: 10.1159/000064638. [DOI] [PubMed] [Google Scholar]
- 54.Guerrero-Martínez JA, Reyes JC. 2018. High expression of SMARCA4 or SMARCA2 is frequently associated with an opposite prognosis in cancer. Sci Rep 8:2043. doi: 10.1038/s41598-018-20217-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hivin P, Frédéric M, Arpin-André C, Basbous J, Gay B, Thébault S, Mesnard J-M. 2005. Nuclear localization of HTLV-I bZIP factor (HBZ) is mediated by three distinct motifs. J Cell Sci 118:1355–1362. doi: 10.1242/jcs.01727. [DOI] [PubMed] [Google Scholar]
- 56.Ma G, Yasunaga J, Matsuoka M. 2016. Multifaceted functions and roles of HBZ in HTLV-1 pathogenesis. Retrovirology 13:16. doi: 10.1186/s12977-016-0249-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trotter KW, Archer TK. 2008. The BRG1 transcriptional coregulator. Nucl Recept Signal 6:e004. doi: 10.1621/nrs.06004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raval GU, Bidoia C, Forlani G, Tosi G, Gessain A, Accolla RS. 2015. Localization, quantification and interaction with host factors of endogenous HTLV-1 HBZ protein in infected cells and ATL. Retrovirology 12:59. doi: 10.1186/s12977-015-0186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hivin P, Basbous J, Raymond F, Henaff D, Arpin-Andre C, Robert-Hebmann V, Barbeau B, Mesnard JM. 2007. The HBZ-SP1 isoform of human T-cell leukemia virus type I represses JunB activity by sequestration into nuclear bodies. Retrovirology 4:14. doi: 10.1186/1742-4690-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vargova J, Vargova K, Skoultchi AI, Stopka T. 2009. Nuclear localization of ISWI ATPase Smarca5 (Snf2h) in mouse. Front Biosci (Elite Ed) 1:553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reyes JC, Muchardt C, Yaniv M. 1997. Components of the human SWI/SNF complex are enriched in active chromatin and are associated with the nuclear matrix. J Cell Biol 137:263–274. doi: 10.1083/jcb.137.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clerc I, Polakowski N, Andre-Arpin C, Cook P, Barbeau B, Mesnard JM, Lemasson I. 2008. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of tax-dependent viral transcription by HBZ. J Biol Chem 283:23903–23913. doi: 10.1074/jbc.M803116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Easley R, Carpio L, Dannenberg L, Choi S, Alani D, Van Duyne R, Guendel I, Klase Z, Agbottah E, Kehn-Hall K, Kashanchi F. 2010. Transcription through the HIV-1 nucleosomes: effects of the PBAF complex in Tat activated transcription. Virology 405:322–333. doi: 10.1016/j.virol.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lemasson I, Polakowski NJ, Laybourn PJ, Nyborg JK. 2006. Tax-dependent displacement of nucleosomes during transcriptional activation of human T-cell leukemia virus type 1. J Biol Chem 281:13075–13082. doi: 10.1074/jbc.M512193200. [DOI] [PubMed] [Google Scholar]
- 65.Luo RX, Dean DC. 1999. Chromatin remodeling and transcriptional regulation. J Natl Cancer Inst 91:1288–1294. doi: 10.1093/jnci/91.15.1288. [DOI] [PubMed] [Google Scholar]
- 66.Peterson CL, Workman JL. 2000. Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr Opin Genet Dev 10:187–192. doi: 10.1016/S0959-437X(00)00068-X. [DOI] [PubMed] [Google Scholar]
- 67.Sudarsanam P, Winston F. 2000. The Swi/Snf family nucleosome-remodeling complexes and transcriptional control. Trends Genet 16:345–351. doi: 10.1016/S0168-9525(00)02060-6. [DOI] [PubMed] [Google Scholar]
- 68.Lin S, Jiang T, Ye L, Han Z, Liu Y, Liu C, Yuan C, Zhao S, Chen J, Wang J, Tang H, Lu S, Yang L, Wang X, Yan D, Peng Z, Fan J. 2016. The chromatin-remodeling enzyme BRG1 promotes colon cancer progression via positive regulation of WNT3A. Oncotarget 7:86051–86063. doi: 10.18632/oncotarget.13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marquez-Vilendrer SB, Rai SK, Gramling SJ, Lu L, Reisman DN. 2016. Loss of the SWI/SNF ATPase subunits BRM and BRG1 drives lung cancer development. Oncoscience 3:322–336. doi: 10.18632/oncoscience.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reisman D, Glaros S, Thompson EA. 2009. The SWI/SNF complex and cancer. Oncogene 28:1653–1668. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 71.Kaufmann B, Wang B, Zhong S, Laschinger M, Patil P, Lu M, Assfalg V, Cheng Z, Friess H, Huser N, von Figura G, Hartmann D. 2017. BRG1 promotes hepatocarcinogenesis by regulating proliferation and invasiveness. PLoS One 12:e0180225. doi: 10.1371/journal.pone.0180225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hendricks KB, Shanahan F, Lees E. 2004. Role for BRG1 in cell cycle control and tumor suppression. Mol Cell Biol 24:362–376. doi: 10.1128/mcb.24.1.362-376.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kang H, Cui K, Zhao K. 2004. BRG1 controls the activity of the retinoblastoma protein via regulation of p21CIP1/WAF1/SDI. Mol Cell Biol 24:1188–1199. doi: 10.1128/mcb.24.3.1188-1199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xi Q, He W, Zhang XH, Le HV, Massague J. 2008. Genome-wide impact of the BRG1 SWI/SNF chromatin remodeler on the transforming growth factor beta transcriptional program. J Biol Chem 283:1146–1155. doi: 10.1074/jbc.M707479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu Q, Madany P, Dobson JR, Schnabl JM, Sharma S, Smith TC, van Wijnen AJ, Stein JL, Lian JB, Stein GS, Muthuswami R, Imbalzano AN, Nickerson JA. 2016. The BRG1 chromatin remodeling enzyme links cancer cell metabolism and proliferation. Oncotarget 7:38270–38281. doi: 10.18632/oncotarget.9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao T. 2016. The role of HBZ in HTLV-1-induced oncogenesis. Viruses 8:E34. doi: 10.3390/v8020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iyer NG, Ozdag H, Caldas C. 2004. p300/CBP and cancer. Oncogene 23:4225–4231. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- 78.Naidu SR, Love IM, Imbalzano AN, Grossman SR, Androphy EJ. 2009. The SWI/SNF chromatin remodeling subunit BRG1 is a critical regulator of p53 necessary for proliferation of malignant cells. Oncogene 28:2492–2501. doi: 10.1038/onc.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.He W, Dorn DC, Erdjument-Bromage H, Tempst P, Moore MAS, Massagué J. 2006. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell 125:929–941. doi: 10.1016/j.cell.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 80.Zhao T, Satou Y, Sugata K, Miyazato P, Green PL, Imamura T, Matsuoka M. 2011. HTLV-1 bZIP factor enhances TGF-beta signaling through p300 coactivator. Blood 118:1865–1876. doi: 10.1182/blood-2010-12-326199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ward-Hartstonge KA, Kemp RA. 2017. Regulatory T-cell heterogeneity and the cancer immune response. Clin Transl Immunol 6:e154. doi: 10.1038/cti.2017.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boultwood J, Fidler C, Lewis S, MacCarthy A, Sheridan H, Kelly S, Oscier D, Buckle VJ, Wainscoat JS. 1993. Allelic loss of IRF1 in myelodysplasia and acute myeloid leukemia: retention of IRF1 on the 5q- chromosome in some patients with the 5q- syndrome. Blood 82:2611–2616. [PubMed] [Google Scholar]
- 83.Willman CL, Sever CE, Pallavicini MG, Harada H, Tanaka N, Slovak ML, Yamamoto H, Harada K, Meeker TC, List AF, et al. . 1993. Deletion of IRF-1, mapping to chromosome 5q31.1, in human leukemia and preleukemic myelodysplasia. Science 259:968–971. doi: 10.1126/science.8438156. [DOI] [PubMed] [Google Scholar]
- 84.Mukai R, Ohshima T. 2011. Dual effects of HTLV-1 bZIP factor in suppression of interferon regulatory factor 1. Biochem Biophys Res Commun 409:328–332. doi: 10.1016/j.bbrc.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 85.Wurster AL, Precht P, Becker KG, Wood WH III, Zhang Y, Wang Z, Pazin MJ. 2012. IL-10 transcription is negatively regulated by BAF180, a component of the SWI/SNF chromatin remodeling enzyme. BMC Immunol 13:9. doi: 10.1186/1471-2172-13-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sawada L, Nagano Y, Hasegawa A, Kanai H, Nogami K, Ito S, Sato T, Yamano Y, Tanaka Y, Masuda T, Kannagi M. 2017. IL-10-mediated signals act as a switch for lymphoproliferation in human T-cell leukemia virus type-1 infection by activating the STAT3 and IRF4 pathways. PLoS Pathog 13:e1006597. doi: 10.1371/journal.ppat.1006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sohn DH, Lee KY, Lee C, Oh J, Chung H, Jeon SH, Seong RH. 2007. SRG3 interacts directly with the major components of the SWI/SNF chromatin remodeling complex and protects them from proteasomal degradation. J Biol Chem 282:10614–10624. doi: 10.1074/jbc.M610563200. [DOI] [PubMed] [Google Scholar]
- 88.Van Duyne R, Guendel I, Klase Z, Narayanan A, Coley W, Jaworski E, Roman J, Popratiloff A, Mahieux R, Kehn-Hall K, Kashanchi F. 2012. Localization and sub-cellular shuttling of HTLV-1 tax with the miRNA machinery. PLoS One 7:e40662. doi: 10.1371/journal.pone.0040662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang L, Liu M, Merling R, Giam CZ. 2006. Versatile reporter systems show that transactivation by human T-cell leukemia virus type 1 Tax occurs independently of chromatin remodeling factor BRG1. J Virol 80:7459–7468. doi: 10.1128/JVI.00130-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cullen SJ, Ponnappan S, Ponnappan U. 2009. Catalytic activity of the proteasome fine-tunes Brg1-mediated chromatin remodeling to regulate the expression of inflammatory genes. Mol Immunol 47:600–605. doi: 10.1016/j.molimm.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gross C, Thoma-Kress AK. 2017. Reporter systems to study HTLV-1 transmission. Methods Mol Biol 1582:33–46. doi: 10.1007/978-1-4939-6872-5_3. [DOI] [PubMed] [Google Scholar]
- 92.Murphy JL, Hall WW, Sheehy N. 2014. Investigation of the role of nuclear factors associated with double-stranded RNA protein members in the pathogenesis of HTLV-2. Retrovirology 11:P113. doi: 10.1186/1742-4690-11-S1-P113. [DOI] [Google Scholar]
- 93.Seo S, Herr A, Lim JW, Richardson GA, Richardson H, Kroll KL. 2005. Geminin regulates neuronal differentiation by antagonizing Brg1 activity. Genes Dev 19:1723–1734. doi: 10.1101/gad.1319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McCabe A, Hashimoto K, Hall WW, Sheehy N. 2013. The four and a half LIM family members are novel interactants of the human T-cell leukemia virus type 1 Tax oncoprotein. J Virol 87:7435–7444. doi: 10.1128/JVI.00070-13. [DOI] [PMC free article] [PubMed] [Google Scholar]