

Recently, we evaluated the effects of HVEM and its ligands (LIGHT, CD160, and BTLA) on HSV-1 infectivity. However, the effect of LTα, another member of the TNF superfamily, on HSV-1 latency and eye disease is not known. Here, we demonstrate increased latency and corneal scarring in LTα−/− infected mice, independent of the presence of LAT. In addition, infected mice were highly susceptible to HSV-1 infection, and survival was partially but not significantly restored by adoptive T cell transfer. These results suggest that the absence of LTα affects HSV-1 infectivity differently than the absence of HVEM, LIGHT, CD160, and BTLA.
KEYWORDS: ocular, virus replication, corneal scarring, latency, survival
ABSTRACT
Previously, we reported that the absence of herpesvirus entry mediator (HVEM) decreases latency but not primary infection in ocularly infected mice. Recently, we reported that similar to the absence of HVEM, the absence of HVEM ligands (i.e., LIGHT, CD160, and B and T lymphocyte attenuator [BTLA]) also decreased latency but not primary infection. Similar to LIGHT, CD160, and BTLA, another member of tumor necrosis factor (TNF) superfamily, lymphotoxin-α (LTα), also interacts with HVEM. To determine whether LTα decreases latency in infected mice, we ocularly infected LTα−/− mice with latency-associated transcript-positive [LAT(+)] and LAT(−) viruses using similarly infected wild-type (WT) mice as controls. In contrast to WT C57BL/6 mice, LTα−/− mice were highly susceptible to ocular herpes simplex virus 1 (HSV-1) infection, independent of the presence or absence of LAT. Survival was partially restored by adoptive transfer of CD4+, CD8+, or total T cells. Infected LTα−/− mice had significantly higher corneal scarring than WT mice, and adoptive T cell transfer did not alter the severity of eye disease. In contrast to results in WT mice, the amount of latency was not affected by the absence of LAT. The amount of LAT RNA in LTα−/− mice infected with LAT(+) virus was similar to that in WT mice, and adoptive T cell transfer did not alter LAT RNA levels in LTα−/− infected mice. Increased latency in the absence of LTα correlated with upregulation of HVEM, LIGHT, CD160, and BTLA transcripts as well as with an increase in markers of T cell exhaustion. The results of our study suggest that LTα has antipathogenic and anti-inflammatory functions and may act to protect the host from infection.
IMPORTANCE Recently, we evaluated the effects of HVEM and its ligands (LIGHT, CD160, and BTLA) on HSV-1 infectivity. However, the effect of LTα, another member of the TNF superfamily, on HSV-1 latency and eye disease is not known. Here, we demonstrate increased latency and corneal scarring in LTα−/− infected mice, independent of the presence of LAT. In addition, infected mice were highly susceptible to HSV-1 infection, and survival was partially but not significantly restored by adoptive T cell transfer. These results suggest that the absence of LTα affects HSV-1 infectivity differently than the absence of HVEM, LIGHT, CD160, and BTLA.
INTRODUCTION
Herpes simplex virus-1 (HSV-1) has at least seven known cellular receptors that facilitate its ability to infect most cell types (1–11). Among these receptors, the herpesvirus entry mediator (HVEM) was the first to be identified and is the receptor for the virion envelope glycoprotein D (gD) of HSV-1 (6). Previously, we reported that the latency-associated transcript (LAT) upregulates HVEM in vivo and in vitro but not the other six HSV-1 cellular receptors (12). We have shown that HSV-1 latency/reactivation but not primary infection was significantly reduced in HVEM−/− mice. In addition to gD, B and T lymphocyte attenuator (BTLA), LIGHT (TNFSF14), CD160, lymphotoxin-α (LTα), and synaptic adhesion-like molecule 5 (SALM5) also bind to HVEM (13–18). We recently reported that similar to HVEM−/− mice (12), mice lacking LIGHT, BTLA, or CD160 showed less latency reactivation than wild-type (WT) mice infected with LAT-negative [LAT(−)] virus (19). Our published results show that the inability of BTLA, LIGHT, and CD160 to compete with gD for binding to HVEM reduced latency reactivation but not primary infection.
Similar to LIGHT and CD160, LTα is derived from the tumor necrosis factor (TNF) lineage (15, 16). These molecules bind to distinct sites on HVEM (18), providing costimulatory signals (20). LTα, which exists as a soluble homotrimer and in membrane-bound heterotrimeric complexes with LTβ (21), plays an important role in secondary lymphoid organ formation during development (22). LTα is produced by a variety of lymphocytes, especially activated CD4 and CD8 T cells (23). Mice deficient in LTα lack Peyer patches, all lymph nodes, follicular dendritic cells (FDC), and germinal centers (24–26).
HSV-1 gD competes with the cellular ligands for binding to HVEM, consistent with our recent finding that T cell activation and latency reactivation are reduced via the LIGHT-BTLA-CD160-HVEM pathway and that latency reactivation is reduced in the presence of LAT (19). Although LTα is structurally related to LIGHT and although both bind to HVEM (18, 20), whether LTα and LIGHT have similar effects on HSV-1 latency is not known. To determine whether LTα and LIGHT have similar functions, we compared ocular infection of LTα −/− mice with LAT(+) and LAT(−) viruses. We found the following: (i) LTα−/− mice are highly susceptible to HSV-1 infection, and adoptive transfer of CD4+, CD8+, or total T cells did not completely restore protection; (ii) absence of LTα enhanced corneal scarring (CS), and T cell transfer did not change the level of corneal scarring; (iii) LTα−/− mice had higher levels of latency, which was independent of LAT expression; (iv) expression levels of BTLA, CD160, HVEM, and LIGHT were upregulated in trigeminal ganglia (TG) of infected LTα−/− mice, and these higher levels were further increased in the presence of LAT; and (v) increased latency in LTα−/− mice was associated with significantly higher expression of exhaustion markers. Thus, although our previous studies showed a link between HVEM, its ligands, and LAT (12, 19), here we show that the LTα response to HSV-1 infection differs from that of its HVEM binding counterparts.
RESULTS
Virus replication in the eyes of LTα−/− infected mice.
LTα binds to HVEM and may compete with HSV-1 gD for binding to HVEM as we reported recently for other HVEM ligands (i.e., LIGHT, BTLA, and CD160) (19). Therefore, we used LTα−/− mice to assess the effect of LTα binding to HVEM on primary virus replication in the eye of ocularly infected mice in the presence and absence of LAT. WT mice were used as controls. LTα−/− and WT mice were ocularly infected with 2 × 105 PFU/eye of LAT(+) or LAT(−) virus as described in Materials and Methods. To determine if LTα affects HSV-1 replication during primary ocular infection, we collected tear films from 20 eyes/group daily on days 1 to 5 postinfection (p.i.). The amount of virus in each eye was determined using a standard plaque assay. We found previously that except for LIGHT−/− infected mice, replication of HSV-1 in the eyes of ocularly infected HVEM−/−, BTLA−/−, and CD160−/− mice was not affected by the presence or absence of LAT (12, 19). To determine if LAT affects ocular virus replication in the absence of LTα, we compared viral titers following primary infection with LAT(+) or LAT(−) virus. The viral titers after LAT(+) or LAT(−) virus infection did not differ significantly between WT and LTα−/− mice (Fig. 1) (P > 0.05) on days 1, 3, and 5. However, on days 2 and 4 p.i., WT mice infected with LAT(−) virus had higher virus titers than the other three groups (Fig. 1) (P < 0.05), but by day 5 p.i., virus titers in all groups declined and were similar (Fig. 1) (P > 0.05). Higher virus replication in the eye of LAT(−) virus-infected mice on days 2 and 4 could be due to the effect of LAT on expression of type I interferon (IFN) genes in the eyes of infected mice. Recently, we reported that LAT downregulates components of the type I IFN pathway in ocularly infected mice (27). As we reported previously (12, 19), we did not detect any difference in virus titers between LTα−/− mice infected with LAT(+) and LAT(−) virus (Fig. 1) (P > 0.05). These results confirm that, similar to HVEM, BTLA, and CD160 and in contrast to the HEVM binding partner LIGHT, LTα is not affected by the absence of LAT.
FIG 1.

Virus titers in eyes following ocular infection of mice. WT and LTα−/− mice were infected ocularly with LAT(+) or LAT(−) virus, and the amount of infectious HSV-1 in tear films was determined daily by standard plaque assay as described in Materials and Methods. At each time point, the virus titer represents the average of the titers from 20 eyes ± standard error of the mean. Viral titers did not differ significantly between WT and LTα−/− mice after LAT(+) or LAT(−) virus infection on days 1, 3, and 5, while on days 2 and 4 p.i., WT mice infected with LAT(−) virus had significantly higher virus titers than the other three groups.
Survival of LTα−/− infected mice.
C57BL/6 mice are refractory to ocular infection with 2 × 105 PFU/eye of HSV-1 strain McKrae, LAT(+) virus, in the absence of corneal scarification. Mice on a C57BL/6 genetic background with knockout of HVEM (HVEM−/−) and its ligands (i.e., BTLA, LIGHT, and CD160) also are refractory to HSV-1 infection (12, 19). Thus, since LTα is an HVEM family member, survival of LTα−/− mice was monitored following ocular infection in both eyes with 2 × 105 PFU/eye of LAT(+) or LAT(−) virus as described above, and survival data from four separate experiments were combined. Nine of 31 (29% survival) LTα−/− mice infected with LAT(+) virus survived ocular infection, and 13 of 32 (41% survival) survived ocular infection with LAT(−) virus; these differences were not statistically significant (Fig. 2) (P > 0.05; Fisher exact test). In contrast, and as expected, 53 of 55 (96% survival) WT mice infected with LAT(+) virus and 53 of 56 (95% survival) WT mice infected with LAT(−) virus survived ocular infection (Fig. 2). Survival differences between LTα−/− mice infected with LAT(+) or LAT(−) virus and WT mice infected with either virus were highly significant (Fig. 2) (P < 0.0001; Fisher exact test). These results suggest that LTα−/− mice are susceptible to HSV-1 infection and that the absence of LAT slightly, but not statistically significantly, enhances neurovirulence in infected mice.
FIG 2.

Survival of ocularly infected mice. WT and LTα−/− mice were ocularly infected with LAT(+) or LAT(−) virus as described in the legend of Fig. 1. An additional group of LTα−/− mice received CD4+, CD8+, or total T cells, as described in Materials and Methods, 2 weeks before infection with LAT(+) virus. Survival of infected mice was determined 28 days p.i. Fifty-three of 55 WT mice infected with LAT(+) virus survived infection, 53 of 56 WT mice infected with LAT(−) virus survived infection, 9 of 31 LTα−/− mice infected with LAT(+) virus survived infection, 13 of 32 LTα−/− mice infected with LAT(−) virus survived infection, 14 of 22 LTα−/− mice that received CD4+ T cells and that were infected with LAT(+) virus survived infection, 11 of 22 LTα−/− mice that received CD8+ T cells and that were infected with LAT(+) virus survived infection, and 6 of 10 LTα−/− mice that received total T cells and that were infected with LAT(+) virus survived infection. The difference in survival rates between LTα−/− mice infected with the LAT(+) and LAT(−) viruses was not statistically significant. Similarly, the difference in survival rates between WT mice infected with the LAT(+) and LAT(−) viruses was not statistically significant. In contrast, survival rate differences between LTα−/− mice infected with LAT(+) or LAT(−) virus and WT mice infected with either virus were highly significant. Finally, transfer of CD4+ T cells, CD8+ T cells, or total T cells partially restored protection compared to levels of either infected WT mice or to infected LTα−/− mice without adoptive transfer, but the differences were not statistically significant.
These results further suggest that in contrast to WT mice and our previous studies with HVEM−/−, BTLA−/−, LIGHT−/−, and CD160−/− mice (12, 19), LTα−/− mice are highly susceptible to HSV-1 infection. Thus, we next asked whether transfer of either CD4+ or CD8+ T cells alone could restore protection to ocularly infected recipient LTα−/− mice, as described in Materials and Methods. Recipient LTα−/− mice were ocularly infected as described above with 2 × 105 PFU/eye of LAT(+) virus, and survival was monitored for 28 days. Fourteen of 22 (64%) of LTα−/− mice that received CD4+ T cells survived ocular infection while 11 of 22 (50%) of LTα−/− mice that received CD8+ T cells survived ocular infection, but differences between the two groups were not statistically significant (Fig. 2) (for mice receiving CD4 versus those receiving CD8, P > 0.05). Although transfer of CD4+ or CD8+ T cells alone partially restored protection compared to that of either infected WT mice (Fig. 2) (P > 0.05) or infected LTα−/− mice without adoptive transfer (Fig. 2) (P > 0.05), these differences did not reach statistical significance.
Transfer of CD4+ or CD8+ T cells alone as described above did not completely restore protection to recipient LTα−/− mice. Thus, we next tested the ability of total T cells to restore protection to recipient LTα−/− mice. Total T cells were transferred to recipient LTα−/− mice, and at 2 weeks posttransfer, recipient mice were infected with LAT(+) virus as described above. Six of 10 (60%) LTα−/− recipient mice survived ocular infection (Fig. 2, +T cells). Survival in mice that received total T cells was similar to that of mice that received CD4+ T cells, and survival rates for both were higher than the rate for mice that received CD8+ T cells. However, survival differences between recipient mice with each other and with no transfer LTα−/− mice were not statistically significant (Fig. 2) (P > 0.05). Overall, our results suggest that LTα−/− mice are highly susceptible to HSV-1 infection and that T cell transfer partially restores protection against ocular infection.
Effects of LTα on corneal scarring in ocularly infected mice.
During primary infection we found more blepharitis and dermatitis in LTα−/− infected mice than in WT mice, and adoptive transfer of CD4+, CD8+, or total T cells to LTα−/− mice did not enhance blepharitis and dermatitis compared to levels in their naive but infected counterparts (data not shown). The severity of corneal scarring in surviving mice on day 28 p.i. was scored in a masked fashion using a scale of 0 to 4, as described in Materials and Methods. As shown in Fig. 3, corneal scarring in WT mice infected with LAT(+) virus was significantly less than that in LTα−/− infected mice (Fig. 3, LTα−/−) (P < 0.01). Similarly, transfer of CD4+, CD8+, or total T cells to LTα−/− mice did not change corneal scarring compared with that in LTα−/− mice that did not receive any T cells (Fig. 3) (P > 0.05). Thus, the absence of LTα has a major effect on HSV-1-induced corneal scarring, and transfer of CD4+, CD8+, or total T cells did not change the severity of corneal scarring compared with that in WT or LTα mice that did not receive any T cells.
FIG 3.

Corneal scarring (CS) in surviving mice infected with LAT(+) virus. From 10 to 55 WT and LTα−/− mice, as described in the legend of Fig. 2, were ocularly infected with LAT(+) virus. Some LTα−/− mice received CD4+, CD8+, or total T cells before infection with LAT(+) virus. CS in all infected mice that survived ocular infection was assessed on day 28 p.i. on a scale of 0 to 4. CS is based on mean ± standard error of the mean of 64 eyes for WT mice, 18 eyes for LTα−/− mice, 28 eyes for LTα−/− mice that received CD4+ T cells, 22 eyes for LTα−/− mice that received CD8+ T cells, and 12 eyes for LTα−/− mice that received total T cells.
Viral latency in LTα−/− mice infected with LAT(+) and LAT(−) viruses.
Previously we reported that the absence of BTLA, CD160, HVEM, and LIGHT is associated with a significant reduction in the amount of latency in the TG of infected mice, independent of the viral LAT status (12, 19). Because LTα is an HVEM ligand, we investigated the potential effect of LTα on latency. TG from surviving WT and LTα−/− mice infected with LAT(+) and LAT(−) viruses as described in the legend of Fig. 2 were harvested on day 28 p.i., and levels of latency were determined based on expression of HSV-1 gB by quantitative PCR (qPCR), as described in Materials and Methods. Similar to published studies (12, 28), we found significantly more HSV-1 gB DNA in the TG from WT mice infected with LAT(+) than with LAT(−) virus (Fig. 4; WT, P < 0.0001). However, in contrast to our results with BTLA, CD160, HVEM, and LIGHT (12, 19), we found similar levels of latent genomes in the TG of LTα−/− mice infected with LAT(+) or LAT(−) virus (Fig. 4; LTα−/−, P > 0.05). Indeed, the amount of latency in LTα−/− mice infected with LAT(+) or LAT(−) virus was similar to that in LAT(+) virus-infected WT mice (Fig. 4) (WT verus LTα−/−, P > 0.05) but significantly higher than that in WT mice infected with LAT(−) virus (Fig. 4) (P < 0.01). Thus, LTα appears to play a role in controlling the amount of latency in the TG of infected mice, independent of the presence or absence of LAT.
FIG 4.

Effects of LTα on the level of gB in TG of latently infected mice. WT and LTα−/− mice were ocularly infected with LAT(+) or LAT(−) virus as described in the legend of Fig. 1. On day 28 p.i., TG were harvested from the surviving latently infected mice. qPCR was performed on each individual mouse TG. In each experiment, the estimated relative copy number of gB was calculated from standard curves generated using pAC-gB1 (79). Briefly, the DNA template first was serially diluted 10-fold so that 5 μl contained from 103 to 1011 copies of gB and then subjected to TaqMan PCR with the same set of primers. The copy number for each reaction product was determined by comparing the normalized threshold cycle of each sample to the threshold cycle of the standard. GAPDH expression was used to normalize the relative expression of gB DNA in the TG. Each point represents the mean ± standard error of the mean of 20 TG from WT mice infected with LAT(+) or LAT(−) virus, 18 TG from LTα−/− mice infected with LAT(+) virus, and 20 TG from LTα−/− mice infected with LAT(−) virus.
LAT expression in TG of LTα−/− mice infected with LAT(+) virus with and without T cell transfer.
The above results suggest that gB expression levels are similar in TG of LTα−/− mice infected with LAT(+) or LAT(−) virus and in WT mice infected with LAT(+) virus; however, in WT mice gB expression was dependent on LAT (Fig. 5). Because LAT RNA is abundant during latency (29–31), we analyzed the TG obtained 28 days after ocular infection with LAT(+) virus for LAT RNA expression. Some LTα−/− infected mice also received adoptive transfer of CD4+, CD8+, or both T cell types, as described in Materials and Methods. Total RNA from the TG of surviving mice was isolated and analyzed by quantitative reverse transcription-PCR (qRT-PCR) using LAT primers, as described in Materials and Methods. LAT RNA levels were similar in LTα−/− mice latently infected with LAT(+) virus with no T cell transfer to those in WT mice similarly infected with LAT(+) virus (Fig. 5, WT versus LTα−/− no transfer; P > 0.05, Fisher’s exact test). LAT RNA levels were also similar in WT mice and LTα−/− mice latently infected with LAT(+) virus with or without transfer of CD4+, CD8+, or both types of T cells (Fig. 5) (P > 0.05). This is consistent with the above data showing similar levels of latency, as judged by the number of viral genomes (Fig. 4), and with reports that LAT levels during latency are related to the amount of viral genome (28, 32, 33). However, our results with LTα−/− infected mice are in contrast to levels of latency in HVEM−/−, BTLA−/−, LIGHT−/−, and CD160−/− mice that all had lower levels of LAT RNA than WT mice latently infected with LAT(+) virus (12, 19). Thus, in contrast to our finding of decreased latency in the absence of HVEM, BTLA, LIGHT, CD160, or LAT (12, 19), the absence of LTα increases latency, influencing the amount of latency that is established and/or maintained.
FIG 5.
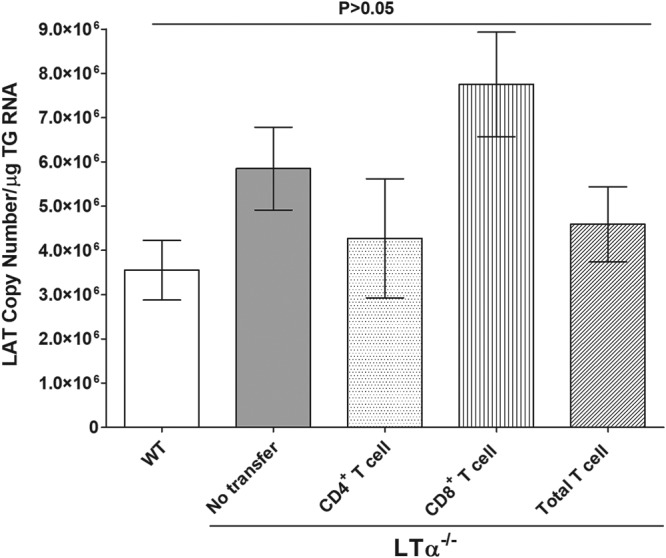
Effects of LTα on levels of LAT in TG of latently infected mice with and without T cell transfer. LTα−/− and WT mice were ocularly infected with LAT(+) virus as described in the legend of Fig. 1. Some LTα−/− mice received CD4+, CD8+, or total T cells, as described in Materials and Methods. On day 28 p.i., TG were harvested from surviving latently infected mice. qRT-PCR was performed on each mouse TG. In each experiment, the estimated relative copy number of LAT was calculated using standard curves generated from pGem-LAT5317. Briefly, the DNA template was serially diluted as described in the legend of Fig. 4, and the copy number for each reaction product was determined. GAPDH expression was used to normalize relative expression of LAT RNA in the TG. Each point represents the mean ± standard error of the mean of 20 TG from WT mice, 20 TG f from LTα−/− mice that received CD4+ T cells, 20 TG from LTα−/− mice that received CD8+ T cells, and 12 TG from LTα−/− mice that received total T cells.
Effect of LTα absence on expression of the other members of the TNF superfamily in TG of uninfected mice.
To determine whether the absence of LTα affects expression of BTLA, CD160, HVEM, and LIGHT molecules, TG from uninfected naive LTα−/− and WT mice were isolated. BTLA, CD160, HVEM, and LIGHT mRNA expression levels were determined using qRT-PCR analysis. The results are presented as fold change compared to baseline mRNA levels in the TG from uninfected WT mice (Fig. 6). There were no significant differences in BTLA, CD160, HVEM, and LIGHT mRNA expression levels in LTα−/− mice (Fig. 6). Although BTLA mRNA expression was highest while HVEM was lowest in LTα−/− mice, these differences were not significant compared with levels in WT mice (Fig. 6) (P > 0.05). Overall, these results indicate that the absence of LTα expression did not affect mRNA expression of BTLA, CD160, HVEM, and LIGHT in naive mice.
FIG 6.

Expression of HVEM, LIGHT, BTLA and CD160 in TG of uninfected LTα−/− mice. TG from WT and LTα−/− mice were harvested, RNA from each TG was isolated, and qRT-PCR was performed on each individual mouse TG. Levels of HVEM, LIGHT, BTLA, and CD160 transcript expression in TG of LTα−/− mice were normalized to the level of each transcript in the WT mouse TG and are shown as fold increase or decrease compared to the levels in the WT mice. GAPDH expression was used to normalize relative expression of each transcript in the TG of infected mice. Each point represents the mean ± standard error of the mean of 10 TG.
Effects of LAT and LTα on BTLA, LIGHT, CD160, and HVEM expression in TG of latently infected WT and LTα−/− mice.
We have shown that upregulation of HVEM but not BTLA, CD160, or LIGHT is LAT dependent (12, 19). Thus, to determine if the absence of LTα and LAT affects BTLA, CD160, HVEM, or LIGHT expression, the TG from LAT(+) or LAT(−) virus-infected LTα−/− and WT mice were harvested at day 28 p.i. as described in the legend of Fig. 1. Total RNA was extracted, and qRT-PCR was used to quantitate BTLA, CD160, HVEM, and LIGHT mRNA expression levels. The results are presented as fold change compared to baseline mRNA levels in TG from uninfected naive mice of each strain (Fig. 7). In WT mice, levels of BTLA, CD160, and LIGHT were the same in LAT(+) and LAT(−) virus-infected WT mice (Fig. 7, WT; P > 0.05), while HVEM levels were affected by the presence of LAT (Fig. 7, WT HVEM; P < 0.01) as we reported previously (12, 19). In contrast, levels of BTLA, CD160, HVEM, and LIGHT were significantly higher in LTα−/− mice infected with either LAT(+) or LAT(−) virus (Fig. 7; P < 0.01). In addition, levels of BTLA, CD160, HVEM, and LIGHT were significantly higher in the TG of mice infected with LAT(+) virus than with LAT(−) virus (Fig. 7; P < 0.01). Overall, the absence of LTα significantly affected expression of BTLA, CD160, HVEM, and LIGHT in infected mice, and this upregulation was further affected by the presence of LAT. Thus, LTα is required for control of BTLA, CD160, HVEM, and LIGHT expression in infected mice.
FIG 7.

Effect of LAT and LTα on HVEM, BTLA, LIGHT, and CD160 expression in TG of latently infected mice. WT and LTα−/− mice were ocularly infected with LAT(+) or LAT(−) virus. TG were isolated individually on day 28 p.i., and qRT-PCR was performed using total RNA. The BTLA, LIGHT, HVEM, and CD160 expression levels in naive mice for each strain were used to estimate the relative expression level of each transcript in TG. GAPDH expression was used to normalize the relative expression of each transcript in TG of latently infected mice. Each point represents the normalized fold gene expression calculated from the mean ± standard error of the mean of 10 TG. Differences between groups were analyzed using ANOVA.
CD4, CD8, PD-1, Tim-3, IL-2, IL-21, IFN-γ, and TNF-α mRNA levels during HSV-1 latency in LTα−/− mice.
Previously, we showed that the degree of latency affects T cell exhaustion (12, 19, 28). The above results suggest similar levels of latency in TG of LTα−/− and WT mice although the absence of LAT decreased latency in WT mice while upregulating latency in LTα−/− mice in a LAT-independent manner. Thus, we determined the effect of LTα on expression of mRNAs that are considered markers of T cell subtypes (CD4 and CD8) or exhaustion (PD-1, Tim-3, interleukin-21 [IL-21], IL-2, IFN-γ, and TNF-α). TG were harvested on day 28 p.i. from infected WT and LTα−/− mice, and total RNA was extracted and analyzed using qRT-PCR, as described in Materials and Methods. The results are presented as fold change compared to baseline mRNA levels in TG from uninfected naive mice for each mouse strain (Fig. 8). By comparing mRNA levels in the TG of infected LTα−/− mice with those of infected WT mice, we found significantly higher levels of all markers in LTα−/− mice during latency than in WT infected mice (Fig. 8) (P < 0.001). Thus, despite similar levels of latency in WT and LTα−/− mice, the exhaustion marker levels were significantly higher in LTα−/− mice than in WT mice. Therefore, differences between infected LTα−/− and WT mice could be an immune function rather than a receptor-ligand function, and LTα is required to reduce the immune infiltrates in the TG to better control of disease.
FIG 8.

Effect of LAT and LTα on T cell exhaustion markers in the TG of latently infected mice. TG from latently infected WT and LTα−/− mice as described in the legend of Fig. 1 were individually isolated on day 28 p.i., and qRT-PCR was performed using total RNA, as described in Materials and Methods. CD4, CD8, PD-1, Tim-3, IL-2, IFN-γ, IL-21, and TNF-α expression levels in naive mice from each line were used as baselines to estimate the relative expression levels of each transcript in TG of latently infected mice. GAPDH expression was used to normalize the relative expression of each transcript. Each point represents the normalized fold gene expression calculated from the mean ± standard error of the mean from 10 TG. For all statistical tests, P values of ≤0.01 were considered statistically significant and are marked by a single asterisk (*).
DISCUSSION
Activated T, B, and NK cells express lymphotoxins (LTs) (34, 35), which consist of LTα and LTβ molecules (36, 37). LTα−/− and LTβ−/− mice have some phenotypic similarities, but specific differences have also been noted between these mouse strains, indicating that LTα and LTβ have independent functions (38). A unique function of LTα is its ability to bind to HVEM (15). Recently, we reported that the absence of interaction between HVEM and three of its ligands (i.e., BTLA, CD160, and LIGHT) reduced latency reactivation but not primary infection (19). However, the role, if any, of LTα in HVEM signaling during HSV-1 infection remains unknown. In this study, we investigated the roles of LTα during the course of HSV-1 infection using LTα−/− mice. Previously, we showed that HVEM is upregulated by LAT both in vitro and in vivo (12). In this study, we also demonstrated that BTLA, CD160, HVEM, and LIGHT are upregulated in TG of latently infected LTα−/− mice; however, upregulation of these molecules was higher in the presence of LAT. Levels of HSV-1 latency were similar in LTα−/− mice infected with LAT(+) and LAT(−) virus, similar to that in WT mice infected with LAT(+) virus, and significantly higher than that in WT mice infected with LAT(−) virus, suggesting that LTα has a protective role during HSV-1 infection. LTα induces apoptosis (21, 39) while LAT has antiapoptotic function (40); thus, the absence of an apoptosis function of LTα could have contributed to the similarity of the levels of latency in WT and LTα−/− mice and independent of the presence or absence of LAT.
LTα and LIGHT are members of the tumor necrosis factor superfamily (TNFSF), and both bind to HVEM (15). While LTα enhances binding between HVEM and BTLA (41, 42), LTα and LIGHT have distinct binding effects on HVEM, with HVEM having a stronger binding affinity to LIGHT than to LTα (15). This lower binding affinity of HVEM for LTα may be the reason for LAT(+) and LAT(−) viruses lacking a phenotype with regard to the level of latency in infected LTα−/− mice. The absence of an LAT phenotype in LTα−/− mice compared with that in LIGHT−/− mice may be due to limited expression of LTα in activated T, B, NK, and LTi cells compared with a broader range of LIGHT expression. Recently, we showed that the absence of LAT significantly reduced latency in LIGHT−/− mice below that seen in WT mice (19). Overall, and in contrast to the absence of BTLA, CD160, HVEM or LIGHT, the absence of LTα and independence of LAT negatively impacted latency, suggesting that LTα may be essential to control latency but not primary infection.
Similar to BTLA−/−, CD160−/−, HVEM−/−, and WT mice and in contrast to LIGHT−/− mice (19), virus replication levels in the eye of LTα−/− mice infected with LAT(+) virus were similar to those of their counterparts infected with LAT(−) virus on days 1 to 5 p.i. or to levels in WT control mice. However, the absence of LTα had a significant effect on the level of corneal scarring (CS) in ocularly infected mice. Previously, we showed that HVEM−/−, BTLA−/−, CD160−/−, and LIGHT−/− mice in C57BL/6 backgrounds are refractory to HSV-1-induced CS (19), and C57BL/6 mice are resistant to CS following infection with virulent LAT(+) or LAT(−) virus (43). Because the LTα−/− mice used in this study are on a C57BL/6 background, the presence of significant CS in these mice was unexpected even though these mice had the same level of primary virus replication as WT mice. However, this is consistent with our previous study showing that primary virus titers in the eye and TG did not correlate with levels of viral DNA in latent TG and eye disease (44). Both CD4+ and CD8+ T cells have been implicated in HSV-1-induced CS (45–48). In this study, levels of CS were similar in LTα−/− mice with and without adoptive transfer of CD4+, CD8+, or total T cells from WT mice. Although it is well established that HSV-1-induced CS is due to immune response to the virus, our transfer studies support the idea that exacerbation of CS in LTα−/− mice could be due to cells such as macrophages or NK cells, as reported previously (43, 49–52).
Similar to their resistance to CS, WT C57BL/6 mice are resistant to the lethal dose of virus that we used in this study. For example, of 111 C57BL/6 mice that we infected with LAT(+) or LAT(−) virus, only 5 mice died, a 96% survival rate, while 41 of the 63 LTα−/− mice infected with LAT(+) and LAT(−) viruses died, a 35% survival rate. We have previously showed that, similar to WT C57BL/6 mice, BTLA−/−, LIGHT−/−, CD160−/−, and HVEM−/− mice are also refractory to HSV-1 infection. In this study, we found that transfer of CD4+, CD8+, or total T cells partially but not completely restored protection LTα−/− mice. This is consistent with previous reports showing that reconstitution of irradiated LTα−/− mice with wild-type (WT) mouse bone marrow cells restored the ability to form FDC, germinal centers, and adjuvant-independent IgG responses, but these functions were not restored by transfer of spleen T cells (53). Furthermore, B cells alone can restore protection to LTα−/− mice (34), and generation or expression of memory T cells from these mice was not affected (54). Thus, the absence of complete protection after T cell transfer in this study is not unexpected. In previous studies, LTα−/− mice developed CD8+ T cells normally, but these cells were functionally impaired, making the infected animals susceptible to HSV-1 infection (55). CD8+ T cells from LTα−/− mice failed to mature into cytotoxic T cells (CTL) or to express intracellular IFN-γ upon ex vivo stimulation with antigen (Ag) (55). We have also shown that transfer of CD4+, CD8+, or total T cells from WT mice to LTα−/− mice did not provide complete protection, suggesting that even T cells from WT mice are not completely functional in the absence of LTα expression in LTα−/− mice. In contrast to LTα−/− mice, RAG mice were completely protected against lethal HSV-1 infection following adoptive T cell transfer (56).
Similar to this study, a previous study has shown that LTα−/− mice have increased susceptibility to infections (57). The level of susceptibility to infection in LTα−/− mice varied with different viral infections. For example, LTα−/− mice were able to successfully clear lymphocytic choriomeningitis virus (LCMV) infection (57). Similarly, respiratory challenge of LTα−/− mice with murine gammaherpesvirus 68 (MHV-68) led to an acute productive infection and latent infection. However, despite reduced immune responses, infected mice were able to clear the infection and control the latent infection but with delayed kinetics compared to those of WT mice (58). In contrast, LTα−/− mice are more susceptible to influenza virus infection than WT mice, with delayed but effective B and T cell responses to infection (59). Similar to WT mice, LTα−/− mice cleared systemic rotavirus infection within 10 days, while clearance of intestinal infection in LTα−/− mice depended on local generation of mucosal IgA (60). Previously it was reported that humoral immune responses to different antigens is impaired in LTα−/− mice (61). Similarly, LTα−/− mice immunized with attenuated HSV-2 virus showed delayed virus clearance following lethal challenge with a virulent HSV-2 virus, but all challenged mice were protected from disease and death (62).
In this study, we detected significantly higher expression levels of BTLA, CD160, HVEM, and LIGHT mRNAs in the TG of LTα−/− mice latently infected with LAT(+) than with LAT(−) virus, but these transcripts were more highly expressed in LTα−/− mice infected with LAT(−) virus than in WT mice infected with either virus. In contrast, and as we reported previously (19), we did not detect any differences in BTLA, LIGHT, CD160, or HVEM mRNA levels in the TG of BTLA−/−, LIGHT−/−, CD160−/−, and HVEM−/− mice latently infected with LAT(+) or LAT(−) virus. Levels of BTLA, CD160, HVEM, and LIGHT were higher in TG of LAT(+) virus -infected LTα−/− mice than in LTα−/− mice infected with LAT(−) virus. These higher expression levels in the presence of LAT could be due to higher antiapoptotic function of LAT in the absence of LTα. In addition to the upregulation of BTLA, LIGHT, CD160, or HVEM in the TG of LTα−/− infected mice, markers of T cell exhaustion (CD4, CD8, PD-1, Tim-3, IL-2, IFN-γ, IL-21, and TNF-α) are also upregulated in infected mice (63–69). Previously, we found that increased latency is associated with increased levels of T cell exhaustion (70) and that higher latency in the presence of LAT was associated with higher levels of T cell exhaustion (28, 71). However, during HSV-1 latency not all of the T cells are exhausted (72). In this study, we show a correlation between higher latency in LTα−/− infected mice with higher levels of CD4, CD8, PD-1, Tim-3, IL-2, IFN-γ, IL-21, and TNF-α that was not observed in WT mice. In contrast to our recent study (19), BTLA−/−, LIGHT−/−, CD160−/−, and HVEM−/− infected mice had lower levels of CD4, CD8, PD-1, Tim-3, IL-2, IFN-γ, IL-21, and TNF-α than WT infected mice. This may be due to lower levels of latency in the infected mice. In general, we consistently find that markers of T cell exhaustion are higher in WT mice than in their knockout counterparts due to higher levels of latency (19, 28). But in this study, infected WT mice had lower expression levels of CD4, CD8, PD-1, Tim-3, IL-2, IFN-γ, IL-21, and TNF-α transcripts than LTα−/− infected mice. The elevated expression of these transcripts in TG of infected mice is independent of the presence of LAT and could be associated with the proinflammatory nature of this gene. LTα was originally identified as a proinflammatory molecule (73). It can induce apoptosis, necroptosis, differentiation, proliferation, and the expression of a variety of inflammatory cytokines and chemokines, depending on its environment (21, 39). Thus, the absence of some or all of these LTα functions may contribute to increased viral pathogenesis in LTα−/− mice.
In summary, the results of this study suggest the following: (i) that LTα is required to achieve WT levels of CS and survival; (ii) that LTα affects latency in an LAT-independent manner; and (iii) that LTα inhibits the induction of HVEM and its ligands as well as T cell exhaustion markers. Furthermore, we have shown that the function of the HVEM ligand, LTα, is completely different from the functions of other HVEM ligands (i.e., BTLA, CD160, and LIGHT) and that further studies are warranted to dissect the role of LTα in HSV-1 pathogenesis.
MATERIALS AND METHODS
Virus and mice.
Plaque-purified HSV-1 strains McKrae [WT; LAT-positive, or LAT(+)] and dLAT2903 [LAT-negative, or LAT(−)], derived from McKrae, were grown in rabbit skin (RS) cell monolayers in minimal essential medium (MEM) containing 5% fetal calf serum (FCS), as described previously (33, 74). Throughout the study, McKrae and dLAT2903 viruses are referred to as LAT(+) and LAT(−) viruses, respectively.
WT C57BL/6 and LTα−/− mice were purchased from The Jackson Laboratories and bred in-house. Both male and female mice were used in the study. The animal research protocol was approved by the Institutional Animal Care and Use Committee of Cedars-Sinai Medical Center (Protocol no. 5030).
Ocular infection.
Mice were infected ocularly with 2 × 105 PFU of LAT(+) or LAT(−) virus in 2 μl of MEM tissue culture medium with 5% FCS, administered in an eye drop without corneal scarification.
Titration of virus in tears of infected mice.
Tear films from mice infected as described above were collected from both eyes of 10 mice per group on days 1 to 5 postinfection (p.i.), using a Dacron-tipped swab (75). Each swab was placed in 1 ml of MEM tissue culture medium and squeezed, and the amount of virus was determined using a standard plaque assay on RS cells as we described previously (76).
Adoptive transfer of T cell to LTα−/− mice.
In an effort to improve survival of LTα−/− mice after ocular infection, CD4+, CD8+, or total T cells isolated from WT mice were adoptively transferred into LTα−/− mice before infection with HSV-1. Briefly, spleens from WT C57BL/6 mice were pooled, and single-cell suspensions were prepared as described previously (77). CD4+ T cells alone, CD8+ T cells alone, or total T cells were isolated using magnetic beads as described by the manufacturer (Miltenyi Biotec, San Diego, CA). Recipient mice received a single intraperitoneal injection of either CD4+ T cells, CD8+ T cells, or total T cells suspended in MEM (300 μl) equivalent to the amount injected into a single donor mouse. The mice were infected ocularly with McKrae LAT(+) virus 2 weeks after the T cell transfer.
CS.
The severity of corneal scarring (CS) in surviving mice was scored in a masked fashion on a scale of 0 to 4 (0, no disease; 1, 25% corneal staining or involvement; 2, 50%; 3, 75%; and 4, 100%) as we described previously (78).
DNA extraction and PCR analysis for HSV-1 gB DNA.
DNA was isolated from homogenized individual TG using a commercially available DNeasy blood and tissue kit (Qiagen, Stanford, CA) according to the manufacturer's instructions. PCR analyses were carried out using gB-specific primers (forward, 5′-AACGCGACGCACATCAAG-3′; reverse, 5′-CTGGTACGCGATCAGAAAGC-3′; probe, 5′-FAM-CAGCCGCAGTACTACC-3′, where FAM is 6-carboxyfluorescein). The amplicon length for this primer set is 72 bp. Relative gB copy numbers were calculated using standard curves generated from the plasmid pAc-gB1 (79). In all experiments, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to normalize transcripts.
RNA extraction, cDNA synthesis, and TaqMan RT-PCR.
TG from individual mice were collected on day 28 p.i., immersed in RNAlater RNA stabilization reagent (Thermo Fisher Scientific, Waltham, MA), and stored at −80°C until processing. In some experiments, TG from uninfected mice of the same age as infected mice were also collected as described above. Total RNA extraction and RT-PCR were carried out as we have described previously (80, 81). In mice infected with LAT(+) virus, the level of LAT RNA from latent TG was determined using a custom-made primer and probe set for LAT as follows: forward primer, 5′-GGGTGGGCTCGTGTTACAG-3′; reverse primer, 5′-GGACGGGTAAGTAACAGAGTCTCTA-3′; and probe, 5′-FAM-ACACCAGCCCGTTCTTT-3′ (amplicon length, 81 bp). The LAT amplicon primer set corresponds to LAT nucleotides (nt) 119553 to 119634. Relative LAT copy numbers were calculated using standard curves generated from the pGem-LAT5317 plasmid.
The levels of various transcripts (CD4, CD8, PD-1, Tim-3, IL-2, IL-21, IFN-γ, TNF-β, HVEM, LIGHT, BTLA, and CD160) in TG were evaluated using commercially available TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA) with optimized primer and probe concentrations. Primer probe sets consisted of two unlabeled PCR primers and a FAM dye-labeled TaqMan MGB (minor groove binder) probe formulated into a single mixture. Additionally, all cellular amplicons included an intron-exon junction to eliminate signals from genomic DNA contamination. The following assays were used in this study: (1)for HVEM, ABI assay Mm00619239_m1, with amplicon length of 65 bp; (2) for LIGHT, ABI Mm00444567_m1, with amplicon length of 68 bp; (3) for BTLA, ABI Mm00616981_m1, with amplicon length of 71 bp; 4 for CD160, ABI Mm00444461_m1, with amplicon length of 61 bp; (5) for CD4, ABI Mm00442754_m1, with amplicon length of 72 bp; (6) for CD8 (α chain), ABI Mn01182108_m1, with amplicon length of 67 bp; (7) for CD8 (β chain), ABI Mm_00438116_m1, with amplicon length of 89 bp; (8) for PD-1 (programmed death 1; also known as CD279), ABI Mm00435532_m1, with amplicon length of 65 bp; (9) for Tim-3, ABI Mm00454540_m1, with amplicon length of 98 bp; (10) for IL-21, ABI Mm00517640_m1, with amplicon length of 67 bp; (11) for IL-2, ABI Mm00434256_m1, with amplicon length of 82 bp; (12) for IFN-γ, ABI Mm00801778_m1, with amplicon length of 101 bp; (13) for TNF-α, ABI Mm00443258_m1, with amplicon length of 81 bp; and (14) for GAPDH (used to normalize transcripts), ABI Mm999999.15_G1, with amplicon length of 107 bp.
Statistical analysis.
Student's t test, Fisher’s exact test, and analysis of variance (ANOVA) were performed using the computer program Instat (GraphPad, San Diego, CA). Results were considered statistically significant when the P value was <0.05.
ACKNOWLEDGMENTS
This study was supported by Public Health Service NIH grants RO1 EY13615, RO1 EY026944, and RO1 EY029160.
REFERENCES
- 1.Koujah L, Suryawanshi RK, Shukla D. 2019. Pathological processes activated by herpes simplex virus-1 (HSV-1) infection in the cornea. Cell Mol Life Sci 76:405–419. doi: 10.1007/s00018-018-2938-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spear PG, Eisenberg RJ, Cohen GH. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1–8. doi: 10.1006/viro.2000.0529. [DOI] [PubMed] [Google Scholar]
- 3.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. doi: 10.1038/nature09420. [DOI] [PubMed] [Google Scholar]
- 4.Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci U S A 107:866–871. doi: 10.1073/pnas.0913351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon M, Zago A, Shukla D, Spear PG. 2003. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J Virol 77:9221–9231. doi: 10.1128/JVI.77.17.9221-9231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436. doi: 10.1016/S0092-8674(00)81363-X. [DOI] [PubMed] [Google Scholar]
- 7.Taylor JM, Lin E, Susmarski N, Yoon M, Zago A, Ware CF, Pfeffer K, Miyoshi J, Takai Y, Spear PG. 2007. Alternative entry receptors for herpes simplex virus and their roles in disease. Cell Host Microbe 2:19–28. doi: 10.1016/j.chom.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22. doi: 10.1016/S0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 9.O'Donnell CD, Kovacs M, Akhtar J, Valyi-Nagy T, Shukla D. 2010. Expanding the role of 3-O sulfated heparan sulfate in herpes simplex virus type-1 entry. Virology 397:389–398. doi: 10.1016/j.virol.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karaba AH, Kopp SJ, Longnecker R. 2011. Herpesvirus entry mediator and nectin-1 mediate herpes simplex virus 1 infection of the murine cornea. J Virol 85:10041–10047. doi: 10.1128/JVI.05445-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agelidis AM, Shukla D. 2015. Cell entry mechanisms of HSV: what we have learned in recent years. Future Virol 10:1145–1154. doi: 10.2217/fvl.15.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen SJ, Rhode-Kurnow A, Mott KR, Jiang X, Carpenter D, Rodriguez-Barbosa JI, Jones C, Wechsler SL, Ware CF, Ghiasi H. 2014. Regulatory interactions between herpesvirus entry mediator (TNFRSF14) and latency associated transcript (LAT) during HSV-1 latency. J Virol 88:1961–1971. doi: 10.1128/JVI.02467-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pfeffer K, Ware CF, Murphy TL, Murphy KM. 2005. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol 6:90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 14.Cai G, Anumanthan A, Brown JA, Greenfield EA, Zhu B, Freeman GJ. 2008. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol 9:176–185. doi: 10.1038/ni1554. [DOI] [PubMed] [Google Scholar]
- 15.Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, Spear PG, Ware CF. 1998. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 8:21–30. doi: 10.1016/S1074-7613(00)80455-0. [DOI] [PubMed] [Google Scholar]
- 16.del Rio ML, Lucas CL, Buhler L, Rayat G, Rodriguez-Barbosa JI. 2010. HVEM/LIGHT/BTLA/CD160 cosignaling pathways as targets for immune regulation. J Leukoc Biol 87:223–235. doi: 10.1189/jlb.0809590. [DOI] [PubMed] [Google Scholar]
- 17.Zhu Y, Yao S, Augustine MM, Xu H, Wang J, Sun J, Broadwater M, Ruff W, Luo L, Zhu G, Tamada K, Chen L. 2016. Neuron-specific SALM5 limits inflammation in the CNS via its interaction with HVEM. Sci Adv 2:e1500637. doi: 10.1126/sciadv.1500637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarrias MR, Whitbeck JC, Rooney I, Ware CF, Eisenberg RJ, Cohen GH, Lambris JD. 2000. The three HveA receptor ligands, gD, LT-alpha and LIGHT bind to distinct sites on HveA. Mol Immunol 37:665–673. doi: 10.1016/S0161-5890(00)00089-4. [DOI] [PubMed] [Google Scholar]
- 19.Wang S, Ljubimov AV, Jin L, Pfeffer K, Kronenberg M, Ghiasi H. 2018. Herpes simplex virus 1 latency and the kinetics of reactivation are regulated by a complex network of interactions between the herpesvirus entry mediator, its ligands (gD, BTLA, LIGHT, and CD160), and the latency-associated transcript. J Virol 92:e01451-18. doi: 10.1128/JVI.01451-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai G, Freeman GJ. 2009. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol Rev 229:244–258. doi: 10.1111/j.1600-065X.2009.00783.x. [DOI] [PubMed] [Google Scholar]
- 21.Smith CA, Farrah T, Goodwin RG. 1994. The TNF receptor superfamily of cellular and viral proteins: activation, costimulation, and death. Cell 76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 22.Matsumoto M. 1999. Role of TNF ligand and receptor family in the lymphoid organogenesis defined by gene targeting. J Med Invest 46:141–150. [PubMed] [Google Scholar]
- 23.Gramaglia I, Mauri DN, Miner KT, Ware CF, Croft M. 1999. Lymphotoxin αβ is expressed on recently activated naive and Th1-like CD4 cells but is down-regulated by IL-4 during Th2 differentiation. J Immunol 162:1333–1338. [PubMed] [Google Scholar]
- 24.Matsumoto M, Lo SF, Carruthers CJ, Min J, Mariathasan S, Huang G, Plas DR, Martin SM, Geha RS, Nahm MH, Chaplin DD. 1996. Affinity maturation without germinal centres in lymphotoxin-alpha-deficient mice. Nature 382:462–466. doi: 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto M, Mariathasan S, Nahm MH, Baranyay F, Peschon JJ, Chaplin DD. 1996. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science 271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 26.De Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, Smith SC, Carlson R, Shornick LP, Strauss-Schoenberger J. 1994. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science 264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 27.Tormanen K, Allen S, Mott KR, Ghiasi H. 2019. The latency-associated transcript inhibits apoptosis via downregulation of components of the type I interferon pathway during latent herpes simplex virus 1 ocular infection. J Virol 93:e00103-19. doi: 10.1128/JVI.00103-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen SJ, Hamrah P, Gate DM, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, Benmohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J Virol 85:4184–4197. doi: 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL. 1987. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol 61:3820–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wechsler SL, Nesburn AB, Watson R, Slanina S, Ghiasi H. 1988. Fine mapping of the major latency-related RNA of herpes simplex virus type 1 in humans. J Gen Virol 69:3101–3106. doi: 10.1099/0022-1317-69-12-3101. [DOI] [PubMed] [Google Scholar]
- 31.Wechsler SL, Nesburn AB, Watson R, Slanina SM, Ghiasi H. 1988. Fine mapping of the latency-related gene of herpes simplex virus type 1: alternative splicing produces distinct latency-related RNAs containing open reading frames. J Virol 62:4051–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phelan D, Barrozo ER, Bloom DC. 2017. HSV1 latent transcription and non-coding RNA: a critical retrospective. J Neuroimmunol 308:65–101. doi: 10.1016/j.jneuroim.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu YX, Huang G, Wang Y, Chaplin DD. 1998. B lymphocytes induce the formation of follicular dendritic cell clusters in a lymphotoxin alpha-dependent fashion. J Exp Med 187:1009–1018. doi: 10.1084/jem.187.7.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ware CF. 2005. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol 23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 36.Browning JL, Sizing ID, Lawton P, Bourdon PR, Rennert PD, Majeau GR, Ambrose CM, Hession C, Miatkowski K, Griffiths DA, Ngam-Ek A, Meier W, Benjamin CD, Hochman PS. 1997. Characterization of lymphotoxin-alpha beta complexes on the surface of mouse lymphocytes. J Immunol 159:3288–3298. [PubMed] [Google Scholar]
- 37.Matsumoto M, Fu YX, Molina H, Chaplin DD. 1997. Lymphotoxin-alpha-deficient and TNF receptor-I-deficient mice define developmental and functional characteristics of germinal centers. Immunol Rev 156:137–144. doi: 10.1111/j.1600-065X.1997.tb00965.x. [DOI] [PubMed] [Google Scholar]
- 38.Alimzhanov MB, Kuprash DV, Kosco-Vilbois MH, Luz A, Turetskaya RL, Tarakhovsky A, Rajewsky K, Nedospasov SA, Pfeffer K. 1997. Abnormal development of secondary lymphoid tissues in lymphotoxin beta-deficient mice. Proc Natl Acad Sci U S A 94:9302–9307. doi: 10.1073/pnas.94.17.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Etemadi N, Holien JK, Chau D, Dewson G, Murphy JM, Alexander WS, Parker MW, Silke J, Nachbur U. 2013. Lymphotoxin alpha induces apoptosis, necroptosis and inflammatory signals with the same potency as tumour necrosis factor. FEBS J 280:5283–5297. doi: 10.1111/febs.12419. [DOI] [PubMed] [Google Scholar]
- 40.Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 41.Cheung TC, Humphreys IR, Potter KG, Norris PS, Shumway HM, Tran BR, Patterson G, Jean-Jacques R, Yoon M, Spear PG, Murphy KM, Lurain NS, Benedict CA, Ware CF. 2005. Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc Natl Acad Sci U S A 102:13218–13223. doi: 10.1073/pnas.0506172102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzalez LC, Loyet KM, Calemine-Fenaux J, Chauhan V, Wranik B, Ouyang W, Eaton DL. 2005. A coreceptor interaction between the CD28 and TNF receptor family members B and T lymphocyte attenuator and herpesvirus entry mediator. Proc Natl Acad Sci U S A 102:1116–1121. doi: 10.1073/pnas.0409071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghiasi H, Cai S, Perng GC, Nesburn AB, Wechsler SL. 2000. The role of natural killer cells in protection of mice against death and corneal scarring following ocular HSV-1 infection. Antiviral Res 45:33–45. doi: 10.1016/S0166-3542(99)00075-3. [DOI] [PubMed] [Google Scholar]
- 44.Matundan HH, Mott KR, Allen SJ, Wang S, Bresee CJ, Ghiasi YN, Town T, Wechsler SL, Ghiasi H. 2016. Interrelationship of primary virus replication, level of latency, and time to reactivation in the trigeminal ganglia of latently infected mice. J Virol 90:9533–9542. doi: 10.1128/JVI.01373-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hendricks RL, Janowicz M, Tumpey TM. 1992. Critical role of corneal Langerhans cells in the CD4- but not CD8- mediated immunopathology in herpes simplex virus-1-infected mouse corneas. J Immunol 148:2522–2529. [PubMed] [Google Scholar]
- 46.Ghiasi H, Roopenian DC, Slanina S, Cai S, Nesburn AB, Wechsler SL. 1997. The importance of MHC-I and MHC-II responses in vaccine efficacy against lethal herpes simplex virus type 1 challenge. Immunology 91:430–435. doi: 10.1046/j.1365-2567.1997.00261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mercadal CM, Bouley DM, DeStephano D, Rouse BT. 1993. Herpetic stromal keratitis in the reconstituted scid mouse model. J Virol 67:3404–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doymaz MZ, Rouse BT. 1992. Herpetic stromal keratitis: an immunopathologic disease mediated by CD4+ T lymphocytes. Invest Ophthalmol Vis Sci 33:2165–2173. [PubMed] [Google Scholar]
- 49.Tumpey TM, Cheng H, Cook DN, Smithies O, Oakes JE, Lausch RN. 1998. Absence of macrophage inflammatory protein-1α prevents the development of blinding herpes stromal keratitis. J Virol 72:3705–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tumpey TM, Cheng H, Yan XT, Oakes JE, Lausch RN. 1998. Chemokine synthesis in the HSV-1-infected cornea and its suppression by interleukin-10. J Leukoc Biol 63:486–492. doi: 10.1002/jlb.63.4.486. [DOI] [PubMed] [Google Scholar]
- 51.Oakes JE, Monteiro CA, Cubitt CL, Lausch RN. 1993. Induction of interleukin-8 gene expression is associated with herpes simplex virus infection of human corneal keratocytes but not human corneal epithelial cells. J Virol 67:4777–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brandt CR, Salkowski CA. 1992. Activation of NK cells in mice following corneal infection with herpes simplex virus type-1. Invest Ophthalmol Vis Sci 33:113–120. [PubMed] [Google Scholar]
- 53.Fu YX, Molina H, Matsumoto M, Huang G, Min J, Chaplin DD. 1997. Lymphotoxin-alpha (LTα) supports development of splenic follicular structure that is required for IgG responses. J Exp Med 185:2111–2120. doi: 10.1084/jem.185.12.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu YX, Huang G, Wang Y, Chaplin DD. 2000. Lymphotoxin-alpha-dependent spleen microenvironment supports the generation of memory B cells and is required for their subsequent antigen-induced activation. J Immunol 164:2508–2514. doi: 10.4049/jimmunol.164.5.2508. [DOI] [PubMed] [Google Scholar]
- 55.Kumaraguru U, Davis IA, Deshpande S, Tevethia SS, Rouse BT. 2001. Lymphotoxin alpha−/− mice develop functionally impaired CD8+ T cell responses and fail to contain virus infection of the central nervous system. J Immunol 166:1066–1074. doi: 10.4049/jimmunol.166.2.1066. [DOI] [PubMed] [Google Scholar]
- 56.Banerjee K, Biswas PS, Kumaraguru U, Schoenberger SP, Rouse BT. 2004. Protective and pathological roles of virus-specific and bystander CD8+ T cells in herpetic stromal keratitis. J Immunol 173:7575–7583. doi: 10.4049/jimmunol.173.12.7575. [DOI] [PubMed] [Google Scholar]
- 57.Eugster HP, Muller M, Karrer U, Car BD, Schnyder B, Eng VM, Woerly G, Le Hir M, di Padova F, Aguet M, Zinkernagel R, Bluethmann H, Ryffel B. 1996. Multiple immune abnormalities in tumor necrosis factor and lymphotoxin-alpha double-deficient mice. Int Immunol 8:23–36. doi: 10.1093/intimm/8.1.23. [DOI] [PubMed] [Google Scholar]
- 58.Lee BJ, Santee S, Von Gesjen S, Ware CF, Sarawar SR. 2000. Lymphotoxin-alpha-deficient mice can clear a productive infection with murine gammaherpesvirus 68 but fail to develop splenomegaly or lymphocytosis. J Virol 74:2786–2792. doi: 10.1128/jvi.74.6.2786-2792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lund FE, Partida-Sanchez S, Lee BO, Kusser KL, Hartson L, Hogan RJ, Woodland DL, Randall TD. 2002. Lymphotoxin-alpha-deficient mice make delayed, but effective, T and B cell responses to influenza. J Immunol 169:5236–5243. doi: 10.4049/jimmunol.169.9.5236. [DOI] [PubMed] [Google Scholar]
- 60.Lopatin U, Blutt SE, Conner ME, Kelsall BL. 2013. Lymphotoxin alpha-deficient mice clear persistent rotavirus infection after local generation of mucosal IgA. J Virol 87:524–530. doi: 10.1128/JVI.01801-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Banks TA, Rouse BT, Kerley MK, Blair PJ, Godfrey VL, Kuklin NA, Bouley DM, Thomas J, Kanangat S, Mucenski ML. 1995. Lymphotoxin-alpha-deficient mice. Effects on secondary lymphoid organ development and humoral immune responsiveness. J Immunol 155:1685–1693. [PubMed] [Google Scholar]
- 62.Roth KL, Bhavanam S, Jiang H, Gillgrass A, Ho K, Ferreira VH, Kaushic C. 2013. Delayed but effective induction of mucosal memory immune responses against genital HSV-2 in the absence of secondary lymphoid organs. Mucosal Immunol 6:56–68. doi: 10.1038/mi.2012.48. [DOI] [PubMed] [Google Scholar]
- 63.Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, Wong JC, Satkunarajah M, Schweneker M, Chapman JM, Gyenes G, Vali B, Hyrcza MD, Yue FY, Kovacs C, Sassi A, Loutfy M, Halpenny R, Persad D, Spotts G, Hecht FM, Chun TW, McCune JM, Kaul R, Rini JM, Nixon DF, Ostrowski MA. 2008. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med 205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Golden-Mason L, Palmer BE, Kassam N, Townshend-Bulson L, Livingston S, McMahon BJ, Castelblanco N, Kuchroo V, Gretch DR, Rosen HR. 2009. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol 83:9122–9130. doi: 10.1128/JVI.00639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Elsaesser H, Sauer K, Brooks DG. 2009. IL-21 is required to control chronic viral infection. Science 324:1569–1572. doi: 10.1126/science.1174182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yi JS, Du M, Zajac AJ. 2009. A vital role for interleukin-21 in the control of a chronic viral infection. Science 324:1572–1576. doi: 10.1126/science.1175194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frohlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, Marsland BJ, Oxenius A, Kopf M. 2009. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science 324:1576–1580. doi: 10.1126/science.1172815. [DOI] [PubMed] [Google Scholar]
- 68.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 77:4911–4927. doi: 10.1128/jvi.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. 1998. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mott KR, Bresee CJ, Allen SJ, BenMohamed L, Wechsler SL, Ghiasi H. 2009. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. J Virol 83:2246–2254. doi: 10.1128/JVI.02234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chentoufi AA, Kritzer E, Tran MV, Dasgupta G, Lim CH, Yu DC, Afifi RE, Jiang X, Carpenter D, Osorio N, Hsiang C, Nesburn AB, Wechsler SL, BenMohamed L. 2011. The herpes simplex virus 1 latency-associated transcript promotes functional exhaustion of virus-specific CD8+ T cells in latently infected trigeminal ganglia: a novel immune evasion mechanism. J Virol 85:9127–9138. doi: 10.1128/JVI.00587-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.St Leger AJ, Jeon S, Hendricks RL. 2013. Broadening the repertoire of functional herpes simplex virus type 1-specific CD8+ T cells reduces viral reactivation from latency in sensory ganglia. J Immunol 191:2258–2265. doi: 10.4049/jimmunol.1300585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paul NL, Ruddle NH. 1988. Lymphotoxin. Annu Rev Immunol 6:407–438. doi: 10.1146/annurev.iy.06.040188.002203. [DOI] [PubMed] [Google Scholar]
- 74.Osorio Y, Ghiasi H. 2003. Comparison of adjuvant efficacy of herpes simplex virus type 1 recombinant viruses expressing TH1 and TH2 cytokine genes. J Virol 77:5774–5783. doi: 10.1128/JVI.77.10.5774-5783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghiasi H, Bahri S, Nesburn AB, Wechsler SL. 1995. Protection against herpes simplex virus-induced eye disease after vaccination with seven individually expressed herpes simplex virus 1 glycoproteins. Invest Ophthalmol Vis Sci 36:1352–1360. [PubMed] [Google Scholar]
- 76.Ghiasi H, Kaiwar R, Nesburn AB, Slanina S, Wechsler SL. 1994. Expression of seven herpes simplex virus type 1 glycoproteins (gB, gC, gD, gE, gG, gH, and gI): comparative protection against lethal challenge in mice. J Virol 68:2118–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ahmed R, King CC, Oldstone MB. 1987. Virus-lymphocyte interaction: T cells of the helper subset are infected with lymphocytic choriomeningitis virus during persistent infection in vivo. J Virol 61:1571–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Osorio Y, Sharifi BG, Perng GC, Ghiasi NS, Ghiasi H. 2002. The role of TH1 and TH2 cytokines in HSV-1-induced corneal scarring. Ocular Immunol Inflamm 10:105–116. doi: 10.1076/ocii.10.2.105.13982. [DOI] [PubMed] [Google Scholar]
- 79.Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1992. Expression of herpes simplex virus type 1 glycoprotein B in insect cells. Initial analysis of its biochemical and immunological properties. Virus Res 22:25–39. doi: 10.1016/0168-1702(92)90087-P. [DOI] [PubMed] [Google Scholar]
- 80.Mott KR, Perng GC, Osorio Y, Kousoulas KG, Ghiasi H. 2007. A recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J Virol 81:12962–12972. doi: 10.1128/JVI.01442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mott KR, Osorio Y, Brown DJ, Morishige N, Wahlert A, Jester JV, Ghiasi H. 2007. The corneas of naive mice contain both CD4+ and CD8+ T cells. Mol Vis 13:1802–1812. [PubMed] [Google Scholar]